Role of the G Protein-Coupled Receptor, mGlu1, in Melanoma Development

{kind=link}
Abstract
Introduction
GPCRs
Role of GPCRs in Human Diseases
GPCRs as Oncogenes
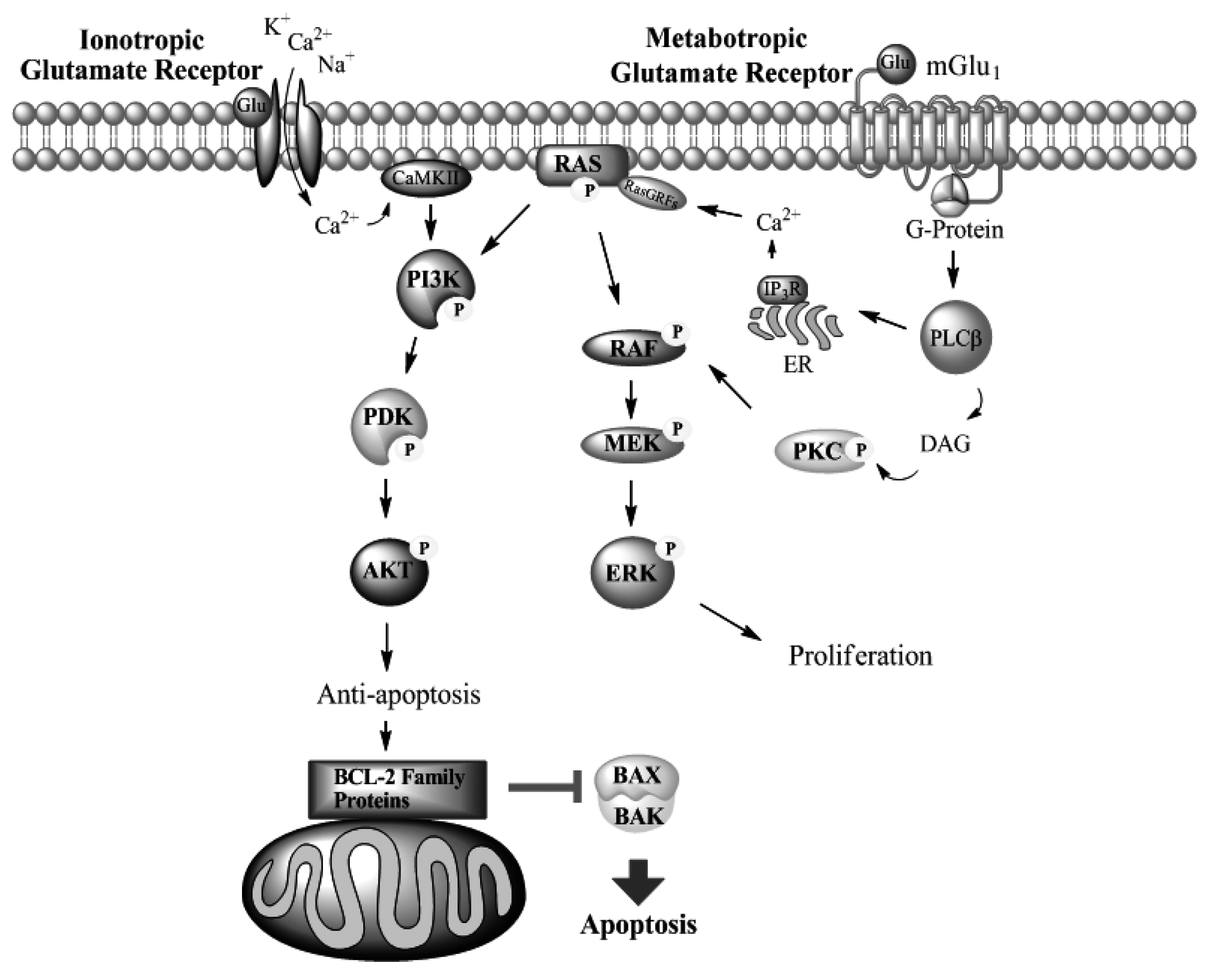
Metabotropic Glutamate Receptors
mGlu1 in Melanoma
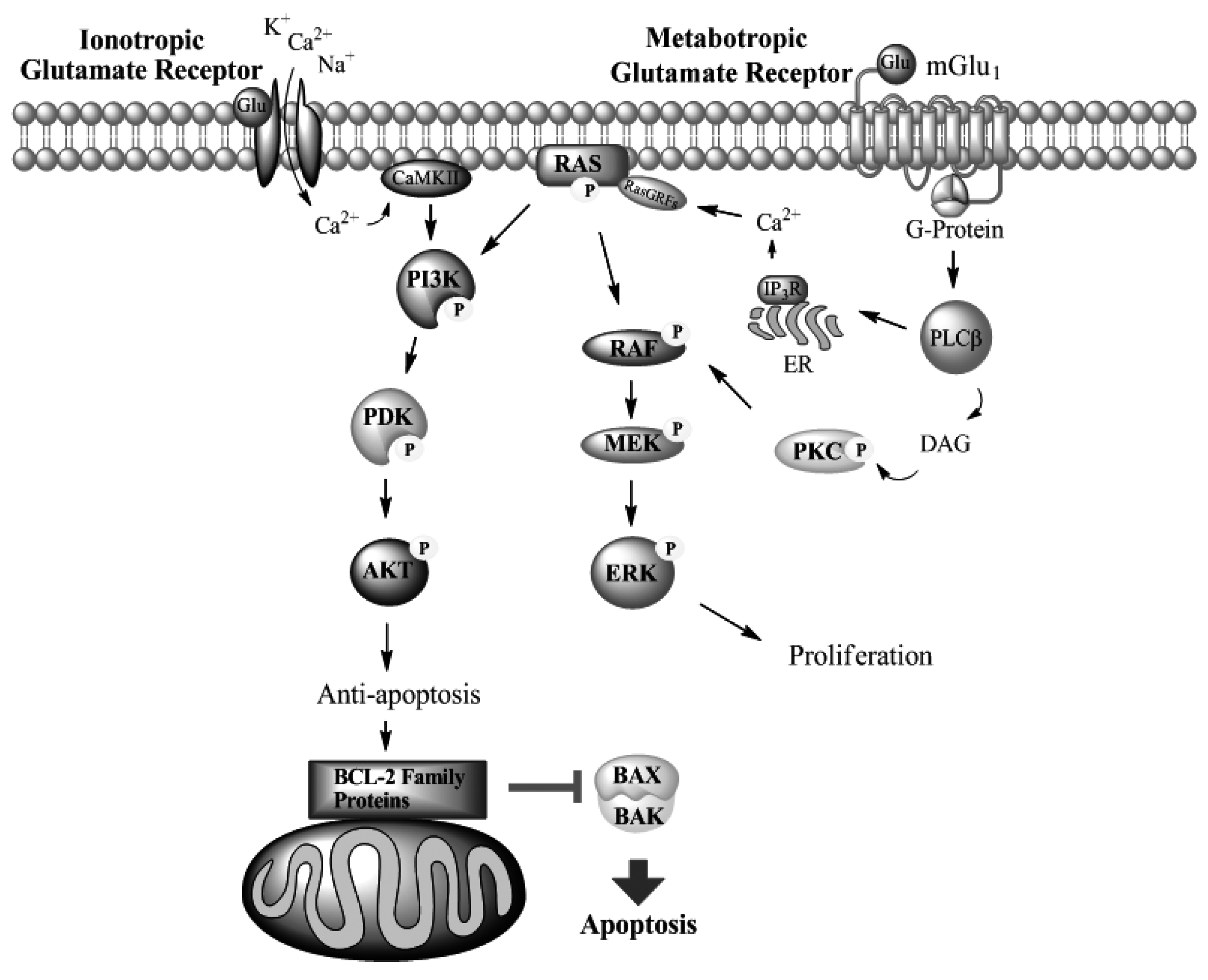
Glutamate Receptors in Other Cancers
Conclusions
References
- Rigel, D.S. Malignant melanoma: Perspectives on incidence and its effects on awareness, diagnosis, and treatment. Ca-A Cancer J. Clin. 1996, 46, 195–198. [Google Scholar]
- Chang, A.E.; Karnell, L.H.; Menck, H.R. The National Cancer Data Base report on cutaneous and noncutaneous melanoma: A summary of 84,836 cases from the past decade. The American College of Surgeons Commission on Cancer and the American Cancer Society. Cancer 1998, 83, 1664–1678. [Google Scholar] [PubMed]
- American Cancer Society, Cancer facts & figures; American Cancer Society: Atlanta, GA, USA, 2010.
