Mechanism of Allosteric Modulation of the Cys-loop Receptors

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction

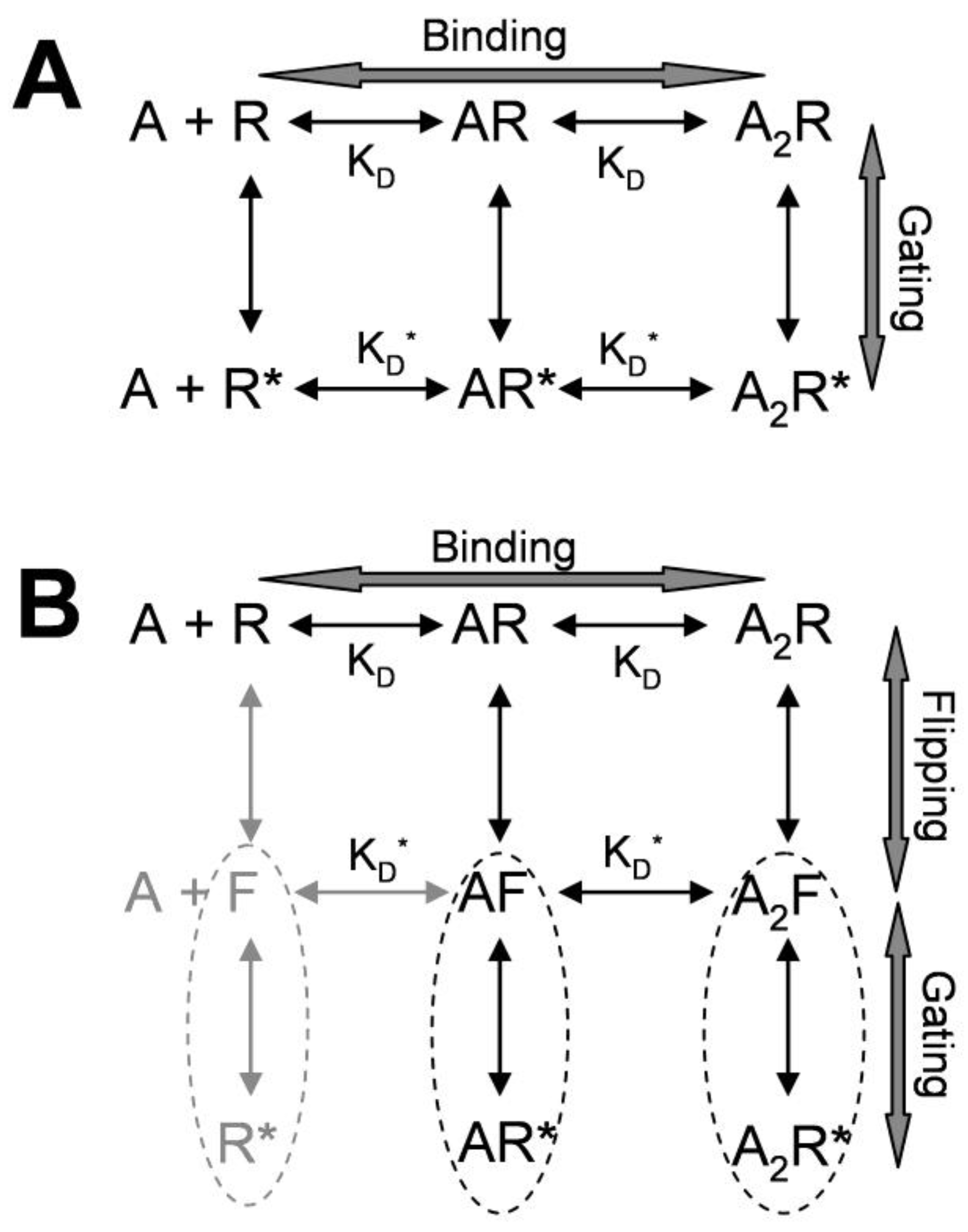
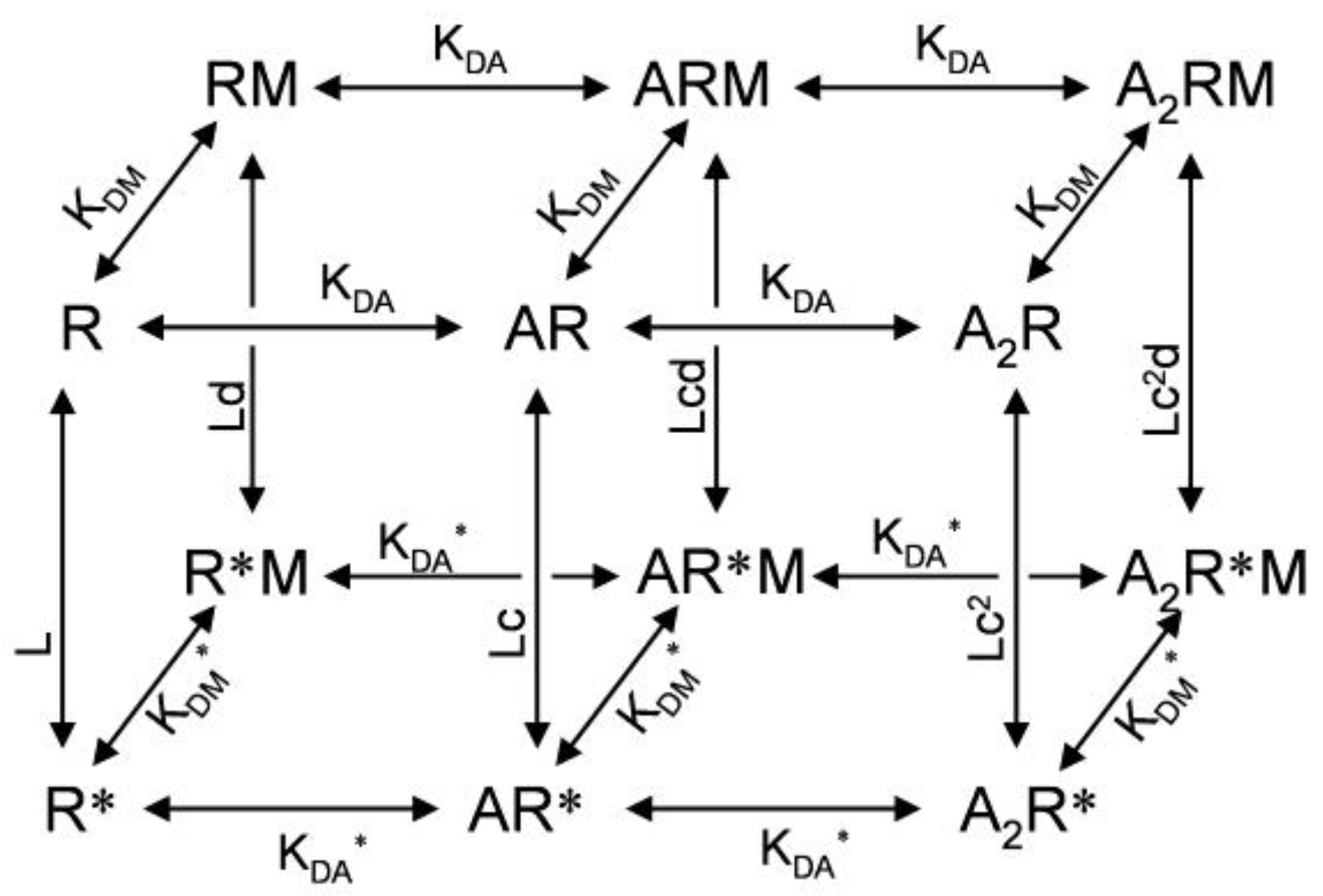
2. Kinetic Models of Allosteric Activation
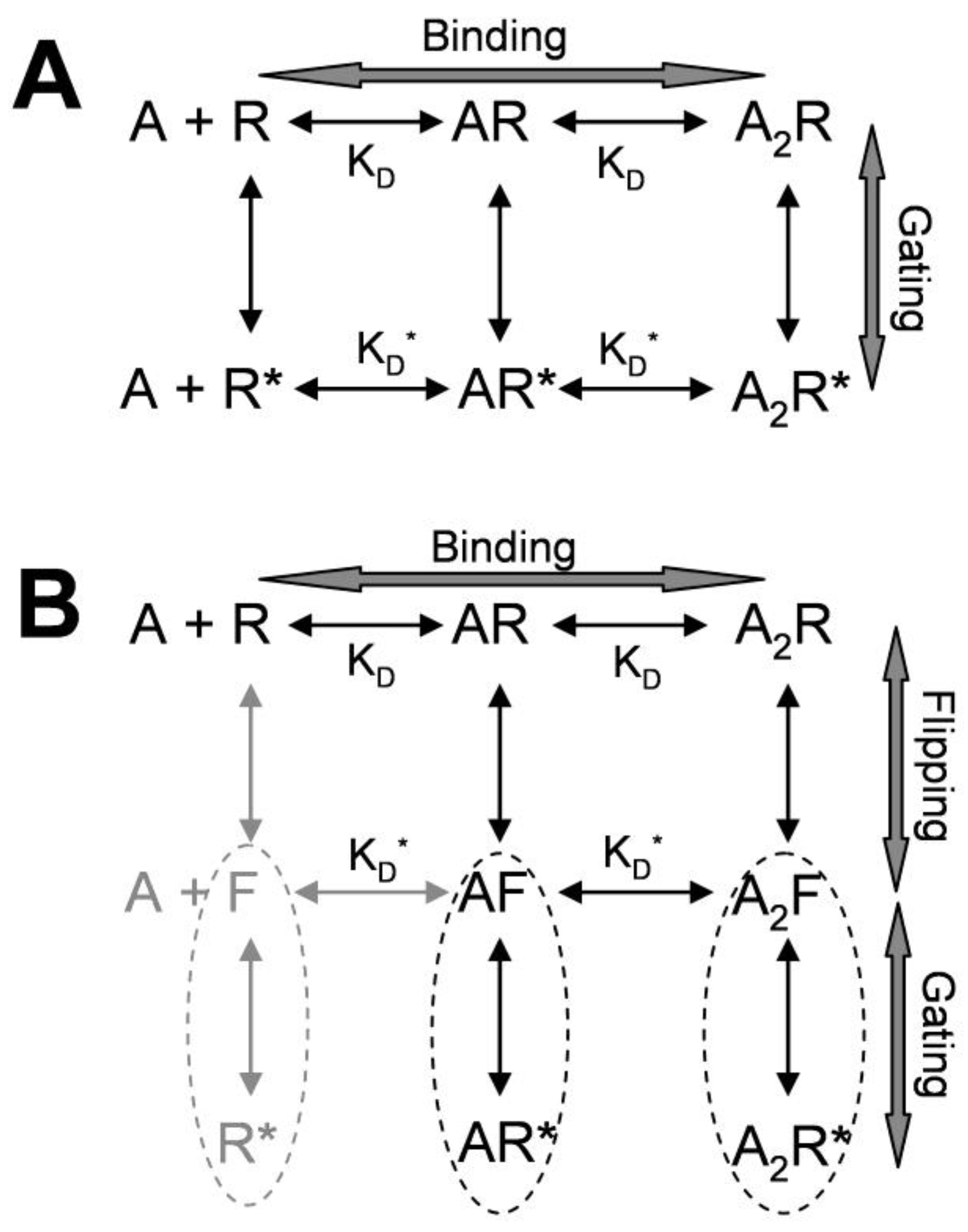
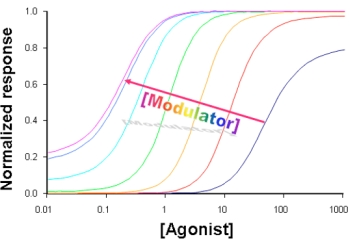
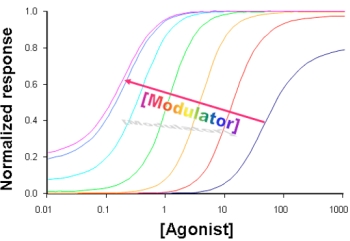
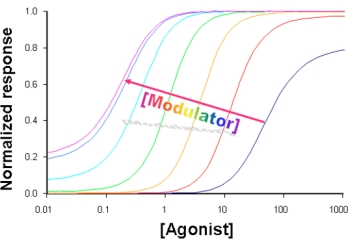
3. Kinetic Models and Mechanisms of Allosteric Modulation
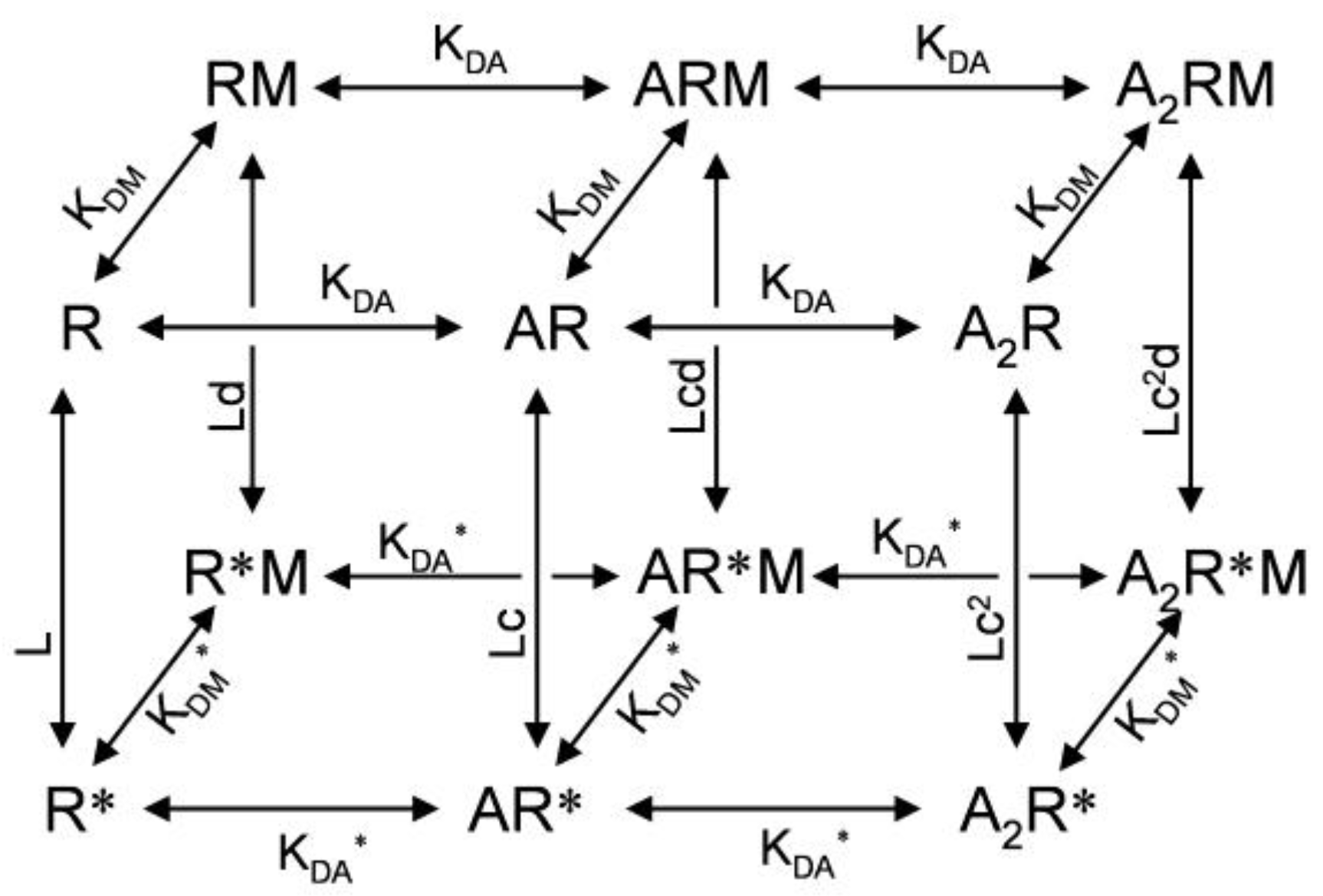
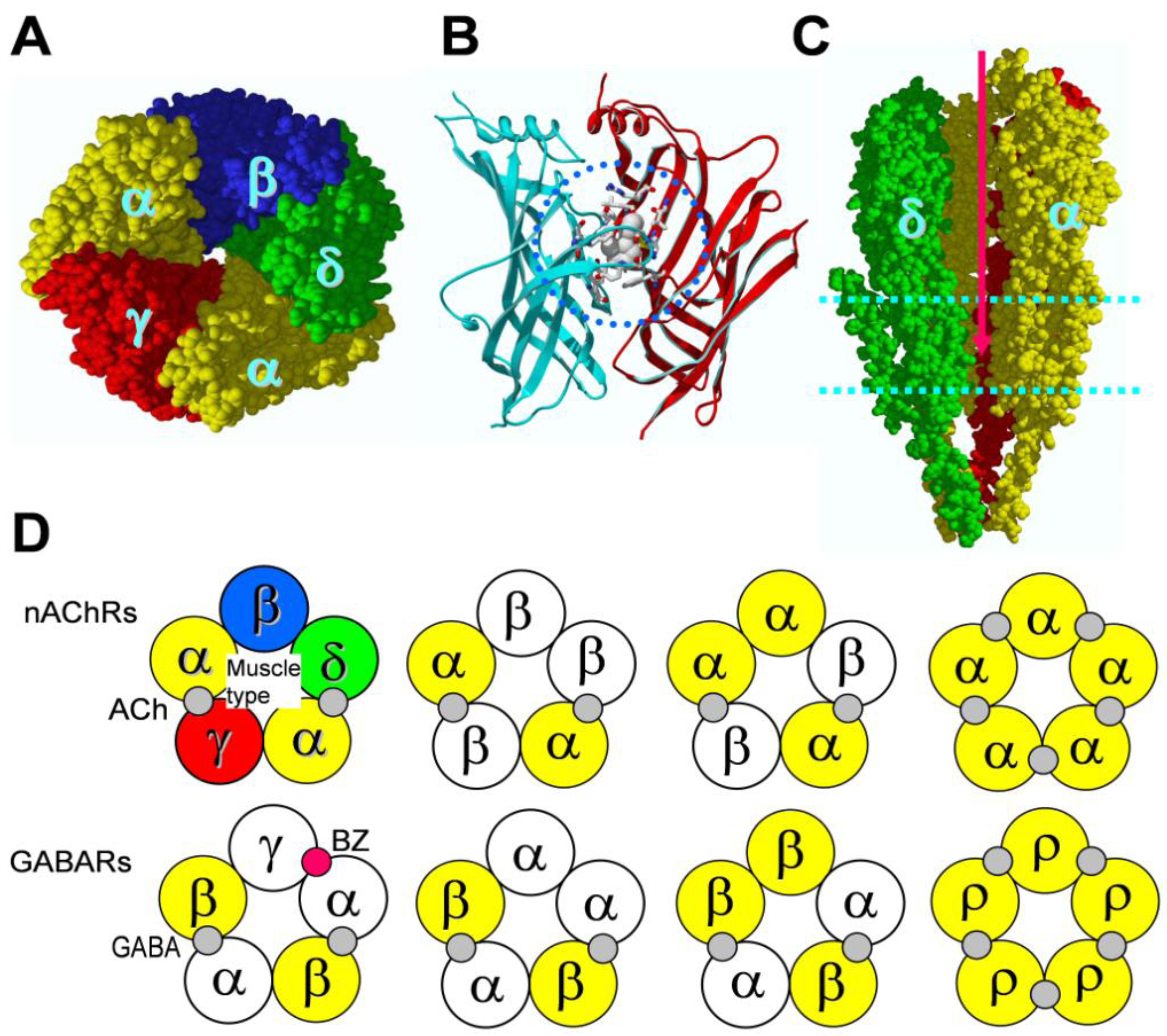
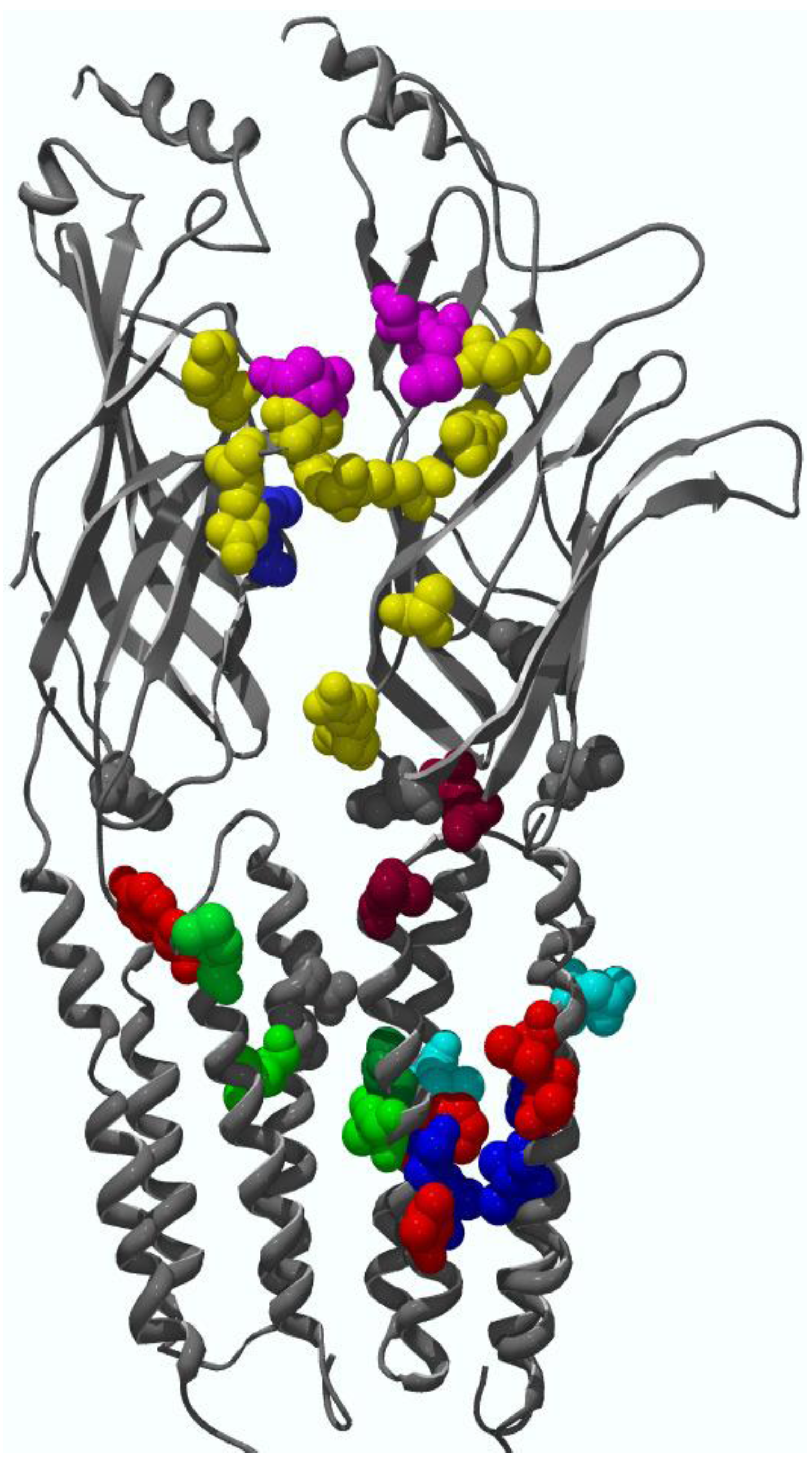
4. Binding Sites for Allosteric Modulators
4.1. Amino terminal binding sites for allosteric modulators
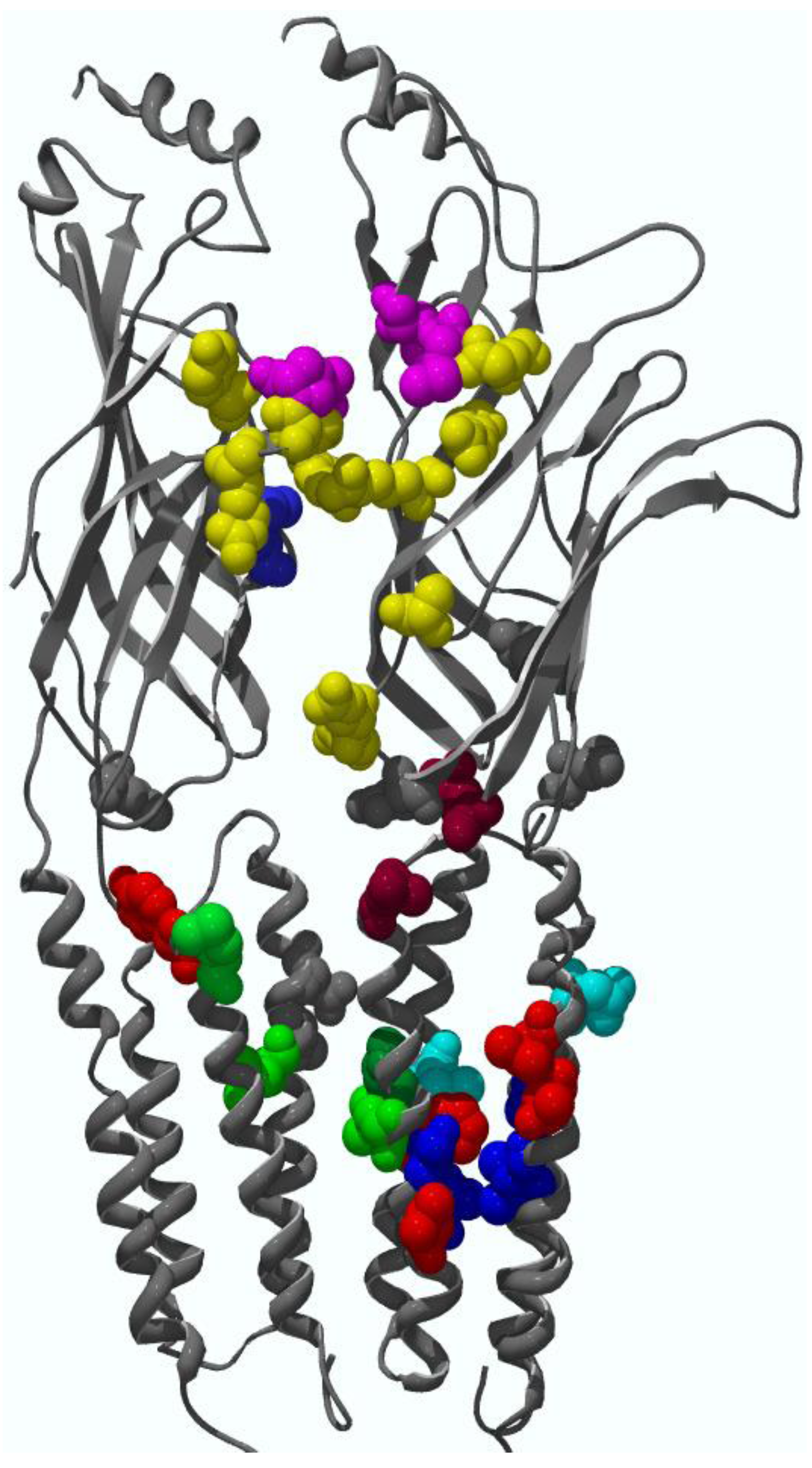
4.2. Transmembrane domain binding sites for allosteric modulators
Acknowledgements
References
- Albuquerque, E.; Pereira, E.; Alkondon, M.; Rogers, S. Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol. Rev. 2009, 89, 73–120. [Google Scholar]
- Thompson, A.; Lummis, S. The 5-HT3 receptor as a therapeutic target. Expert Opin. Ther. Targets 2007, 11, 527–540. [Google Scholar]
- Olsen, R.; Sieghart, W. International Union of Pharmacology. LXX. Subtypes of γ-aminobutyric acid (A) receptors: classification on the basis of subunit composition, pharmacology, and function. Update. Pharmacol. Rev. 2008, 60, 243–260. [Google Scholar]
- Olsen, R.; Sieghart, W. GABAA receptors: subtypes provide diversity of function and pharmacology. Neuropharmacology 2009, 56, 273–284. [Google Scholar]
- Johnston, G.; Chebib, M.; Hanrahan, J.; Mewett, K. GABAC receptors as drug targets. Curr. Drug Targets CNS Neurol. Disord. 2003, 2, 260. [Google Scholar]
- Lynch, J. Native glycine receptor subtypes and their physiological roles. Neuropharmacology 2009, 56, 303–309. [Google Scholar]
- Davies, P.; Wang, W.; Hales, T.; Kirkness, E. A novel class of ligand-gated ion channel is activated by Zn2+. J. Biol. Chem. 2003, 278, 712–717. [Google Scholar]
- Changeux, J.; Edelstein, S. Allosteric receptors after 30 years. Neuron 1998, 21, 959–980. [Google Scholar]
- Lester, H.; Dibas, M.; Dahan, D.; Leite, J.; Dougherty, D. Cys-loop receptors: new twists and turns. Trends. Neurosci. 2004, 27, 329–336. [Google Scholar]
- Ortells, M.; Lunt, G. Evolutionary history of the ligand-gated ion-channel superfamily of receptors. Trends. Neurosci. 1995, 18, 121–127. [Google Scholar]
- Bocquet, N.; Nury, H.; Baaden, M.; Poupon, C.; Changeux, J.; Delarue, M.; Corringer, P. X-ray structure of a pentameric ligand-gated ion channel in an apparently open conformation. Nature 2009, 457, 111–114. [Google Scholar]
