Use of p38 MAPK Inhibitors for the Treatment of Werner Syndrome


{kind=link}
{kind=link}
{kind=link}
{kind=link}
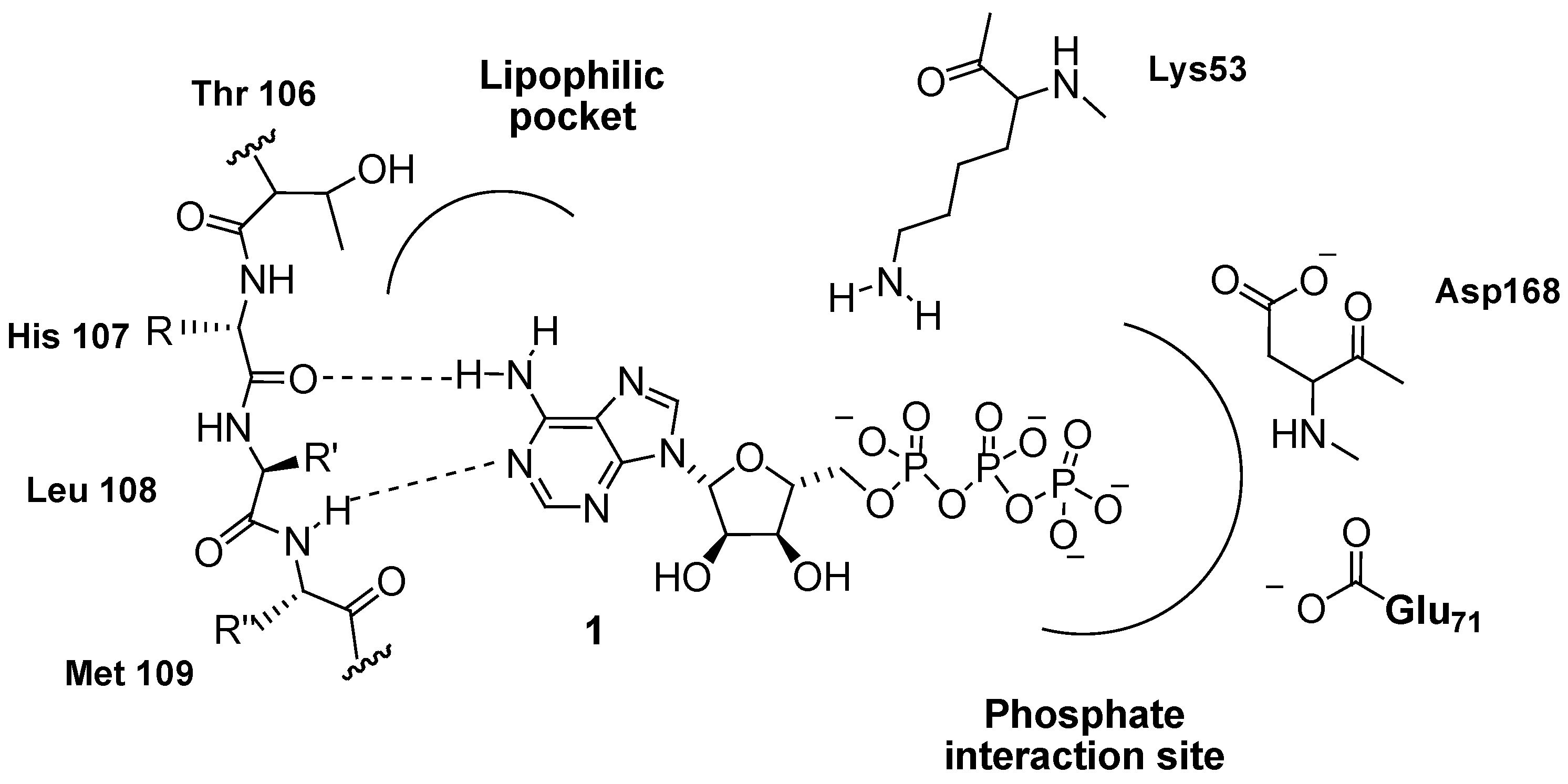{kind=link}
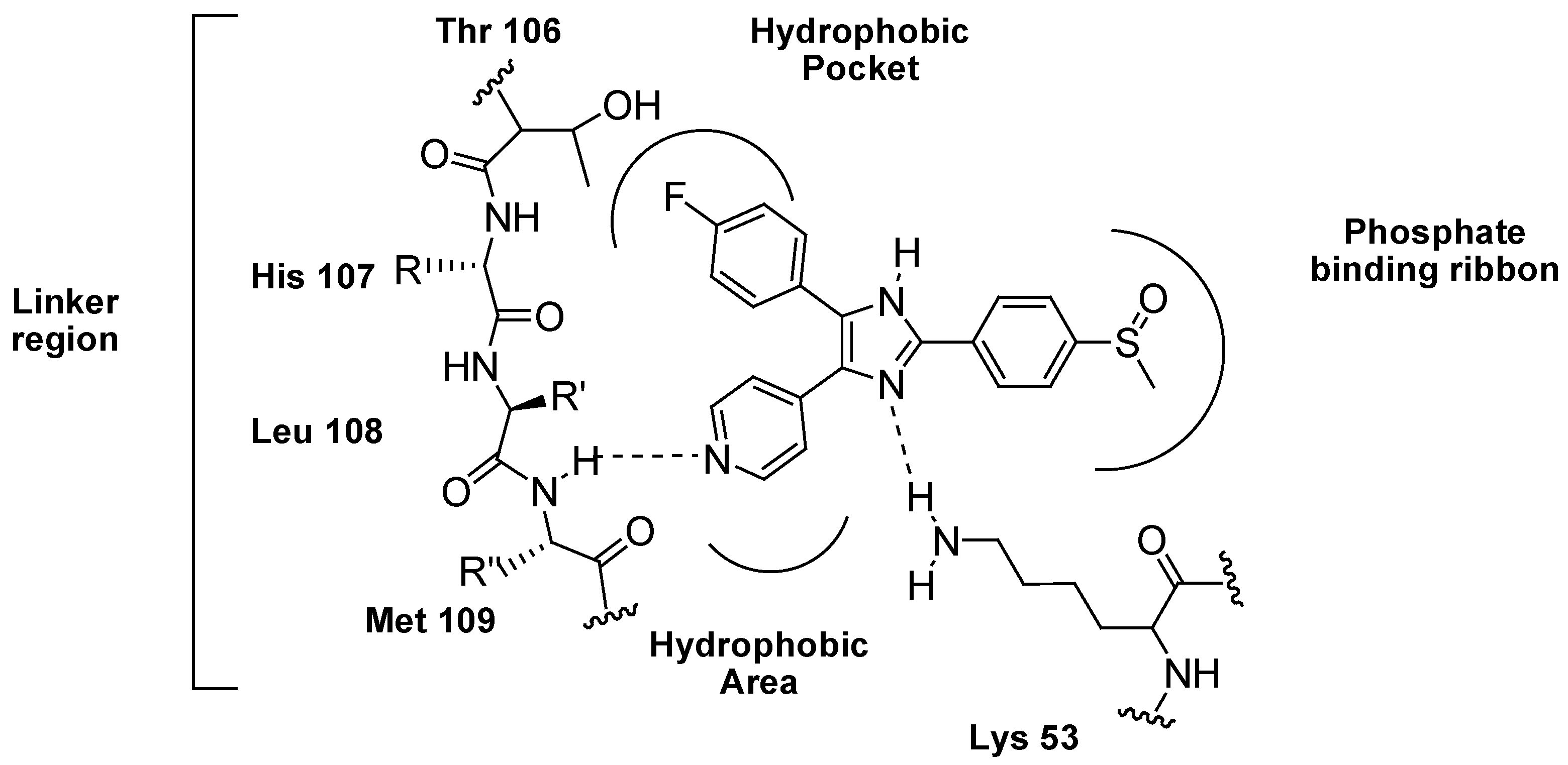{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
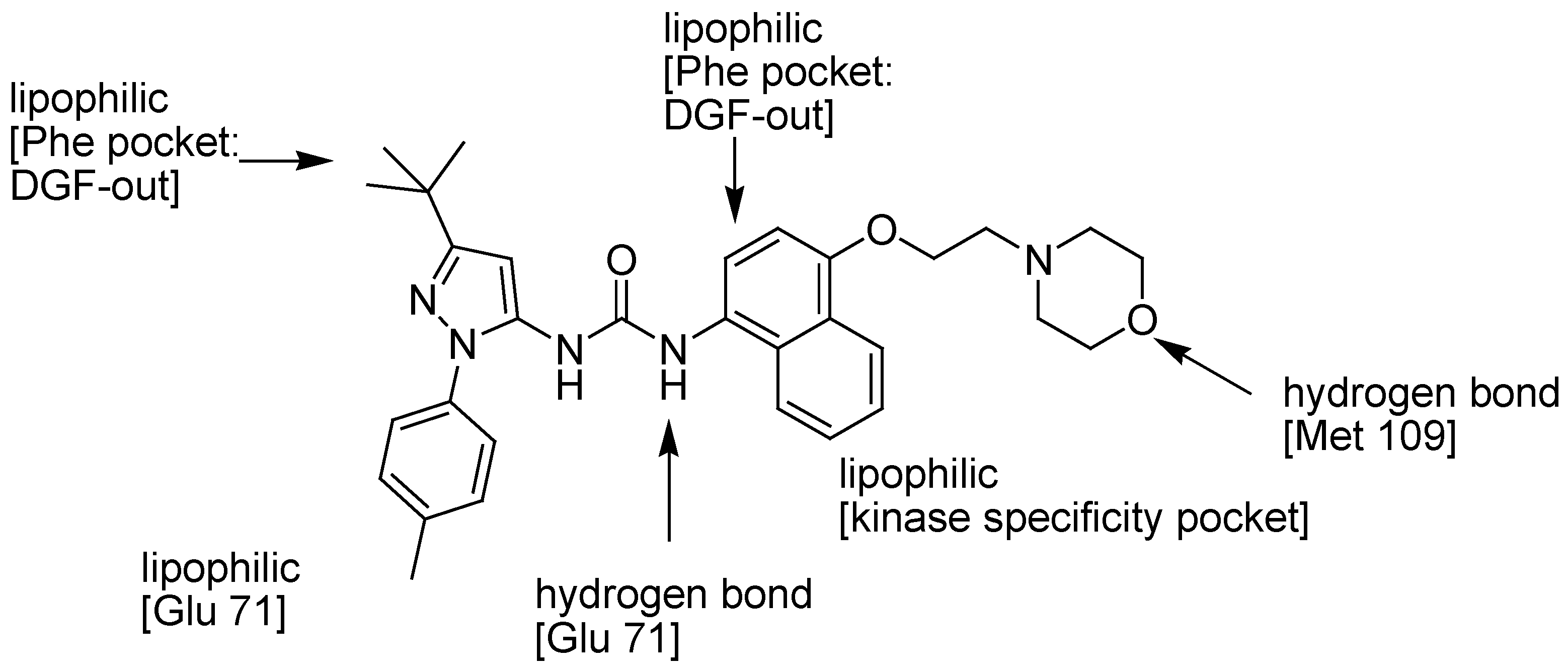{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Premature Ageing Disorders and Werner Syndrome
1.1. Pathophysiology of Werner Syndrome
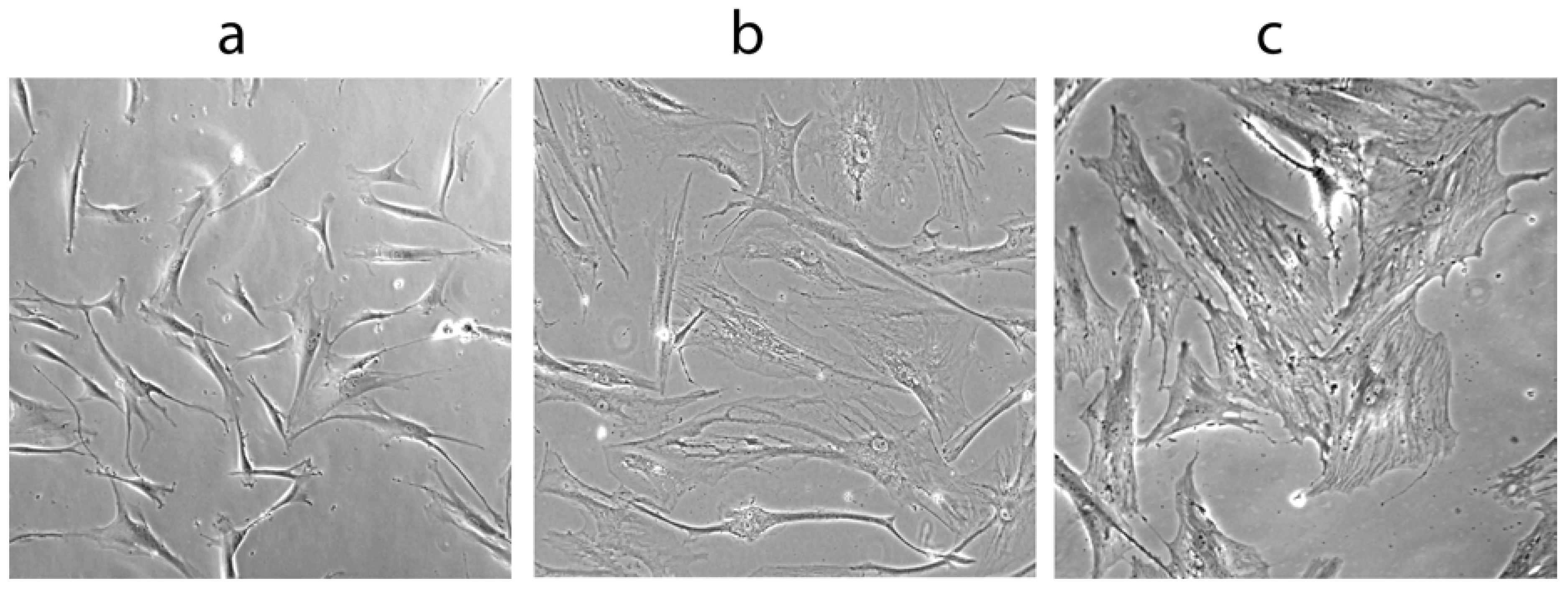
1.2. The WS Cellular Phenotype
1.3. The Werner Protein (WRNp) and Genome Instability in WS
1.4. Telomere-Dependent and Telomere-Independent Mechanisms of Cell Ageing in WS Cells
1.5. Werner Syndrome and Inflammation
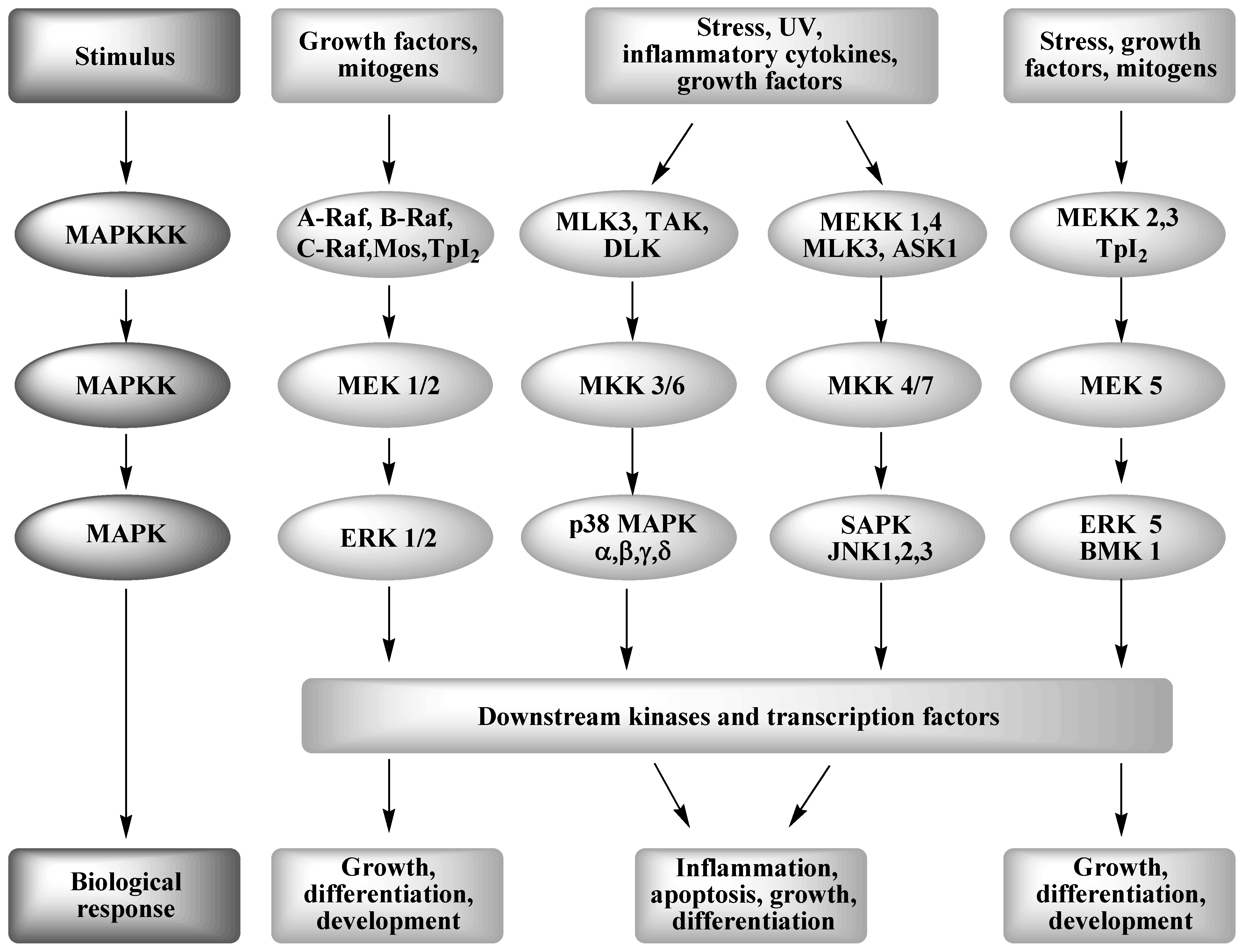
2. Mitogen Activated Protein Kinase Signal Transduction
2.1. The JNK (SAPK) Pathway
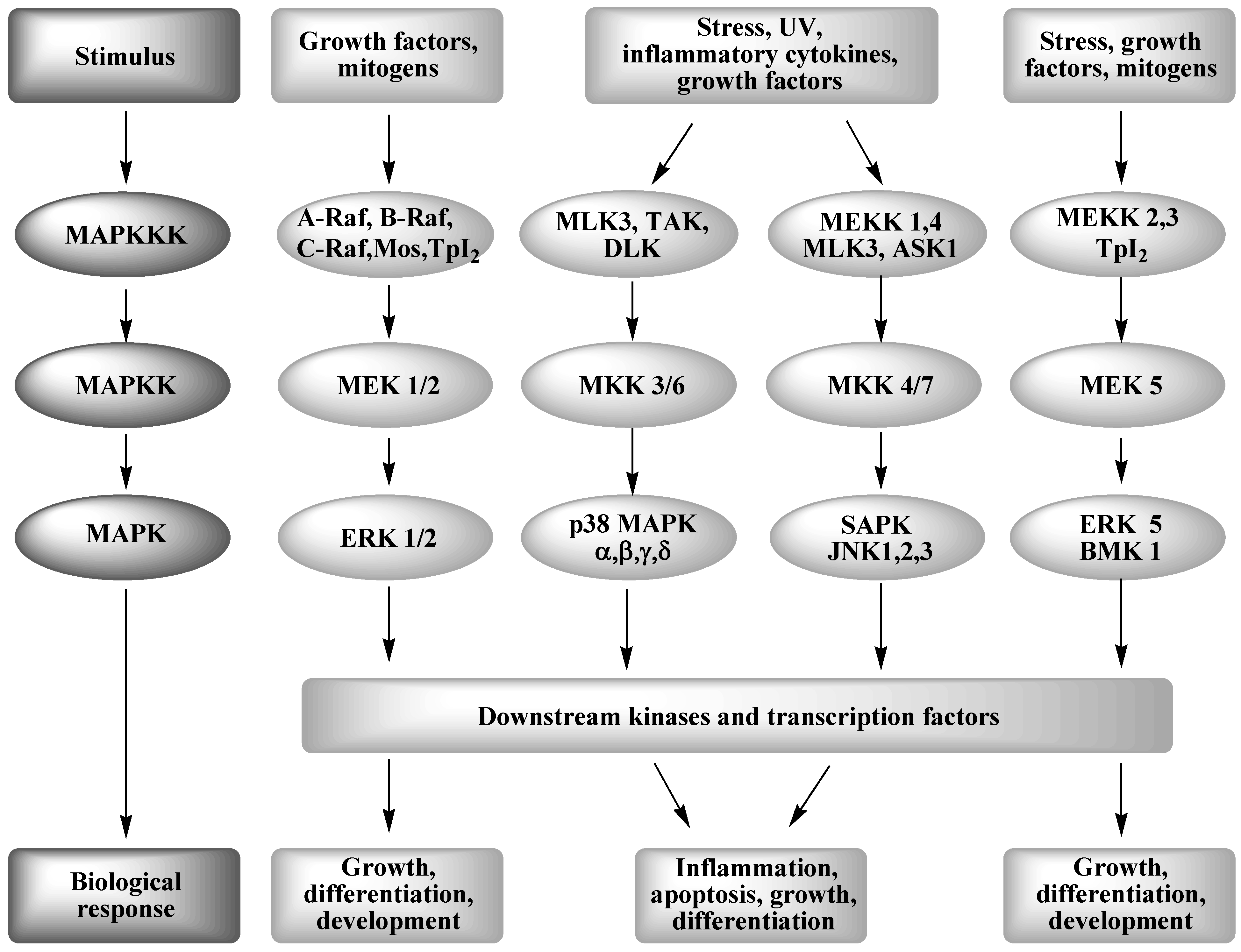
2.2. The ERK Pathway
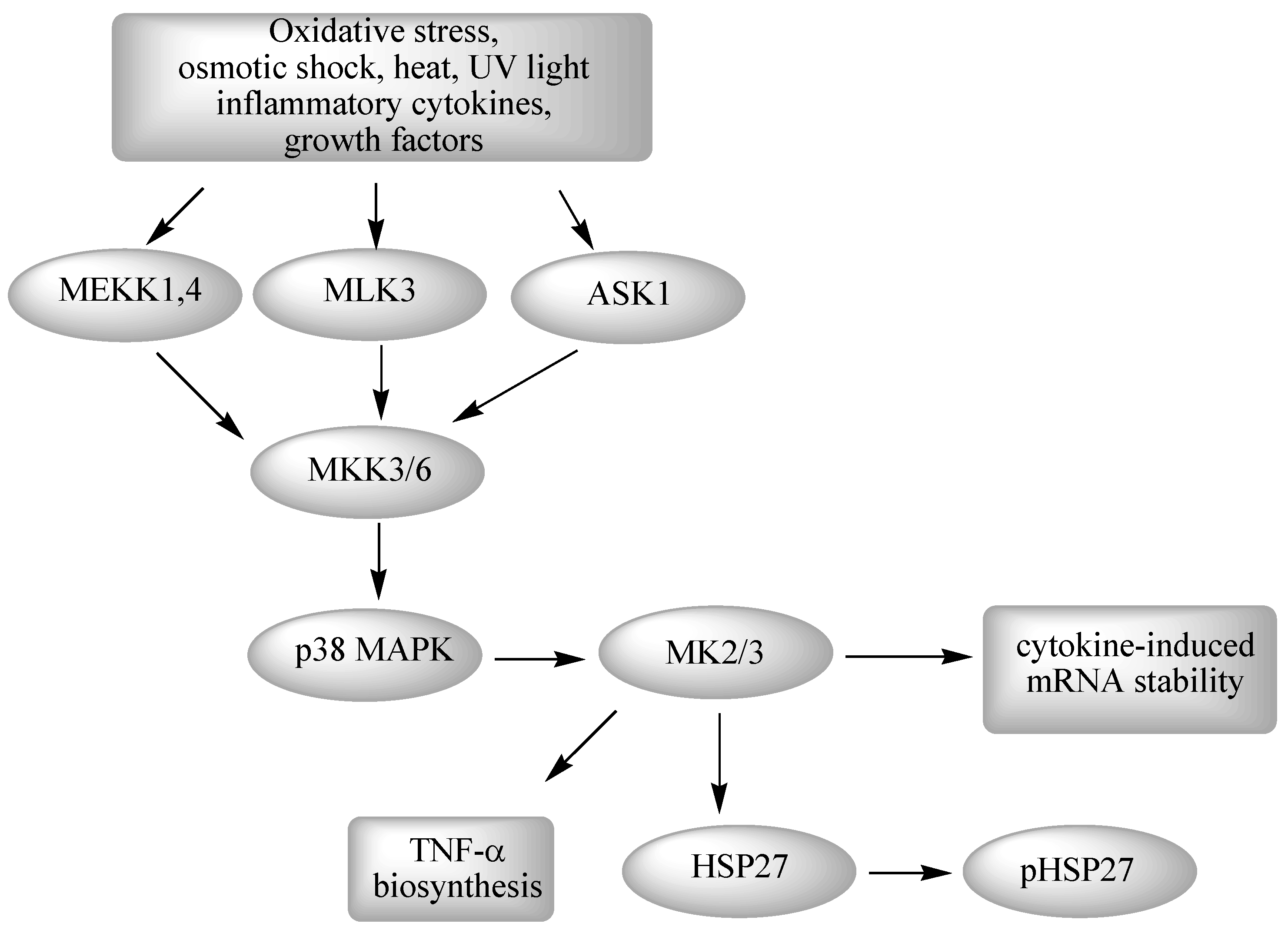
2.3. The p38 MAPK Pathway

2.4. WS and MAPK signaling
3. Use of MAPK Inhibitors for the Treatment of Werner Syndrome
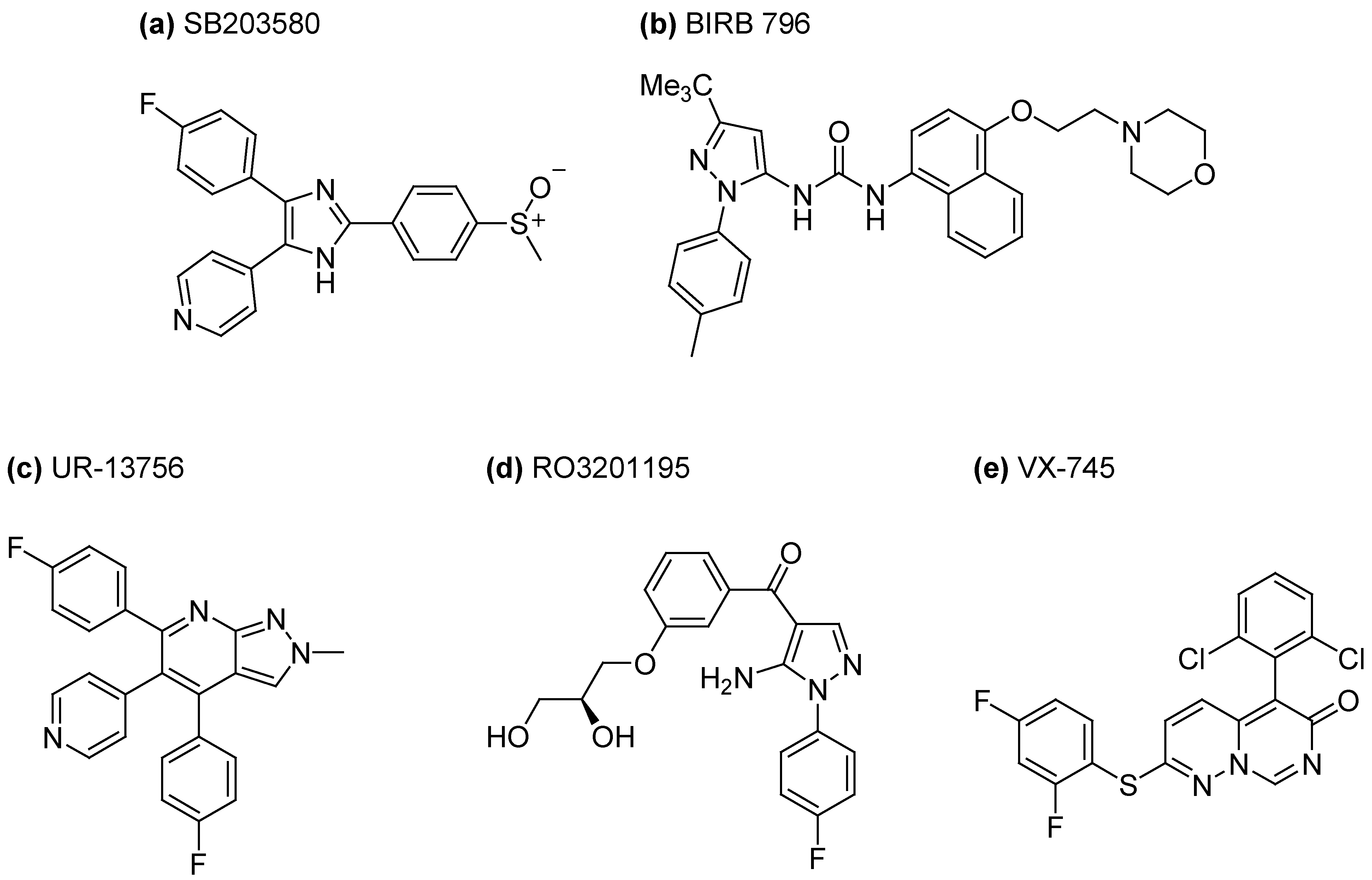
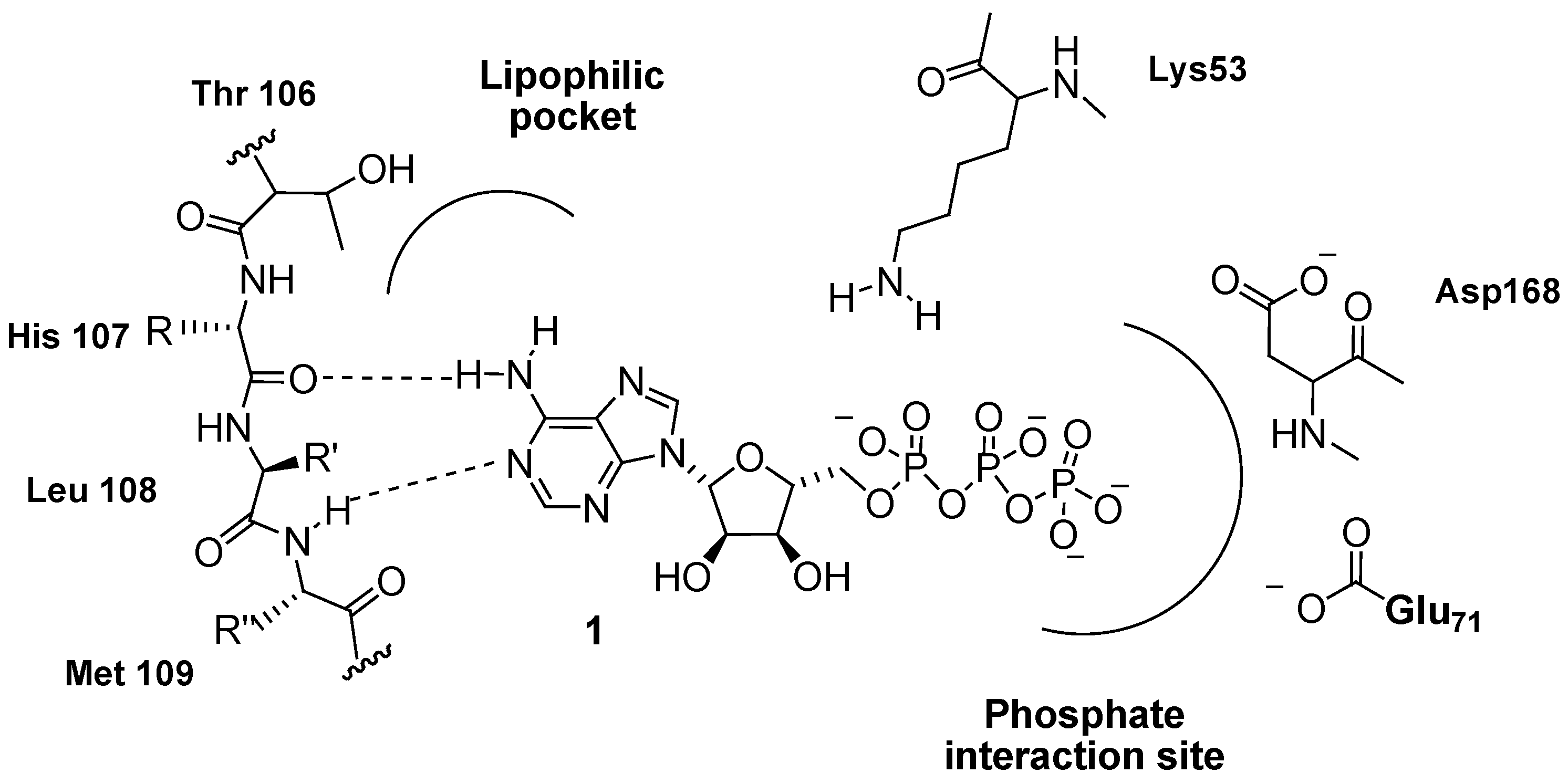
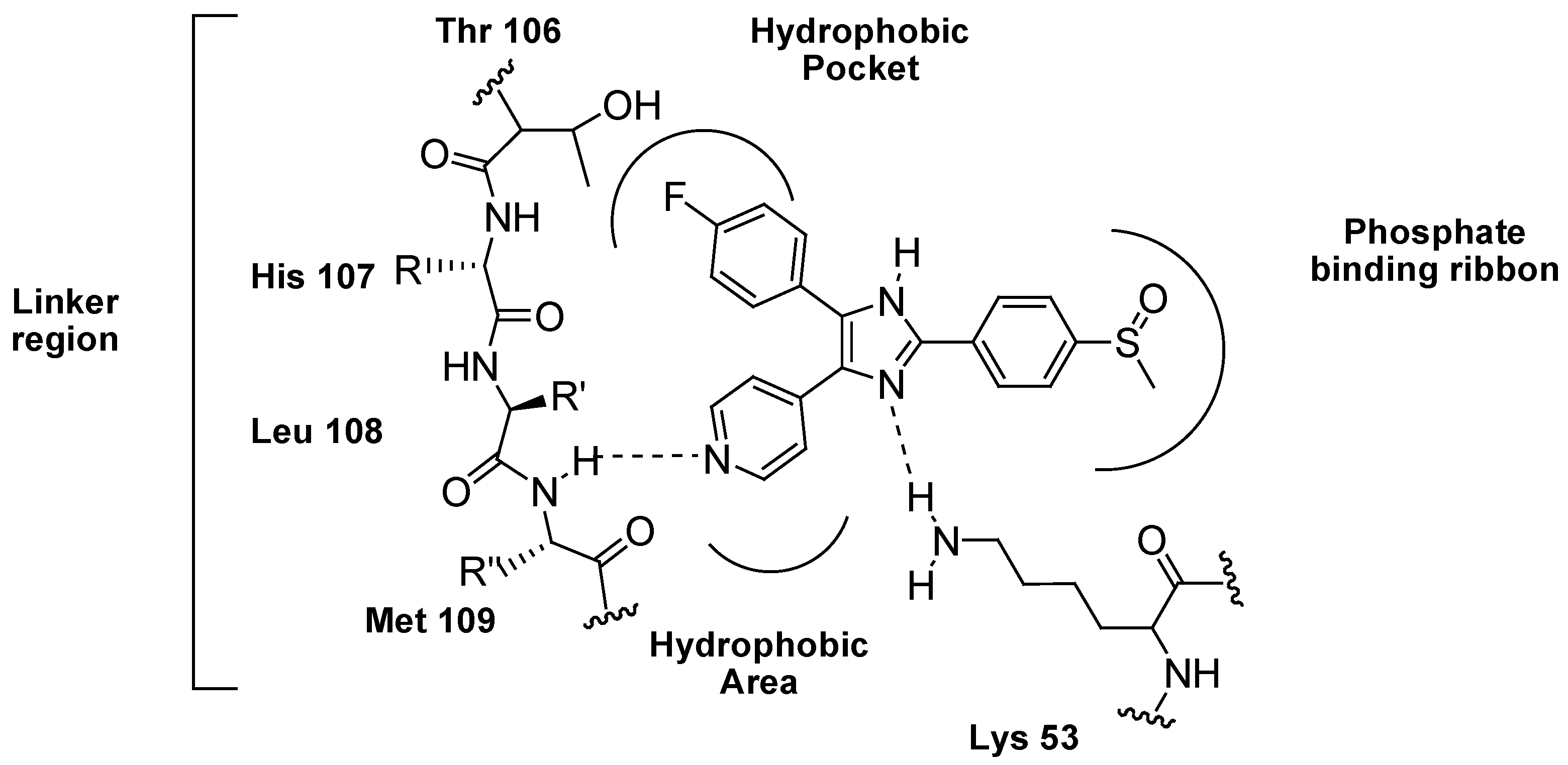
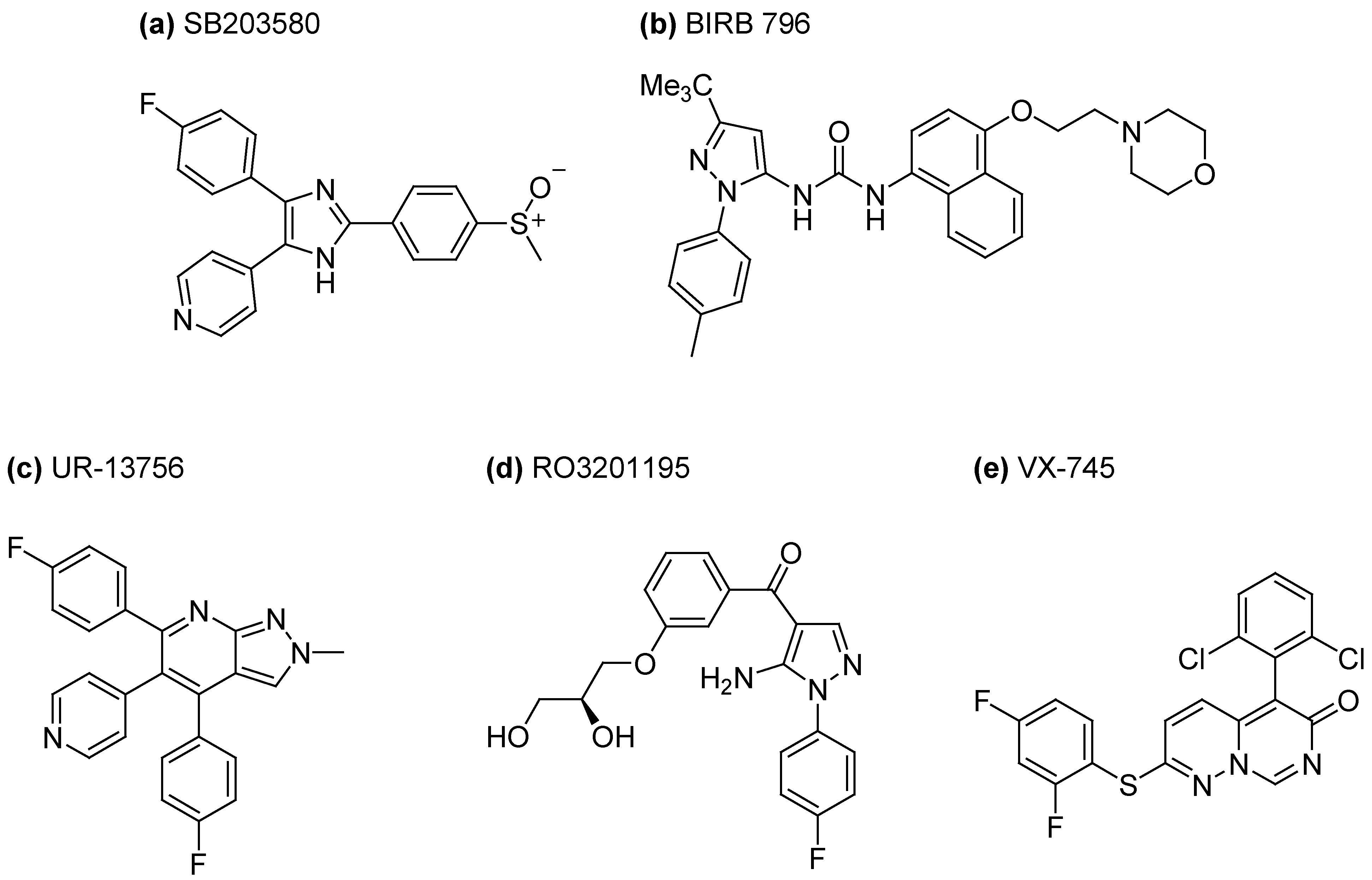
3.1. Inhibition of P38 MAPK Signaling in WS Cells by the Prototypical Inhibitor SB203580
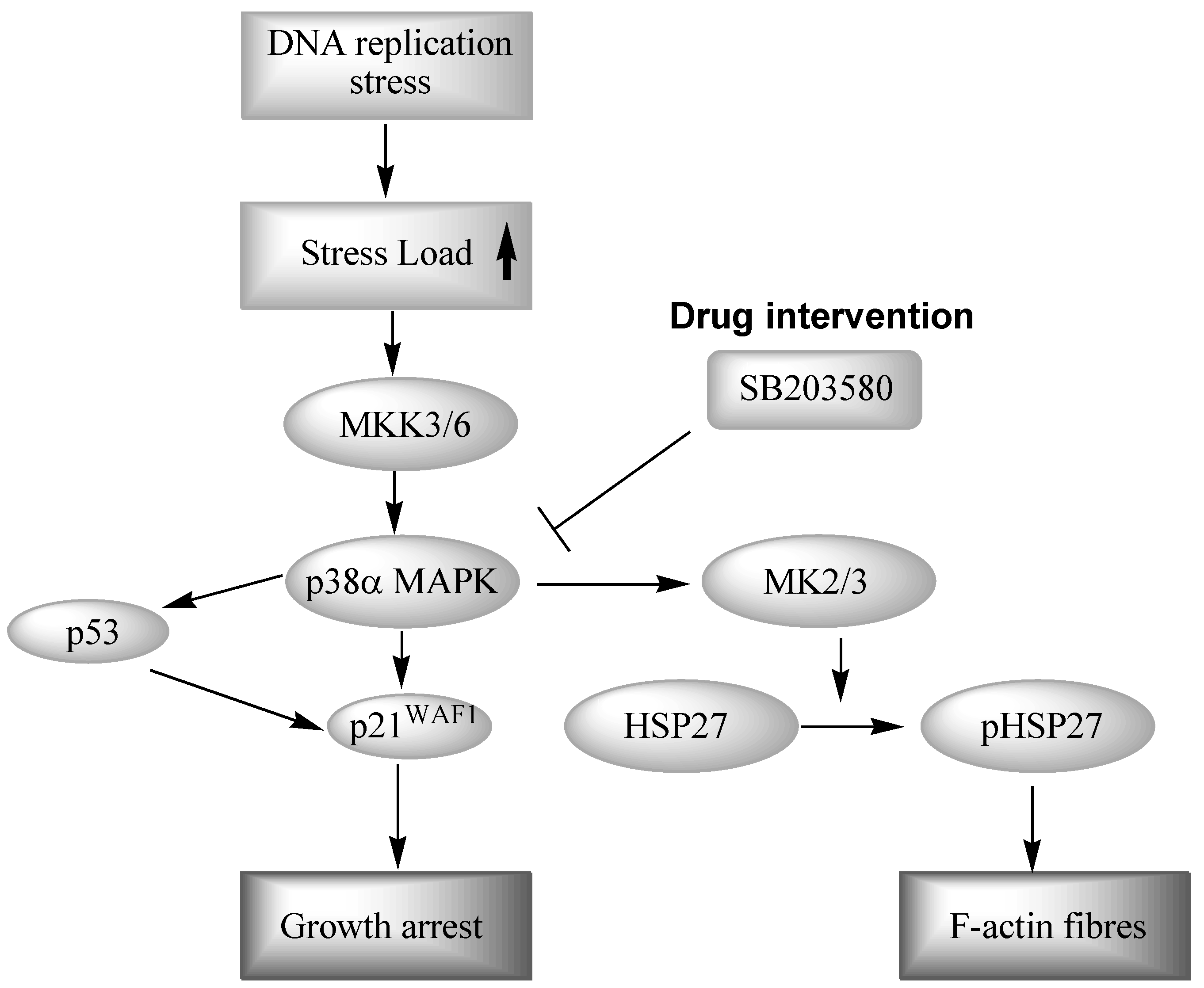
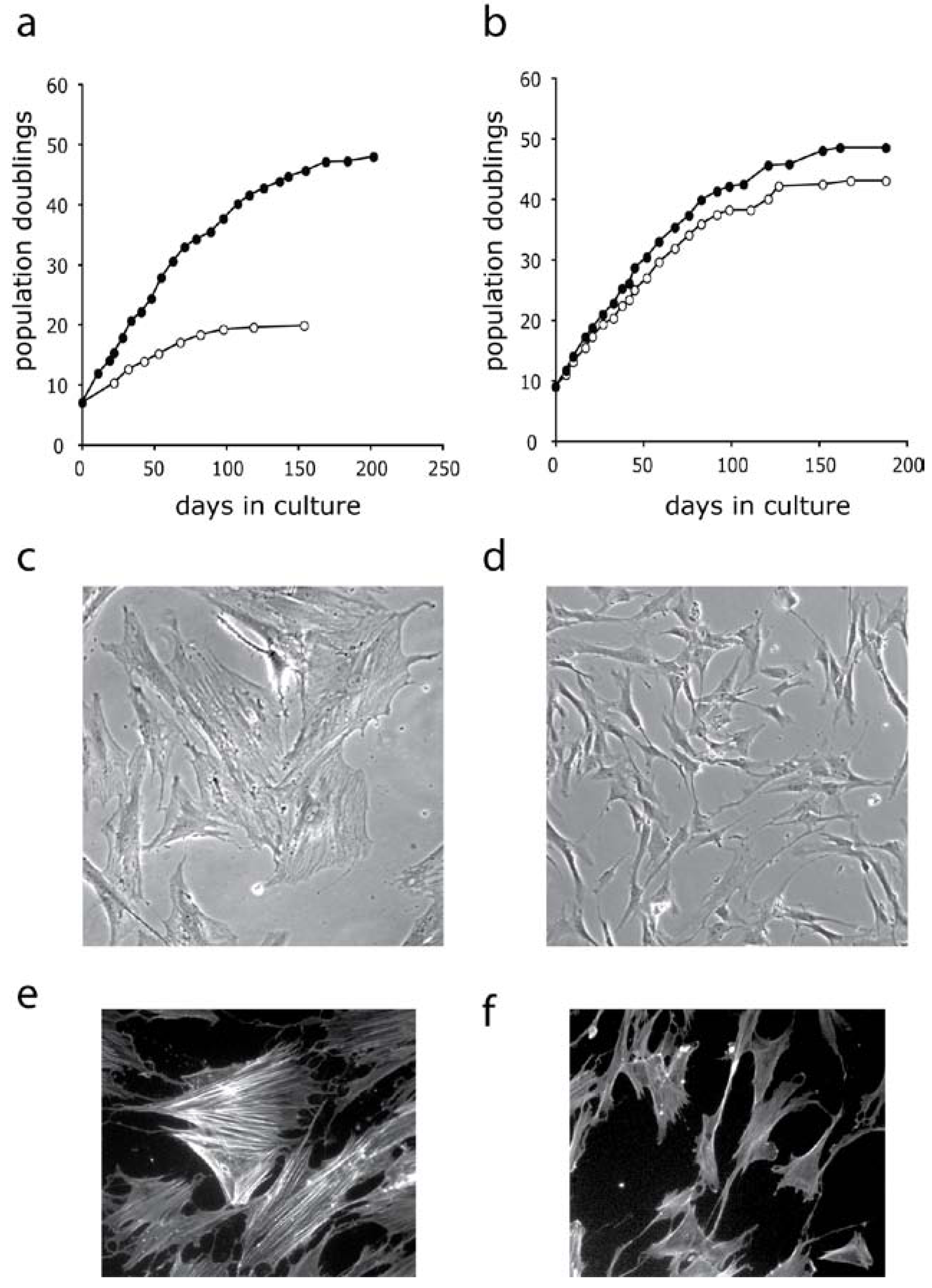
3.2. Inhibition of MAPK Signaling in WS Cells Using Other P38 Inhibitors
