Mechanisms of Broad-Spectrum Antiemetic Efficacy of Cannabinoids against Chemotherapy-Induced Acute and Delayed Vomiting

{kind=link}
Abstract
:1. Introduction
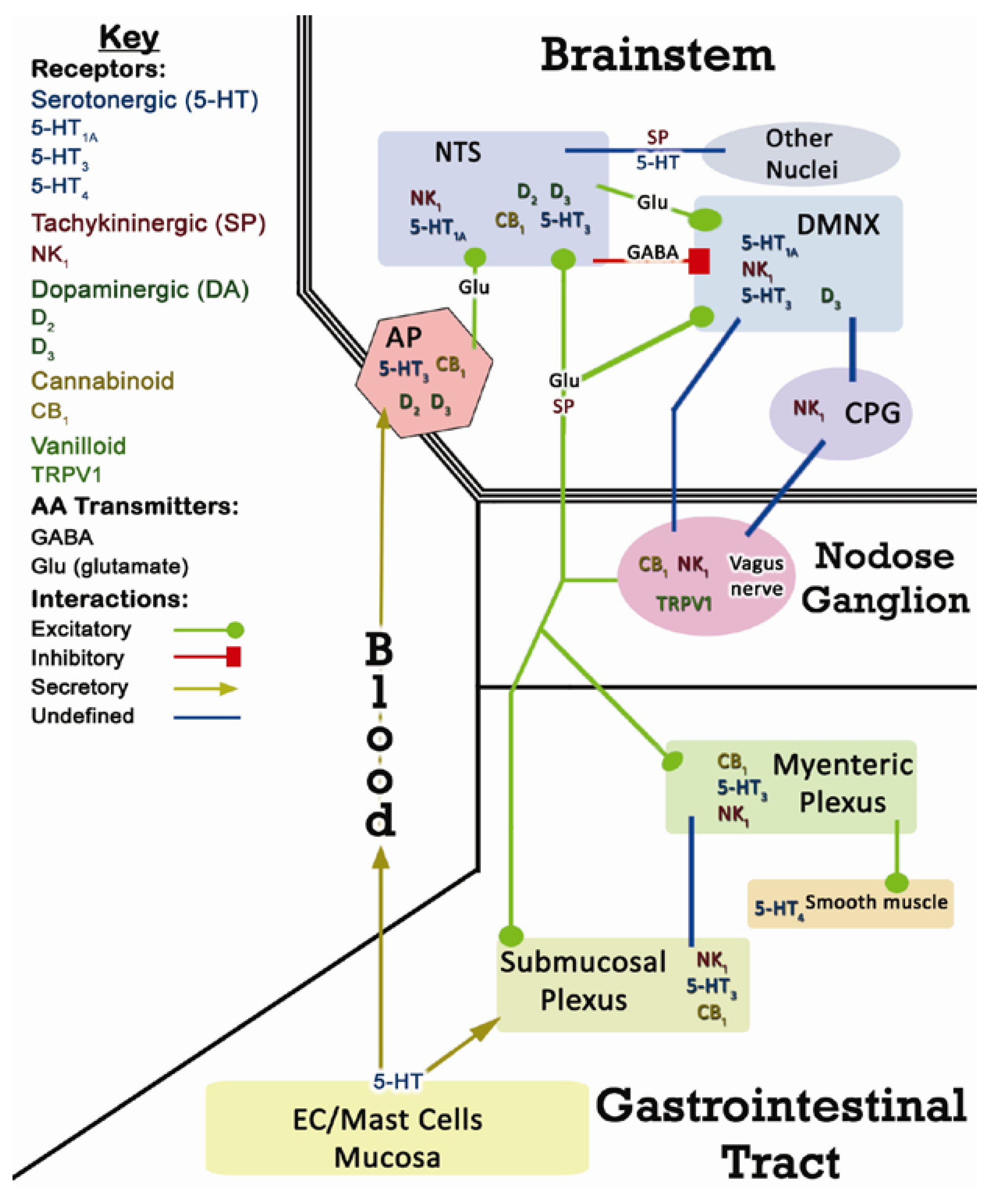
2. CINV Emetic Circuits
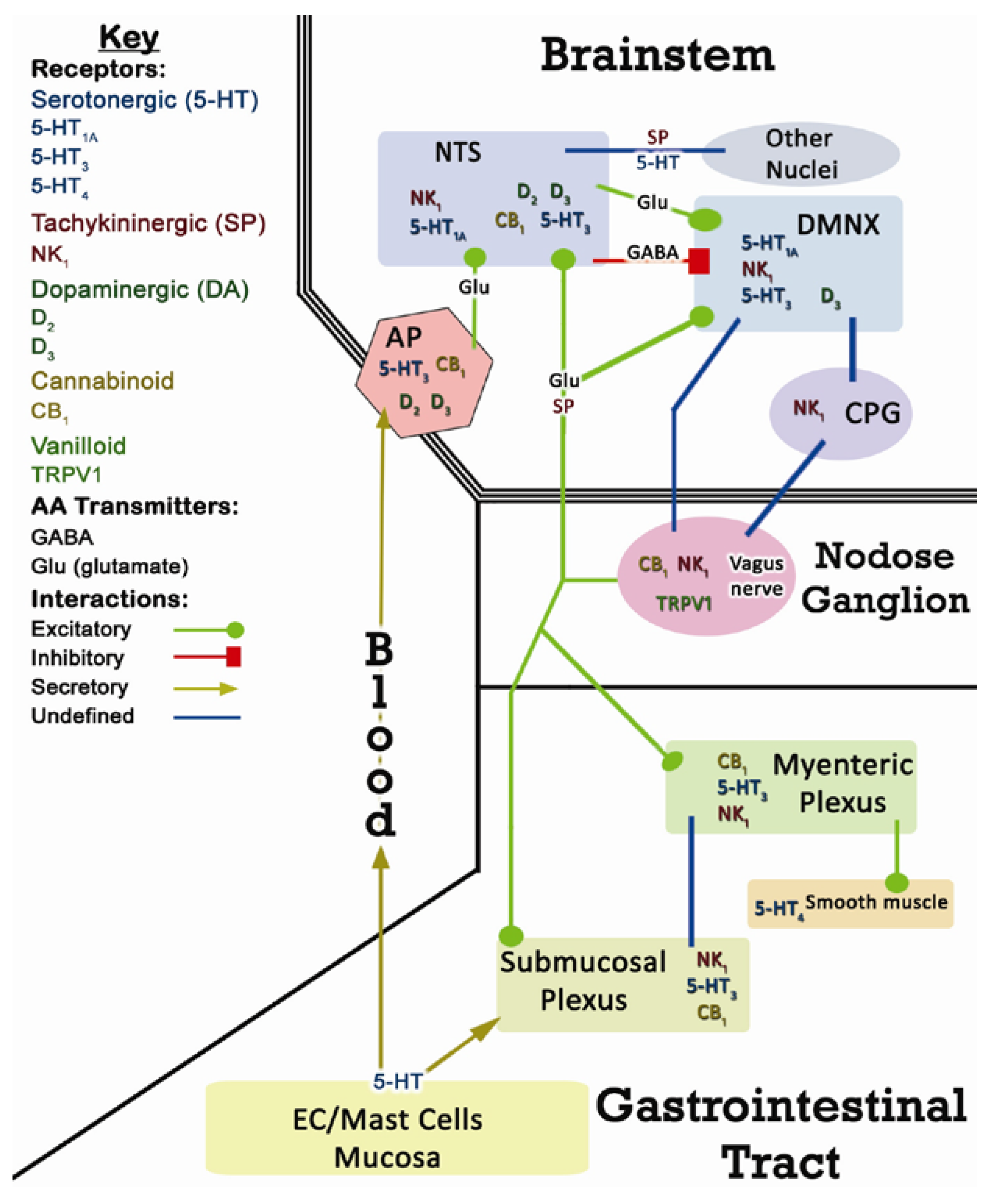
2.1. Terminology
2.2. Peripheral Components of CINV
2.3. Central Components of CINV
3. Cannabinoids and Endocannabinoids
4. Cannabinoid Targets in Emetic Circuits
4.1. Dorsal Vagal Complex (DVC)
4.2. Vagal Afferents
4.3. Enteric Nervous System (ENS)
4.4. Gastrointestinal Tissue
5. Mechanisms of the Antiemetic Actions of Phyto and Synthetic Cannabinoids’ against Acute and Delayed CINV
5.1. Antiemetic Activity of Cannabinoid CB1 Receptors
5.2. The Broad-spectrum Antiemeic Nature of Cannabinoid CB1 Receptor Agonists Involves Both Central and Peripheral Emetic Loci
6. Mechanisms via Which Cannabinoids Prevent Chemotherapy-Induced Acute and Delayed Phase Emesis
7. The Nature of Pro- and Antiemetic Actions of Endocannabinoids and Endovanilloids
8. Maijuana and Hyperemesis Syndrome
Acknowledgements
References
- Darmani, N.A.; Ray, A.P. Evidence for a re-evaluation of the neurochemical and anatomical bases of chemotherapy-induced vomiting. Chem. Rev. 2009, 109, 3158–3199. [Google Scholar]
- Darmani, N.A. Antiemetic action of Δ9-tetrahydrocannabinol and synthetic cannabinoids. in chemotherapy-induced nausea and vomiting. In Biology of Marijuana: From Gene to Behavior; Onaivi, E.S., Ed.; Taylor and Francis Books Ltd.: London, UK, 2002; pp. 356–389. [Google Scholar]
- Slatkin, N.E. Cannabinoids in the treatment of chemotherapy-induced nausea and vomiting: beyond prevention of acute emesis. J. Support Oncol. 2007, 5, 1–9. [Google Scholar]
- Darmani, N.A. Delta(9)-tetrahydrocannabinol and synthetic cannabinoids prevent emesis produced by the cannabinoid CB(1) receptor antagonist/inverse agonist SR 141716A. Neuropsychopharmacology 2001, 24, 198–203. [Google Scholar]
- Ray, A.P.; Griggs, L.; Darmani, N.A. Delta 9-tetrahydrocannabinol suppresses vomiting behavior and Fos expression in both acute and delayed phases of cisplatin-induced emesis in the least shrew. Behav. Brain Res. 2009, 196, 30–36. [Google Scholar]
- Van Sickle, M.D.; Oland, L.D.; Ho, W.; Hillard, C.J.; Mackie, K.; Davison, J.S.; Sharkey, K.A. Cannabinoids inhibit emesis through CB1 receptors in the brainstem of the ferret. Gastroenterology 2001, 121, 767–774. [Google Scholar]
- Van Sickle, M.D.; Oland, L.D.; Mackie, K.; Davison, J.S.; Sharkey, K.A. Delta9-tetrahydrocannabinol selectively acts on CB1 receptors in specific regions of dorsal vagal complex to inhibit emesis in ferrets. Am. J. Physio. Gastrointest Liver Physiol. 2003, 285, G566–G576. [Google Scholar]
- Di Marzo, V. CB(1) receptor antagonism: biological basis for metabolic effects. Drug Discov. Today 2008, 23-24, 1026–1041. [Google Scholar] [CrossRef]
- Sharkey, K.A.; Cristino, L.; Oland, L.D.; Van Sickle, M.D.; Starowicz, K.; Pittman, Q.J.; Guglielmotti, V.; Davison, J.S.; Di Marzo, V. Arvanil, anandamide and N-arachidonoyl-dopamine (NADA) inhibit emesis through cannabinoid CB1 and vanilloid TRPV1 receptors in the ferret. Eur. J. Neurosci. 2007, 25, 2773–2782. [Google Scholar]
- Parker, L.A.; Limebeer, C.L.; Rock, E.M.; Litt, D.L.; Kwaitkowska, M.; Piomelli, D. The FAAH inhibitor URB-597 interferes with cisplatin- and nicotine-induced vomiting in the Suncus murinus (house musk shrew). Physiol. Behav. 2009, 97, 121–124. [Google Scholar]
- Darmani, N.A. The potent emetogenic effects of the endocannabinoid, 2-AG (2-arachidonoylglycerol) are blocked by delta(9)-tetrahydrocannabinol and other cannabinoids. J. Pharmacol. Exp. Ther. 2002, 300, 34–42. [Google Scholar]
