Functional Consequences of GPCR Heterodimerization: GPCRs as Allosteric Modulators

Abstract
: G Protein Coupled Receptors (GPCRs) represent the largest family of membrane proteins in the human genome, are the targets of approximately 25% of all marketed pharmaceuticals, and the focus of intensive research worldwide given that this superfamily of receptors is as varied in function as it is ubiquitously expressed among all cell types. Increasing evidence has shown that the classical two part model of GPCR signaling (one GPCR, one type of heterotrimeric G protein) is grossly oversimplified as many GPCRs can couple to more than one type of G protein, each subunit of the heterotrimeric G protein can activate different downstream effectors, and, surprisingly, other GPCRs can affect receptor behavior in G protein-independent ways. The concept of GPCR heterodimerization, or the physical association of two different types of GPCRs, presents an unexpected mechanism for GPCR regulation and function, and provides a novel target for pharmaceuticals. Here we present a synopsis of the functional consequences of GPCR heterodimerization in both in vitro and in vivo studies, focusing on the concept of GPCRs as allosteric modulators. Typically, an allosteric modulator is a ligand or molecule that alters a receptor's innate functional properties, but here we propose that in the case of GPCR heterodimers, it is the physical coupling of two receptors that leads to changes in cognate receptor signaling.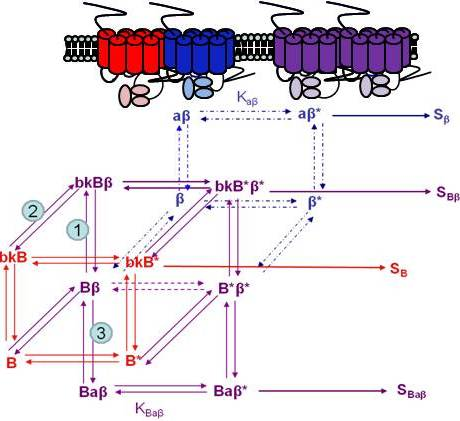
1. Introduction
Given that of the 266 human targets for approved drugs, 27% correspond to family A GPCRs [1], it is not surprising that over the last fifteen years extensive efforts have been devoted to refining existing pharmaceuticals, and identifying new drugs that target and modulate GPCRs. This endeavor was further complicated by the discovery that different GPCRs were capable of physically associating or heterodimerizing, often changing the conventional signaling pathway of a given receptor. Receptor heterodimerization can have substantial consequences on the binding of ligands, as discussed recently by Birdsall [2]. Here, we will provide examples of GPCR heterodimerization and the functional consequences thereof within the framework of allosteric modulation, focusing more heavily on the consequences of heterodimerization on receptor function downstream of ligand binding. We propose that instead of solely investigating the consequences of GPCR-GPCR interaction in terms of changes in signaling or trafficking of a single receptor, one should also consider the physical association with a second receptor as a potential modulator of GPCR activity.
1.1. Allosteric Modulation of GPCRs
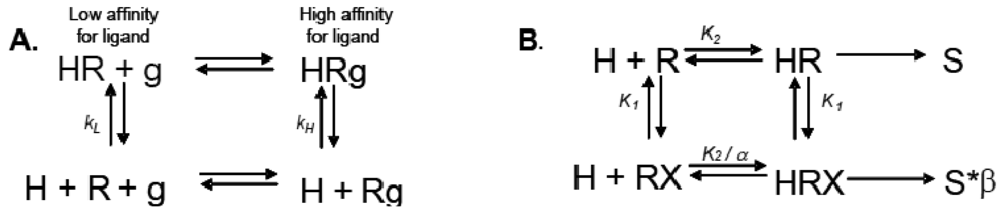
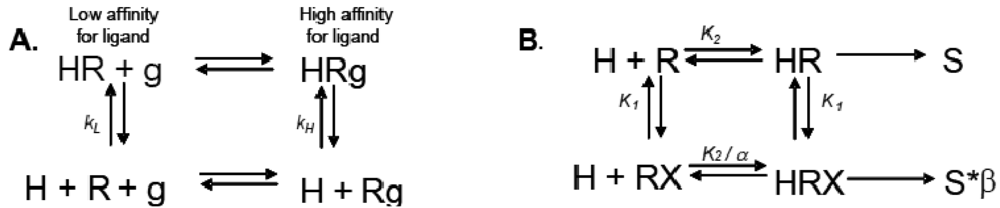
Allosteric modulation of GPCRs is a topic of great interest, and has been the subject of a substantial body of literature; unfortunately, due to space limitations, we are unable to reference every paper and, instead, will focus only on seminal studies that are relevant to the concept of modulation of one receptor by another. Early observations made from agonist binding analysis of many GPCRs including the beta-2 adrenoceptor (β2AR), muscarinic receptors, dopamine receptors and glucagon receptors all demonstrated the existence of both high and low affinity states of a GPCR. Thus, the simplistic binary model of inactive receptor R → active receptor R* would not work as a sufficient model for describing GPCR signaling. Keeping in mind this notion of a receptor existing in more than one conformation, the shift from high to low affinity states is likely related to increases in second messenger activity leading to feedback onto the receptor itself [3]. The Lefkowitz group first suggested that the signaling cascade of the β2AR leading to increases in adenylyl cyclase (AC) activity is the result of a ternary complex that reflects the state of the receptor, R, the ligand or hormone, H, and an additional membrane component (in this instance, the G protein), g. Ligand H can bind saturably and reversibly to the receptor R, and the rate of the reaction is driven by the concentration of the ligand, the equilibrium dissociation constants, and the additional component g (Figure 1A) [3].