- Tasneem, A.; Iyer, L.M.; Jakobsson, E.; Aravind, L. Identification of the prokaryotic ligand-gated ion channels and their implications for the mechanisms and origins of animal Cys-loop ion channels. Genome. Biol. 2004, 6, R4. [Google Scholar]
- Hilf, R.; Dutzler, R. X-ray structure of a prokaryotic pentameric ligand-gated ion channel. Nature 2008, 452, 375–379. [Google Scholar]
- Unwin, N. Nicotinic acetylcholine receptor at 9 Å resolution. J. Mol. Biol. 1993, 229, 1101–1124. [Google Scholar]
- Miyazawa, A.; Fujiyoshi, Y.; Unwin, N. Structure and gating mechanism of the acetylcholine receptor pore. Nature 2003, 423, 949–955. [Google Scholar]
- Unwin, N. Refined structure of the nicotinic acetylcholine receptor at 4 Å resolution. J. Mol. Biol. 2005, 346, 967–989. [Google Scholar]
- Celie, P.; Rossum-Fikkert, S.; Dijk, W.; Brejc, K.; Smit, A.; Sixma, T. Nicotine and carbamylcholine binding to nicotinic acetylcholine receptors as studied in AChBP crystal structures. Neuron 2004, 41, 907–914. [Google Scholar]
- Brejc, K.; Dijk, W.; Klaassen, R.; Schuurmans, M.; Oost, J.; Smit, A.; Sixma, T. Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature 2001, 411, 269–276. [Google Scholar]
- Ulens, C.; Hogg, R.; Celie, P.; Bertrand, D.; Tsetlin, V.; Smit, A.; Sixma, T. Structural determinants of selective α-conotoxin binding to a nicotinic acetylcholine receptor homolog AChBP. Proc. Natl. Acad. Sci. USA 2006, 103, 3615–3620. [Google Scholar]
- Hansen, S.; Sulzenbacher, G.; Huxford, T.; Marchot, P.; Taylor, P.; Bourne, Y. Structures of Aplysia AChBP complexes with nicotinic agonists and antagonists reveal distinctive binding interfaces and conformations. EMBO J. 2005, 24, 3635–3646. [Google Scholar]
- Chang, Y.; Wu, W.; Zhang, J.; Huang, Y. Allosteric activation mechanism of the cys-loop receptors. Acta Pharmacol. Sin. 2009, 30, 663–672. [Google Scholar]
- Edelstein, S.; Changeux, J. Allosteric proteins after thirty years: the binding and state functions of the neuronal α7 nicotinic acetylcholine receptors. Experientia 1996, 52, 1083–1090. [Google Scholar]
- Chen, Y.; Reilly, K.; Chang, Y. Evolutionarily conserved allosteric network in the cys-loop family of ligand-gated ion channels revealed by statistical covariance analyses. J. Biol. Chem. 2006, 281, 18184–18192. [Google Scholar]
- Millar, P.; Smart, T. Binding; activation and modulation of Cys-loop receptors. Trends. Pharmacol. Sci. 2010, 31, 161–174. [Google Scholar]
- Bartos, M.; Corradi, J.; Bouzat, C. Structural basis of activation of cys-loop receptors: the extracellular-transmembrane interface as a coupling region. Mol. Neurobiol. 2009, 40, 236–252. [Google Scholar]
- Yakel, J. Gating of nicotinic ACh receptors: latest insights into ligand binding and function. J. Physiol. 2010, 588, 597–602. [Google Scholar]