- Andrews, P.L.R.; Rudd, J.A. The role of tachykinins and the tachykinin NK1 receptor in nausea and emesis. J. Exp. Pharmacol. 2004, 164, 359–340. [Google Scholar]
- Endo, T.; Minami, M.; Hirafuji, M.; Ogawa, T.; Akita, K.; Nemoto, M.; Saito, H.; Yoshioka, M.; Parvez, S.H. Neurochemistry and neuropharmacology of emesis - the role of serotonin. Toxicology 2000, 153, 189–201. [Google Scholar]
- Koch, K.L. Approach to the patient with nausea and vomiting. In Textbook of Gastroenterology; Yamada, T., Ed.; Lippincott: Philadelphia, PA, USA, 1995; pp. 83–388. [Google Scholar]
- Tanihata, S.; Oda, S.; Kakuta, S.; Uchiyama, T. Antiemetic effect of a tachykinin NK1 receptor antagonist GR205171 on cisplatin-induced early and delayed emesis in the pigeon. Eur. J. Pharmacol. 2003, 461, 197–206. [Google Scholar]
- Darmani, N.A.; Crim, J.L.; Janoyan, J.J.; Abad, J.; Ramirez, J. A re-evaluation of the neurotransmitter basis of chemotherapy-induced immediate and delayed vomiting: evidence from the least shrew. Brain Res. 2009, 1248, 40–58. [Google Scholar] [PubMed]
- Jordan, K.; Schmoll, H.J.; Aapro, M.S. Comparative activity of antiemetic drugs. Crit. Rev. Oncol. Hematol. 2007, 61, 162–175. [Google Scholar]
- Rudd, J.A.; Jordan, C.C.; Naylor, R.J. The action of the NK1 tachykinin receptor antagonist, CP 99,994, in antagonizing the acute and delayed emesis induced by cisplatin in the ferret. Br. J. Pharmacol. 1996, 119, 931–936. [Google Scholar] [PubMed]
- Sam, T.S.; Cheng, J.T.; Johnston, K.D.; Kan, K.K.; Ngan, M.P.; Rudd, J.A.; Wai, M.K.; Yeung, J.H. Action of 5-HT3 receptor antagonists and dexamethasone to modify cisplatin-induced emesis in Suncus murinus (house musk shrew). Eur. J. Pharmacol. 2003, 472, 135–145. [Google Scholar]
- Hesketh, P.J.; Van Belle, S.; Aapro, M.; Tattersall, F.D.; Naylor, R.J.; Hargreaves, R.; Carides, A.D.; Evans, J.K.; Horgan, K.J. Differential involvement of neurotransmitters through the time course of cisplatin-induced emesis as revealed by therapy with specific receptor antagonists. Eur. J. Cancer 2003, 39, 1074–1080. [Google Scholar]
- Milano, S.; Blower, P.; Romain, D.; Grelot, L. The piglet as a suitable animal model for studying the delayed phase of cisplatin-induced emesis. J. Pharmacol. Exp. Ther. 1995, 274, 951–961. [Google Scholar]
- Rudd, J.A.; Tse, J.Y.H.; Wai, M.K. Cisplatin-induced emesis in the cat: effect of granisetron and dexamethasone. Eur. J. Pharmacol. 2000, 391, 145–150. [Google Scholar]
- Minami, M.; Endo, T.; Hirafuji, M.; Hamaue, N.; Liu, Y.; Hiroshige, T.; Nemoto, M.; Saito, H.; Yoshioka, M. Pharmacological aspects of anticancer drug-induced emesis with emphasis on serotonin release and vagal nerve activity. Pharmacol. Ther. 2003, 99, 149–165. [Google Scholar]
- Hansen, M.B; Witte, A.B. The role of serotonin in intestinal luminal sensing and secretion. Acta Physiol. (Oxf) 2008, 193, 311–323. [Google Scholar] [PubMed]
- Sjolund, K.; Sanden, G.; Hakanson, R.; Sundler, F. Endocrine cells in human intestine: an immunocytochemical study. Gastroenterology 1983, 85, 1120–1130. [Google Scholar]
- Fukui, H.; Yamamoto, M.; Ando, T.; Sasaki, S.; Sato, S. Increase in serotonin levels in the dog ileum and blood by cisplatin as measured by microdialysis. Neuropharmacology 1993, 32, 959–968. [Google Scholar]
- Gershon, M.D. 5-HT (serotonin) physiology and related drugs. Curr. Opin. Gastroenterol. 2000, 16, 113–120. [Google Scholar]
- Li, Z.S.; Pham, T.D.; Tamir, H.; Chen, J.J.; Gershon, M.D. Enteric dopaminergic neurons: definition, developmental lineage, and effects of extrinsic denervation. J. Neurosci. 2004, 24, 1330–1339. [Google Scholar] [PubMed]
- Holzer, P.; Holzer-Petsche, U. Tachykinins in the gut. Part I. Expression, release and motor function. Pharmacol. Ther. 1997, 73, 173–217. [Google Scholar] [CrossRef] [PubMed]