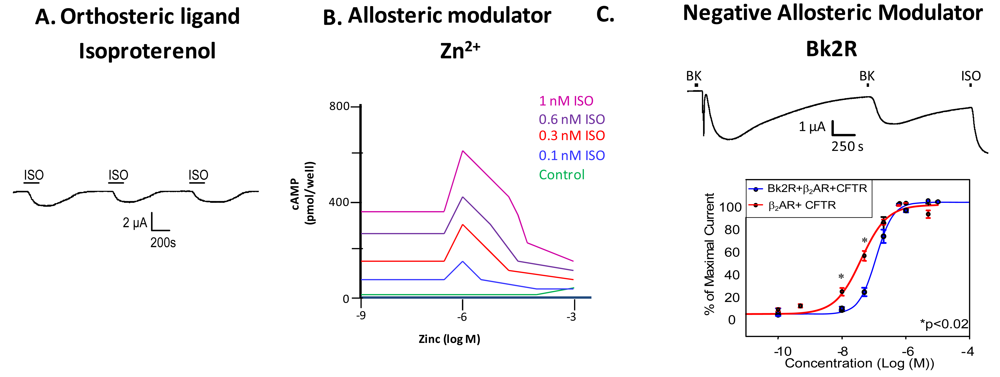
Just as the canonical example of hemoglobin illustrated that proteins often have more than one binding site, and that these sites can work cooperatively (that binding at one site has influence on the binding at other sites), most GPCRs also have more than one ligand binding site. Most GPCRs possess an orthosteric site where the endogenous ligand binds, and an allosteric site, where another ligand or molecule can bind to modulate receptor activity. Binding of an allosteric modulator can influence receptor signaling and behavior in either a positive or negative direction. All GPCRs have at least one allosteric site; binding of G protein incurs allostery in the most general sense of the term, and can shift the affinity for the orthosteric ligand. Recent studies have shown that many GPCRs bear one or more allosteric sites in addition to the G protein binding site. Allosteric modulators of GPCRs are defined as ligands that bind to an allosteric site on the GPCR to modulate the binding and/or signaling properties of the orthosteric site [4]. Allosteric modulators can increase or decrease the propensity of the receptor to bind the orthosteric ligand by stabilizing the active or the inactive state of the receptor, or by directly altering ligand selectivity. In addition to engineered ligands that serve as allosteric modulators of GPCRs, naturally occurring molecules at physiological concentrations such as ions (as in the case of zinc modulation of β2AR) or small proteins (heparin binding to the neurokinin NK1 receptor) also serve as allosteric modulators of GPCRs (Figures 2A, B) [5,6]. GPCRs can also be modulated by other GPCRs (Figure 2C); we will discuss this in depth in a later section.
1.2. Types of Allosteric Modulators
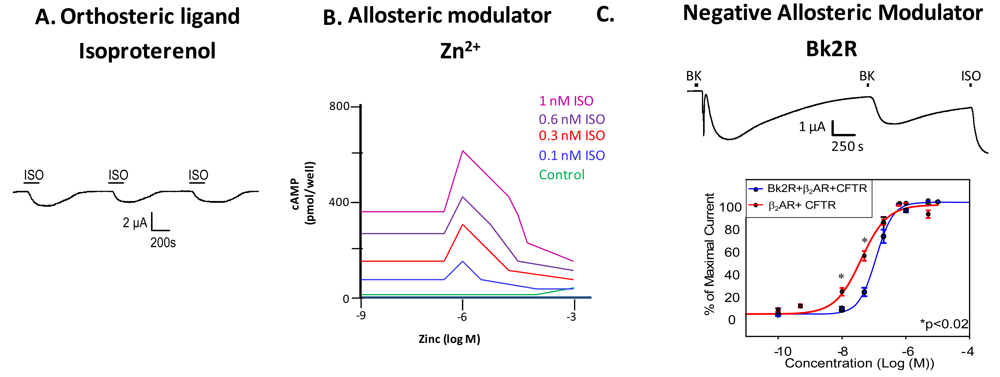
Examination of GPCR ligand binding and signaling within the framework of allostery requires further definition of different types of ligands that bind a GPCR. An agonist is a ligand that binds to the orthosteric site to shift the equilibrium to the receptor being in an active conformation (Figure 2A), inverse agonists shift the equilibrium towards the receptor's inactive state, decreasing the basal/constitutive receptor activity, and an antagonist prevents other ligands from binding without a shift in equilibrium. For example, β2AR binding of isoproterenol (ISO, in Figure 2A, or H, in Figure 1) would lead to one stimulus (S). Figure 2A is a representative recording of currents elicited in a Xenopus oocyte expressing β2AR and the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR). When isoproterenol, a non-selective β2AR agonist, signals through its cognate G protein leading to subsequent increases in cAMP, PKA phosphorylates CFTR, and opens the channel. Hence, CFTR can be used to monitor the activity of the β2AR.
A molecule that binds at the allosteric site does not necessarily require any agonist or inverse agonist functionality of its own, only that it be able to shift the equilibrium of the receptor towards a more active or inactive state by stabilizing or destabilizing a certain receptor conformation. A positive allosteric modulator is a molecule that increases the receptor's affinity or efficacy at binding its ligand at the orthosteric site, or the receptor's ability to enter the active conformation, and a negative allosteric modulator would do the opposite. For example, cinacalcet, a positive allosteric modulator of the Class C Calcium Sensing Receptor, changes the receptor's sensitivity to circulating calcium, thereby being beneficial in disease states such as hyperparathyroidism in which there is a calcium sensing receptor deficiency [8]. Maraviroc, a negative allosteric modulator of the Class A CCR5 chemokine receptor, has been shown to block CCR5-mediated entry of HIV into cells [8]. Additionally, allosteric modulators can possess “pharmacological silence”, as in the case of M-5MPEP, an allosteric ligand for the MGluR5 (Metabotropic Glutamate receptor 5) receptor. M-5MPEP does not inactivate or activate MGluR5, but it partially blocks the response upon agonist binding, leading to partial mGluR5 inhibition [9].
Ions can also modulate GPCRs. For example, zinc has been shown to alter the ligand binding properties of a few different GPCRs at physiologically relevant concentrations (Figure 2B) [10]. Binding of either an agonist or an antagonist to 5-hydroxytryptamine/serotonin receptor 1A (5HT1A-R) is completely prevented by zinc [11]. In the continued presence of zinc, D1 and D2 dopamine receptors do not lose all ligand binding properties like the 5HT1A-R does, but they do have decreased affinity for ligand [12]. Referring back to our example of the β2AR in Figure 1B and Figure 2B, addition of zinc (X in Figure 1B) would shift the equilibrium constant such that binding of ISO would lead to a modified stimulus (S*β) compared to the stimulus S in the absence of the allosteric modulator.
We propose that a second GPCR can also act as an allosteric modulator of a GPCR. In the case of the β2AR, our previous work has shown that both in vitro and in vivo, binding of bradykinin (BK) to B2R transactivates β2AR [7]. Control experiments show that this transactivation is second messenger independent, and is a result of conformational changes in both receptors [7]. Figure 2C shows that upon coexpression of B2R and β2AR, stimulation with BK leads to activation of both a calcium activated chloride channel (as indicated by the sharp current spike) and CFTR (as indicated by the slow current wave); the former is via B2R-mediated activation of Gαq signaling, and the latter is via heterodimer-mediated activation of Gαs signaling [7]. Stimulation with BK a second time also leads to β2AR-mediated CFTR opening. These results show that B2R is able to transactivate β2AR. Other results suggest that B2R is a negative allosteric modulator of β2AR, as shown in the concentration response curve in the bottom panel of Figure 2C. In this case, the concentration response curve generated in Xenopus oocytes in response to increasing concentrations of terbutaline, a selective β2AR agonist, exhibited a rightward shift in cells also expressing B2R, indicating that B2R dampens the ability of β2AR to signal through the Gαs pathway. Because we hypothesize that one GPCR can modulate another, it is important to review the consequences of GPCR/GPCR interaction.