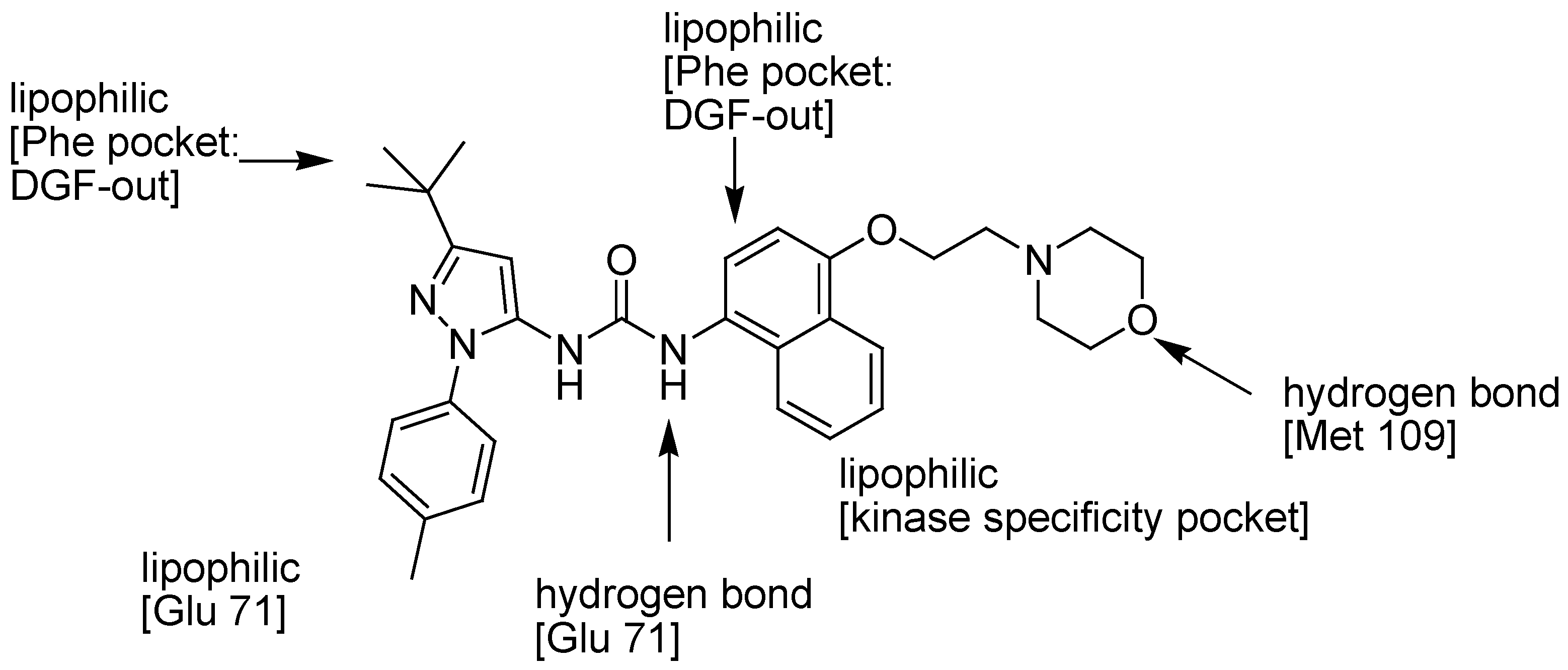

4. Conclusions and Outlook
Acknowledgements
References and Notes
- Kipling, D.; Davis, T.; Ostler, E.L.; Faragher, R.G. What can progeroid syndromes tell us about human aging? Science 2004, 305, 1426–1431. [Google Scholar] [CrossRef] [PubMed]
- Navarro, C.L.; Cau, P.; Levy, N. Molecular bases of progeroid syndromes. Hum. Mol. Genet. 2006, 15, R151–R161. [Google Scholar]
- Kudlow, B.A.; Kennedy, B.K.; Monnat, R.J., Jr. Werner and Hutchinson-Gilford progeria syndromes: mechanistic basis of human progeroid diseases. Nat. Rev. Mol. Cell Biol. 2007, 8, 394–404. [Google Scholar]
- Brown, W.T.; Kieras, F.J.; Houck, G.E., Jr.; Dutkowski, R.; Jenkins, E.C. A comparison of adult and childhood progerias: Werner syndrome and Hutchinson-Gilford progeria syndrome. Adv. Exp. Med. Biol. 1985, 190, 229–244. [Google Scholar]
- Martin, G.M.; Oshima, J.; Gray, M.D.; Poot, M. What geriatricians should know about the Werner syndrome. J. Am. Geriatr. Soc. 1999, 47, 1136–1144. [Google Scholar]
- Miller, R.A. 'Accelerated aging': a primrose path to insight? Aging Cell 2004, 3, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Davis, T.; Tivey, H.S.E.; Kipling, D. Telomere dynamics and biology in human progeroid syndromes. In Telomeres: Function, Shortening and Lengthening; Mancini, L., Ed.; Nova: New York, NY, USA, 2009; pp. 1–75. [Google Scholar]