- Minami, M.; Endo, T.; Yokota, H.; Ogawa, T.; Nemoto, M.; Hamaue, N.; Hirafuji, M.; Yoshioka, M.; Nagahisa, A.; Andrews, P.L. Effects of CP-99, 994, a tachykinin NK(1) receptor antagonist, on abdominal afferent vagal activity in ferrets: evidence for involvement of NK(1) and 5-HT(3) receptors. Eur. J. Pharmacol. 2001, 428, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.X.; Zhu, X.Y.; Owyang, C.; Li, Y. Intestinal serotonin acts as a paracrine substance to mediate vagal signal transmission evoked by luminal factors in the rat. J. Physiol. 2001, 530, 431–442. [Google Scholar]
- Cordoba-Rodriguez, R.; Moore, K.A.; Kao, J.P.; Weinreich, D. Calcium regulation of a slow post-spike hyperpolarization in vagal afferent neurons. Proc. Natl. Acad. Sci. USA 1999, 96, 7650–7657. [Google Scholar]
- Munoz, N.M.; Shioya, T.; Murphy, T.M.; Primack, S.; Dame, C.; Sands, M.F.; Leff, A.R. Potentiation of vagal contractile response by thromboxane mimetic U-46619. J. App. Physiol. 1986, 61, 1173–1179. [Google Scholar]
- Rudd, J.A.; Qian, Y.; Tsui, K.K.; Jones, R.L. Non-prostanoid prostacyclin mimetics as neuronal stimulants in the rat: comparison of vagus nerve and NANC innervation of the colon. Br. J. Pharmacol. 2000, 129, 782–790. [Google Scholar]
- Matsumoto, S.; Ikeda, M.; Yoshida, S.; Tanimoto, T.; Takeda, M.; Nasu, M. Prostaglandin E2-induced modification of tetrodotoxin-resistant Na+ currents involves activation of both EP2 and EP4 receptors in neonatal rat nodose ganglion neurons. Br. J. Pharmacol. 2005, 145, 503–513. [Google Scholar]
- Nakamura, K.; Kaneko, T.; Yamashita, Y.; Hasegawa, H.; Katoh, H.; Negishi, M. Immunohistochemical localization of prostaglandin EP3 receptor in the rat nervous system. J. Comp. Neurol. 2000, 421, 543–569. [Google Scholar]
- Kummer, W.; Bachmann, S.; Neuhuber, W.L.; Hanze, J.; Lang, R.E. Tyrosine-hydroxylase-containing vagal afferent neurons in the rat nodose ganglion are independent from neuropeptide-Y-containing populations and project to esophagus and stomach. Cell Tissue Res. 1993, 271, 135–144. [Google Scholar]
- Lawrence, A.J.; Krstew, E.; Jarrott, B. Functional dopamine D2 receptors on rat vagal afferent neurons. Br. J. Pharmacol. 1995, 114, 1329–1334. [Google Scholar]
- Minami, M.; Nemoto, M.; Endo, T.; Hamaue, N.; Kohno, Y. Effects of talipexole on emesis-related changes in abdominal afferent vagal activity and ileal serotonin metabolism in rats. Res. Commun. Mol. Pathol. Pharmacol. 1997, 95, 67–82. [Google Scholar]
- Darmani, N.A.; Wang, Y.; Abad, J.; Ray, A.P.; Thrush, G.R.; Ramirez, J. Utilization of the least shrew as a rapid and selective screening model for the antiemetic potential and brain penetration of substance P and NK1 receptor antagonists. Brain Res. 2008, 1214, 58–72. [Google Scholar] [CrossRef] [PubMed]
- Arluison, M.; Derer, P. Forebrain connections of the rat paraventricular thalamic nucleus as demonstrated using the carbocyanide dye DiI. Neurobiology (Bp) 1993, 1, 337–350. [Google Scholar] [PubMed]
- Browning, K.N.; Travagli, R.A. Short-term receptor trafficking in the dorsal vagal complex: an overview. Auton. Neurosci. 2006, 126-127, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Hyde, T.M.; Knable, M.B.; Murray, A.M. Distribution of dopamine D1-D4 receptor subtypes in human dorsal vagal complex. Synapse 1996, 24, 224–232. [Google Scholar]
- Ito, H.; Seki, M. Ascending projections from the area postrema and the nucleus of the solitary tract of Suncus murinus: anterograde tracing study using Phaseolus vulgaris leucoagglutinin. Okajimas Folia Anat. Jpn. 1998, 75, 9–31. [Google Scholar]