2. GPCR Dimerization: A Brief History and Introduction
Until recently, it was thought that two components make up the classical GPCR signaling pathway: the monomeric GPCR and the heterotrimeric G proteins with which it associates. Increasing evidence has shown that this two part model is grossly oversimplified as many GPCRs couple to more than one type of G protein, each subunit of the heterotrimeric G protein can activate different downstream effectors, many modulators exist to alter the signaling of the GPCR, and GPCRs even can alter other effectors in G protein-independent ways. This complexity is further entangled by the emergence of the concept of GPCR dimerization. In 1978, Watanabe and coworkers discovered that methacholine, a non-selective muscarinic receptor agonist, can restore the affinity of the β2AR for its agonist isoproterenol in the presence of GTP [13]. This result was later hypothesized to be due to receptor-receptor interaction [14], referred to as dimerization. For our purposes here, dimerization is defined as the physical association of two GPCRs. The possibility of two molecules of the same GPCR being physically coupled (homodimerization) or two different GPCRs being coupled (heterodimerization), presents an unexpected mechanism for GPCR regulation and function. Given the number of other ligand binding proteins that form dimers in order to function, including receptor tyrosine kinases and intracellular steroid receptors, dimerization of GPCRs seemed like a feasible possibility.
The first studies to investigate GPCR dimers used radiation inactivation or target size analysis to study the differences in sizes of molecular species by using high energy particles to disrupt polypeptides, thereby identifying the molecular weight of singular components. This technique was used to illustrate that β2AR, opioid receptors (OPRs), muscarinic receptors, Gonadotropin releasing hormone receptors, thyrotropin receptors, and dopamine D1 receptors all formed homodimers [15-17]. The advantages of this technique originally were that the difficulty of both solubilizing and having a great enough yield of receptor membrane protein to detect via other biochemical methods were not limitations to the detection of oligomeric species [18,19].
Although additional methods to study GPCR dimers have grown in the last decade, given the difficulty with resolving endogenous membrane-spanning proteins that are not expressed in relative abundance, most studies rely on transient transfection of cDNA constructs encoding receptors and pharmacological methods such as radioligand competition binding studies to collect indirect evidence for dimerization. These methods are used in conjunction with biochemical methods such as co-immunoprecipitation and bioluminescence or Förster resonance energy transfer (BRET, FRET). Recently, a number of papers have also used bioinformatics and modeling to predict dimer interfaces [20-23]. Excitingly, recent work by Wu and others have illustrated via crystallography that GPCRs can exist as homodimers; the CXCR4 homodimer was crystallized at a resolution of 2.5–3.2 angstroms [24]. All of these techniques have the potential for artifact, so there has been persistent skepticism in proving GPCR dimerization as a verifiable phenomenon. This, in turn, requires that new dimer pairs that are discovered must be found to dimerize both in heterologous and endogenous expression systems. Table 1 provides a representation of a small number of GPCRs that have been shown to heterodimerize either in vitro, in vivo, or both [1,3,4,25-33].
The canonical examples of GPCR dimerization are the Class C GABABR1 and GABABR2 receptors and the umami and sweet taste receptors [36]. GABABR1 and GABABR2, both Gαi coupled proteins, form naturally occurring obligate dimers meaning that in order to function as a receptor, they must dimerize. This pair was identified in transiently transfected HEK cells by the finding that when either GABABR1 or GABABR2 were expressed singly, the response to ligand was minimal; however, cotransfection robustly increased the response to GABA [36]. These experiments were then repeated in Xenopus oocytes injected with cRNAs encoding these receptors and G-protein mediated inwardly rectifying potassium (GIRK) channels to provide a measure of receptor activity [37]. Colocalization and physical association of GABABR1 and GABABR2 were confirmed using immunoprecipitation and confocal microscopy in transiently transfected HEK cells as well as in situ hybridization and radiolabeling in rat brains [36]. Interestingly, this seminal work is one of the only examples where GPCR dimerization was studied by electrophysiological methods and uses both heterologous and endogenous expression systems.
Other canonical examples of GPCR heterodimers are the Class C Taste receptors T1R1, T1R2, and T1R3. Detection of sweet taste is mediated by of the T1R2-T1R3 heterodimer, and detection of umami taste is mediated by the T1R1-T1R3 heterodimer. The selectivity of these heterodimers lies in the venus flytrap domain (VFD), where the active site for ligand binding is located. Chimeric experiments have shown that swapping of the VFDs switches the ligand selectivity entirely [38].
A number of studies have examined other examples of GPCR heterodimerization both in vitro and in vivo, including those summarized in Table 1. Although it is fascinating to examine the physical association of two GPCRs, the potential functionality of that interaction remains the more pertinent question.
3. Functional Consequences of GPCR Dimerization
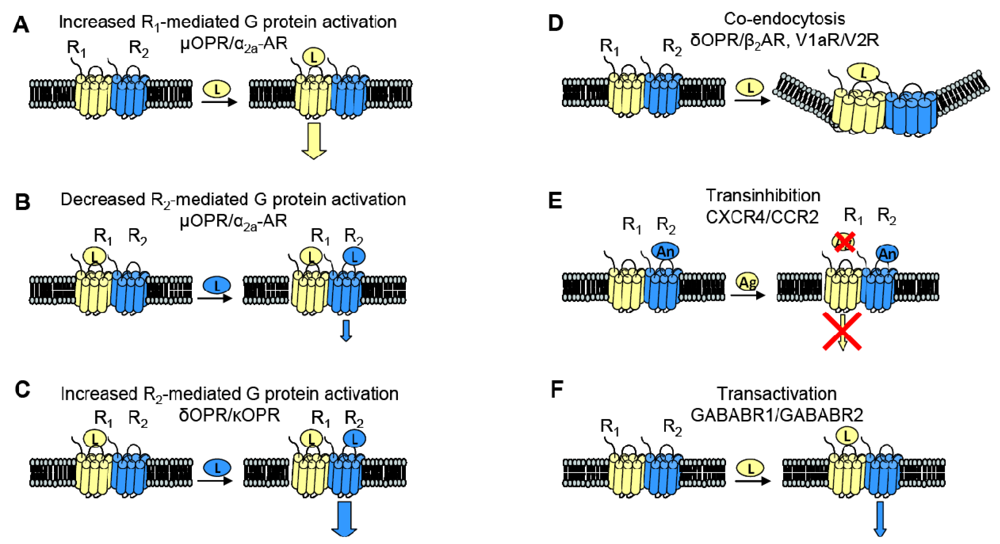
When examining the interaction between two receptors, potential changes in receptor behavior can include changes in pharmacology, signaling and trafficking. Figure 3 summarizes some of the functional consequences of heterodimerization outlined here.