- Cederholm, J.; Schofield, P.; Lewis, T. Gating mechanisms in Cys-loop receptors. Eur. Biophys. J. 2009, 39, 37–49. [Google Scholar]
- Taly, A.; Corringer, P.; Guedin, D.; Lestage, P.; Changeux, J. Nicotinic receptors: allosteric transitions and therapeutic targets in the nervous system. Nature Rev. Drug Dicovery 2009, 8, 733–750. [Google Scholar]
- Burzomato, V.; Beato, M.; Groot-Kormelink, P.; Colquhoun, D.; Sivilotti, L. Single-channel behavior of heteromeric α1β glycine receptors: an attempt to detect a conformational change before the channel opens. J. Neurosci. 2004, 24, 10924–10940. [Google Scholar]
- Moroni, M.; Zwart, R.; Sher, E.; Cassels, B.; Bermudez, I. α4β2 nicotinic receptors with high and low acetylcholine sensitivity: pharmacology, stoichiometry, and sensitivity to long-term exposure to nicotine. Mol. Pharmacol. 2006, 70, 755–768. [Google Scholar]
- Monod, J.; Wyman, J.; Changeux, J. On the nature of allosteric proteins: a plausible model. J. Mol. Biol. 1965, 12, 88–118. [Google Scholar]
- Jackson, M. Spontaneous openings of the acetylcholine receptor channel. Proc. Natl. Acad. Sci. USA 1984, 81, 3901–3904. [Google Scholar]
- Chang, Y.; Weiss, D. Allosteric activation mechanism of the α1β2γ2 γ-aminobutyric acid type A receptor revealed by mutation of the conserved M2 leucine. Biophys. J. 1999, 77, 2542–2551. [Google Scholar]
- Lape, R.; Colquhoun, D.; Sivilotti, L. On the nature of partial agonism in the nicotinic receptor superfamily. Nature 2008, 454, 722–727. [Google Scholar]
- Campo-Soria, C.; Chang, Y.; Weiss, D. Mechanism of action of benzodiazepines on GABAA receptors. Br. J. Pharmacol. 2006, 148, 984–990. [Google Scholar]
- Rüsch, D.; Forman, S. Classic benzodiazepines modulate the open–close equilibrium in α1β2γ2L γ-aminobutyric acid type A receptors. Anesthesiology 2005, 102, 783–792. [Google Scholar]
- Bau, R.; Sigel, E. Benzodiazepines affect channel opening of GABAA receptors induced by either agonist binding site. Mol. Pharmacol. 2005, 67, 1005–1008. [Google Scholar]
- Kloda, H.; Czajkowski, C. Agonist-, antagonist-, and benzodiazepine-Induced structural changes in the a1Met113-Leu132 region of the GABAA receptor. Mol. Pharmacol. 2007, 71, 483–493. [Google Scholar]
- Rüsch, D.; Zhong, H.; Forman, S. Gating allosterism at a single class of etomidate sites on α1β2γ2L GABAA receptors accounts for both direct activation and agonist modulation. J. Biol. Chem. 2004, 279, 20982–20992. [Google Scholar]
- Li, G.; Chiara, D.; Sawyer, G.; Husain, S.; Olsen, R.; Cohen, J. Identification of a GABAA receptor anesthetic binding site at subunit interfaces by photolabeling with an etomidate analog. J. Neurosci. 2006, 26, 11599–11605. [Google Scholar]