- Koga, T.; Fukuda, H. Neurons in the nucleus of the solitary tract mediating inputs from emetic vagal afferents and the area postrema to the pattern generator for the emetic act in dog. Neurosci. Res. 1992, 14, 166–179. [Google Scholar]
- McRitchie, D.A.; Töurk, I. Distribution of substance P-like immunoreactive neurons and terminals throughout the nucleus of the solitary tract in the human brainstem. J. Comp. Neurol. 1994, 343, 83–101. [Google Scholar]
- Miller, A.D.; Nonaka, S.; Jakus, J. Brain areas essential or non-essential for emesis. Brain Res. 1994, 647, 255–264. [Google Scholar]
- Ricardo, J.A.; Koh, E.T. Anatomical evidence of direct projections from the nucleus of the solitary tract to the hypothalamus, amygdala, and other forebrain structures in the rat. Brain Res. 1978, 153, 1–26. [Google Scholar] [PubMed]
- Travagli, R.A.; Hermann, G.E.; Browning, K.N.; Rogers, R.C. Brainstem circuits regulating gastric function. Ann. Rev. Physiol. 2006, 68, 279–305. [Google Scholar]
- Onishi, T.; Mori, T.; Yanagihara, M.; Furukawa, N.; Fukuda, H. Similarities of the neuronal circuit for the induction of fictive vomiting between ferrets and dogs. Auton. Neurosci. 2007, 136, 20–30. [Google Scholar]
- Hornby, P.J. Central neurocircuitry associated with emesis. Am. J. Med. 2001, 111 (Suppl. 8A), 106S–112S. [Google Scholar] [CrossRef] [PubMed]
- Hornby, P.J.; Abrahams, T.P. Central control of lower esophageal sphincter relaxation. Am. J. Med. 2000, 108, 90S–98S. [Google Scholar]
- Urayama, Y.; Yamada, Y.; nakamura, E.; Koga, T.; Fukuda, H. Electrical and chemical stimulation of the nucleus raphe magnus inhibits induction of retching by afferent vagal fibers. Auton. Neurosci. 2010, 152, 35–40. [Google Scholar]
- Partosoedarso, E.R.; Abrahams, T.P.; Scullion, R.T.; Moerschbaecher, J.M.; Hornby, P.J. Cannabinoid1 receptor in the dorsal vagal complex modulates lower oesophageal sphincter relaxation in ferrets. J. Physiol. 2003, 550, 149–158. [Google Scholar] [PubMed]
- Browning, K.N.; Travagli, R.A. Characterization of the in vitro effects of 5-hydroxytryptamine (5-HT) on identified neurones of the rat dorsal motor nucleus of the vagus (DMV). Br. J. Pharmacol. 1999, 128, 1307–1315. [Google Scholar] [PubMed]
- Carpenter, D.O.; Briggs, D.B.; Strominger, N. Behavioral and electrophysiological studies of peptide-induced emesis in dogs. Fed. Proc. 1984, 43, 2952–2954. [Google Scholar] [PubMed]
- Di Marzo, V. Endocannabinoids: synthesis and degradation. Endocannabinoids: synthesis and degradation. Rev. Physiol. Biochem. Pharmacol. 2008, 160, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Batkai, S.; Kunos, G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol. Rev. 2006, 58, 389–462. [Google Scholar]
- Van Sickle, M.D.; Duncan, M.; Kingsley, P.J.; Mouihate, A.; Urbani, P.; Mackie, K.; Stella, N.; Makriyannis, A.; Piomelli, D.; Davison, J.S.; Marnett, L.J.; Di Marzo, V.; Pittman, Q.J.; Patel, K.D.; Sharkey, K.A. Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science 2005, 310, 329–332. [Google Scholar]
- Chebolu, S.; Wang, Y.; Ray, A.P.; Darmani, N.A. Pranlukast prevents cysteinyl leukotriene-induced emesis in the least shrew (Cryptotis parva). Eur. J. Pharmacol. 2010, 628, 195–201. [Google Scholar]
- Izzo, A.A.; Sharkey, K.A. Cannabinoids and the gut: new developments and emerging concepts. Pharmacol. Therap. 2010, 126, 21–38. [Google Scholar]
- Duncan, M.; Davison, J.S.; Sharkey, K.A. Review article: endocannabinoids and their receptors in the enteric nervous system. Aliment. Pharmacol. Ther. 2005, 22, 667–683. [Google Scholar]
- Duncan, M.; Thomas, A.D.; Cluny, N.L.; Patel, A.; Patel, K.D.; Lutz, B.; Piomelli, D.; Alexander, S.P.; Sharkey, K.A. Distribution and function of monoacylglycerol lipase in the gastrointestinal tract. Am. J. Physiol. Gastrointes. LiverPhysiol. 2008, 295, G1255–G1265. [Google Scholar]