3.1. Changes in Pharmacology
GPCR heterodimerization can lead to changes in a cognate receptor's ability to signal upon ligand binding. Given our previous definition of an allosteric modulator (an entity that binds a receptor at a site other than the orthosteric site thereby changing the properties of that receptor), then GPCRs can also serve as allosteric modulators of one another. In the specific examples detailed below, coexpression of a second receptor, R2, changes the properties of the first receptor, R1, thereby altering the response to R1's cognate ligand. Thus, R2 is an allosteric modulator of R1 (Figure 3).
The interaction of the family of Gαi-coupled OPRs is a classical example of GPCR heterodimerization leading to changes in pharmacology. It has been shown that κOPR heterodimerizes with δOPR but not μOPR [32]. Although the κOPR/ δOPR heterodimer does not exhibit altered selectivity for either receptor's specific agonist or antagonist, the heterodimer can bind partial agonists for either receptor with increased affinity (Figure 3A). Heterodimerization of these two OPRs also leads to enhanced agonism in the presence of both ligands; addition of the δOPR agonist DPDPE in the continued presence of the κOPR agonist U69563 increases binding affinity and receptor activation (Figure 3C). This synergy is also seen in the presence of both receptor's antagonists, suggesting the presentation of a novel ligand binding pocket in the heterodimer [39]. This κOPR/ δOPR heterodimer was later found to exist in vivo because the ligand 6′GNTI, an agonist that has tissue-specific analgesic effects, selectively targets the heterodimer; in contrast, this ligand is able to target the κOPR expressed singly at a very low efficacy and targets δOPR expressed singly not at all [27].
One GPCR in a heterodimer also can have an effect on the other receptor's cognate signaling pathways. The α2A-AR and μOPR, both Gαi coupled receptors, when coexpressed in HEK cells were shown to heterodimerize by FRET measurements. Formation of the heterodimer enhances μOPR signaling when stimulated with morphine only, but simultaneous addition of an α2A-AR agonist greatly decreased the μOPR signaling response, thus representing both a change in pharmacology and in signaling (Figure 3B). In the inverse experiments, there was also a dampening of norepinephrine-mediated G protein activation in the continuing presence of morphine, but not to the same extent [40].
3.2. Transinhbition
Given the aforementioned example of α2A-AR and μOPR heterodimers, it is now clear that heterodimerization also can lead to transinhibition of either R1 or R2 upon addition of antagonist against one of the two receptors (Figure 3E). The Gi-coupled chemokine receptors CCR2 and CXCR4 heterodimerize, and are well studied because of their role in Human Immunodeficiency Virus (HIV) entry into cells and in other inflammatory diseases (CCR2 is expressed on the surface of T lymphocytes and is involved in the recruitment of monocytes to atherosclerotic lesions). Antagonists against either CCR2 or CXCR4 transinhibit the other receptor, preventing binding of chemokines to the opposite receptor in both leukocytes and heterologous cell lines. The CCR2/CCR5 heterodimer also exhibits the same transinhibition [41].
3.3. Altered Trafficking
Heterodimerization also can alter trafficking of either receptor compared to when they are expressed singly. For example, when coexpressed in HEK293 cells, V1a and V2 vasopressin receptors can heterodimerize. Although both receptors associate with β-arrestin, V1aR when singly expressed rapidly dissociates from β-arrestin in order to be rapidly recycled to the plasma membrane whereas V2R singly expressed remains stably associated with β-arrestin, thereby accumulating intracellularly. The V1aR/V2R heterodimer followed the V2R endocytic/recycling pattern when stimulated with a nonselective agonist. Selective agonism of the V1aR led to the co-endocytosis of the V2R, suggesting that the heterodimer is influenced by the activated receptor (Figure 3D) [28]. Similarly, cotransfection of β2AR, a Gαs coupled receptor, and δOPR, a Gαi coupled receptor, in CHO and HEK cells led to heterodimerization. The heterodimer underwent both isoproterenol-mediated and opioid-mediated endocytosis (Figure 3D). Interestingly, the δOPR/ β2AR heterodimer did not exhibit any changes in ligand selectivity compared to each cognate receptor [29]. As mentioned previously, α1B-AR and α1D-AR also heterodimerize; not only does this interaction change ligand binding characteristics as discussed above, but co-expression of α1D-AR greatly increases the trafficking of α1B-AR to the cell surface. Without α1D-AR present, only negligible amounts of α1B-AR are expressed at the cell surface [31].
Despite the increasing amount of knowledge about GPCRs, and the likelihood that they often exist as homo- or heterodimers, there is still a gap in the number of dimer pairs that have been shown to interact in both heterologous and endogenous systems (Table 1). Because transfection into a cultured cell line typically creates overexpression of a given protein, it is often suspected that the apparent dimerization and functional consequences are artifacts of non-physiological conditions. Furthermore, methods such as BRET and FRET, often used to study heterodimerization, assume that the introduction of the fluorescent tags attached to the proteins of interest does not induce any false positive interaction. A recent paper from the Sheikh group shows in three separate commonly used cell lines that previously published data on the Angiotensin type II receptor (ATR1)/B2R heterodimer are the result of artifact [42]. Thus, when examining potential interactions, both physical and functional, between two GPCRs, it is of utmost importance to be thorough in technique and cell type using both heterologous and endogenous expression systems to lend credence to statements regarding a putative heterodimer pair.
