Allosteric Modulation of Muscarinic Acetylcholine Receptors

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Role of Muscarinic Receptors in Psychiatric and Neurological Disorders
3. Therapeutic Potential of Muscarinic Allosteric Modulators
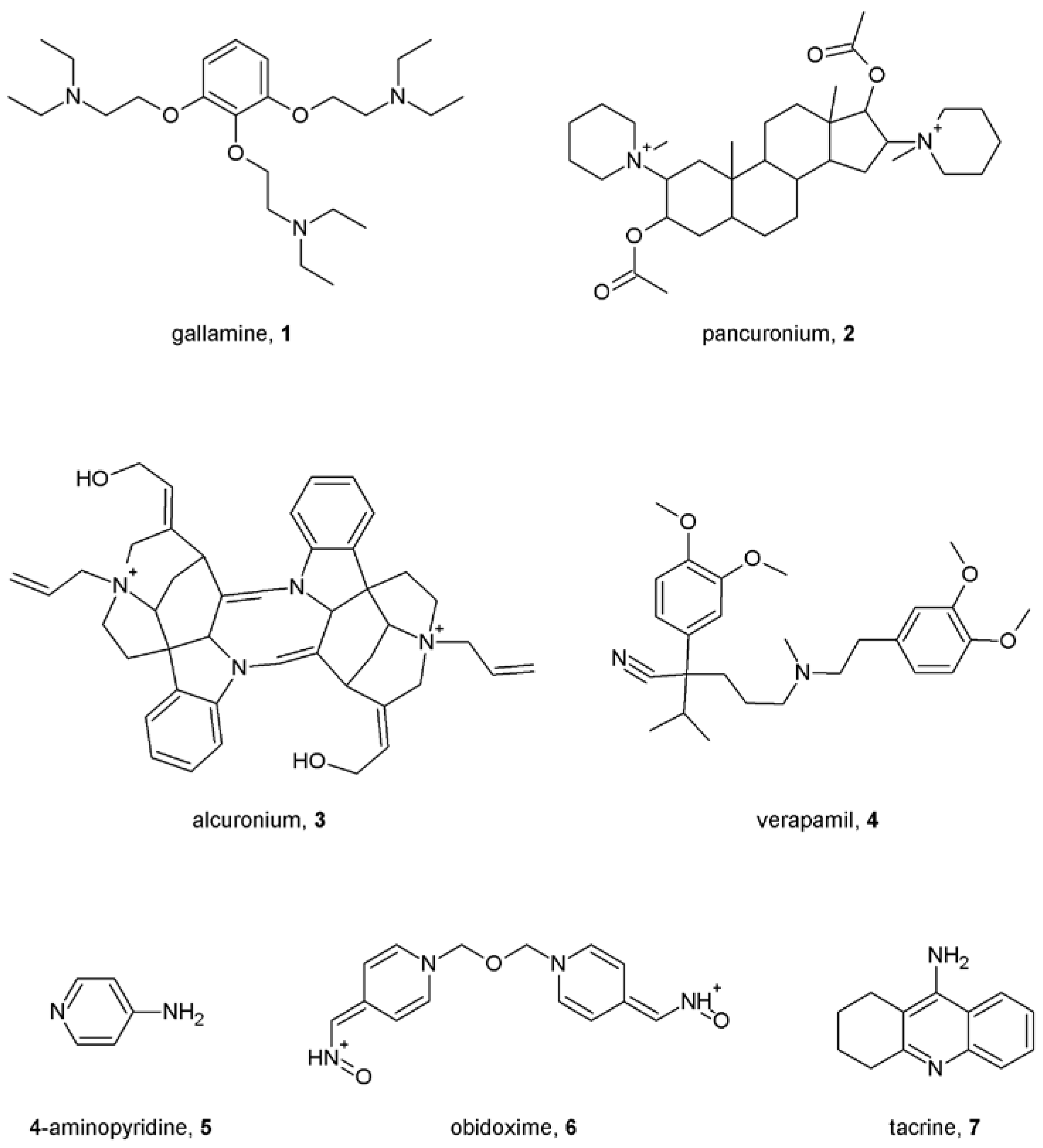
4. First Allosteric Modulators of Muscarinic Receptors
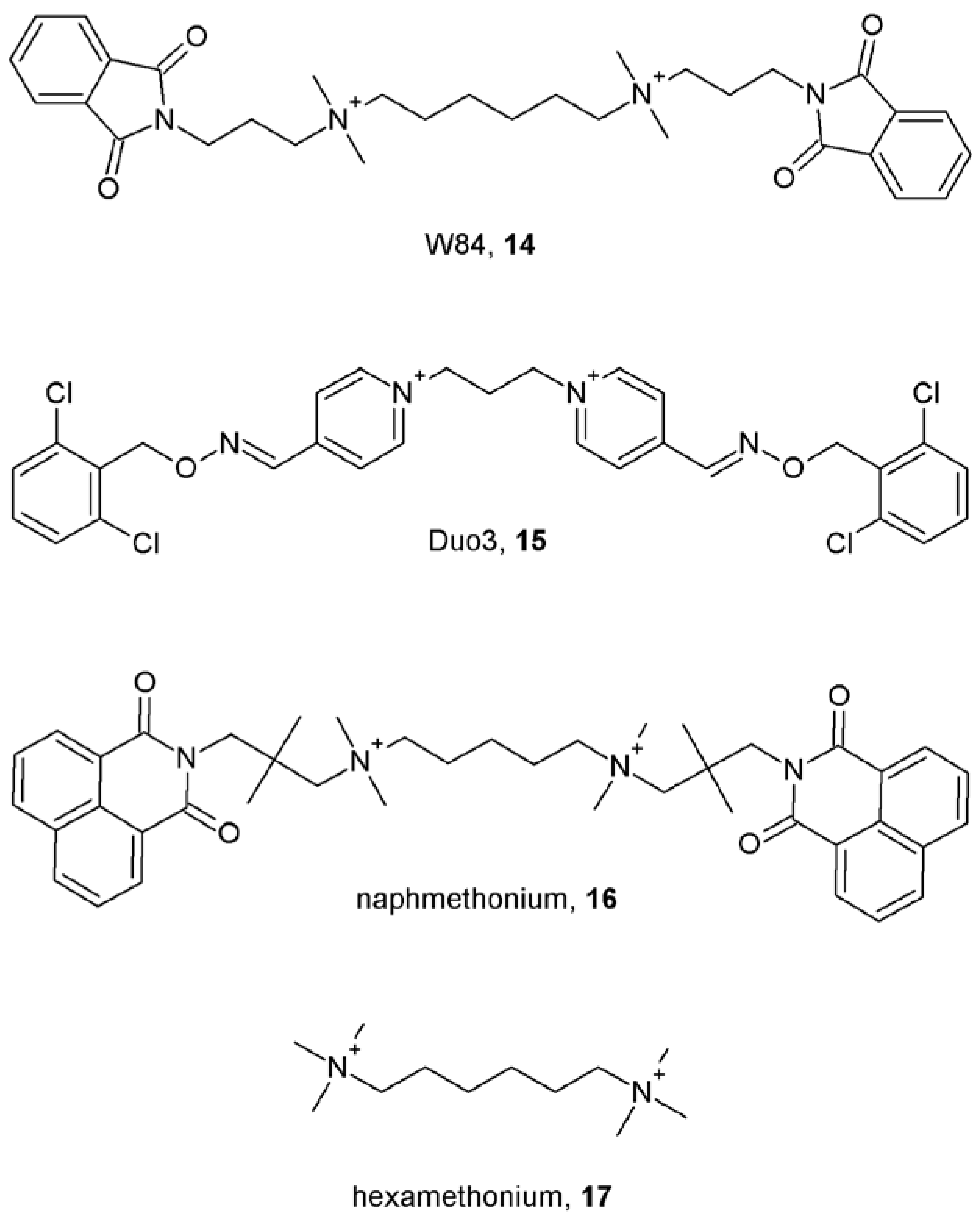
5. Nature of Allosteric Modulators

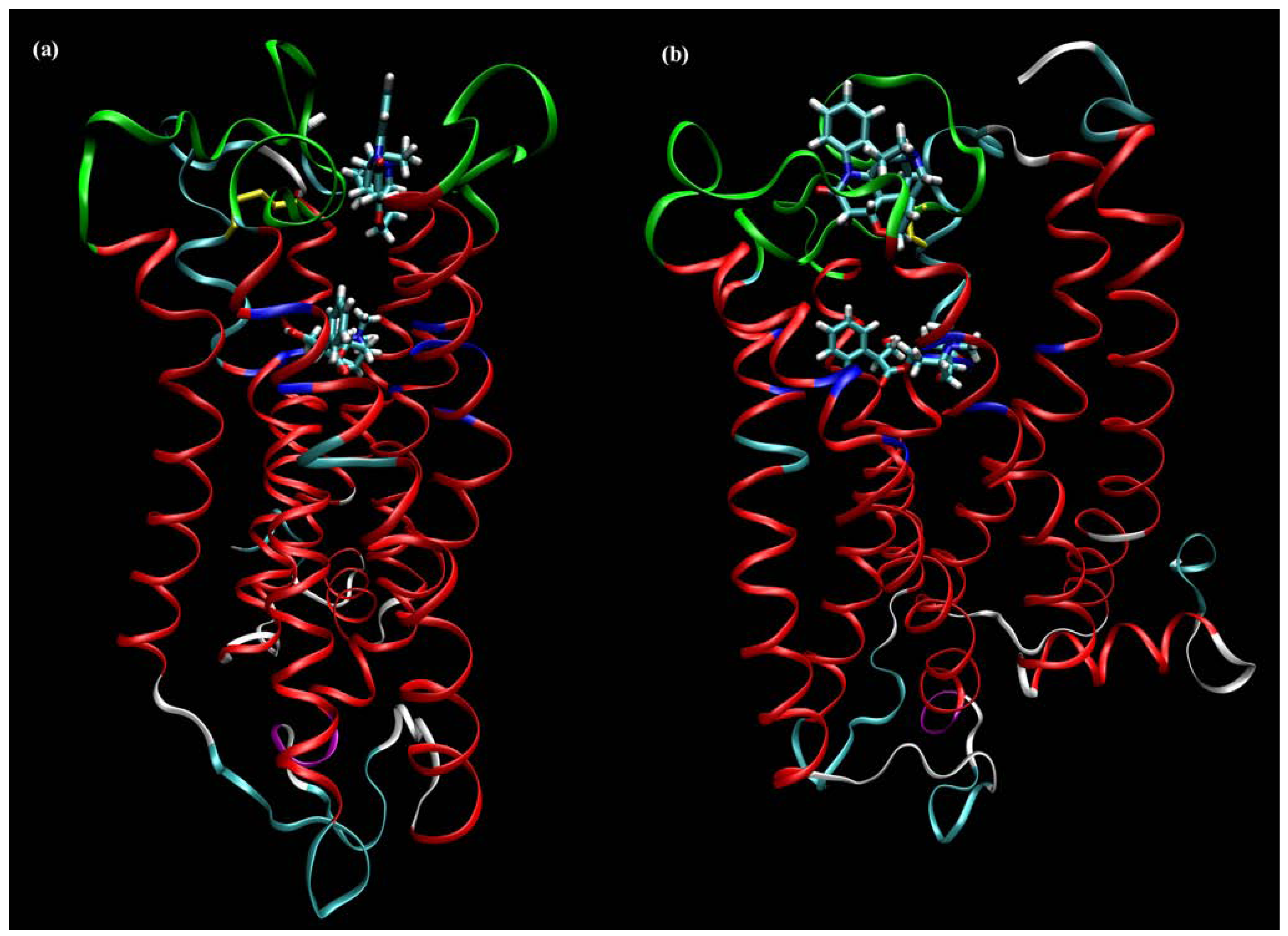
6. Binding Site of Allosteric Modulators at Muscarinic Receptors
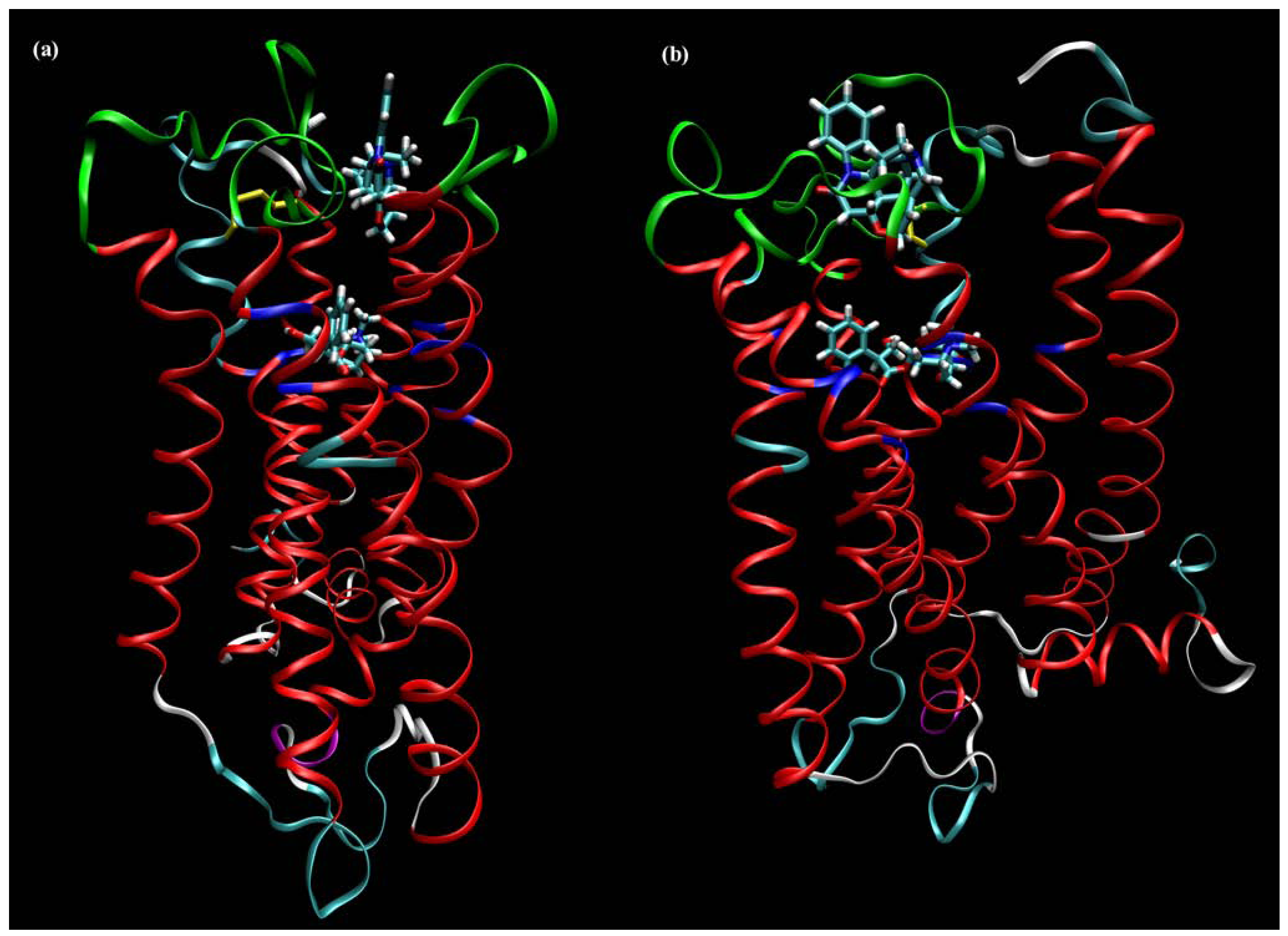
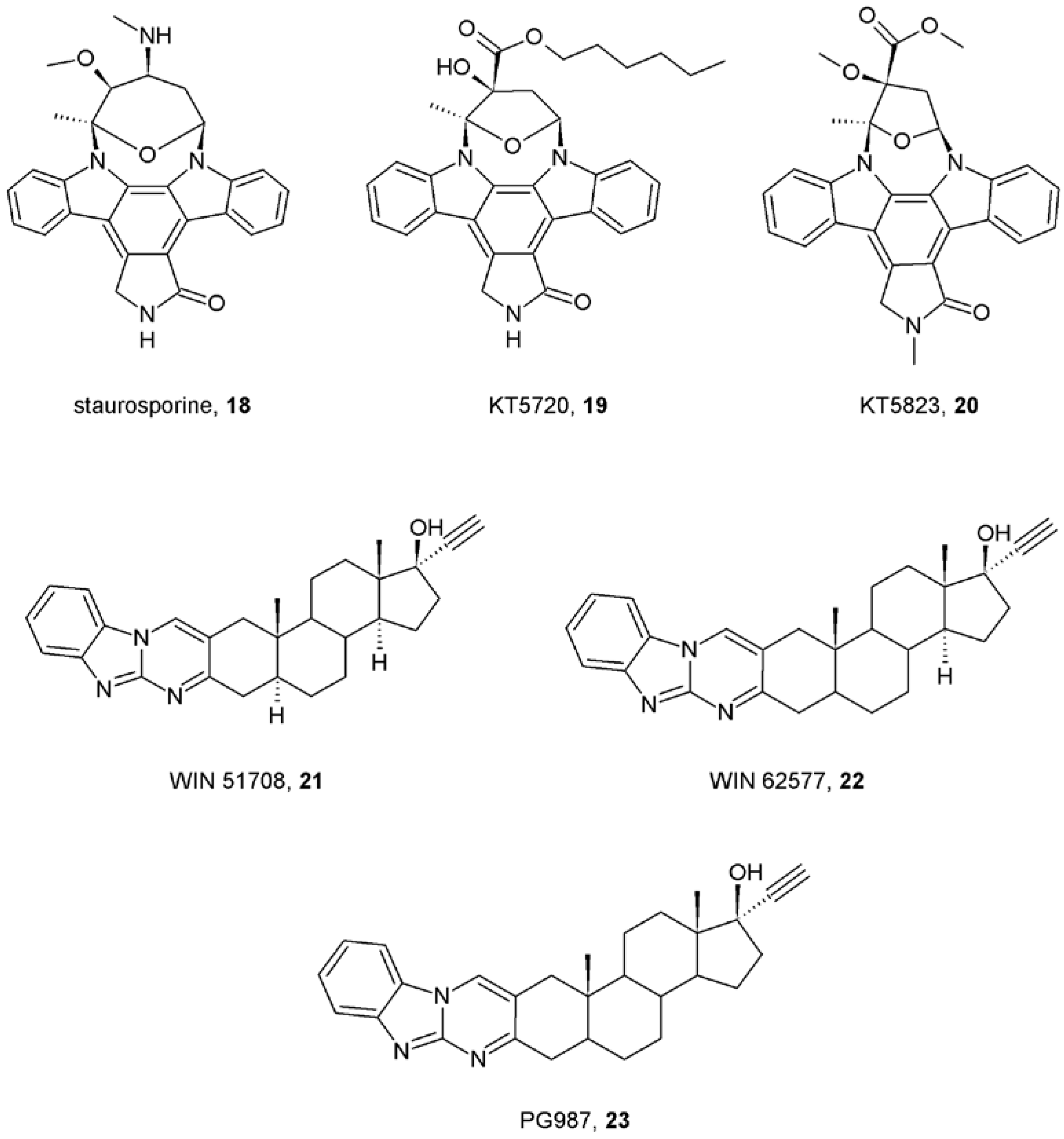
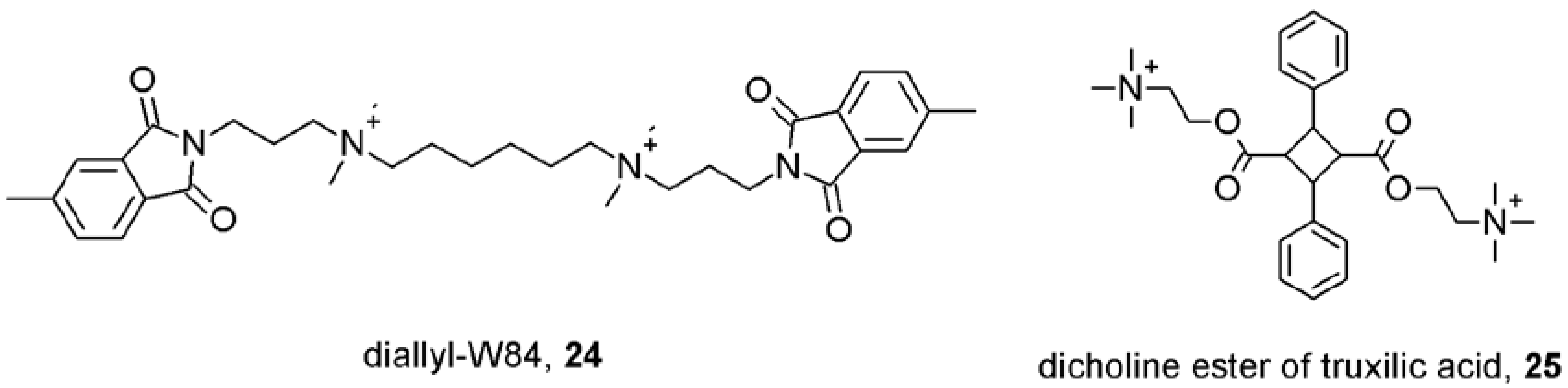
7. Mechanisms of Conformational Changes Induced by Allosteric Modulators
8. Endogenous Allosteric Modulators of Muscarinic Receptors
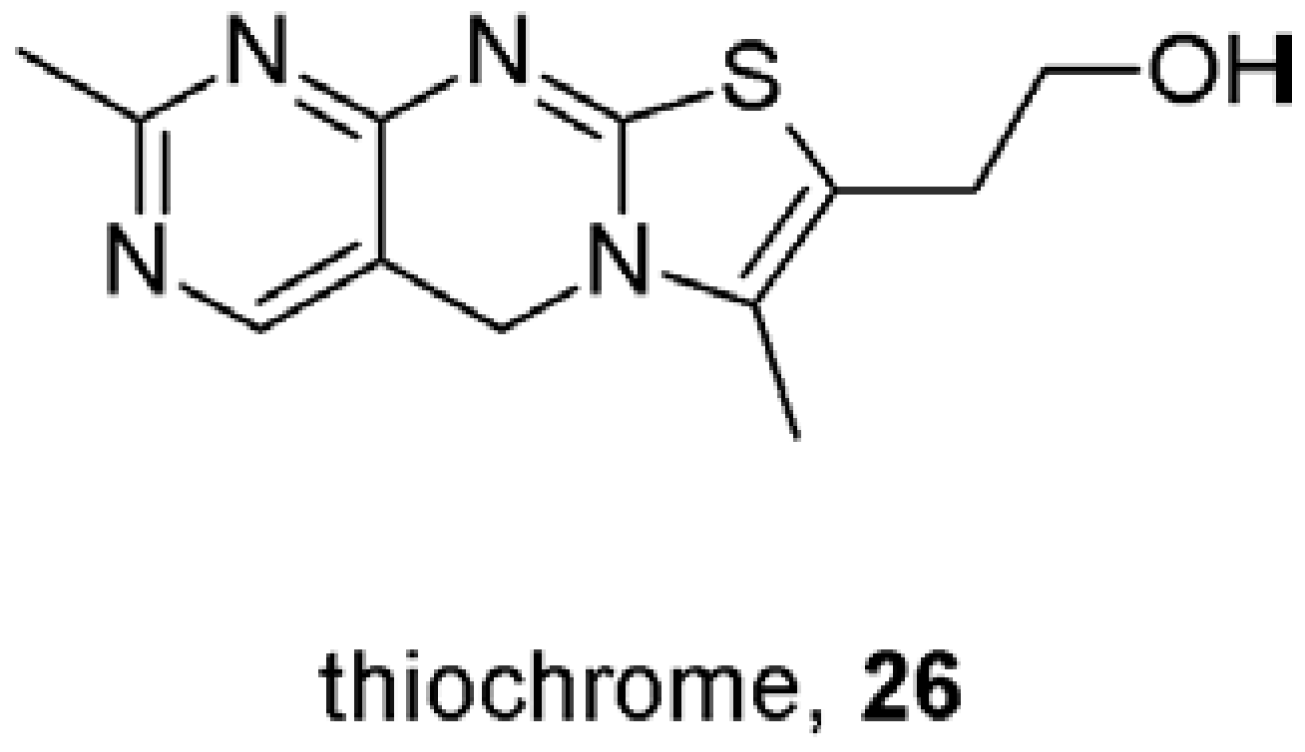
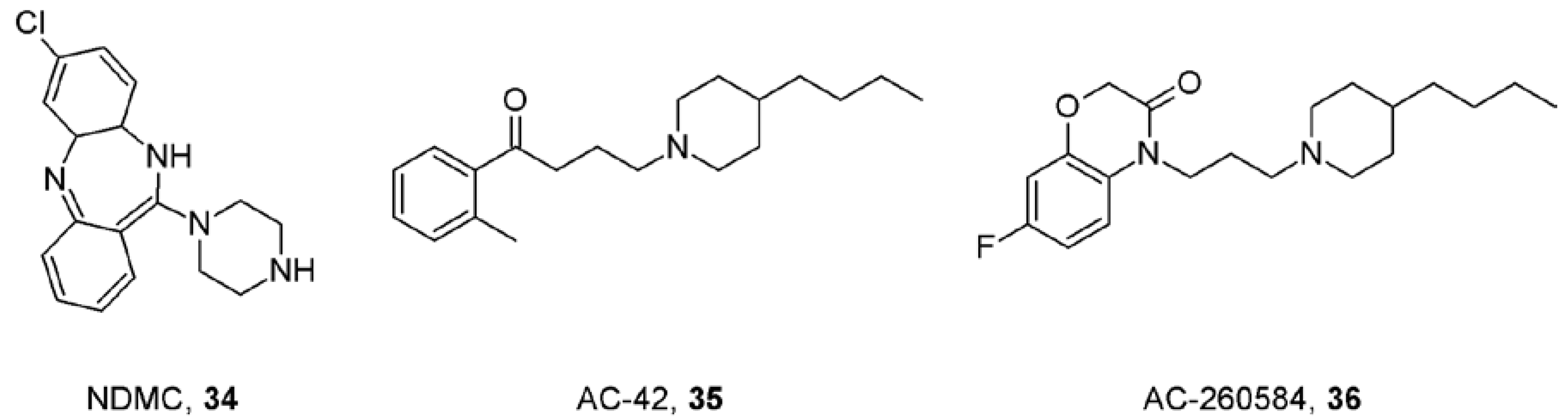
9. First Positive Allosteric Modulators of Acetylcholine
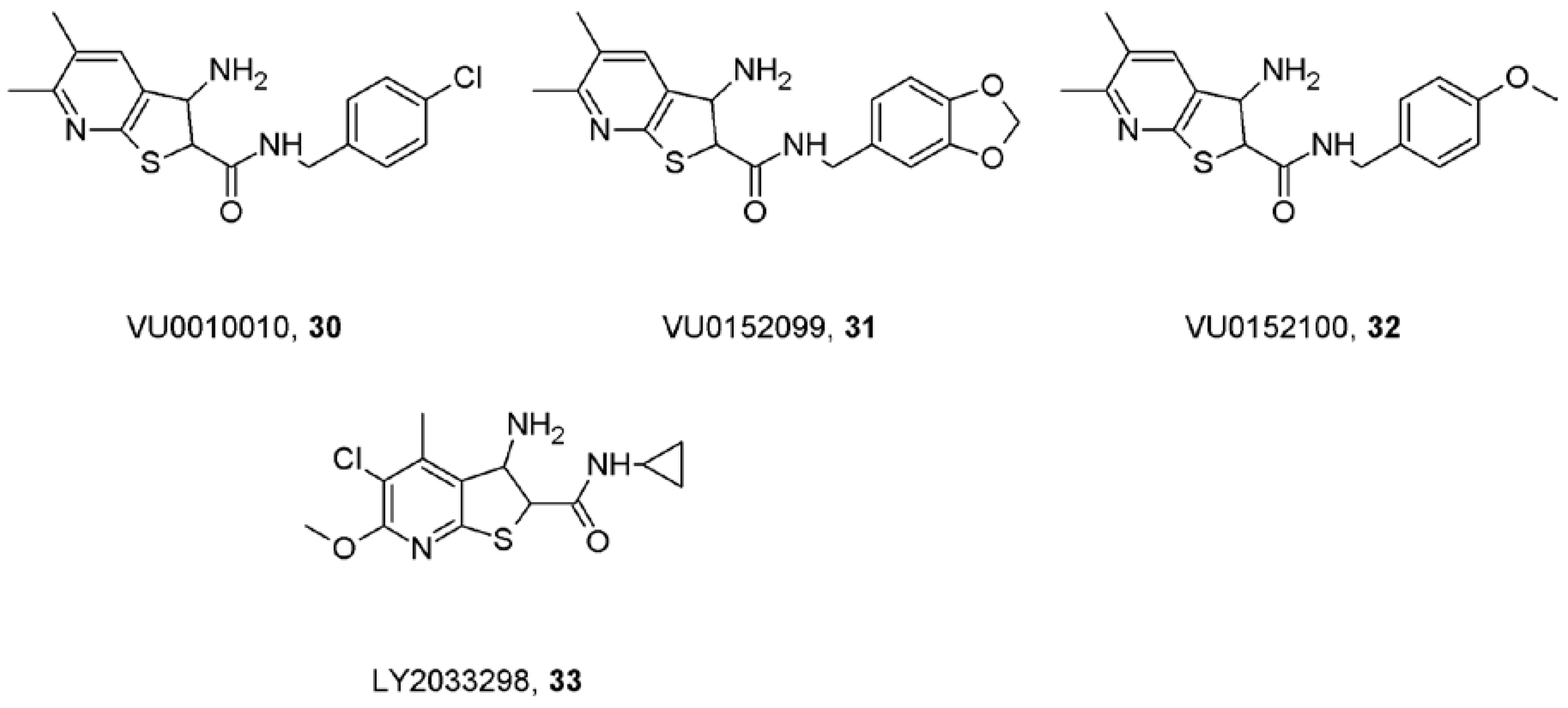
10. Truly Selective Positive Allosteric Modulators of Acetylcholine

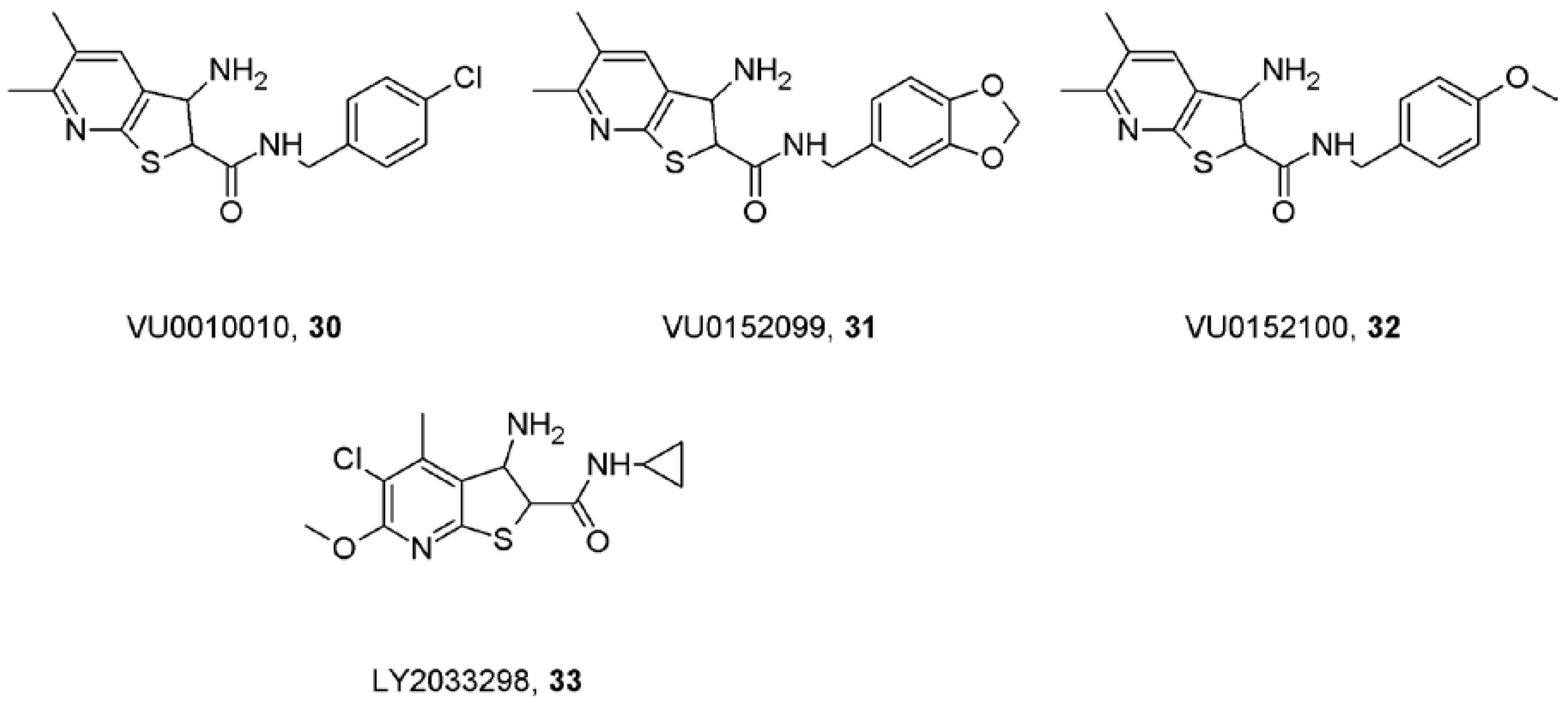
11. Caveats about Allosteric Modulators

12. Conclusions
Acknowledgements
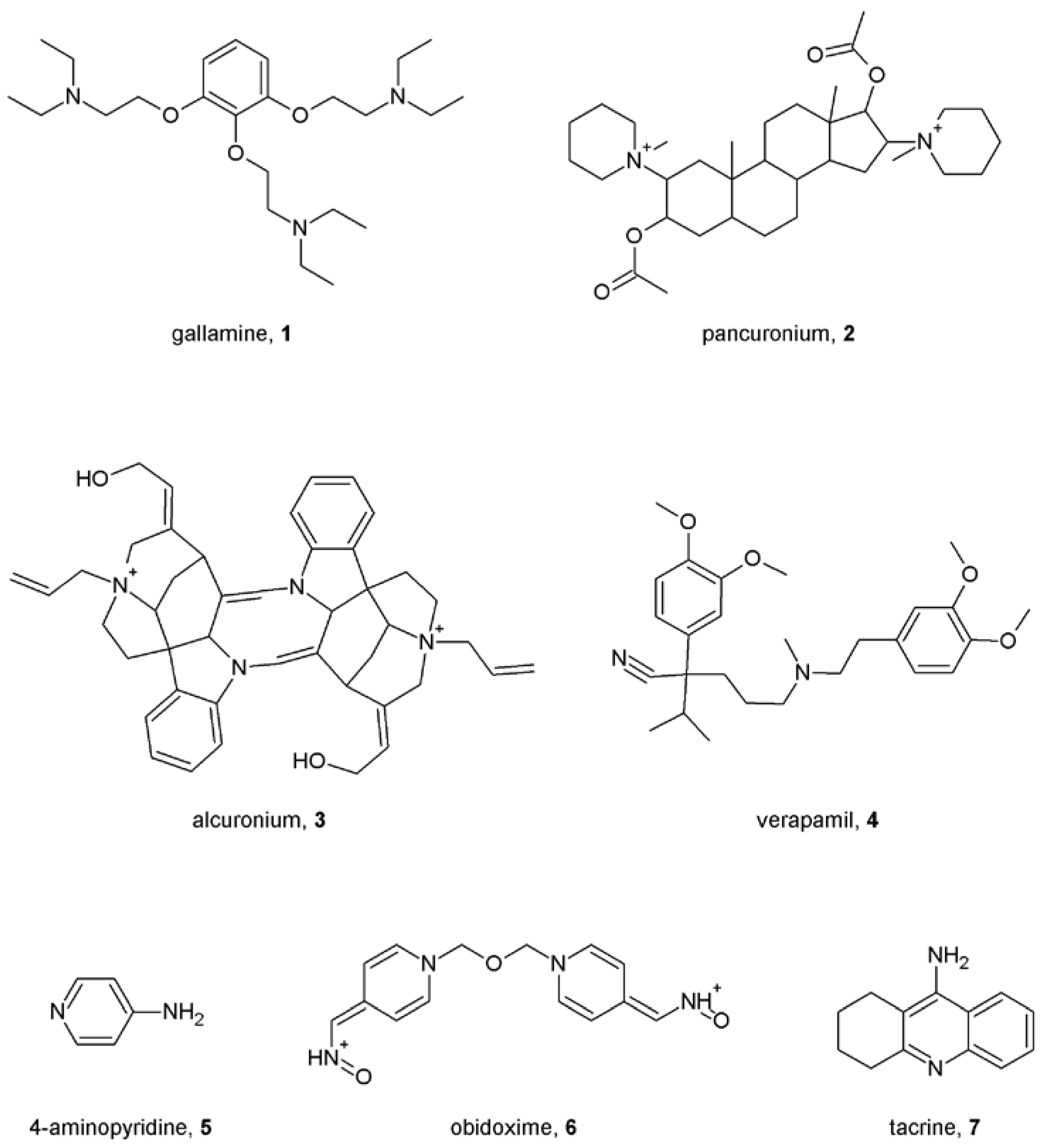
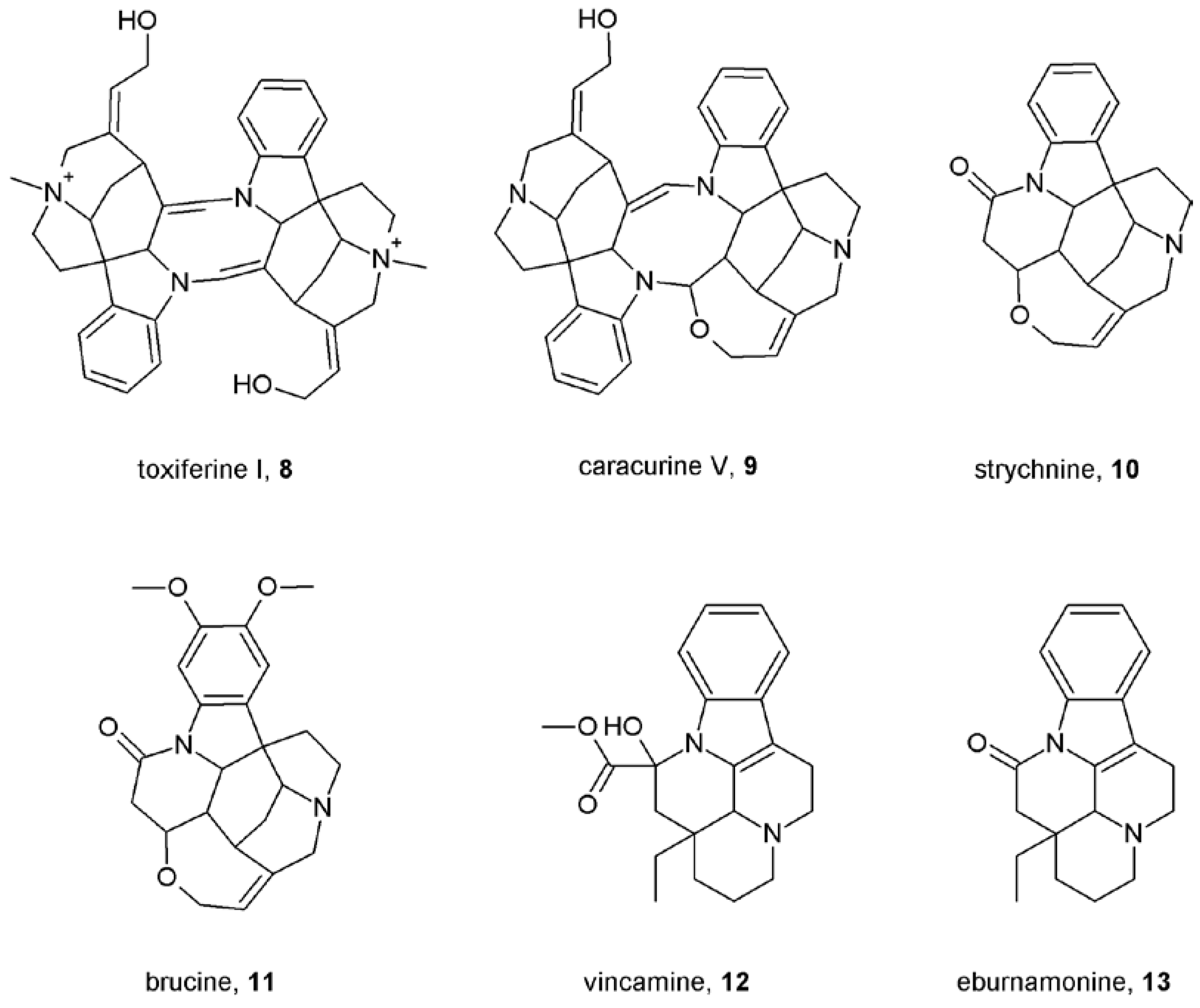
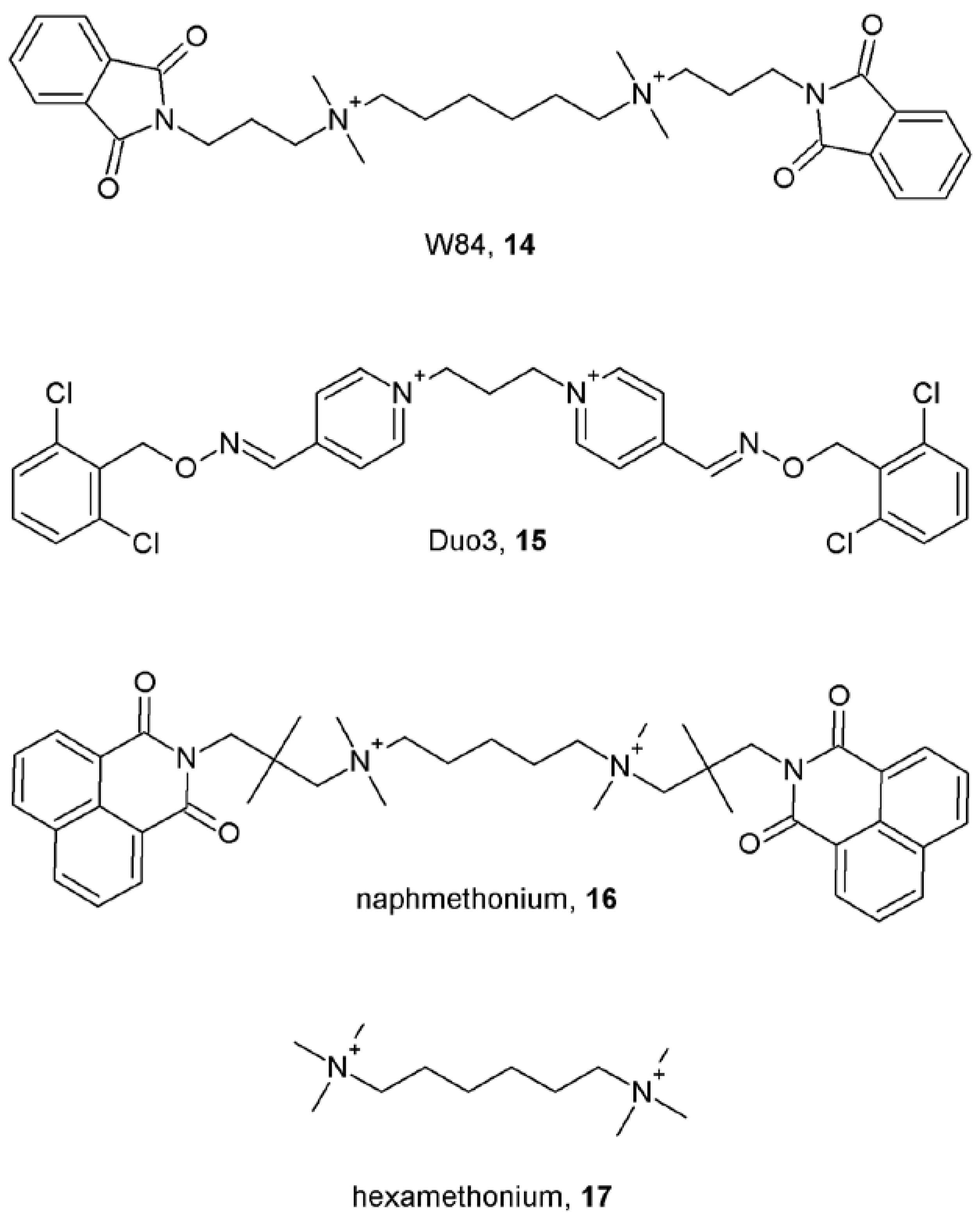
Notes (List of compounds and their chemical names)
References
- Dale, H.H. The action of certain esters and ethers of choline and their relation to muscarine. J. Pharmacol. 1914, 6, 147–190. [Google Scholar]
- Loewi, O. Über humorale Übertragbarkeit der Herznervenwirkung. I. Pflügers Archiv. 1921, 189, 239–242. [Google Scholar] [CrossRef]
- Bonner, T.I.; Buckley, N.J.; Young, A.C.; Brann, M.R. Identification of a family of muscarinic acetylcholine receptor genes. Science 1987, 237, 527–532. [Google Scholar]
- Caulfield, M.P. Muscarinic receptors--characterization, coupling and function. Pharmacol. Ther. 1993, 58, 319–379. [Google Scholar]
- Levey, A.I. Immunological localization of m1-m5 muscarinic acetylcholine receptors in peripheral tissues and brain. Life Sci. 1993, 52, 441–448. [Google Scholar]
- Felder, C.C.; Porter, A.C.; Skillman, T.L.; Zhang, L.; Bymaster, F.P.; Nathanson, N.M.; Hamilton, S.E.; Gomeza, J.; Wess, J.; McKinzie, D.L. Elucidating the role of muscarinic receptors in psychosis. Life Sci. 2001, 68, 2605–2613. [Google Scholar]
- Langmead, C.J.; Watson, J.; Reavill, C. Muscarinic acetylcholine receptors as CNS drug targets. Pharmacol.Ther. 2008, 117, 232–243. [Google Scholar]
- Brito, G.N.; Davis, B.J.; Stopp, L.C.; Stanton, M.E. Memory and the septo-hippocampal cholinergic system in the rat. Psychopharmacology (Berl) 1983, 81, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Meck, W.H.; Church, R.M.; Wenk, G.L.; Olton, D.S. Nucleus basalis magnocellularis and medial septal area lesions differentially impair temporal memory. J. Neurosci. 1987, 7, 3505–3511. [Google Scholar]
- Christie, J.E.; Shering, A.; Ferguson, J.; Glen, A.I. Physostigmine and arecoline, effects of intravenous infusions in Alzheimer presenile dementia. Br. J. Psychiatry. 1981, 138, 46–50. [Google Scholar]