- Bisogno, T.; Berrendero, F.; Ambrosino, G.; Cebeira, M.; Ramos, J.A.; Fernandez-Ruiz, J.J.; Di Marzo, V. Brain regional distribution of endocannabinoids: implications for their biosynthesis and biological function. Biochem. Biophys. Res. Commun. 1999, 256, 377–380. [Google Scholar]
- Seagard, J.L.; Dean, C.; Patel, S.; Rademacher, D.J.; Hopp, F.A.; Schmeling, W.T.; Hillard, C.J. Anadamide content and interaction of endocannabinoid/GABA modulatory effects in the NTS on baroreflex-evoked sympathoinhibition. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H992–H1000. [Google Scholar]
- Darmani, N.A.; Sim-Selley, L.J.; Martin, B.R.; Janoyan, J.J.; Crim, J.L.; Parekh, B.; Breivogel, C.S. Antiemetic and motor-depressive actions of CP55,940: cannabinoid CB1 receptor characterization, distribution, and G-protein activation. Eur. J. Pharmacol. 2003, 459, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Herkenham, M.; Lynn, A.B.; Johnson, M.R.; Melvin, L.S.; de Costa, B.R.; Rice, K.C. Characterization and localization of cannabinoid receptors in rat brain: a quantitative in vitro autoradiographic study. J. Neurosci 1991, 11, 563–583. [Google Scholar] [PubMed]
- Mailleux, P.; Parmentier, M.; Vanderhaeghen, J.J. Distribution of cannabinoid receptor messenger RNA in the human brain: an in situ hybridization histochemistry with oligonucleotides. Neurosci. Lett. 1992, 143, 200–204. [Google Scholar]
- Derbenev, A.V.; Stuart, T.C.; Smith, B.N. Cannabinoids suppress synaptic input to neurones of the rat dorsal motor nucleus of the vagus nerve. J. Physiol. 2004, 559, 923–938. [Google Scholar] [PubMed]
- Shiroshita, Y.; Koga, T.; Fukuda, H. Capsaicin in the 4th ventricle abolishes retching and transmission of emetic vagal afferents to solitary nucleus neurons. Eur. J. Pharmacol. 1997, 339, 183–192. [Google Scholar]
- Burdyga, G.; Lal, S.; Varro, A.; Dimaline, R.; Thompson, D.G.; Dockray, G.J. Expression of cannabinoid CB1 receptors by vagal afferent neurons is inhibited by cholecystokinin. J. Neurosci. 2004, 24, 2708–2715. [Google Scholar]
- Krowwicki, Z.K.; Moerschbaecher, J.M.; Winsauer, P.J.; Digvalli, S.V.; Hornby, P.J. Delta9-tetrahydrobannabinol inhibits gastric motility in the rat through cannabinoidCB1 receptors. Eur. J. Pharmacol. 1999, 371, 187–196. [Google Scholar]
- Anand, U.; Otto, W.R.; Sanchez-Herrera, D.; Facer, P.; Yiangou, Y.; Korchev, Y.; Birch, R.; Benham, C.; Bountra, C.; Chessell, I.P.; Anand, P. Cannabinoid receptor CB2 localisation and agonist-mediated inhibition of capsaicin responses in human sensory neurons. Pain 2008, 138, 667–680. [Google Scholar]
- Andrews, P.L.; Bhandari, P. Resinferatoxin, an ultrapotent capsaicin analogue, has anti-emetic properties in the ferret. Neuropharmacology 1993, 32, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Rudd, J.A.; Wai, M.K. Genital grooming and emesis induced by vanilloids in Suncus murinus, the house musk shrew. Eur. J. Pharmacol. 2001, 422, 185–195. [Google Scholar]
- Yamakuni, H.; Sawai-Nakayama, H.; Imazumi, K.; Maeda, Y.; Matsuo, M.; Manda, T.; Mutoh, S. Resiniferatoxin antagonizes cisplatin-induced emesis in dogs and ferrets. Eur. J. Pharmacol. 2002, 442, 273–278. [Google Scholar]
- Schicho, R.; Florian, W.; Liebmann, I.; Holzer, P.; Lippe, I.T. Increased expression of TRPV1 receptor in dorsal root ganglia by acid insult of the rat gastric mucosa. Eur. J. Neurosci. 2004, 19, 1811–1818. [Google Scholar]
- Szallasi, A.; Blumberg, P.M. Vanilloid (Capsaicin) receptors and mechanisms. Pharmacol. Rev. 1999, 51, 159–212. [Google Scholar]