- Werner, O. On cataract associated in conjunction with scleroderma. Ph.D. Dissertation, Schmidt and Klaunig, Kiel, Germany, 1904. [Google Scholar]
- Thannhauser, S.J. Werner's syndrome (progeria of the adult) and Rothmund's syndrome: two types of closely related heredofamilial atrophic dermatosis with juvenile cataracts and endocrine features. A critcal study of five new cases. Ann. Int. Med. 1945, 23, 559–625. [Google Scholar]
- Postiglione, A.; Soricelli, A.; Covelli, E.M.; Iazzetta, N.; Ruocco, A.; Milan, G.; Santoro, L.; Alfano, B.; Brunetti, A. Premature aging in Werner's syndrome spares the central nervous system. Neurobiol. Aging 1996, 17, 325–330. [Google Scholar]
- Hofer, A.C.; Tran, R.T.; Aziz, O.Z.; Wright, W.; Novelli, G.; Shay, J.; Lewis, M. Shared phenotypes among segmental progeroid syndromes suggest underlying pathways of aging. J. Gerontol. A Biol. Sci. Med. Sci. 2005, 60, 10–20. [Google Scholar]
- Epstein, C.J.; Martin, G.M.; Schultz, A.L.; Motulsky, A.G. Werner's syndrome a review of its symptomatology, natural history, pathologic features, genetics and relationship to the natural aging process. Medicine (Baltimore) 1966, 45, 177–221. [Google Scholar] [PubMed]
- Goto, M.; Miller, R.W.; Ishikawa, Y.; Sugano, H. Excess of rare cancers in Werner syndrome (adult progeria). Cancer Epidemiol. Biomarkers Prev. 1996, 5, 239–246. [Google Scholar]