- Jemal, A.; Siegel, R.; Ward, E.; Hao, Y.; Xu, J.; Murray, T.; Thun, M.J. Cancer statistics, 2008. CA-Cancer J. Clin. 2008, 58, 71–96. [Google Scholar] [CrossRef] [PubMed]
- Cannon-Albright, L.A.; Goldgar, D.E.; Meyer, L.J.; Lewis, C.M.; Anderson, D.E.; Fountain, J.W.; Hegi, M.E.; Wiseman, R.W.; Petty, E.M.; Bale, A.E.; et al. Assignment of a locus for familial melanoma, MLM, to chromosome 9p13-p22. Science 1992, 258, 1148–1152. [Google Scholar] [PubMed]
- Quelle, D.E.; Cheng, M.; Ashmun, R.A.; Sherr, C.J. Cancer-associated mutations at the INK4a locus cancel cell cycle arrest by p16INK4a but not by the alternative reading frame protein p19ARF. Proc. Natl. Acad. Sci. USA 1997, 94, 669–673. [Google Scholar]
- Quelle, D.E.; Zindy, F.; Ashmun, R.A.; Sherr, C.J. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell 1995, 83, 993–1000. [Google Scholar]
- Becker, T.M.; Rizos, H.; Kefford, R.F.; Mann, G.J. Functional impairment of melanoma-associated p16(INK4a) mutants in melanoma cells despite retention of cyclin-dependent kinase 4 binding. Clin. Cancer Res. 2001, 7, 3282–3288. [Google Scholar]
- Sotillo, R.; Garcia, J.F.; Ortega, S.; Martin, J.; Dubus, P.; Barbacid, M.; Malumbres, M. Invasive melanoma in Cdk4-targeted mice. Proc. Natl. Acad. Sci. USA 2001, 98, 13312–13317. [Google Scholar]
- McKenzie, H.; Becker, T.M.; Scurr, L.L.; Kefford, R.F.; Rizos, H. Wild type and melanoma-associated mutant p16(IN4a) proteins do not oligomerize in vivo. Pigment Cell Melanoma Res. 2009, 22, 131–133. [Google Scholar] [PubMed]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; Davis, N.; Dicks, E.; Ewing, R.; Floyd, Y.; Gray, K.; Hall, S.; Hawes, R.; Hughes, J.; Kosmidou, V.; Menzies, A.; Mould, C.; Parker, A.; Stevens, C.; Watt, S.; Hooper, S.; Wilson, R.; Jayatilake, H.; Gusterson, B.A.; Cooper, C.; Shipley, J.; Hargrave, D.; Pritchard-Jones, K.; Maitland, N.; Chenevix-Trench, G.; Riggins, G.J.; Bigner, D.D.; Palmieri, G.; Cossu, A.; Flanagan, A.; Nicholson, A.; Ho, J.W.; Leung, S.Y.; Yuen, S.T.; Weber, B.L.; Seigler, H.F.; Darrow, T.L.; Paterson, H.; Marais, R.; Marshall, C.J.; Wooster, R.; Stratton, M.R.; Futreal, P.A. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar]
- Collisson, E.; De, A.; Suzuki, H.; Gambhir, S.; Kolodney, M. Treatment of metastatic melanoma with an orally available inhibitor of the Ras-Raf-MAPK cascade. Can. Res. 2003, 63, 5669–5673. [Google Scholar]
- Libra, M.; Malaponte, G.; Navolanic, P.M.; Gangemi, P.; Bevelacqua, V.; Proietti, L.; Bruni, B.; Stivala, F.; Mazzarino, M.C.; Travali, S.; McCubrey, J.A. Analysis of BRAF mutation in primary and metastatic melanoma. Cell Cycle 2005, 4, 1382–1384. [Google Scholar]
- Dahl, C.; Guldberg, P. The genome and epigenome of malignant melanoma. APMIS 2007, 115, 1161–1176. [Google Scholar]
- Tuveson, D.A.; Weber, B.L.; Herlyn, M. BRAF as a potential therapeutic target in melanoma and other malignancies. Cancer Cell 2003, 4, 95–98. [Google Scholar]
- Pollock, P.; Harper, U.; Hansen, K.; Yudt, L.; Stark, M.; Robbins, C.; Moses, T.; Hostetter, G.; Wagner, U.; Kakareka, J.; Salem, G.; Pohida, T.; Heenan, P.; Duray, P.; Kallioniemi, O.; Hayward, N.; Trent, J.; Meltzer, P. High frequency of BRAF mutations in nevi. Nat. Genet. 2003b, 33, 19–20. [Google Scholar] [PubMed]
- Whitwam, T.; Vanbrocklin, M.W.; Russo, M.E.; Haak, P.T.; Bilgili, D.; Resau, J.H.; Koo, H.M.; Holmen, S.L. Differential oncogenic potential of activated RAS isoforms in melanocytes. Oncogene 2007, 26, 4563–4570. [Google Scholar]
- Barbacid, M. Ras oncogenes: Their role in neoplasia. Eur. J. Clin. Invest. 1990, 20, 225–235. [Google Scholar]