- Landi, M.; Croci, T.; Rinaldi-Carmona, M.; Maffrand, J.P.; Le Fur, G.; Manara, L. Modulation of gastric emptying and gastrointestinal transit in rats through intestinal cannabinoid CB(1) receptors. Eur. J. Pharmacol. 2002, 450, 77–83. [Google Scholar]
- Darmani, N.A.; Johnson, J.C. Central and peripheral mechanisms contribute to the antiemetic actions of delta-9-tetrahydrocannabinol against 5-hydroxytryptophan-induced emesis. Eur. J. Pharmacol. 2004, 488, 201–212. [Google Scholar]
- Lehmann, A.; Blackshaw, L.A.; Branden, L.; Carlsson, A.; Jensen, J.; Nygren, E.; Smid, S.D. Cannabinoid receptor agonism inhibits transient lower esophageal sphincter relaxations and reflux in dogs. Gastroenterology 2002, 123, 1129–1134. [Google Scholar]
- Duncan, M.; Mouihate, A.; Mackie, K.; Keenan, C.M.; Buckley, N.E.; Davison, J.S.; Patel, K.D.; Pittman, Q.J.; Sharkey, K.A. Cannabinoid CB2 receptors in the enteric nervous system modulate gastrointestinal contractility in lipopolysaccharide-treated rats. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 295, G78–G87. [Google Scholar]
- Izzo, A.A. The cannabinoid CB(2) receptor: a good friend in the gut. Neurogastroenterol. Motil. 2007, 19, 704–708. [Google Scholar]
- Storr, M.A.; Keenan, C.M.; Emmerdinger, D.; Zhang, H.; Yuce, B.; Sibaev, A.; Massa, F.; Buckley, N.E.; Lutz, B.; Goke, B.; Brand, S.; Patel, K.D.; Sharkey, K.A. Targeting endocannabinoid degradation protects against experimental colitis in mice: involvement of CB1 and CB2 receptors. J. Mol. Med. 2008, 86, 925–936. [Google Scholar]
- Hillsley, K.; McCaul, C.; Aerssens, J.; Peeters, P.J.; Gijsen, H.; Moechars, D.; Coulie, B.; Grundy, D.; Stead, R.H. Activation of the cannabinoid 2 (CB2) receptor inhibits murine mesenteric afferent nerve activity. Neurogastroenterol. Motil. 2007, 19, 769–777. [Google Scholar]
- Starowicz, K.; Nigam, S.; Di Marzo, V. Biochemistry and pharmacology of endovanilloids. Pharmacol. Ther. 2007, 114, 13–33. [Google Scholar]
- Coutts, A.A.; Izzo, A.A. The gastrointestinal pharmacology of cannabinoids: an update. Curr. Opin. Pharmacol. 2004, 4, 572–579. [Google Scholar]
- McVey, D.C.; Schmid, P.C.; Schmid, H.H.; Vigna, S.R. Endocannabinoids induce ileitis in rats via the capsaicin receptor (VR1). J. Pharmacol. Exp. Ther. 2003, 304, 713–722. [Google Scholar]
- Darmani, N.A. Endocannabinoids and gastrointestinal function. In The Brain and Body's Marijuana and Beyond; Onaivi, E.S., Sugiura, T., Di Marzo, V., Eds.; CRC: Boca Raton, USA, 2005; pp. 393–418. [Google Scholar]
- Pinto, L.; Capasso, R.; Di Carlo, G.; Izzo, A.A. Prostaglandins Leukot. Essent. Fatty Acids 2002, 66, 333–341. [Google Scholar]
- Darmani, N.A.; McClanahan, B.A.; Trinh, C.; Petrosino, S.; Valenti, M.; Di Marzo, V. Cisplatin increases brain 2-arachidonoylglycerol (2-AG) and concomitantly reduces intestinal 2-AG and anandamide levels in the least shrew. Neuropharmacology 2005, 49, 502–513. [Google Scholar]
- Storr, M.A.; Sharkey, K.A. The endocannabinoid system and gut-brain signaling. Curr. Opin. Pharmacol. 2007, 7, 575–582. [Google Scholar]
- Machado Rocha, F.C.; Stefano, S.C.; De Cassia Haiek, R.; Rosa Oliveira, L.M.; Da Silveira, D.X. Therapeutic use of Cannabis sativa on chemotherapy-induced nausea and vomiting among cancer patients: systematic review and meta-analysis (Engl). Eur. J. Cancer Care 2008, 17, 431–443. [Google Scholar]
- Tramer, M.R.; Carroll, D.; Campbell, F.A.; Reynolds, D.J.; Moore, R.A.; McQuay, H.J. Cannabinoids for control of chemotherapy induced nausea and vomiting: quantitative systematic review. BMJ 2001, 323, 16–21. [Google Scholar]
- Wang, Y.; McClanahan, B.A.; Darmani, N.A. Interactions of Δ9 – THC with classically used antiemetics against the acute phase of cispaltin-induced emesis. Soc. Neurocience Abs. 2006. No.766.3.. [Google Scholar]