- Goto, M.; Tanimoto, K.; Horiuchi, Y.; Sasazuki, T. Family analysis of Werner's syndrome: a survey of 42 Japanese families with a review of the literature. Clin. Genet. 1981, 19, 8–15. [Google Scholar]
- Davis, T.; Wyllie, F.S.; Rokicki, M.J.; Bagley, M.C.; Kipling, D. The role of cellular senescence in Werner syndrome: toward therapeutic intervention in human premature aging. Ann. N.Y. Acad. Sci. 2007, 1100, 455–469. [Google Scholar] [CrossRef] [PubMed]
- Faragher, R.G.; Kill, I.R.; Hunter, J.A.; Pope, F.M.; Tannock, C.; Shall, S. The gene responsible for Werner syndrome may be a cell division "counting" gene. Proc. Natl. Acad. Sci. USA 1993, 90, 12030–12034. [Google Scholar]
- James, S.E.; Faragher, R.G.; Burke, J.F.; Shall, S.; Mayne, L.V. Werner's syndrome T lymphocytes display a normal in vitro life-span. Mech. Ageing Dev. 2000, 121, 139–149. [Google Scholar] [PubMed]
- Ostler, E.L.; Wallis, C.V.; Sheerin, A.N.; Faragher, R.G. A model for the phenotypic presentation of Werner's syndrome. Exp. Gerontol. 2002, 37, 285–292. [Google Scholar]
- Davis, T.; Faragher, R.G.; Jones, C.J.; Kipling, D. Investigation of the signaling pathways involved in the proliferative life span barriers in werner syndrome fibroblasts. Ann. N.Y. Acad. Sci. 2004, 1019, 274–277. [Google Scholar] [CrossRef] [PubMed]