- Pollock, P.M.; Cohen-Solal, K.; Sood, R.; Namkoong, J.; Martino, J.J.; Koganti, A.; Zhu, H.; Robbins, C.; Makalowska, I.; Shin, S.S.; Marin, Y.; Roberts, K.G.; Yudt, L.M.; Chen, A.; Cheng, J.; Incao, A.; Pinkett, H.W.; Graham, C.L.; Dunn, K.; Crespo-Carbone, S.M.; Mackason, K.R.; Ryan, K.B.; Sinsimer, D.; Goydos, J.; Reuhl, K.R.; Eckhaus, M.; Meltzer, P.S.; Pavan, W.J.; Trent, J.M.; Chen, S. Melanoma mouse model implicates metabotropic glutamate signaling in melanocytic neoplasia. Nat. Genet. 2003, 34, 108–112. [Google Scholar]
- Gudermann, T.; Nurnberg, B.; Schultz, G. Receptors and G-Proteins as primary components of transmembrane signal transduction. G-protein-coupled receptors -structure and function. J. Molec. Med.-JMM 1995, 73, 51–63. [Google Scholar] [CrossRef]
- Bourne, H.R.; Sanders, D.A.; McCormick, F. The GTPase superfamily: a conserved switch for diverse cell functions. Nature 1990, 348, 125–132. [Google Scholar]
- Bourne, H.R.; Sanders, D.A.; McCormick, F. The GTPase superfamily: conserved structure and molecular mechanism. Nature 1991, 349, 117–127. [Google Scholar]
- Fredriksson, R.; Schioth, H.B. The repertoire of G-protein-coupled receptors in fully sequenced genomes. Mol. Pharmacol. 2005, 67, 1414–1425. [Google Scholar]
- Insel, P.A.; Tang, C.M.; Hahntow, I.; Michel, M.C. Impact of GPCRs in clinical medicine: Monogenic diseases, genetic variants and drug targets. Biochim.Biophys. Acta 2007, 1768, 994–1005. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.D.; Burnham, W.M.; Cole, D.E. The G protein-coupled receptors: Pharmacogenetics and disease. Crit. Rev. Clin. Lab. Sci. 2005, 42, 311–392. [Google Scholar]
- Spiegel, A.M.; Weinstein, L.S. Inherited diseases involving g proteins and g protein-coupled receptors. Annu.Rev. Med. 2004, 55, 27–39. [Google Scholar]
- Muradov, K.G.; Artemyev, N.O. Loss of the effector function in a transducin-alpha mutant associated with Nougaret night blindness. J. Biol. Chem. 2000, 275, 6969–6974. [Google Scholar]
- Wuller, S.; Wiesner, B.; Loffler, A.; Furkert, J.; Krause, G.; Hermosilla, R.; Schaefer, M.; Schulein, R.; Rosenthal, W.; Oksche, A. Pharmacochaperones post-translationally enhance cell surface expression by increasing conformational stability of wild-type and mutant vasopressin V2 receptors. J. Biol. Chem. 2004, 279, 47254–47263. [Google Scholar]
- Small, K.M.; Wagoner, L.E.; Levin, A.M.; Kardia, S.L.; Liggett, S.B. Synergistic polymorphisms of beta1- and alpha2C-adrenergic receptors and the risk of congestive heart failure. N. Engl. J. Med. 2002, 347, 1135–1142. [Google Scholar]
- Na, G.Y.; Lee, K.H.; Kim, M.K.; Lee, S.J.; Kim, D.W.; Kim, J.C. Polymorphisms in the melanocortin-1 receptor (MC1R) and agouti signaling protein (ASIP) genes in Korean vitiligo patients. Pigment Cell Res. 2003, 16, 383–387. [Google Scholar]
- Liu, R.; Paxton, W.A.; Choe, S.; Ceradini, D.; Martin, S.R.; Horuk, R.; MacDonald, M.E.; Stuhlmann, H.; Koup, R.A.; Landau, N.R. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell 1996, 86, 367–377. [Google Scholar]
- Marinissen, M.J.; Gutkind, J.S. G-protein-coupled receptors and signaling networks: emerging paradigms. Trends Pharmacol.Sci. 2001, 22, 368–376. [Google Scholar]
- Rozengurt, E. Early signals in the mitogenic response. Science 1986, 234, 161–166. [Google Scholar]
- Gutkind, J.S.; Novotny, E.A.; Brann, M.R.; Robbins, K.C. Muscarinic acetylcholine receptor subtypes as agonist-dependent oncogenes. Proc. Natl. Acad. Sci. USA 1991, 88, 4703–4707. [Google Scholar]
- Gutkind, J.S. Cell growth control by G protein-coupled receptors: From signal transduction to signal integration. Oncogene 1998, 17, 1331–1342. [Google Scholar]
- Cuttitta, F.; Carney, D.N.; Mulshine, J.; Moody, T.W.; Fedorko, J.; Fischler, A.; Minna, J.D. Bombesin-like peptides can function as autocrine growth factors in human small-cell lung cancer. Nature 1985, 316, 823–826. [Google Scholar]