3.4. Allosteric Modulation: Transactivation
Two sets of studies have shown that interactions between two GPCRs also can lead to transactivation, where the ligand-induced conformational change in the R1 receptor leads to activation of the R2 receptor (Figure 3F). The first was the recognition that GABABR1 and GABABR2 must heterodimerize to form a functional receptor. In this case, GABABR1 can bind ligand but cannot activate signaling, while GABABR2 cannot bind ligand, but can activate Gαi-mediated signaling. Hence, binding of ligand to GABABR1 is thought to induce the adoption of an activated conformation by GABABR2. Our own work demonstrated that upon binding BK, B2R is able to transactivate β2AR (Figure 2C, top). Additionally, coexpression of B2R is sufficient to hinder β2AR's ability to respond to its selective ligand (Figure 2C, bottom). Thus, in this case the B2R is a negative allosteric modulator of β2AR [7]. We know that B2R and β2AR are physically associated in Xenopus oocytes, and in these cells, B2R coexpression alters β2AR pharmacology as well as activating this receptor directly in the absence of a β2AR-selective ligand (Figure 2C, top). Schwartz and Holst define an allosteric modulator as: “a ligand that functions as both an agonist on its own and as an allosteric modulator of the efficacy and/or potency of the orthosteric ligand” [43]. Although B2R is not a ligand, it fits the definition of an allosteric modulator: BK-bound B2R acts as an agonist at β2AR, yet B2R negatively affects the sensitivity of β2AR to activation by its cognate ligand.
4. A New Model of Allosteric Regulation of GPCRs by Receptor Heterodimerization
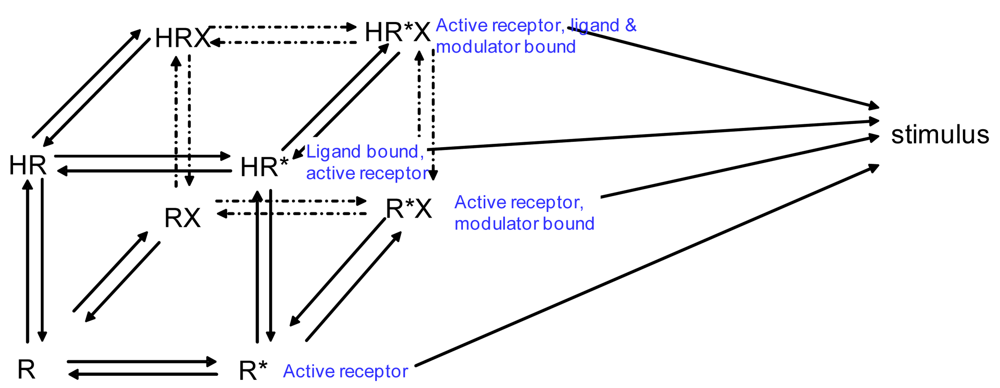
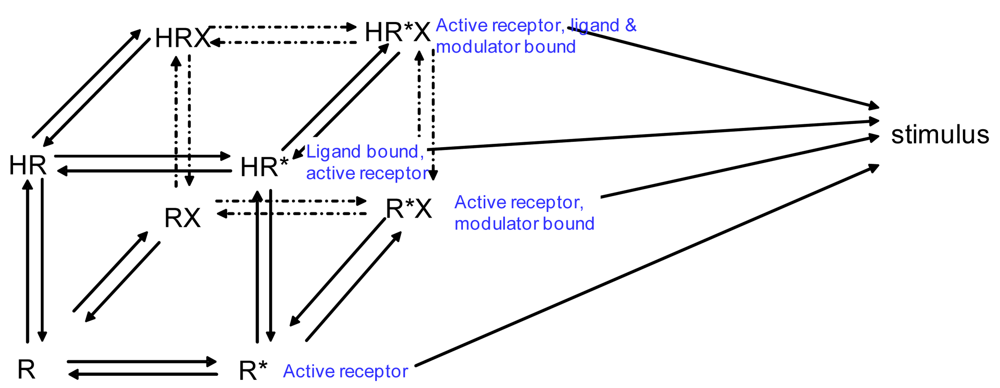
If we wanted to build a model to represent the interaction of two receptors of a GPCR heterodimer, we must first reflect further on the ternary complex model of GPCRs. A limitation of the previously described ternary complex model is that it cannot factor in the effects on affinity, efficacy, and the number of receptors in the active or the inactive state as determined by specific conformations stabilized by the binding of specific orthosteric or allosteric modulators and their affinity for different receptor states. The allosteric two state model developed by Hall in 2000 factors in the binding of a molecule at an allosteric site on a receptor that switches between inactive and active conformations (Figure 4) [44]. Although Hall's model referred to ligands as allosteric modulators, we focus here on the notion that other receptors also may serve as allosteric modulators of GPCRs via receptor heterodimerization.
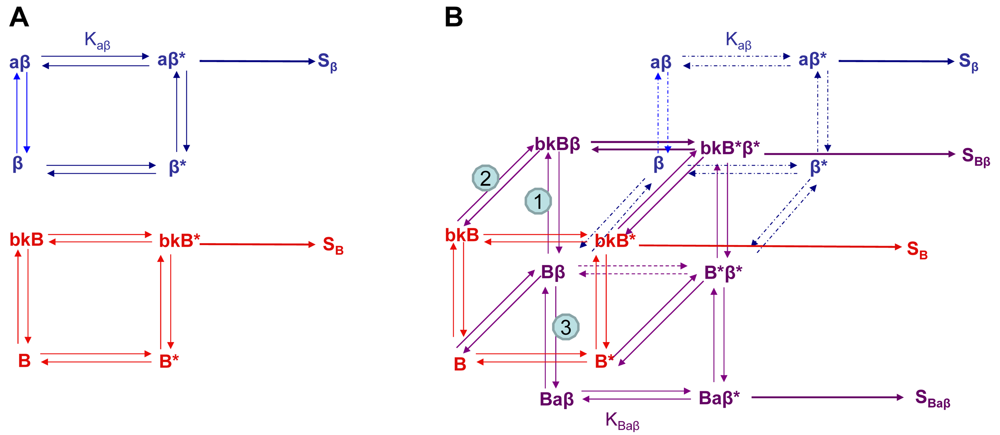
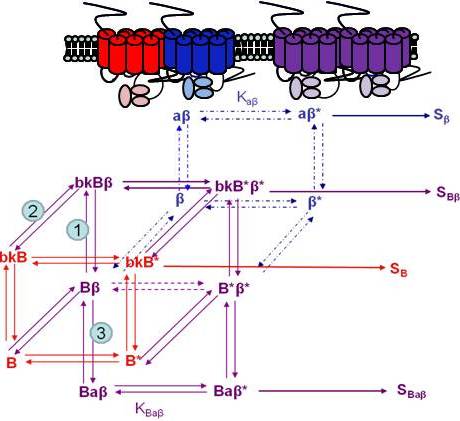
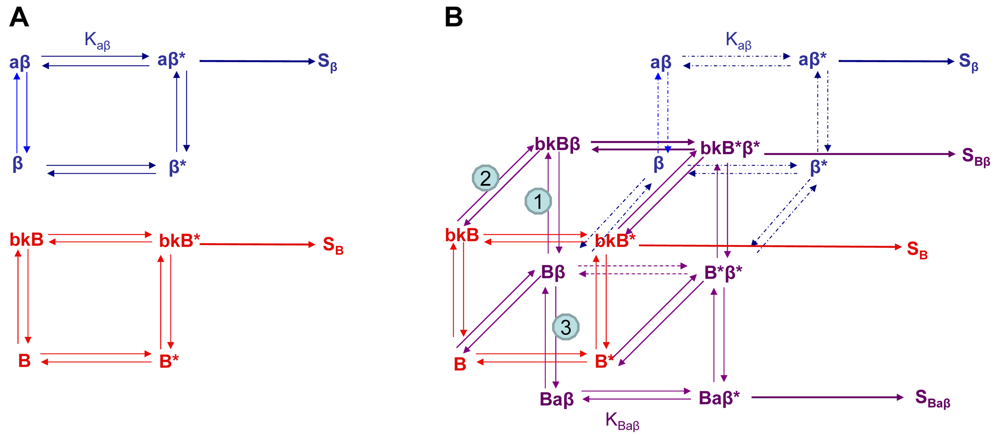
Using the B2R and β2AR as an example of a GPCR heterodimer, we propose a model that takes into account three different pools of receptors: B2R (monomers and homodimers combined), β2AR (monomers and homodimers combined), and B2R/β2AR heterodimers; monomers and homodimers are treated the same in order to avoid overly complicating the scheme. As shown in Figure 5A, we can first model either B2R (B) or β2AR (β) using the DeLean ternary complex model, in which the receptors fluctuating back and forth between inactive and active (*) conformations can bind their ligands bradykinin (bk in this figure) or isoproterenol (a in this figure), respectively, and thus activate their G protein pathway cascades, leading to stimuli SB or Sβ.