- Davis, T.; Singhrao, S.K.; Wyllie, F.S.; Haughton, M.F.; Smith, P.J.; Wiltshire, M.; Wynford-Thomas, D.; Jones, C.J.; Faragher, R.G.; Kipling, D. Telomere-based proliferative lifespan barriers in Werner-syndrome fibroblasts involve both p53-dependent and p53-independent mechanisms. J. Cell Sci. 2003, 116, 1349–1357. [Google Scholar]
- Yu, C.E.; Oshima, J.; Fu, Y.H.; Wijsman, E.M.; Hisama, F.; Alisch, R.; Matthews, S.; Nakura, J.; Miki, T.; Ouais, S.; Martin, G.M.; Mulligan, J.; Schellenberg, G.D. Positional cloning of the Werner's syndrome gene. Science 1996, 272, 258–262. [Google Scholar]
- Gray, M.D.; Wang, L.; Youssoufian, H.; Martin, G.M.; Oshima, J. Werner helicase is localized to transcriptionally active nucleoli of cycling cells. Exp. Cell Res. 1998, 242, 487–494. [Google Scholar]
- Marciniak, R.A.; Lombard, D.B.; Johnson, F.B.; Guarente, L. Nucleolar localization of the Werner syndrome protein in human cells. Proc. Natl. Acad. Sci. USA 1998, 95, 6887–9682. [Google Scholar]
- Shiratori, M.; Sakamoto, S.; Suzuki, N.; Tokutake, Y.; Kawabe, Y.; Enomoto, T.; Sugimoto, M.; Goto, M.; Matsumoto, T.; Furuichi, Y. Detection by epitope-defined monoclonal antibodies of Werner DNA helicases in the nucleoplasm and their upregulation by cell transformation and immortalization. J. Cell Biol. 1999, 144, 1–9. [Google Scholar]
- Bohr, V.A.; Brosh, R.M., Jr.; von Kobbe, C.; Opresko, P.; Karmakar, P. Pathways defective in the human premature aging disease Werner syndrome. Biogerontology 2002, 3, 89–94. [Google Scholar]