- Cardona, C.; Rabbitts, P.H.; Spindel, E.R.; Ghatei, M.A.; Bleehen, N.M.; Bloom, S.R.; Reeve, J.G. Production of neuromedin B and neuromedin B gene expression in human lung tumor cell lines. Cancer Res. 1991, 51, 5205–5211. [Google Scholar]
- Namkoong, J.; Shin, S.S.; Lee, H.J.; Marin, Y.E.; Wall, B.A.; Goydos, J.S.; Chen, S. Metabotropic glutamate receptor 1 and glutamate signaling in human melanoma. Cancer Res. 2007, 67, 2298–2305. [Google Scholar]
- Hoogduijn, M.J.; Hitchcock, I.S.; Smit, N.P.; Gillbro, J.M.; Schallreuter, K.U.; Genever, P.G. Glutamate receptors on human melanocytes regulate the expression of MiTF. Pigment Cell Res. 2006, 19, 58–67. [Google Scholar]
- Young, D.; Waitches, G.; Birchmeier, C.; Fasano, O.; Wigler, M. Isolation and characterization of a new cellular oncogene encoding a protein with multiple potential transmembrane domains. Cell 1986, 45, 711–719. [Google Scholar]
- Julius, D.; Livelli, T.J.; Jessell, T.M.; Axel, R. Ectopic expression of the serotonin 1c receptor and the triggering of malignant transformation. Science 1989, 244, 1057–1062. [Google Scholar]
- Sodhi, A.; Montaner, S.; Patel, V.; Zohar, M.; Bais, C.; Mesri, E.A.; Gutkind, J.S. The Kaposi's sarcoma-associated herpes virus G protein-coupled receptor up-regulates vascular endothelial growth factor expression and secretion through mitogen-activated protein kinase and p38 pathways acting on hypoxia-inducible factor 1alpha. Cancer Res. 2000, 60, 4873–4880. [Google Scholar]
- Bais, C.; Santomasso, B.; Coso, O.; Arvanitakis, L.; Raaka, E.G.; Gutkind, J.S.; Asch, A.S.; Cesarman, E.; Gershengorn, M.C.; Mesri, E.A. G-protein-coupled receptor of Kaposi's sarcoma-associated herpesvirus is a viral oncogene and angiogenesis activator. Nature 1998, 391, 86–89. [Google Scholar]
- Montaner, S.; Sodhi, A.; Pece, S.; Mesri, E.A.; Gutkind, J.S. The Kaposi's sarcoma-associated herpesvirus G protein-coupled receptor promotes endothelial cell survival through the activation of Akt/protein kinase B. Cancer Res. 2001, 61, 2641–2648. [Google Scholar]
- Mazzuco, T.L.; Chabre, O.; Feige, J.J.; Thomas, M. Aberrant GPCR expression is a sufficient genetic event to trigger adrenocortical tumorigenesis. Mol. Cell Endocrinol. 2007, 265-266, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Mazzuco, T.L.; Chabre, O.; Sturm, N.; Feige, J.J.; Thomas, M. Ectopic expression of the gastric inhibitory polypeptide receptor gene is a sufficient genetic event to induce benign adrenocortical tumor in a xenotransplantation model. Endocrinology 2006, 147, 782–790. [Google Scholar]
- Sladeczek, F.; Pin, J.P.; Recasens, M.; Bockaert, J.; Weiss, S. Glutamate stimulates inositol phosphate formation in striatal neurones. Nature 1985, 317, 717–719. [Google Scholar]
- Tanabe, Y.; Masu, M.; Ishii, T.; Shigemoto, R.; Nakanishi, S. A family of metabotropic glutamate receptors. Neuron 1992, 8, 169–179. [Google Scholar]
- Masu, M.; Tanabe, Y.; Tsuchida, K.; Shigemoto, R.; Nakanishi, S. Sequence and expression of a metabotropic glutamate receptor. Nature 1991, 349, 760–765. [Google Scholar]
- Houamed, K.M.; Kuijper, J.L.; Gilbert, T.L.; Haldeman, B.A.; O'Hara, P.J.; Mulvihill, E.R.; Almers, W.; Hagen, F.S. Cloning, expression, and gene structure of a G protein-coupled glutamate receptor from rat brain. Science 1991, 252, 1318–1321. [Google Scholar] [PubMed]
- Conn, P.J.; Pin, J.-P. Pharmacology and functions of metabotropic glutamate receptors. Annu.Rev. Pharmacol. Toxicol. 1997, 37, 205–237. [Google Scholar]
- Hermans, E.; Challiss, R.A. Structural, signalling and regulatory properties of the group I metabotropic glutamate receptors: Prototypic family C G-protein-coupled receptors. Biochem J 2001, 359, 465–484. [Google Scholar]
- Goudet, C.; Magnaghi, V.; Landry, M.; Nagy, F.; Gereau, R.W.t.; Pin, J.P. Metabotropic receptors for glutamate and GABA in pain. Brain Res. Rev. 2009, 60, 43–56. [Google Scholar]