Figure 5B takes into account the formation of functional heterodimers. In this model, the B2R/β2AR heterodimer has signaling characteristics different from those of either singly expressed receptor. When B2R of the B2R/β2AR heterodimer binds BK (bkBβ) (reaction ➀ in Figure 5B) or when the BK-bound B2R heterodimerizes with β2AR (reaction ➁), both receptors will switch to an active conformation (bkB*β*). This complex will result in activation of both Gαq- and Gαs-coupled signaling pathways resulting in stimulus SBβ. Although this may not be the case for all other GPCR heterodimers, our model can also take into account the negative allosteric effect that B2R coexpression has on β2AR signaling. In this case, when agonist a binds β2AR of the B2R/β2AR heterodimer (reaction ➂), the equilibrium constant, KBaβ, is less than Kaβ, and the resultant stimulus SBaβ is less than Sβ. It is also possible that heterodimerization with B2R alters the binding of agonist to the β2AR; this, too, would affect apparent potency. Our model provides a framework within which to consider the different species of receptors (monomers/homodimers and heterodimers), and the effect of the active and inactive conformations each receptor of the heterodimer can occupy.
5. Concluding Remarks
Because there is a possibility of two different GPCRs being physically associated, existing drugs with deleterious side effects are now being re-examined for their ability to target more than one GPCR. For example, levodopa, a dopamine precursor capable of crossing the blood-brain barrier used to treat Parkinson's disease, often has a dyskinesia side effect, thought to be from levodopa-induced co-activation of the A2A adenosine receptor [45]. Thus, it has been proposed that administration of an A2A adenosine receptor antagonist in conjunction with levodopa may decrease some of these side effects [46,47]. This may not be unexpected considering that other work has indicated that these two receptors have both physical and functional interaction in the basal ganglia [48,49]. Taken together, it is quite possible that the existence of GPCR heterodimers should be considered when examining side effects of existing drugs and the presence of GPCR heterodimers may also serve as an axis for developing new drugs. Because GPCR ligands are common drug targets, targeting a heterodimer directly for its allostery could enforce or strengthen the effect of an orthosteric agonist, increase pharmacological diversity, and potentially provide stimulation of a target receptor in cases in which the orthosteric agonist is not present [43]. Conversely, if the site of dimerization is known, then targeting this interaction site for allosteric potentiator or inhibitors as a new class of drug therapies or therapeutic antibody therapies could have specific benefits. It is apparent that the complex signaling pathways and the various modes of regulation that GPCRs undergo make them, to paraphrase a recently published article, a “panacea or Pandora's box for novel drug discovery” [21].


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| β1AR | β2AR | V2R | dOPR | kOPR | mOPR | SST2a | α2AR | D2 | CB1 | Bk2R | ATR1 | V1a | 5HT2CR | α1aAR | α1bAR | mGluR2 | mGluR4 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| β1AR | ||||||||||||||||||
| β2AR | ||||||||||||||||||
| V2R | ||||||||||||||||||
| dOPR | ||||||||||||||||||
| kOPR | ||||||||||||||||||
| mOPR | ||||||||||||||||||
| SST2a | ||||||||||||||||||
| α2AR | ||||||||||||||||||
| D2 | ||||||||||||||||||
| CB1 | ||||||||||||||||||
| Bk2R | ||||||||||||||||||
| ATR1 | ||||||||||||||||||
| V1a | ||||||||||||||||||
| 5HT2CR | ||||||||||||||||||
| α1aAR | ||||||||||||||||||
| α1bAR | ||||||||||||||||||
| mGluR2 | ||||||||||||||||||
| mGluR4 |
| heterodimer in both in vitro and in vivo | |
| heterodimer in heterologous expression system | |
| unknown | |
| do not dimerize |
Acknowledgements
The authors thank H. Radhakrishna for input to this work, and the Emory-Egleston Children's Research Center for support.