- Bymaster, F.P.; McKinzie, D.L.; Felder, C.C.; Wess, J. Use of M1-M5 muscarinic receptor knockout mice as novel tools to delineate the physiological roles of the muscarinic cholinergic system. Neurochem.Res. 2003, 28, 437–442. [Google Scholar]
- Wess, J.; Eglen, R.M.; Gautam, D. Muscarinic acetylcholine receptors, mutant mice provide new insights for drug development. Nat. Rev. Drug Discov. 2007, 6, 721–733. [Google Scholar]
- Caccamo, A.; Oddo, S.; Billings, L.M.; Green, K.N.; Martinez-Coria, H.; Fisher, A.; LaFerla, F.M. M1 receptors play a central role in modulating AD-like pathology in transgenic mice. Neuron 2006, 49, 671–682. [Google Scholar]
- Jones, C.K.; Brady, A.E.; Davis, A.A.; Xiang, Z.; Bubser, M.; Tantawy, M.N.; Kane, A.S.; Bridges, T.M.; Kennedy, J.P.; Bradley, S.R.; Peterson, T.E.; Ansari, M.S.; Baldwin, R.M.; Kessler, R.M.; Deutch, A.Y.; Lah, J.J.; Levey, A.I.; Lindsley, C.W.; Conn, P.J. Novel selective allosteric activator of the M1 muscarinic acetylcholine receptor regulates amyloid processing and produces antipsychotic-like activity in rats. J. Neurosci. 2008, 28, 10422–10433. [Google Scholar]
- Perry, E.K.; Kilford, L.; Lees, A.J.; Burn, D.J.; Perry, R.H. Increased Alzheimer pathology in Parkinson's disease related to antimuscarinic drugs. Ann. Neurol. 2003, 54, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Machová, E.; Jakubík, J.; Michal, P.; Oksman, M.; Iivonen, H.; Tanila, H.; Doležal, V. Impairment of muscarinic transmission in transgenic APPswe/PS1dE9 mice. Neurobiol.Aging 2008, 29, 368–378. [Google Scholar]
- Machová, E.; Rudajev, V.; Smycková, H.; Koivisto, H.; Tanila, H.; Dolezal, V. Functional cholinergic damage develops with amyloid accumulation in young adult APPswe/PS1dE9 transgenic mice. Neurobiol.Dis. 2010, 38, 27–35. [Google Scholar]
- Biel, J.H.; Nuhfer, P.A.; Hoya, W.K.; Leiser, H.A.; Abood, L.G. Cholinergic blockade as an approach to the development of new psychotropic agents. Ann. N. Y. Acad. Sci. 1962, 96, 251–262. [Google Scholar]
- Mego, D.M.; Omori, J.M.; Hanley, J.F. Transdermal scopolamine as a cause of transient psychosis in two elderly patients. South.Med. J. 1988, 81, 394–395. [Google Scholar]
- Dean, B.; Crook, J.M.; Opeskin, K.; Hill, C.; Keks, N.; Copolov, D.L. The density of muscarinic M1 receptors is decreased in the caudate-putamen of subjects with schizophrenia. Mol. Psychiatry 1996, 1, 54–58. [Google Scholar]
- Dean, B.; Crook, J.M.; Pavey, G.; Opeskin, K.; Copolov, D.L. Muscarinic1 and 2 receptor mRNA in the human caudate-putamen, no change in m1 mRNA in schizophrenia. Mol. Psychiatry 2000, 5, 203–207. [Google Scholar]
- Crook, J.M.; Dean, B.; Pavey, G.; Copolov, D. The binding of [3H]AF-DX 384 is reduced in the caudate-putamen of subjects with schizophrenia. Life Sci. 1999, 64, 1761–1771. [Google Scholar]
- Crook, J.M.; Tomaskovic-Crook, E.; Copolov, D.L.; Dean, B. Decreased muscarinic receptor binding in subjects with schizophrenia, a study of the human hippocampal formation. Biol. Psychiatry 2000, 48, 381–388. [Google Scholar]
- Crook, J.M.; Tomaskovic-Crook, E.; Copolov, D.L.; Dean, B. Low muscarinic receptor binding in prefrontal cortex from subjects with schizophrenia, a study of Brodmann's areas 8, 9, 10, and 46 and the effects of neuroleptic drug treatment. Am. J. Psychiatry 2001, 158, 918–925. [Google Scholar] [PubMed]