- Shen, J.; Loeb, L.A. Unwinding the molecular basis of the Werner syndrome. Mech. Ageing Dev. 2001, 122, 921–944. [Google Scholar]
- Kamath-Loeb, A.S.; Loeb, L.A.; Johansson, E.; Burgers, P.M.; Fry, M. Interactions between the Werner syndrome helicase and DNA polymerase delta specifically facilitate copying of tetraplex and hairpin structures of the d(CGG)n trinucleotide repeat sequence. J. Biol. Chem. 2001, 276, 16439–16446. [Google Scholar]
- Brosh, R.M., Jr.; Bohr, V.A. Roles of the Werner syndrome protein in pathways required for maintenance of genome stability. Exp. Gerontol. 2002, 37, 491–506. [Google Scholar]
- Mohaghegh, P.; Karow, J.K.; Brosh, R.M., Jr.; Bohr, V.A.; Hickson, I.D. The Bloom's and Werner's syndrome proteins are DNA structure-specific helicases. Nucleic Acids Res. 2001, 29, 2843–2849. [Google Scholar]
- Opresko, P.L.; von Kobbe, C.; Laine, J.P.; Harrigan, J.; Hickson, I.D.; Bohr, V.A. Telomere-binding protein TRF2 binds to and stimulates the Werner and Bloom syndrome helicases. J. Biol. Chem. 2002, 277, 41110–41119. [Google Scholar]
- Orren, D.K.; Theodore, S.; Machwe, A. The Werner syndrome helicase/exonuclease (WRN) disrupts and degrades D-loops in vitro. Biochemistry 2002, 41, 13483–13488. [Google Scholar]
- Constantinou, A.; Tarsounas, M.; Karow, J.K.; Brosh, R.M.; Bohr, V.A.; Hickson, I.D.; West, S.C. Werner's syndrome protein (WRN) migrates Holliday junctions and co-localizes with RPA upon replication arrest. EMBO Rep. 2000, 1, 80–84. [Google Scholar]