- Pin, J.P.; Kniazeff, J.; Goudet, C.; Bessis, A.S.; Liu, J.; Galvez, T.; Acher, F.; Rondard, P.; Prezeau, L. The activation mechanism of class-C G-protein coupled receptors. Biol. Cell 2004, 96, 335–342. [Google Scholar]
- Bradl, M.; Klein-Szanto, A.; Porter, S.; Mintz, B. Malignant melanoma in transgenic mice. Proc. Natl. Acad. Sci. USA 1991, 88, 164–168. [Google Scholar]
- Broome Powell, M.; Gause, P.R.; Hyman, P.; Gregus, J.; Lluria-Prevatt, M.; Nagle, R.; Bowden, G.T. Induction of melanoma in TPras transgenic mice. Carcinogenesis 1999, 20, 1747–1753. [Google Scholar]
- Chin, L.; Merlino, G.; DePinho, R.A. Malignant melanoma: Modern black plague and genetic black box. Genes. Dev. 1998, 12, 3467–3481. [Google Scholar]
- Iwamoto, T.; Takahashi, M.; Ito, M.; Hamatani, K.; Ohbayashi, M.; Wajjwalku, W.; Isobe, K.; Nakashima, I. Aberrant melanogenesis and melanocytic tumour development in transgenic mice that carry a metallothionein/ret fusion gene. EMBO J. 1991, 10, 3167–3175. [Google Scholar]
- Otsuka, T.; Takayama, H.; Sharp, R.; Celli, G.; LaRochelle, W.J.; Bottaro, D.P.; Ellmore, N.; Vieira, W.; Owens, J.W.; Anver, M.; Merlino, G. c-Met autocrine activation induces development of malignant melanoma and acquisition of the metastatic phenotype. Cancer Res. 1998, 58, 5157–5167. [Google Scholar]
- Sharpless, N.E.; Bardeesy, N.; Lee, K.H.; Carrasco, D.; Castrillon, D.H.; Aguirre, A.J.; Wu, E.A.; Horner, J.W.; DePinho, R.A. Loss of p16Ink4a with retention of p19Arf predisposes mice to tumorigenesis. Nature 2001, 413, 86–91. [Google Scholar]
- Krimpenfort, P.; Quon, K.C.; Mooi, W.J.; Loonstra, A.; Berns, A. Loss of p16Ink4a confers susceptibility to metastatic melanoma in mice. Nature 2001, 413, 83–86. [Google Scholar]
- Zhu, H.; Reuhl, K.; Zhang, X.; Botha, R.; Ryan, K.; Wei, J.; Chen, S. Development of heritable melanoma in transgenic mice. J. Invest. Dermatol. 1998, 110, 247–252. [Google Scholar]
- Colon-Teicher, L.; Wise, L.S.; Martino, J.J.; Baskin, L.; Sakoulas, G.; Pollack, R.; Chen, S. Genomic sequences capable of committing mouse and rat fibroblasts to adipogenesis. Nucl. Acid. Res. 1993, 21, 2223–2228. [Google Scholar]
- Chen, S.; Teicher, L.C.; Kazim, D.; Pollack, R.E.; Wise, L.S. Commitment of mouse fibroblasts to adipocyte differentiation by DNA transfection. Science 1989, 244, 582–585. [Google Scholar]
- Zhu, H.; Reuhl, K.; Botha, R.; Ryan, K.; Wei, J.; Chen, S. Development of early melanocytic lesions in transgenic mice predisposed to melanoma. Pigm.Cell Res. 2000, 13, 158–164. [Google Scholar]
- Trent, J.; Stanbridge, E.; McBride, H.; Meese, E.; Casey, G.; Araujo, D.; Witkowski, C.; Nagle, R. Tumorigenicity in human melanoma cell lines controlled by introduction of human chromosome 6. Science 1990, 247, 568–571. [Google Scholar]
- Thompson, F.H.; Emerson, J.; Olson, S.; Weinstein, R.; Leavitt, S.A.; Leong, S.P.; Emerson, S.; Trent, J.M.; Nelson, M.A.; Salmon, S.E.; et al. Cytogenetics of 158 patients with regional or disseminated melanoma. Subset analysis of near-diploid and simple karyotypes. Cancer Genet. Cytogenet. 1995, 83, 93–104. [Google Scholar] [PubMed]
- Ohtani, Y.; Harada, T.; Funasaka, Y.; Nakao, K.; Takahara, C.; Abdel-Daim, M.; Sakai, N.; Saito, N.; Nishigori, C.; Aiba, A. Metabotropic glutamate receptor subtype-1 is essential for in vivo growth of melanoma. Oncogene 2008, 27, 7162–7170. [Google Scholar]
- Funusaka, Y.; Harada, T.; Aiba, A.; Nishigori, C. Expression of metabotropic glutamate receptor 1 and phosphorylated extracellular signal-regulated kinase 1/2 proteins in human melanocytic lesions. Pigm. Cell Res. 2006, 19, 256. [Google Scholar]