- Van Gaal, L.F.; Rissanen, A.M.; Scheen, A.J.; Ziegler, O.; Rossner, S. Effects of the cannabinoid-1 receptor blocker rimonabant on weight reduction and cardiovascular risk factors in overweight patients: 1-year experience from the RIO-Europe study. Lancet 2005, 365, 1389–1397. [Google Scholar]
- Lichtman, A.H.; Wiley, J.L.; LaVecchia, K.L.; Neviaser, S.T.; Arthur, D.B.; Wilson, D.M.; Martin, B.R. Effects of SR 141716A after acute or chronic cannabinoid administration in dogs. Eur. J. Pharmacol. 1998, 357, 139–148. [Google Scholar]
- Salamone, J.D.; McLaughlin, P.J.; Sink, K.; Makriyannis, A.; Parker, L.A. Cannabinoid CB1 receptor inverse agonists and neutral antagonists: effects on food intake, food-reinforced behavior and food aversions. Phys. Behav. 2007, 91, 383–388. [Google Scholar] [CrossRef]
- McLaughlin, P.J.; Winston, K.M.; Limbeer, C.K.; Parker, L.A.; Makriyamis, A; Salamone, J.D. The cannabinoid CB1 antagonist AM251 produces food avoidance and behaviors associated with nausea but does not impair feeding efficiency in rats. Psychopharmacology 2005, 180, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Darmani, N.A.; Janoyan, J.J.; Kumar, N.; Crim, J.L. Behaviorally active doses of the CB1 receptor antagonist SR 141716A increase brain serotonin and dopamine levels and turnover. Pharmacol. Biochem. Behav. 2003, 75, 777–787. [Google Scholar]
- Schlicker, E.; Kathman, M. Modulation of transmitter release via presynaptic cannabinoid receptors. Trends Pharmacol. Sci. 2001, 22, 565–572. [Google Scholar]
- Darmani, N.A. The cannabinoid CB1 receptor antagonist SR141716A reverses the antiemetic and motor depressant actions of WIN55, 212-2. Eur. J. Pharmacol. 2001, 430, 49–58. [Google Scholar]
- Darmani, N.A. Delta-9-tetrahydrocannabinol differentially suppresses cisplatin-induced emesis and indices of motor function via cannabinoid CB(1) receptors in the least shrew. Pharmacol. Biochem. Behav. 2001, 69, 239–249. [Google Scholar]
- London, S.W.; McCarthy, L.E.; Borison, H.L. Suppression of cancer-induced vomiting in the cat by nabilone, a synthetic cannabinoid. Proc. Soc. Exp. Biol. Med. 1979, 160, 437–440. [Google Scholar]
- Kwiatkowska, M.; Parker, L.A.; Burton, P.; Mechoulam, R. A comparative analysis of the potential of cannabinoids and ondansetron to suppress cisplatin-induced emesis in the Suncus murinus (house musk shrew). Psychopharmacology (Berl) 2004, 174, 254–259. [Google Scholar] [PubMed]
- Ferrari, F.; Ottani, A.; Giuliani, D. Cannabimimetic activity in rats and pigeons of Hu 210, a potent antiemetic drug. Pharmacol. Biochem. Behav. 1999, 62, 75–80. [Google Scholar]
- Abrahamov, A.; Abrahamov, A.; Mechoulam, R. An efficient new cannabinoid antiemetic in pediatric oncology. Life Sci. 1995, 56, 2097–2102. [Google Scholar] [PubMed]
- Darmani, N.A.; Crim, J.; McClanahan, B.A.; Wang, Y. Δ9 – THC prevents emesis produced byl-dopa in the least shrew. Soc. Neuroscience Abs. 2006. Number 765.26.. [Google Scholar]
- Darmani, N.A.; Crim, J.L. Delta-9-tetrahydrocannabinol differentially suppresses emesis versus enhanced locomotor activity produced by chemically diverse dopamine D2/D3 receptor agonists in the least shrew (Cryptotis parva). Pharmacol. Biochem. Behav. 2005, 80, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Darmani, N.A.; Janoyan, J.J.; Crim, J.; Ramirez, J. Receptor mechanism and antiemetic activity of structurally-diverse cannabinoids against radiation-induced emesis in the least shrew. Eur. J. Pharmacol. 2007, 563, 187–196. [Google Scholar]
- Darmani, N.A.; Gerdes, D.; Trinh, C. Structurally Diverse Cannabinoids Prevent Substance P-induced Emesis via Cannabinoid CB1 Receptor in Cryptotis Parva. In The 15th Annual Symposium on the Cannabinoids; Clearwater, FL, USA: 2005.