- Raedler, T.J.; Bymaster, F.P.; Tandon, R.; Copolov, D.; Dean, B. Towards a muscarinic hypothesis of schizophrenia. Mol. Psychiatry 2007, 12, 232–246. [Google Scholar]
- Tzavara, E.T.; Bymaster, F.P.; Davis, R.J.; Wade, M.R.; Perry, K.W.; Wess, J.; McKinzie, D.L.; Felder, C.; Nomikos, G.G. M4 muscarinic receptors regulate the dynamics of cholinergic and dopaminergic neurotransmission, relevance to the pathophysiology and treatment of related CNS pathologies. FASEB J. 2004, 18, 1410–1412. [Google Scholar]
- Kenakin, T. Allosteric agonist modulators. J. Recept. Signal. Transduct. Res. 2007, 27, 247–259. [Google Scholar]
- Clark, A.L.; Mitchelson, F. The inhibitory effect of gallamine on muscarinic receptors. Br. J. Pharmacol. 1976, 58, 323–331. [Google Scholar]
- Stockton, J.M.; Birdsall, N.J.; Burgen, A.S.; Hulme, E.C. Modification of the binding properties of muscarinic receptors by gallamine. Mol. Pharmacol. 1983, 23, 551–557. [Google Scholar] [PubMed]
- Nedoma, J.; Dorofeeva, N.A.; Tuček, S.; Shelkovnikov, S.A.; Danilov, A.F. Interaction of the neuromuscular blocking drugs alcuronium, decamethonium, gallamine, pancuronium, ritebronium, tercuronium and d-tubocurarine with muscarinic acetylcholine receptors in the heart and ileum. Naunyn Schmiedebergs Arch. Pharmacol. 1985, 329, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Nedoma, J.; Tucek, S.; Danilov, A.F.; Shelkovnikov, S.A. Stabilization of antagonist binding to cardiac muscarinic acetylcholine receptors by gallamine and other neuromuscular blocking drugs. J. Pharmacol. Exp. Ther. 1986, 236, 219–223. [Google Scholar]
- Waelbroeck, M.; Robberecht, P.; De Neef, P.; Christophe, J. Effects of verapamil on the binding properties of rat heart muscarinic receptors, evidence for an allosteric site. Biochem. Biophys. Res. Commun. 1984, 121, 340–345. [Google Scholar]
- Lai, W.S.; Ramkumar, V.; el-Fakahany, E.E. Possible allosteric interaction of 4-aminopyridine with rat brain muscarinic acetylcholine receptors. J. Neurochem. 1985, 44, 1936–1942. [Google Scholar]
- Kloog, Y.; Sokolovsky, M. Allosteric interactions between muscarinic agonist binding sites and effector sites demonstrated by the use of bisquaternary pyridinium oximes. Life Sci. 1985, 36, 2127–2136. [Google Scholar]
- Flynn, D.D.; Mash, D.C. Multiple in vitro interactions with and differential in vivo regulation of muscarinic receptor subtypes by tetrahydroaminoacridine. J. Pharmacol. Exp. Ther. 1989, 250, 573–581. [Google Scholar] [PubMed]
- Jooste, E.; Klafter, F.; Hirshman, C.A.; Emala, C.W. A mechanism for rapacuronium-induced bronchospasm, M2 muscarinic receptor antagonism. Anesthesiology 2003, 98, 906–911. [Google Scholar]
- Hu, J.; el-Fakahany, E.E. Allosteric interaction of dynorphin and myelin basic protein with muscarinic receptors. Pharmacology 1993, 47, 351–359. [Google Scholar]
- Dalton, D.W.; Tyers, M.B. A comparison of the muscarinic antagonist actions of pancuronium and alcuronium. J. Auton. Pharmacol. 1982, 2, 261–266. [Google Scholar]