- Stewart, D.; Desai, R.; Cheng, Q.; Liu, A.; Forman, S. Tryptophan mutations at Azi-Etomidate photo-incorporation sites on α1 or β2 subunits enhance GABAA receptor gating and reduce etomidate modulation. Mol. Pharmacol. 2008, 74, 1687–1695. [Google Scholar]
- Li, W.; Jin, X.; Covey, D.; Steinbach, J. Neuroactive steroids and human recombinant ρ1 GABAC receptors. J. Pharmacol. Exp. Ther. 2007, 323, 236–247. [Google Scholar]
- Li, P.; Khatri, A.; Bracamontes, J.; Weiss, D.; Steinbach, J.; Akk, G. Site-specific fluorescence reveals distinct structural changes induced in the human ρ1 GABA receptor by inhibitory neurosteroids. Mol. Pharmacol. 2010, 77, 539–546. [Google Scholar]
- Young, G.; Zwart, R.; Walker, A.; Sher, E.; Millar, N. Potentiation of α7 nicotinic acetylcholine receptors via an allosteric transmembrane site. Proc. Natl. Acad. Sci. USA 2008, 105, 14686–14691. [Google Scholar]
- Barron, S.; McLaughlin, J.; See, J.; Richards, V.; Rosenberg, R. An allosteric modulator of α7 nicotinic receptors, N- (5-Chloro-2,4-dimethoxyphenyl)-N'- (5-methyl-3-isoxazolyl)-urea (PNU-120596), causes conformational changes in the extracellular ligand binding domain similar to those caused by acetylcholine. Mol. Pharmacol. 2009, 76, 253–263. [Google Scholar]
- McLaughlin, J.; Barron, S.; See, J.; Rosenberg, R. Conformational changes in α7 acetylcholine receptors underlying allosteric modulation by divalent cations. BMC Pharmacol. 2009, 9, 1–13. [Google Scholar]
- Perkins, D.; Trudell, J.; Crawford, D.; Asatryan, L.; Alkana, R.; Davies, D. Loop 2 structure in glycine and GABAA receptors plays a key role in determining ethanol sensitivity. J. Biol. Chem. 2009, 284, 27304–27314. [Google Scholar]
- Perkins, D.; JR, T.; Crawford, D.; Alkana, R.; Davies, D. Targets for ethanol action and antagonism in loop 2 of the extracellular domain of glycine receptors. J. Neurochem. 2008, 106, 1337–1349. [Google Scholar]
- Yevenes, G.; Moraga-Cid, G.; Peoples, R.; Schmalzing, G.; Aguayo, L. A selective Gβγ-linked intracellular mechanism for modulation of a ligand-gated ion channel by ethanol. Proc. Natl. Acad. Sci. USA 2008, 51, 20523–20528. [Google Scholar]
- Chang, Y.; Weiss, D. Site-specific fluorescence reveals distinct structural changes with GABA receptor activation and antagonism. Nat. Neurosci. 2002, 5, 1163–1168. [Google Scholar]
- Zhang, J.; Xue, F.; Chang, Y. Agonist- and antagonist-induced conformational changes of loop F and their contributions to the ρ1 GABA receptor function. J. Physiol. 2009, 587, 139–153. [Google Scholar]
- Chang, C.; Olcese, R.; Olsen, R. A single M1 residue in the β2 subunit alters channel gating of GABAA receptor in anesthetic modulation and direct activation. J. Biol. Chem. 2003, 278, 42821–42828. [Google Scholar]