- Simoneau, II.; Hamza, M.S.; Mata, H.P.; Siegel, E.M.; Vanderah, T.W.; Porreca, F.; Makriyannis, A.; Malan, T.P., Jr. The cannabinoid agonist WIN55,212-2 suppresses opioid-induced emesis in ferrets. Anesthesiology 2001, 94, 882–887. [Google Scholar]
- Cluny, N.L.; Naylor, R.J.; Whittle, B.A.; Javid, F.A. The effects of cannabidiol and tetrahydrocannabinol on motion-induced emesis in Suncus murinus. Basic Clin. Pharmacol. Toxicol. 2008, 103, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.L.; Zhu, G.; Mori, F.; Omoe, K.; Okada, M.; Wakabayashi, K.; Kaneko, S.; Shinagawa, K.; Nakane, A. Staphylococcal enterotoxin induces emesis through increasing serotonin release in intestine and it is downregulated by cannabinoid receptor. CelL Microbiol. 2007, 9, 2267–2277. [Google Scholar] [CrossRef] [PubMed]
- du Sert, N.P.; Rudd, J.A.; Moss, R.; Andrews, P.L.R. The delayed phase of cisplatin-induced emesis is mediated by the area postrema and not the abdominal visceral innervations in the ferret. Neurosc. Lett. 2009, 465, 16–20. [Google Scholar]
- Horn, C.C.; Ciucci, M.; Chaudhury, A. Brain Fos expression during 48 h after cisplatin treatment: neural pathways for acute and delayed visceral sickness. Auton. Neurosci. Basic Clin. 2007, 132, 44–51. [Google Scholar]
- Meiri, E.; Jhangiani, H.; Vredenburgh, J.J.; Barbato, L.M.; Carter, F.J.; Yang, H.M.; Baranowski, V. Efficacy of dronabinol alone and in combination with ondansetron versus ondansetron alone for delayed chemotherapy-induced nausea and vomiting. Curr. Med. Res. Opin. 2007, 23, 533–543. [Google Scholar]
- Wang, Y.; Ray, A.P.; McClanahan, B.A.; Darmani, N.A. The antiemetic interaction of Delta9-tetrahydrocannabinol when combined with tropisetron or dexamethasone in the least shrew. Pharmacol. Biochem. Behav. 2008, 91, 367–373. [Google Scholar] [PubMed]
- Fink, K.B.; Gothert, M. 5-HT receptor regulation of neurotransmitter release. Pharmacol. Rev. 2007, 59, 360–417. [Google Scholar]
- Barann, M.; Molderings, G.; Bruss, M.; Bonisch, H.; Urban, B.W.; Gothert, M. Direct inhibition by cannabinoids of human 5-HT3A receptors: probable involvement of an allosteric modulatory site. Br. J. Pharmacol. 2002, 137, 589–596. [Google Scholar]
- Xiong, W.; Hosoi, M.; Koo, B.; Zhang, L. Anandamide inhibition of 5-HT3A receptors varies with receptor density and desensitization. Mol. Pharmacol. 2007, 73, 314–322. [Google Scholar]
- Higa, G.M.; Auber, M.L.; Altaha, R.; Piktel, D.; Kurian, S.; Hobbs, G.; Landreth, K. 5-Hydroxyindoleacetic acid and substance P profiles in patients receiving emetogenic chemotherapy. J. Oncol. Pharm. Practice 2006, 12, 201–209. [Google Scholar]
- Ray, A.P.; Chevolu, S.; Ramirez, J.; Damani, N.A. Ablation of least shrew central NK1 receptors reduces GR73632-induced vomiting. Behav. Neurosci. 2009, 123, 701–706. [Google Scholar]
- Niranen, A.; Mattson, K. Antiemetic efficacy of nabilone and dexamethasone: a randomized study of patients with lung cancer receiving chemotherapy. Am. J. Clin. Oncol. 1987, 10, 325–329. [Google Scholar]
- Darmani, N.A.; McClanahan, B.A.; Trinh, C.; Petrosino, S; Valenti, M.; Di Marzo, V. Cisplatin increases 2-arachidonoyylglycerol (2-AG) and concomitantly reduces intestinal 2-AG and anandamide levels in the least shrew. Neuropharmacology 2005, 49, 502–513. [Google Scholar] [CrossRef] [PubMed]
- Girod, V.; Dapzol, J.; Bouvier, M.; Grelot, L. The COX inhibitors indomethacin and meloxicam exhibit anti-emetic activity against cisplatin-induced emesis in piglets. Neuropharmacology 2002, 42, 428–436. [Google Scholar]
- Wislicki, L. Systemic adverse reactions to prostaglandin F2 (PGF2 alpha, dinoprostone, prostin F2 alpha, prostalmon F). Int. J. Biol. Res. Pregnancy 1982, 3, 158–160. [Google Scholar] [PubMed]
- Wechsung, E. The involvement of prostaglandins in the inhibiting effect of endotoxin on the myoelectric activity of the gastrointestinal system in pigs. Verh K. Acad. Geneeskd. Belg. 1996, 58, 711–738. [Google Scholar]
- Gadsby, R.; Barnie-Adshead, A.; Grammatoppoulos, D.; Gadsby, P. Nausea and vomiting in pregnancy: an association between symptoms and maternal prostaglandin E2. Gynecol. Obstet. Invest. 2000, 50, 149–152. [Google Scholar]
- Jett, M.; Brinkley, W.; Neill, R.; Gemski, P.; Hunt, R. Staphylococcus aureus enterotoxin B challenge of monkeys: correlation of plasma levels of arachidonic acid cascade products with occurrence of illness. Infect. Immun. 1990, 58, 3494–3499. [Google Scholar]
- Wang, T.; Collet, J.P.; Shapiro, S.; Ware, M.A. Adverse effects of medical cannabinoids: A systematic review. CMAJ 2008, 178, 1669–1678. [Google Scholar]
- Allen, J.H.; de Moore, G.M.; Heddle, R.; Twantz, J.C. Cannabinoid hyperemesis: cyclical hyperemesis in association with chronic cannabis abuse. Gut 2004, 53, 1566–1570. [Google Scholar] [CrossRef] [PubMed]
- Darmani, N.A. Cannabinoid-induced hyperemesis: A conundrum-from clinical recognition to basic science mechanisms. Pharmaceuticals 2010, 3, 2163–2177. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an Open Access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Darmani, N.A. Mechanisms of Broad-Spectrum Antiemetic Efficacy of Cannabinoids against Chemotherapy-Induced Acute and Delayed Vomiting. Pharmaceuticals 2010, 3, 2930-2955. https://doi.org/10.3390/ph3092930
Darmani NA. Mechanisms of Broad-Spectrum Antiemetic Efficacy of Cannabinoids against Chemotherapy-Induced Acute and Delayed Vomiting. Pharmaceuticals. 2010; 3(9):2930-2955. https://doi.org/10.3390/ph3092930
Chicago/Turabian StyleDarmani, Nissar A. 2010. "Mechanisms of Broad-Spectrum Antiemetic Efficacy of Cannabinoids against Chemotherapy-Induced Acute and Delayed Vomiting" Pharmaceuticals 3, no. 9: 2930-2955. https://doi.org/10.3390/ph3092930
APA StyleDarmani, N. A. (2010). Mechanisms of Broad-Spectrum Antiemetic Efficacy of Cannabinoids against Chemotherapy-Induced Acute and Delayed Vomiting. Pharmaceuticals, 3(9), 2930-2955. https://doi.org/10.3390/ph3092930