- Jakubík, J.; Bačáková, L.; El-Fakahany, E.E.; Tuček, S. Positive cooperativity of acetylcholine and other agonists with allosteric ligands on muscarinic acetylcholine receptors. Mol. Pharmacol. 1997, 52, 172–179. [Google Scholar]
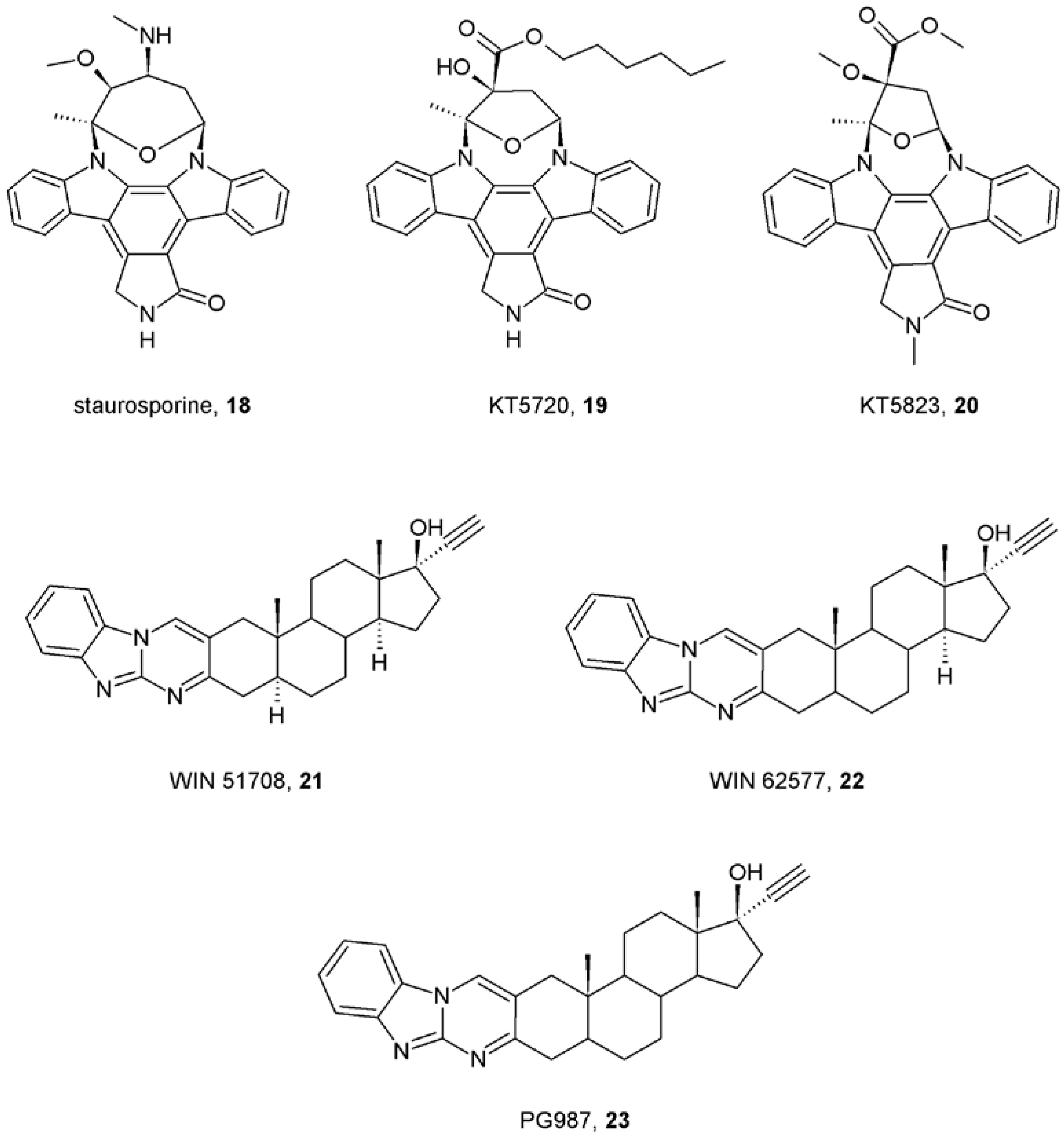
- Lazareno, S.; Popham, A.; Birdsall, N.J. Allosteric interactions of staurosporine and other indolocarbazoles with N-[methyl-(3)H]scopolamine and acetylcholine at muscarinic receptor subtypes, identification of a second allosteric site. Mol. Pharmacol. 2000, 58, 194–207. [Google Scholar] [PubMed]
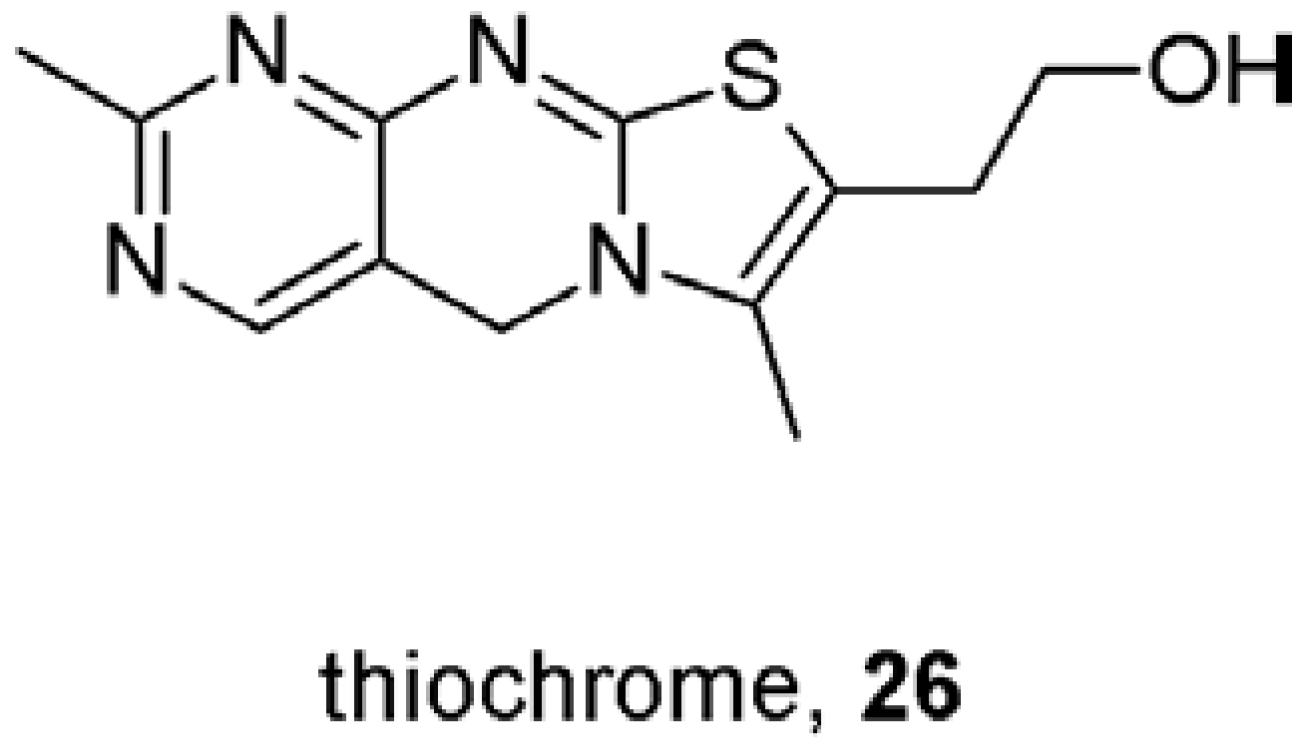
- Lazareno, S.; Doležal, V.; Popham, A.; Birdsall, N.J.M. Thiochrome enhances acetylcholine affinity at muscarinic M4 receptors, receptor subtype selectivity via cooperativity rather than affinity. Mol. Pharmacol. 2004, 65, 257–266. [Google Scholar]
- Zlotos, D.P.; Buller, S.; Stiefl, N.; Baumann, K.; Mohr, K. Probing the pharmacophore for allosteric ligands of muscarinic M2 receptors, SAR and QSAR studies in a series of bisquaternary salts of caracurine V and related ring systems. J. Med. Chem. 2004, 47, 3561–3571. [Google Scholar]
- Proška, J.; Tuček, S. Mechanisms of steric and cooperative actions of alcuronium on cardiac muscarinic acetylcholine receptors. Mol. Pharmacol. 1994, 45, 709–717. [Google Scholar]
- Ellis, J.; Huyler, J.; Brann, M.R. Allosteric regulation of cloned m1-m5 muscarinic receptor subtypes. Biochem.Pharmacol. 1991, 42, 1927–1932. [Google Scholar]
- Jakubík, J.; Bačáková, L.; el-Fakahany, E.E.; Tuček, S. Subtype selectivity of the positive allosteric action of alcuronium at cloned M1-M5 muscarinic acetylcholine receptors. J. Pharmacol. Exp. Ther. 1995, 274, 1077–1083. [Google Scholar]
- Leppik, R.A.; Miller, R.C.; Eck, M.; Paquet, J.L. Role of acidic amino acids in the allosteric modulation by gallamine of antagonist binding at the m2 muscarinic acetylcholine receptor. Mol. Pharmacol. 1994, 45, 983–990. [Google Scholar]
- Krejčí, A.; Tuček, S. Changes of cooperativity between N-methylscopolamine and allosteric modulators alcuronium and gallamine induced by mutations of external loops of muscarinic M(3) receptors. Mol. Pharmacol. 2001, 60, 761–767. [Google Scholar]
- Huang, X.; Prilla, S.; Mohr, K.; Ellis, J. Critical amino acid residues of the common allosteric site on the M2 muscarinic acetylcholine receptor, more similarities than differences between the structurally divergent agents gallamine and bis(ammonio)alkane-type hexamethylene-bis-[dimethyl-(3-phtha)]. Mol. Pharmacol. 2005, 68, 769–778. [Google Scholar]
- Matsui, H.; Lazareno, S.; Birdsall, N.J. Probing of the location of the allosteric site on m1 muscarinic receptors by site-directed mutagenesis. Mol. Pharmacol. 1995, 47, 88–98. [Google Scholar]
- Jakubík, J.; Krejčí, A.; Doležal, V. Asparagine, valine, and threonine in the third extracellular loop of muscarinic receptor have essential roles in the positive cooperativity of strychnine-like allosteric modulators. J. Pharmacol. Exp. Ther. 2005, 313, 688–696. [Google Scholar] [PubMed]