- Hu, X.; Peoples, R. Arginine 246 of the pretransmembrane domain 1 region alters 2,2,2-richloroethanol action in the 5-hydroxytryptamine3A receptor. J. Pharmacol. Exp. Ther. 2008, 324, 1011–1018. [Google Scholar]
- Hosie, A.; Dunne, E.; Harvey, R.; Smart, T. Zinc-mediated inhibition of GABA (A) receptors: discrete binding sites underlie subtype specificity. Nat. Neurosci. 2003, 6, 362–369. [Google Scholar]
- Kash, T.; Jenkins, A.; Kelley, J.; Trudell, J.; Harrison, N. Coupling of agonist binding to channel gating in the GABAA receptor. Nature 2003, 421, 272–275. [Google Scholar]
- Kash, T.; Dizon, M.; Trudell, J.; Harrison, N. Charged residues in the β2 subunit involved in GABAA receptor activation. J. Biol. Chem. 2004, 279, 4887–4893. [Google Scholar]
- Duncalfe, L.; Carpenter, M.; Smillie, L.; Martin, I.; Dunn, S. The major site of photoaffinity labeling of the GABAA receptor by [3H] flunitrazepam is histidine 102 of the a subunit. J. Biol. Chem. 1996, 271, 9209–9214. [Google Scholar]
- Wieland, H.; Luddens, H.; Seeburg, P. A single histidine in GABAA receptors is essential for benzodiazepine agonist binding. J. Biol. Chem. 1992, 267, 1426–1429. [Google Scholar]
- Amin, J.; Brooks-Kayal, A.; Weiss, D. Two tyrosine residues on the a subunit are crucial for benzodiazepine binding and allosteric modulation of γ-aminobutyric acid A receptors. Mol. Pharmacol. 1997, 51, 833–841. [Google Scholar]
- Schaerer, M.; Buhr, A.; Baur, R.; Sigel, E. Amino acid residue 200 on the α1 subunit of GABA (A) receptors affects the interaction with selected benzodiazepine binding site ligands. Eur. J. Pharmacol. 1998, 354, 283–288. [Google Scholar]
- Buhr, A.; Schaerer, M.; Baur, R.; Sigel, E. Residues at positions 206 and 209 of the α1 subunit of γ-aminobutyric acid A receptors influence affinities for benzodiazepine binding site ligands. Mol. Pharmacol. 1997, 52, 676–682. [Google Scholar]
- Buhr, A.; Baur, R.; Sigel, E. Subtle changes in residue 77 of the γ subunit of α1β2γ2 GABAA receptors drastically alter the affinity for ligands of the benzodiazepine binding site and suggest the presence of two sites. J. Biol. Chem. 1997, 272, 11799–11804. [Google Scholar]
- Wingrove, P.; Thompson, S.; Wafford, K.; Whiting, P. Key amino acids in the gamma subunit of the γ-aminobutyric acid A receptor that determine ligand binding and modulation at the benzodiazepine site. Mol. Pharmacol. 1997, 52, 874–881. [Google Scholar]
- Kucken, A.M.; Teissere, J.A.; Seffinga-Clark, J.; Wagner, D.A.; Czajkowski, C. Structural requirements for imidazobenzodiazepine binding to GABA (A) receptors. Mol. Pharmacol. 2003, 63, 289–296. [Google Scholar]