- Marin, Y.E.; Namkoong, J.; Cohen-Solal, K.; Shin, S.S.; Martino, J.J.; Oka, M.; Chen, S. Stimulation of oncogenic metabotropic glutamate receptor 1 in melanoma cells activates ERK1/2 via PKCepsilon. Cell Signal 2006, 18, 1279–1286. [Google Scholar]
- Ishiuchi, S.; Tsuzuki, K.; Yoshida, Y.; Yamada, N.; Hagimura, N.; Okado, H.; Miwa, A.; Kurihara, H.; Nakazato, Y.; Tamura, M.; Sasaki, T.; Ozawa, S. Blockage of Ca(2+)-permeable AMPA receptors suppresses migration and induces apoptosis in human glioblastoma cells. Nat. Med. 2002, 8, 971–978. [Google Scholar]
- Rzeski, W.; Turski, L.; Ikonomidou, C. Glutamate antagonists limit tumor growth. Proc. Natl. Acad. Sci. USA 2001, 98, 6372–6377. [Google Scholar]
- Otsuka, T.; Takayama, H.; Sharp, R.; Celli, G.; LaRochelle, W.J.; Bottaro, D.P.; Ellmore, N.; Vieira, W.; Owens, J.W.; Anver, M.; Merlino, G. c-Met autocrine activation induces development of malignant melanoma and acquisition of the metastatic phenotype. Cancer Res. 1998, 58, 5157–5167. [Google Scholar]
- Bensimon, G.; Lacomblez, L.; Meininger, V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N. Engl. J. Med. 1994, 330, 585–591. [Google Scholar] [PubMed]
- Van Den Bosch, L.; Van Damme, P.; Bogaert, E.; Robberecht, W. The role of excitotoxicity in the pathogenesis of amyotrophic lateral sclerosis. Biochim. Biophys. Acta 2006, 1762, 1068–1082. [Google Scholar] [PubMed]
- Bryson, H.M.; Fulton, B.; Benfield, P. Riluzole. A review of its pharmacodynamic and pharmacokinetic properties and therapeutic potential in amyotrophic lateral sclerosis. Drugs 1996, 52, 549–563. [Google Scholar] [PubMed]
- Doble, A. The pharmacology and mechanism of action of riluzole. Neurology 1996, 47, S233–S241. [Google Scholar] [PubMed]
- Kretschmer, B.D.; Kratzer, U.; Schmidt, W.J. Riluzole, a glutamate release inhibitor, and motor behavior. Naunyn Schmiedebergs Arch Pharmacol 1998, 358, 181–190. [Google Scholar] [PubMed]
- Wang, S.J.; Wang, K.Y.; Wang, W.C. Mechanisms underlying the riluzole inhibition of glutamate release from rat cerebral cortex nerve terminals (synaptosomes). Neuroscience 2004, 125, 191–201. [Google Scholar]
- Yip, D.; Le, M.N.; Chan, J.L.; Lee, J.H.; Mehnert, J.A.; Yudd, A.; Kempf, J.; Shih, W.J.; Chen, S.; Goydos, J.S. A phase 0 trial of riluzole in patients with resectable stage III and IV melanoma. Clin. Cancer Res. 2009, 15, 3896–3902. [Google Scholar]
- Wilhelm, S.M.; Carter, C.; Tang, L.; Wilkie, D.; McNabola, A.; Rong, H.; Chen, C.; Zhang, X.; Vincent, P.; McHugh, M.; Cao, Y.; Shujath, J.; Gawlak, S.; Eveleigh, D.; Rowley, B.; Liu, L.; Adnane, L.; Lynch, M.; Auclair, D.; Taylor, I.; Gedrich, R.; Voznesensky, A.; Riedl, B.; Post, L.E.; Bollag, G.; Trail, P.A. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004, 64, 7099–7109. [Google Scholar]
- Lee, H.; Wall, B.; Wangari-Talbot, J.; Shin, S.; Rosenberg, S.; Chan, J.-K.; Namkoong, J.; Goydos, J.; Chen, S. Targeting Metabotropic Glutamate Signaling in Melanoma; Single Agent and Combinatorial Therapies. 2010; in review. [Google Scholar]
- Kalariti, N.; Pissimissis, N.; Koutsilieris, M. The glutamatergic system outside the CNS and in cancer biology. Expert. Opin. Investig. Drugs 2005, 14, 1487–1496. [Google Scholar]
- Aronica, E.; Yankaya, B.; Jansen, G.H.; Leenstra, S.; van Veelen, C.W.; Gorter, J.A.; Troost, D. Ionotropic and metabotropic glutamate receptor protein expression in glioneuronal tumours from patients with intractable epilepsy. Neuropathol.Appl. Neurobiol. 2001, 27, 223–237. [Google Scholar]
- Naarala, J.; Nykvist, P.; Tuomala, M.; Savolainen, K. Excitatory amino acid-induced slow biphasic responses of free intracellular calcium in human neuroblastoma cells. FEBS Lett. 1993, 330, 222–226. [Google Scholar]
- Stepulak, A.; Luksch, H.; Gebhardt, C.; Uckermann, O.; Marzahn, J.; Sifringer, M.; Rzeski, W.; Staufner, C.; Brocke, K.S.; Turski, L.; Ikonomidou, C. Expression of glutamate receptor subunits in human cancers. Histochem. Cell Biol. 2009, 132, 435–445. [Google Scholar]