- Voigtländer, U.; Jöhren, K.; Mohr, M.; Raasch, A.; Tränkle, C.; Buller, S.; Ellis, J.; Höltje, H.; Mohr, K. Allosteric site on muscarinic acetylcholine receptors, identification of two amino acids in the muscarinic M2 receptor that account entirely for the M2/M5 subtype selectivities of some structurally diverse allosteric ligands in N-methylscopolamine-occupie. Mol. Pharmacol. 2003, 64, 21–31. [Google Scholar]
- Prilla, S.; Schrobang, J.; Ellis, J.; Höltje, H.; Mohr, K. Allosteric interactions with muscarinic acetylcholine receptors, complex role of the conserved tryptophan M2422Trp in a critical cluster of amino acids for baseline affinity, subtype selectivity, and cooperativity. Mol. Pharmacol. 2006, 70, 181–193. [Google Scholar] [PubMed]
- Tränkle, C.; Dittmann, A.; Schulz, U.; Weyand, O.; Buller, S.; Jöhren, K.; Heller, E.; Birdsall, N.J.M.; Holzgrabe, U.; Ellis, J.; Höltje, H.D.; Mohr, K. Atypical muscarinic allosteric modulation, cooperativity between modulators and their atypical binding topology in muscarinic M2 and M2/M5 chimeric receptors. Mol. Pharmacol. 2005, 68, 1597–1610. [Google Scholar]
- Lazareno, S.; Popham, A.; Birdsall, N.J.M. Analogs of WIN 62,577 define a second allosteric site on muscarinic receptors. Mol. Pharmacol. 2002, 62, 1492–1505. [Google Scholar]
- Lanzafame, A.A.; Sexton, P.M.; Christopoulos, A. Interaction studies of multiple binding sites on m4 muscarinic acetylcholine receptors. Mol. Pharmacol. 2006, 70, 736–746. [Google Scholar]
- Tränkle, C.; Mies-Klomfass, E.; Cid, M.H.; Holzgrabe, U.; Mohr, K. Identification of a [3H]Ligand for the common allosteric site of muscarinic acetylcholine M2 receptors. Mol. Pharmacol. 1998, 54, 139–145. [Google Scholar]
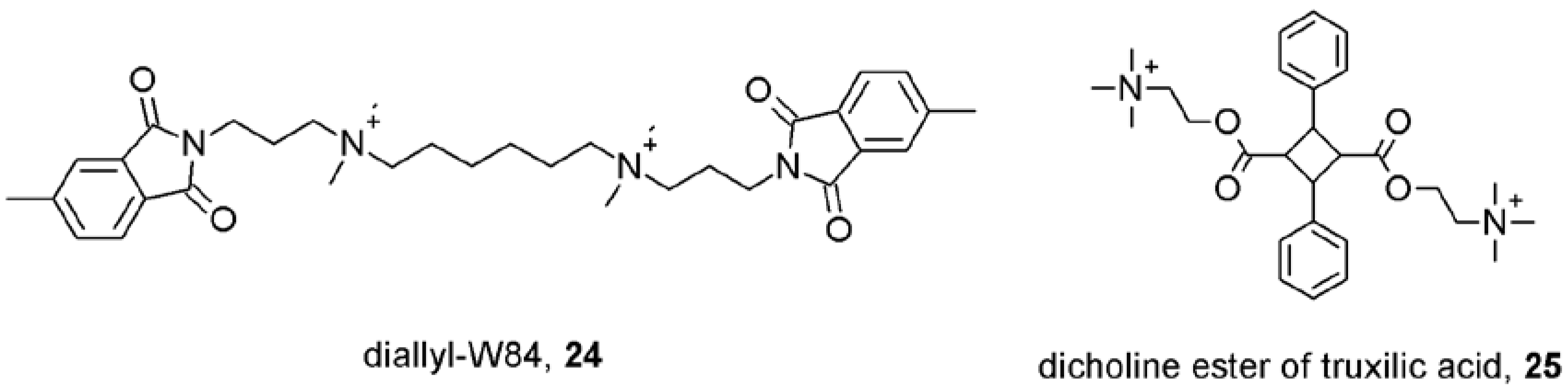
- Lysíková, M.; Fuksová, K.; Elbert, T.; Jakubík, J.; Tuček, S. Subtype-selective inhibition of [methyl-3H]-N-methylscopolamine binding to muscarinic receptors by alpha-truxillic acid esters. Br. J. Pharmacol. 1999, 127, 1240–1246. [Google Scholar]
- Mohr, K.; Tränkle, C.; Holzgrabe, U. Structure/activity relationships of M2 muscarinic allosteric modulators. Receptors Channels 2003, 9, 229–240. [Google Scholar]
- Birdsall, N.J.M.; Lazareno, S. Allosterism at muscarinic receptors, ligands and mechanisms. Mini. Rev. Med. Chem. 2005, 5, 523–543. [Google Scholar] [CrossRef] [PubMed]
- Lazareno, S.; Gharagozloo, P.; Kuonen, D.; Popham, A.; Birdsall, N.J. Subtype-selective positive cooperative interactions between brucine analogues and acetylcholine at muscarinic receptors, radioligand binding studies. Mol. Pharmacol. 1998, 53, 573–589. [Google Scholar]
- Lysíková, M.; Havlas, Z.; Tuček, S. Interactions between allosteric modulators and 4-DAMP and other antagonists at muscarinic receptors, potential significance of the distance between the N and carboxyl C atoms in the molecules of antagonists. Neurochem Res 2001, 26, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Jöhren, K.; Höltje, H. A model of the human M2 muscarinic acetylcholine receptor. J. Comput. Aided Mol. Des. 2002, 16, 795–801. [Google Scholar]
- Jakubík, J.; Tuček, S. Positive allosteric interactions on cardiac muscarinic receptors, effects of chemical modifications of disulphide and carboxyl groups. Eur. J. Pharmacol. 1995, 289, 311–319. [Google Scholar]