References
- Overington, J.P.; Al-Lazikani, B.; Hopkins, A.L. How many drug targets are there? Nat. Rev. Drug Discov. 2006, 5, 993–996. [Google Scholar]
- Birdsall, N.J. Class A GPCR heterodimers: Evidence from binding studies. Trends Pharmacol. Sci. 2010, 31, 499–508. [Google Scholar]
- De Lean, A.; Stadel, J.M.; Lefkowitz, R.J. A ternary complex model explains the agonist-specific binding properties of the adenylate cyclase-coupled beta-adrenergic receptor. J. Biol. Chem. 1980, 255, 7108–7117. [Google Scholar]
- May, L.T.; Leach, K.; Sexton, P.M.; Christopoulos, A. Allosteric modulation of G protein-coupled receptors. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 1–51. [Google Scholar]
- Knaus, G.A.; Knaus, H.G.; Saria, A. Complex allosteric interaction of heparin with neurokinin-1 receptors. Eur. J. Pharmacol. 1991, 207, 267–270. [Google Scholar]
- Swaminath, G.; Steenhuis, J.; Kobilka, B.; Lee, T.W. Allosteric modulation of beta2-adrenergic receptor by Zn(2+). Mol. Pharmacol. 2002, 61, 65–72. [Google Scholar]
- Haack, K.K.; Tougas, M.R.; Jones, K.T.; El-Dahr, S.S.; Radhakrishna, H.; McCarty, N.A. A novel bioassay for detecting GPCR heterodimerization: transactivation of beta 2 adrenergic receptor by bradykinin receptor. J. Biomol. Screen. 2010, 15, 251–260. [Google Scholar]
- Conn, P.J.; Christopoulos, A.; Lindsley, C.W. Allosteric modulators of GPCRs: A novel approach for the treatment of CNS disorders. Nat. Rev. Drug Discov. 2009, 8, 41–54. [Google Scholar]
- Bridges, T.M.; Lindsley, C.W. G-protein-coupled receptors: From classical modes of modulation to allosteric mechanisms. ACS Chem. Biol. 2008, 3, 530–541. [Google Scholar]
- Frielle, T.; Daniel, K.W.; Caron, M.G.; Lefkowitz, R.J. Structural basis of beta-adrenergic receptor subtype specificity studied with chimeric beta 1/beta 2-adrenergic receptors. Proc. Natl. Acad. Sci. USA 1988, 85, 9494–9498. [Google Scholar]
- Barrondo, S.; Salles, J. Allosteric modulation of 5-HT(1A) receptors by zinc: Binding studies. Neuropharmacology 2009, 56, 455–462. [Google Scholar]
- Schetz, J.A.; Sibley, D.R. Zinc allosterically modulates antagonist binding to cloned D1 and D2 dopamine receptors. J. Neurochem. 1997, 68, 1990–1997. [Google Scholar]
- Watanabe, A.M.; McConnaughey, M.M.; Strawbridge, R.A.; Fleming, J.W.; Jones, L.R.; Besch, H.R., Jr. Muscarinic cholinergic receptor modulation of beta-adrenergic receptor affinity for catecholamines. J. Biol. Chem. 1978, 253, 4833–4836. [Google Scholar]
- Birdsall, N.J.M. Can different receptors interact directly with each other? Trends Neurosci. 1982, 5, 138–139. [Google Scholar]
- Nielsen, T.B.; Totsuka, Y.; Kempner, E.S.; Field, J.B. Structure of the thyrotropin receptor and thyroid adenylate cyclase system as determined by target analysis. Biochemistry 1984, 23, 6009–6016. [Google Scholar]
- Conn, P.M.; Venter, J.C. Radiation inactivation (target size analysis) of the gonadotropin-releasing hormone receptor: evidence for a high molecular weight complex. Endocrinology 1985, 116, 1324–1326. [Google Scholar]
- Gredal, O.; Nielsen, M.; Hyttel, J. Target size of dopamine D-1 receptors in rat corpus striatum estimated by binding of the benzazepine 3H-SCH 23390. Pharmacol. Toxicol. 1987, 60, 255–257. [Google Scholar]
- Fraser, C.M.; Venter, J.C. The size of the mammalian lung beta 2-adrenergic receptor as determined by target size analysis and immunoaffinity chromatography. Biochem. Biophys. Res. Commun. 1982, 109, 21–29. [Google Scholar]
- McLawhon, R.W.; Ellory, J.C.; Dawson, G. Molecular size of opiate (enkephalin) receptors in neuroblastoma-glioma hybrid cells as determined by radiation inactivation analysis. J. Biol. Chem. 1983, 258, 2102–2105. [Google Scholar]
- Fanelli, F.; Felline, A. Dimerization and ligand binding affect the structure network of A(2A) adenosine receptor. Biochim. Biophys. Acta 2010. unpublished. [Google Scholar]
- Filizola, M. Increasingly accurate dynamic molecular models of G-protein coupled receptor oligomers: Panacea or Pandora's box for novel drug discovery? Life Sci. 2010, 86, 590–597. [Google Scholar]
- Johnston, J.M.; Aburi, M.; Provasi, D.; Bortolato, A.; Urizar, E.; Lambert, N.A.; Javitch, J.A.; Filizola, M. Making structural sense of dimerization interfaces of delta opioid receptor homodimers. Biochemistry 2011. unpublished. [Google Scholar]
- Filizola, M.; Laakkonen, L.; Loew, G.H. 3D modeling, ligand binding and activation studies of the cloned mouse delta, mu; and kappa opioid receptors. Protein Eng. 1999, 12, 927–942. [Google Scholar]
- Wu, B.; Chien, E.Y.; Mol, C.D.; Fenalti, G.; Liu, W.; Katritch, V.; Abagyan, R.; Brooun, A.; Wells, P.; Bi, F.C.; Hamel, D.J.; Kuhn, P.; Handel, T.M.; Cherezov, V.; Stevens, R.C. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 2010, 330, 1066–1071. [Google Scholar]
- Lavoie, C.; Mercier, J.F.; Salahpour, A.; Umapathy, D.; Breit, A.; Villeneuve, L.R.; Zhu, W.Z.; Xiao, R.P.; Lakatta, E.G.; Bouvier, M.; Hebert, T.E. β1/β2-adrenergic receptor heterodimerization regulates β2-adrenergic receptor internalization and ERK signaling efficacy. J. Biol. Chem. 2002, 277, 35402–35410. [Google Scholar]
- AbdAlla, S.; Lother, H.; Quitterer, U. AT1-receptor heterodimers show enhanced G-protein activation and altered receptor sequestration. Nature 2000, 407, 94–98. [Google Scholar]
- Waldhoer, M.; Fong, J.; Jones, R.M.; Lunzer, M.M.; Sharma, S.K.; Kostenis, E.; Portoghese, P.S.; Whistler, J.L. A heterodimer-selective agonist shows in vivo relevance of G protein-coupled receptor dimers. Proc. Natl. Acad. Sci. USA 2005, 102, 9050–9055. [Google Scholar]