- Iacovelli, L.; Arcella, A.; Battaglia, G.; Pazzaglia, S.; Aronica, E.; Spinsanti, P.; Caruso, A.; De Smaele, E.; Saran, A.; Gulino, A.; D'Onofrio, M.; Giangaspero, F.; Nicoletti, F. Pharmacological activation of mGlu4 metabotropic glutamate receptors inhibits the growth of medulloblastomas. J. Neurosci. 2006, 26, 8388–8397. [Google Scholar]
- de Groot, J.F.; Piao, Y.; Lu, L.; Fuller, G.N.; Yung, W.K. Knockdown of GluR1 expression by RNA interference inhibits glioma proliferation. J. Neurooncol. 2008, 88, 121–133. [Google Scholar]
- Chang, H.J.; Yoo, B.C.; Lim, S.B.; Jeong, S.Y.; Kim, W.H.; Park, J.G. Metabotropic glutamate receptor 4 expression in colorectal carcinoma and its prognostic significance. Clin. Cancer Res. 2005, 11, 3288–3295. [Google Scholar]
- Yoo, B.C.; Jeon, E.; Hong, S.H.; Shin, Y.K.; Chang, H.J.; Park, J.G. Metabotropic glutamate receptor 4-mediated 5-Fluorouracil resistance in a human colon cancer cell line. Clin. Cancer Res. 2004, 10, 4176–4184. [Google Scholar]
- Park, S.Y.; Lee, S.A.; Han, I.H.; Yoo, B.C.; Lee, S.H.; Park, J.Y.; Cha, I.H.; Kim, J.; Choi, S.W. Clinical significance of metabotropic glutamate receptor 5 expression in oral squamous cell carcinoma. Oncol. Rep. 2007, 17, 81–87. [Google Scholar] [Green Version]
- Liu, J.W.; Kim, M.S.; Nagpal, J.; Yamashita, K.; Poeta, L.; Chang, X.; Lee, J.; Park, H.L.; Jeronimo, C.; Westra, W.H.; Mori, M.; Moon, C.; Trink, B.; Sidransky, D. Quantitative hypermethylation of NMDAR2B in human gastric cancer. Int. J. Cancer 2007, 121, 1994–2000. [Google Scholar]
- Abdul, M.; Hoosein, N. N-methyl-D-aspartate receptor in human prostate cancer. J. Membr. Biol. 2005, 205, 125–128. [Google Scholar]
- Luikart, S.D.; Kennealey, G.T.; Kirkwood, J.M. Randomized phase III trial of vinblastine, bleomycin, and cis-dichlorodiammine-platinum versus dacarbazine in malignant melanoma. J. Clin. Oncol. 1984, 2, 164–168. [Google Scholar] [PubMed]
- Huncharek, M.; Caubet, J.F.; McGarry, R. Single-agent DTIC versus combination chemotherapy with or without immunotherapy in metastatic melanoma: a meta-analysis of 3273 patients from 20 randomized trials. Melanoma. Res. 2001, 11, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Kirkwood, J.M.; Strawderman, M.H.; Ernstoff, M.S.; Smith, T.J.; Borden, E.C.; Blum, R.H. Interferon alfa-2b adjuvant therapy of high-risk resected cutaneous melanoma: the Eastern Cooperative Oncology Group Trial EST 1684. J. Clin. Oncol. 1996, 14, 7–17. [Google Scholar]
- Kirkwood, J.M.; Ibrahim, J.G.; Sosman, J.A.; Sondak, V.K.; Agarwala, S.S.; Ernstoff, M.S.; Rao, U. High-dose interferon alfa-2b significantly prolongs relapse-free and overall survival compared with the GM2-KLH/QS-21 vaccine in patients with resected stage IIB-III melanoma: results of intergroup trial E1694/S9512/C509801. J. Clin. Oncol. 2001, 19, 2370–2380. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wangari-Talbot, J.; Goydos, J.; Chen, S. Role of the G Protein-Coupled Receptor, mGlu1, in Melanoma Development. Pharmaceuticals 2010, 3, 2821-2837. https://doi.org/10.3390/ph3092821
Wangari-Talbot J, Goydos J, Chen S. Role of the G Protein-Coupled Receptor, mGlu1, in Melanoma Development. Pharmaceuticals. 2010; 3(9):2821-2837. https://doi.org/10.3390/ph3092821
Chicago/Turabian StyleWangari-Talbot, Janet, James Goydos, and Suzie Chen. 2010. "Role of the G Protein-Coupled Receptor, mGlu1, in Melanoma Development" Pharmaceuticals 3, no. 9: 2821-2837. https://doi.org/10.3390/ph3092821
APA StyleWangari-Talbot, J., Goydos, J., & Chen, S. (2010). Role of the G Protein-Coupled Receptor, mGlu1, in Melanoma Development. Pharmaceuticals, 3(9), 2821-2837. https://doi.org/10.3390/ph3092821