- Huang, X.; Ellis, J. Mutational disruption of a conserved disulfide bond in muscarinic acetylcholine receptors attenuates positive homotropic cooperativity between multiple allosteric sites and has subtype-dependent effects on the affinities of muscarinic allosteric ligands. Mol. Pharmacol. 2007, 71, 759–768. [Google Scholar]
- Diaz-Arrastia, R.; Ashizawa, T.; Appel, S.H. Endogenous inhibitor of ligand binding to the muscarinic acetylcholine receptor. J. Neurochem. 1985, 44, 622–628. [Google Scholar]
- Maslinski, W.; Ryzewski, J.; Bartfai, T. Rat thymocytes release a factor which inhibits muscarinic ligand binding. J. Neuroimmunol. 1988, 17, 275–285. [Google Scholar]
- Fryer, A.D.; el-Fakahany, E.E. An endogenous factor induces heterogeneity of binding sites of selective muscarinic receptor antagonists in rat heart. Membr.Biochem. 1989, 8, 127–132. [Google Scholar]
- Frey, W.H.2.; Emory, C.R.; Wiebenga, M.E.; Saxena, S.; Cardelli, D.; Ala, T.A.; Tollefson, G.D. Inhibitor of antagonist binding to the muscarinic receptor is elevated in Alzheimer's brain. Brain Res. 1994, 655, 153–160. [Google Scholar]
- Fang, Y.I.; Suzuki, T.; Momose, K. Partial purification of an endogenous inhibitor of muscarinic ligand binding. Biochem.Mol. Biol. Int. 1996, 38, 501–507. [Google Scholar] [PubMed]
- Jacoby, D.B.; Gleich, G.J.; Fryer, A.D. Human eosinophil major basic protein is an endogenous allosteric antagonist at the inhibitory muscarinic M2 receptor. J. Clin. Invest. 1993, 91, 1314–1318. [Google Scholar]
- Kjome, J.R.; Swenson, K.A.; Johnson, M.N.; Bordayo, E.Z.; Anderson, L.E.; Klevan, L.C.; Fraticelli, A.I.; Aldrich, S.L.; Fawcett, J.R.; Venters, H.D.J.; Ala, T.A.; Frey, W.H.2. Inhibition of antagonist and agonist binding to the human brain muscarinic receptor by arachidonic acid. J. Mol. Neurosci. 1998, 10, 209–217. [Google Scholar]
- Frey, W.H.2.; Najarian, M.M.; Kumar, K.S.; Emory, C.R.; Menning, P.M.; Frank, J.C.; Johnson, M.N.; Ala, T.A. Endogenous Alzheimer's brain factor and oxidized glutathione inhibit antagonist binding to the muscarinic receptor. Brain Res. 1996, 714, 87–94. [Google Scholar]
- Fawcett, J.R.; Bordayo, E.Z.; Jackson, K.; Liu, H.; Peterson, J.; Svitak, A.; Frey, W.H.2. Inactivation of the human brain muscarinic acetylcholine receptor by oxidative damage catalyzed by a low molecular weight endogenous inhibitor from Alzheimer's brain is prevented by pyrophosphate analogs, bioflavonoids and other antioxidants. Brain Res. 2002, 950, 10–20. [Google Scholar]
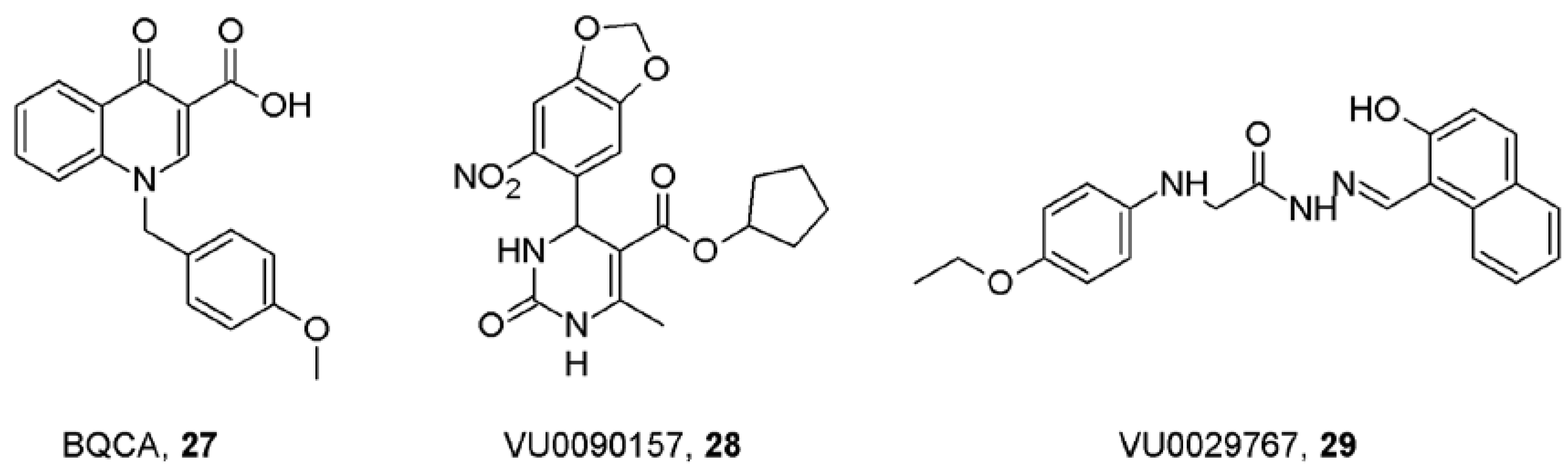
- Ma, L.; Seager, M.A.; Wittmann, M.; Jacobson, M.; Bickel, D.; Burno, M.; Jones, K.; Graufelds, V.K.; Xu, G.; Pearson, M.; McCampbell, A.; Gaspar, R.; Shughrue, P.; Danziger, A.; Regan, C.; Flick, R.; Pascarella, D.; Garson, S.; Doran, S.; Kreatsoulas, C.; Veng, L.; Lindsley, C.W.; Shipe, W.; Kuduk, S.; Sur, C.; Kinney, G.; Seabrook, G.R.; Ray, W.J. Selective activation of the M1 muscarinic acetylcholine receptor achieved by allosteric potentiation. Proc. Natl. Acad. Sci. USA 2009, 106, 15950–15955. [Google Scholar]