- Multani, A.S.; Chang, S. WRN at telomeres: implications for aging and cancer. J. Cell Sci. 2007, 120, 713–721. [Google Scholar]
- Schulz, V.P.; Zakian, V.A.; Ogburn, C.E.; McKay, J.; Jarzebowicz, A.A.; Edland, S.D.; Martin, G.M. Accelerated loss of telomeric repeats may not explain accelerated replicative decline of Werner syndrome cells. Hum. Genet. 1996, 97, 750–754. [Google Scholar]
- Ariyoshi, K.; Suzuki, K.; Goto, M.; Watanabe, M.; Kodama, S. Increased chromosome instability and accumulation of DNA double-strand breaks in Werner syndrome cells. J. Radiat. Res. (Tokyo) 2007, 48, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Crabbe, L.; Jauch, A.; Naeger, C.M.; Holtgreve-Grez, H.; Karlseder, J. Telomere dysfunction as a cause of genomic instability in Werner syndrome. Proc. Natl. Acad. Sci. USA 2007, 104, 2205–2210. [Google Scholar]
- Crabbe, L.; Verdun, R.E.; Haggblom, C.I.; Karlseder, J. Defective telomere lagging strand synthesis in cells lacking WRN helicase activity. Science 2004, 306, 1951–1953. [Google Scholar]
- Pirzio, L.M.; Pichierri, P.; Bignami, M.; Franchitto, A. Werner syndrome helicase activity is essential in maintaining fragile site stability. J. Cell Biol. 2008, 180, 305–314. [Google Scholar]
- Fukuchi, K.; Martin, G.M.; Monnat, R.J., Jr. Mutator phenotype of Werner syndrome is characterized by extensive deletions. Proc. Natl. Acad. Sci. USA 1989, 86, 5893–5897. [Google Scholar]