- Teissere, J.; Czajkowski, C. A β-strand in the γ2 subunit lines the benzodiazepine binding site of the GABAA receptor: structural rearrangements detected during channel gating. J. Neurosci. 2001, 21, 4977–4986. [Google Scholar]
- Buhr, A.; Sigel, E. A point mutation in the γ2 subunit of γ-aminobutyric acid type A receptors results in altered benzodiazepine site specificity. Proc. Natl. Acad. Sci. USA 1997, 94, 8824–8829. [Google Scholar]
- Sancar, F.; Ericksen, S.; Kucken, A.; Teissére, J.; Czajkowski, C. Structural determinants for high-affinity zolpidem binding to GABA-A receptors. Mol. Pharmacol. 2007, 71, 38–46. [Google Scholar]
- Seo, S.; Henry, J.; Lewis, A.; Wang, N.; Levandoski, M. The positive allosteric modulator morantel binds at noncanonical subunit interfaces of neuronal nicotinic acetylcholine receptors. J. Neurosci. 2009, 29, 8734–8742. [Google Scholar]
- Jenkins, A.; Greenblatt, E.; Faulkner, H.; Bertaccini, E.; Light, A.; Lin, A.; Andreasen, A.; Viner, A.; Trudell, J.; Harrison, N. Evidence for a common binding cavity for three general anesthetics within the GABAA receptor. J. Neurosci. 2001, 21, RC136. [Google Scholar]
- Wick, M.; Mihic, S.; Ueno, S.; Mascia, M.; Trudell, J.; Brozowski, S.; Ye, Q.; Harrison, N.; Harris, R. Mutations of gamma-aminobutyric acid and glycine receptors change alcohol cutoff: evidence for an alcohol receptor? Proc. Natl. Acad. Sci. USA 1998, 95, 6504–6509. [Google Scholar]
- Frank, N. General anaesthesia: from molecular targets to neuronal pathways of sleep and arousal. Nat. Rev. Neurosci. 2008, 9, 370–386. [Google Scholar]
- Belelli, D.; Lambert, J.; Peters, J.; Wafford, K.; Whiting, P. The interaction of the general anesthetic etomidate with the γ-aminobutyric acid type A receptor is influenced by a single amino acid. Proc. Natl. Acad. Sci. USA 1997, 94, 11031–11036. [Google Scholar]
- Bali, M.; Akabas, M. Defining the propofol binding site location on the GABAA receptor. Mol. Pharmacol. 2004, 65, 68–76. [Google Scholar]
- Lopreato, G.; Banerjee, P.; Mihic, S. Amino acids in transmembrane domain two influence anesthetic enhancement of serotonin-3A receptor function. Brain Res. Mol. Brain Res. 2003, 118, 45–51. [Google Scholar]
- Li, G.; Chiara, D.; Cohen, J.; Olsen, R. Numerous classes of general anesthetics inhibit etomidate binding to γ-aminobutyric acid type A (GABAA) receptors. J. Biol. Chem. 2010, 285, 8615–8620. [Google Scholar]
- Li, G.; Chiara, D.; Cohen, J.; RW, O. Neurosteroids allosterically modulate binding of the anesthetic etomidate to γ-aminobutyric acid type A receptors. J. Biol. Chem. 2009, 284, 11771–11775. [Google Scholar]
- Belelli, D.; Lambert, J. Neurosteroids: endogenous regulators of the GABAA receptor. Nat. Rev. Neurosci. 2005, 6, 565–575. [Google Scholar]