- Shirey, J.K.; Brady, A.E.; Jones, P.J.; Davis, A.A.; Bridges, T.M.; Kennedy, J.P.; Jadhav, S.B.; Menon, U.N.; Xiang, Z.; Watson, M.L.; Christian, E.P.; Doherty, J.J.; Quirk, M.C.; Snyder, D.H.; Lah, J.J.; Levey, A.I.; Nicolle, M.M.; Lindsley, C.W.; Conn, P.J. A selective allosteric potentiator of the M1 muscarinic acetylcholine receptor increases activity of medial prefrontal cortical neurons and restores impairments in reversal learning. J. Neurosci. 2009, 29, 14271–14286. [Google Scholar] [PubMed]
- Marlo, J.E.; Niswender, C.M.; Days, E.L.; Bridges, T.M.; Xiang, Y.; Rodriguez, A.L.; Shirey, J.K.; Brady, A.E.; Nalywajko, T.; Luo, Q.; Austin, C.A.; Williams, M.B.; Kim, K.; Williams, R.; Orton, D.; Brown, H.A.; Lindsley, C.W.; Weaver, C.D.; Conn, P.J. Discovery and characterization of novel allosteric potentiators of M1 muscarinic receptors reveals multiple modes of activity. Mol. Pharmacol 2009, 75, 577–588. [Google Scholar] [PubMed]
- Bridges, T.M.; Marlo, J.E.; Niswender, C.M.; Jones, C.K.; Jadhav, S.B.; Gentry, P.R.; Plumley, H.C.; Weaver, C.D.; Conn, P.J.; Lindsley, C.W. Discovery of the first highly M5-preferring muscarinic acetylcholine receptor ligand, an M5 positive allosteric modulator derived from a series of 5-trifluoromethoxy N-benzyl isatins. J. Med. Chem. 2009, 52, 3445–3448. [Google Scholar]
- Shirey, J.K.; Xiang, Z.; Orton, D.; Brady, A.E.; Johnson, K.A.; Williams, R.; Ayala, J.E.; Rodriguez, A.L.; Wess, J.; Weaver, D.; Niswender, C.M.; Conn, P.J. An allosteric potentiator of M4 mAChR modulates hippocampal synaptic transmission. Nat. Chem. Biol. 2008, 4, 42–50. [Google Scholar]
- Brady, A.E.; Jones, C.K.; Bridges, T.M.; Kennedy, J.P.; Thompson, A.D.; Heiman, J.U.; Breininger, M.L.; Gentry, P.R.; Yin, H.; Jadhav, S.B.; Shirey, J.K.; Conn, P.J.; Lindsley, C.W. Centrally Active Allosteric Potentiators of the M4 Muscarinic Acetylcholine Receptor Reverse Amphetamine-Induced Hyperlocomotor Activity in Rats. J. Pharm. Expert. Ther. 2008, 327, 941–953. [Google Scholar]
- Chan, W.Y.; McKinzie, D.L.; Bose, S.; Mitchell, S.N.; Witkin, J.M.; Thompson, R.C.; Christopoulos, A.; Lazareno, S.; Birdsall, N.J.M.; Bymaster, F.P.; Felder, C.C. Allosteric modulation of the muscarinic M4 receptor as an approach to treating schizophrenia. Proc. Natl. Acad. Sci. USA 2008, 105, 10978–10983. [Google Scholar]
- Nawaratne, V.; Leach, K.; Suratman, N.; Loiacono, R.E.; Felder, C.C.; Armbruster, B.N.; Roth, B.L.; Sexton, P.M.; Christopoulos, A. New insights into the function of M4 muscarinic acetylcholine receptors gained using a novel allosteric modulator and a DREADD (designer receptor exclusively activated by a designer drug). Mol. Pharmacol. 2008, 74, 1119–1131. [Google Scholar]
- Nawaratne, V.; Leach, K.; Suratman, N.; Loiacono, R.E.; Felder, C.C.; Armbruster, B.N.; Roth, B.L.; Sexton, P.M.; Christopoulos, A. New insights into the function of M4 muscarinic acetylcholine receptors gained using a novel allosteric modulator and a DREADD (designer receptor exclusively activated by a designer drug). Mol. Pharmacol. 2008, 74, 1119–1131. [Google Scholar]