- Terrillon, S.; Barberis, C.; Bouvier, M. Heterodimerization of V1a and V2 vasopressin receptors determines the interaction with beta-arrestin and their trafficking patterns. Proc. Natl. Acad. Sci. USA 2004, 101, 1548–1553. [Google Scholar]
- Jordan, B.A.; Trapaidze, N.; Gomes, I.; Nivarthi, R.; Devi, L.A. Oligomerization of opioid receptors with β2-adrenergic receptors: a role in trafficking and mitogen-activated protein kinase activation. Proc. Natl. Acad. Sci. USA 2001, 98, 343–348. [Google Scholar]
- Jordan, B.A.; Gomes, I.; Rios, C.; Filipovska, J.; Devi, L.A. Functional interactions between μ opioid and alpha 2A-adrenergic receptors. Mol. Pharmacol. 2003, 64, 1317–1324. [Google Scholar]
- Uberti, M.A.; Hall, R.A.; Minneman, K.P. Subtype-specific dimerization of alpha 1-adrenoceptors: Effects on receptor expression and pharmacological properties. Mol. Pharmacol. 2003, 64, 1379–1390. [Google Scholar]
- Gomes, I.; Filipovska, J.; Jordan, B.A.; Devi, L.A. Oligomerization of opioid receptors. Methods 2002, 27, 358–365. [Google Scholar]
- Przybyla, J.A.; Watts, V.J. Ligand-induced regulation and localization of cannabinoid CB1 and dopamine D2L receptor heterodimers. J. Pharmacol. Exp. Ther. 2010, 332, 710–719. [Google Scholar]
- AbdAlla, S.; Abdel-Baset, A.; Lother, H.; el Massiery, A.; Quitterer, U. Mesangial AT1/B2 receptor heterodimers contribute to angiotensin II hyperresponsiveness in experimental hypertension. J. Mol. Neurosci. 2005, 26, 185–192. [Google Scholar]
- Doumazane, E.; Scholler, P.; Zwier, J.M.; Eric, T.; Rondard, P.; Pin, J.P. A new approach to analyze cell surface protein complexes reveals specific heterodimeric metabotropic glutamate receptors. FASEB J. 2011, 25, 66–77. [Google Scholar]
- Jones, K.A.; Borowsky, B.; Tamm, J.A.; Craig, D.A.; Durkin, M.M.; Dai, M.; Yao, W.J.; Johnson, M.; Gunwaldsen, C.; Huang, L.Y.; et al. GABA(B) receptors function as a heteromeric assembly of the subunits GABA(B)R1 and GABA(B)R2. Nature 1998, 396, 674–679. [Google Scholar]
- Margeta-Mitrovic, M.; Jan, Y.N.; Jan, L.Y. Function of GB1 and GB2 subunits in G protein coupling of GABA(B) receptors. Proc. Natl. Acad. Sci. USA 2001, 98, 14649–14654. [Google Scholar]
- Xu, H.; Staszewski, L.; Tang, H.; Adler, E.; Zoller, M.; Li, X. Different functional roles of T1R subunits in the heteromeric taste receptors. Proc. Natl. Acad. Sci. USA 2004, 101, 14258–14263. [Google Scholar]
- Jordan, B.A.; Devi, L.A. G-protein-coupled receptor heterodimerization modulates receptor function. Nature 1999, 399, 697–700. [Google Scholar]
- Vilardaga, J.P.; Nikolaev, V.O.; Lorenz, K.; Ferrandon, S.; Zhuang, Z.; Lohse, M.J. Conformational cross-talk between alpha2A-adrenergic and mu-opioid receptors controls cell signaling. Nat. Chem. Biol. 2008, 4, 126–131. [Google Scholar]
- Sohy, D.; Parmentier, M.; Springael, J.Y. Allosteric transinhibition by specific antagonists in CCR2/CXCR4 heterodimers. J. Biol. Chem. 2007, 282, 30062–30069. [Google Scholar]
- Hansen, J.L.; Hansen, J.T.; Speerschneider, T.; Lyngso, C.; Erikstrup, N.; Burstein, E.S.; Weiner, D.M.; Walther, T.; Makita, N.; Iiri, T.; Merten, N.; Kostenis, E.; Sheikh, S.P. Lack of evidence for AT1R/B2R heterodimerization in COS-7, HEK293, and NIH3T3 cells: How common is the AT1R/B2R heterodimer? J. Biol. Chem. 2009, 284, 1831–1839. [Google Scholar]
- Schwartz, T.W.; Holst, B. Allosteric enhancers, allosteric agonists and ago-allosteric modulators: where do they bind and how do they act? Trends Pharmacol. Sci. 2007, 28, 366–373. [Google Scholar]
- Hall, D.A. Modeling the functional effects of allosteric modulators at pharmacological receptors: an extension of the two-state model of receptor activation. Mol. Pharmacol. 2000, 58, 1412–1423. [Google Scholar]
- Bara-Jimenez, W.; Sherzai, A.; Dimitrova, T.; Favit, A.; Bibbiani, F.; Gillespie, M.; Morris, M.J.; Mouradian, M.M.; Chase, T.N. Adenosine A(2A) receptor antagonist treatment of Parkinson's disease. Neurology 2003, 61, 293–296. [Google Scholar]
- George, S.R.; O'Dowd, B.F.; Lee, S.P. G-protein-coupled receptor oligomerization and its potential for drug discovery. Nat. Rev. Drug Discov. 2002, 1, 808–820. [Google Scholar]
- Bove, J.; Marin, C.; Bonastre, M.; Tolosa, E. Adenosine A2A antagonism reverses levodopa-induced motor alterations in hemiparkinsonian rats. Synapse 2002, 46, 251–257. [Google Scholar]
- Kamiya, T.; Saitoh, O.; Yoshioka, K.; Nakata, H. Oligomerization of adenosine A2A and dopamine D2 receptors in living cells. Biochem. Biophys. Res. Commun. 2003, 306, 544–549. [Google Scholar]
- Ferre, S.; Fredholm, B.B.; Morelli, M.; Popoli, P.; Fuxe, K. Adenosine-dopamine receptorreceptor interactions as an integrative mechanism in the basal ganglia. Trends Neurosci. 1997, 20, 482–487. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Haack, K.K.V.; McCarty, N.A. Functional Consequences of GPCR Heterodimerization: GPCRs as Allosteric Modulators. Pharmaceuticals 2011, 4, 509-523. https://doi.org/10.3390/ph4030509
Haack KKV, McCarty NA. Functional Consequences of GPCR Heterodimerization: GPCRs as Allosteric Modulators. Pharmaceuticals. 2011; 4(3):509-523. https://doi.org/10.3390/ph4030509
Chicago/Turabian StyleHaack, Karla K.V., and Nael A. McCarty. 2011. "Functional Consequences of GPCR Heterodimerization: GPCRs as Allosteric Modulators" Pharmaceuticals 4, no. 3: 509-523. https://doi.org/10.3390/ph4030509
APA StyleHaack, K. K. V., & McCarty, N. A. (2011). Functional Consequences of GPCR Heterodimerization: GPCRs as Allosteric Modulators. Pharmaceuticals, 4(3), 509-523. https://doi.org/10.3390/ph4030509