- Hosie, A.; Wilkins, M.; da Silva, H.; Smart, T. Endogenous neurosteroids regulate GABAA receptors through two discrete transmembrane sites. Nature 2006, 444, 486–489. [Google Scholar]
- Krause, R.; Buisson, B.; Bertrand, S.; Corringer, P.; Galzi, J.; Changeux, J.; Bertrand, D. Ivermectin: a positive allosteric effector of the α7 neuronal nicotinic acetylcholine receptor. Mol. Pharmacol. 1998, 53, 283–294. [Google Scholar]
- Zwart, R.; De Filippi, G.; Broad, L.; McPhie, G.; Pearson, K.; Baldwinson, T.; Sher, E. 5-Hydroxyindole potentiates human α7 nicotinic receptor-mediated responses and enhances acetylcholine-induced glutamate release in cerebellar slices. Neuropharmacology 2002, 43, 374–384. [Google Scholar]
- Charpantier, E.; Wiesner, A.; Huh, K.; Ogier, R.; Hoda, J.; Allaman, G.; Raggenbass, M.; Feuerbach, D.; Bertrand, D.; Fuhrer, C. α7 neuronal nicotinic acetylcholine receptors are negatively regulated by tyrosine phosphorylation and Src-family kinases. J. Neurosci. 2005, 25, 9836–9849. [Google Scholar]
- Ng, H.; Whittemore, E.; Tran, M.; Hogenkamp, D.; Broide, R.; Johnstone, T.; Zheng, L.; Stevens, K.; Gee, K. Nootropic α7 nicotinic receptor allosteric modulator derived from GABAA receptor modulators. Proc. Natl. Acad. Sci. USA 2007, 104, 8059–8064. [Google Scholar]
- Timmermann, D.; Grønlien, J.; Kohlhaas, K.; Nielsen, E.; Dam, E.; Jørgensen, T.; Ahring, P.; Peters, D.; Holst, D.; Christensen, J.; Malysz, J.; Briggs, C.; Gopalakrishnan, M.; Olsen, G. An allosteric modulator of the α7 nicotinic acetylcholine receptor possessing cognition-enhancing properties in vivo. J. Pharmacol. Exp. Ther. 2007, 323, 294–307. [Google Scholar]
- Hu, X.; Lovinger, D. The L293 residue in transmembrane domain 2 of the 5-HT3A receptor is a molecular determinant of allosteric modulation by 5-hydroxyindole. Neuropharmacology 2008, 54, 1153–1165. [Google Scholar]
- Hurst, R.; Hajós, M.; Raggenbass, M.; Wall, T.; Higdon, N.; Lawson, J.; Rutherford-Root, K.; Berkenpas, M.; Hoffmann, W.; Piotrowski, D.; Groppi, V.; Allaman, G.; Ogier, R.; Bertrand, S.; Bertrand, D.; Arneric, S. A novel positive allosteric modulator of the α7 neuronal nicotinic acetylcholine receptor: in vitro and in vivo characterization. J. Neurosci. 2005, 25, 4396–4405. [Google Scholar]
- Smart, T.; Moss, S.; Xie, X.; Huganir, R. GABAA receptors are differentially sensitive to zinc: dependence on subunit composition. Br. J. Pharmacol. 1991, 103, 1837–1839. [Google Scholar]
- Chang, Y.; Amin, J.; Weiss, D. Zinc is a mixed antagonist of homomeric ρ1 γ-aminobutyric acid-activated channels. Mol. Pharmacol. 1995, 47, 595–602. [Google Scholar]
- Guo, X.; Lester, R. Regulation of nicotinic acetylcholine receptor desensitization by Ca2+. J. Neurophysiol. 2007, 97, 93–101. [Google Scholar]