- Jakubík, J.; Randáková, A.; El-Fakahany, E.E.; Doležal, V. Divergence of allosteric effects of rapacuronium on binding and function of muscarinic receptors. BMC Pharmacol. 2009, 9, 15. [Google Scholar]
- Goudsouzian, N.G. Rapacuronium and bronchospasm. Anesthesiology 2001, 94, 727–728. [Google Scholar] [CrossRef] [PubMed]
- Jakubík, J.; Bačáková, L.; Lisá, V.; el-Fakahany, E.E.; Tuček, S. Activation of muscarinic acetylcholine receptors via their allosteric binding sites. Proc. Natl. Acad. Sci. USA 1996, 93, 8705–8709. [Google Scholar]
- Sur, C.; Mallorga, P.J.; Wittmann, M.; Jacobson, M.A.; Pascarella, D.; Williams, J.B.; Brandish, P.E.; Pettibone, D.J.; Scolnick, E.M.; Conn, P.J. N-desmethylclozapine, an allosteric agonist at muscarinic 1 receptor, potentiates N-methyl-D-aspartate receptor activity. Proc. Natl. Acad. Sci. USA 2003, 100, 13674–13679. [Google Scholar]
- Spalding, T.A.; Trotter, C.; Skjaerbaek, N.; Messier, T.L.; Currier, E.A.; Burstein, E.S.; Li, D.; Hacksell, U.; Brann, M.R. Discovery of an ectopic activation site on the M(1) muscarinic receptor. Mol. Pharmacol. 2002, 61, 1297–1302. [Google Scholar]
- Bradley, S.R.; Lameh, J.; Ohrmund, L.; Son, T.; Bajpai, A.; Nguyen, D.; Friberg, M.; Burstein, E.S.; Spalding, T.A.; Ott, T.R.; Schiffer, H.H.; Tabatabaei, A.; McFarland, K.; Davis, R.E.; Bonhaus, D.W. AC-260584, an orally bioavailable M(1) muscarinic receptor allosteric agonist, improves cognitive performance in an animal model. Neuropharmacology 2009, 58, 365–373. [Google Scholar] [PubMed]
- Spalding, T.A.; Ma, J.; Ott, T.R.; Friberg, M.; Bajpai, A.; Bradley, S.R.; Davis, R.E.; Brann, M.R.; Burstein, E.S. Structural requirements of transmembrane domain 3 for activation by the M1 muscarinic receptor agonists AC-42, AC-260584, clozapine, and N-desmethylclozapine, evidence for three distinct modes of receptor activation. Mol. Pharmacol. 2006, 70, 1974–1983. [Google Scholar] [CrossRef] [PubMed]
- Lebon, G.; Langmead, C.J.; Tehan, B.G.; Hulme, E.C. Mutagenic mapping suggests a novel binding mode for selective agonists of M1 muscarinic acetylcholine receptors. Mol. Pharmacol. 2009, 75, 331–341. [Google Scholar]
- Thomas, R.L.; Langmead, C.J.; Wood, M.D.; Challiss, R.A.J. Contrasting effects of allosteric and orthosteric agonists on m1 muscarinic acetylcholine receptor internalization and down-regulation. J. Pharmacol. Exp. Ther. 2009, 331, 1086–1095. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an Open Access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jakubík, J.; El-Fakahany, E.E. Allosteric Modulation of Muscarinic Acetylcholine Receptors. Pharmaceuticals 2010, 3, 2838-2860. https://doi.org/10.3390/ph3092838
Jakubík J, El-Fakahany EE. Allosteric Modulation of Muscarinic Acetylcholine Receptors. Pharmaceuticals. 2010; 3(9):2838-2860. https://doi.org/10.3390/ph3092838
Chicago/Turabian StyleJakubík, Jan, and Esam E. El-Fakahany. 2010. "Allosteric Modulation of Muscarinic Acetylcholine Receptors" Pharmaceuticals 3, no. 9: 2838-2860. https://doi.org/10.3390/ph3092838
APA StyleJakubík, J., & El-Fakahany, E. E. (2010). Allosteric Modulation of Muscarinic Acetylcholine Receptors. Pharmaceuticals, 3(9), 2838-2860. https://doi.org/10.3390/ph3092838