- Chimienti, F.; Hogg, R.; Plantard, L.; Lehmann, C.; Brakch, N.; Fischer, J.; Huber, M.; Bertrand, D.; Hohl, D. Identification of SLURP-1 as an epidermal neuromodulator explains the clinical phenotype of Mal de Meleda. Hum. Mol. Genet. 2003, 12, 3017–3024. [Google Scholar]
- Tipps, M.; Lawshe, J.; Ellington, A.; Mihic, S. Identification of novel specific allosteric modulators of the glycine receptor using phage display. J. Biol. Chem. 2010. Epub ahead of print.. [Google Scholar]
- Trudell, J.; Yue, M.; Bertaccini, E.; Jenkins, A.; Harrison, N. Molecular modeling and mutagenesis reveals a tetradentate binding site for Zn2+ in GABA (A) αβ receptors and provides a structural basis for the modulating effect of the gamma subunit. J. Chem. Inf. Mod. 2008, 48, 344–349. [Google Scholar]
- Le Novère, N.; Grutter, T.; Changeux, J. Models of the extracellular domain of the nicotinic receptors and of agonist- and Ca2+-binding sites. Proc Natl Acad Sci USA 2002, 99, 3210–3215. [Google Scholar]
- Galzi, J.; Bertrand, S.; Corringer, P.; Changeux, J.; Bertrand, D. Identification of calcium binding sites that regulate potentiation of a neuronal nicotinic acetylcholine receptor. EMBO J 1996, 15, 5824–5832. [Google Scholar]
- Yevenes, G.; Moraga-Cid, G.; Guzma´n, L.; Haeger, S.; Oliveira, L.; Olate, J.; Schmalzing, G.; Aguayo, L. Molecular determinants for G protein βγ modulation of ionotropic glycine receptors. J. Biol. Chem. 2006, 281, 39300–39307. [Google Scholar]
- Atack, J. Preclinical and clinical pharmacology of the GABAA receptor α5 subtype-selective inverse agonist alpha5IA. Pharmacol. Ther. 2010, 125, 11–26. [Google Scholar]
- Ballard, T.; Knoflach, F.; Prinssen, E.; Borroni, E.; Vivian, J.; Basile, J.; Gasser, R.; Moreau, J.; Wettstein, J.; Buettelmann, B.; Knust, H.; Thomas, A.; Trube, G.; Hernandez, M. RO4938581, a novel cognitive enhancer acting at GABAA α5 subunit-containing receptors. Psychopharmocology (Berlin) 2009, 202, 207–223. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chang, Y.; Huang, Y.; Whiteaker, P. Mechanism of Allosteric Modulation of the Cys-loop Receptors. Pharmaceuticals 2010, 3, 2592-2609. https://doi.org/10.3390/ph3082592
Chang Y, Huang Y, Whiteaker P. Mechanism of Allosteric Modulation of the Cys-loop Receptors. Pharmaceuticals. 2010; 3(8):2592-2609. https://doi.org/10.3390/ph3082592
Chicago/Turabian StyleChang, Yongchang, Yao Huang, and Paul Whiteaker. 2010. "Mechanism of Allosteric Modulation of the Cys-loop Receptors" Pharmaceuticals 3, no. 8: 2592-2609. https://doi.org/10.3390/ph3082592
APA StyleChang, Y., Huang, Y., & Whiteaker, P. (2010). Mechanism of Allosteric Modulation of the Cys-loop Receptors. Pharmaceuticals, 3(8), 2592-2609. https://doi.org/10.3390/ph3082592