- Comai, L.; Li, B. The Werner syndrome protein at the crossroads of DNA repair and apoptosis. Mech. Ageing Dev. 2004, 125, 521–528. [Google Scholar]
- Meyn, M.S. Chromosome instability syndromes: lessons for carcinogenesis. Curr. Top. Microbiol. Immunol. 1997, 221, 71–148. [Google Scholar]
- Allsopp, R.C.; Harley, C.B. Evidence for a critical telomere length in senescent human fibroblasts. Exp. Cell Res. 1995, 219, 130–136. [Google Scholar]
- Bodnar, A.G.; Ouellette, M.; Frolkis, M.; Holt, S.E.; Chiu, C.P.; Morin, G.B.; Harley, C.B.; Shay, J.W.; Lichtsteiner, S.; Wright, W.E. Extension of life-span by introduction of telomerase into normal human cells. Science 1998, 279, 349–352. [Google Scholar]
- Vaziri, H.; Benchimol, S. Reconstitution of telomerase activity in normal human cells leads to elongation of telomeres and extended replicative life span. Curr. Biol. 1998, 8, 279–282. [Google Scholar]
- Wright, W.E.; Shay, J.W. Historical claims and current interpretations of replicative aging. Nat. Biotechnol. 2002, 20, 682–688. [Google Scholar]
- Baird, D.M.; Davis, T.; Rowson, J.; Jones, C.J.; Kipling, D. Normal telomere erosion rates at the single cell level in Werner syndrome fibroblast cells. Hum. Mol. Genet. 2004, 13, 1515–1524. [Google Scholar]
- Kruk, P.A.; Rampino, N.J.; Bohr, V.A. DNA damage and repair in telomeres: relation to aging. Proc. Natl. Acad. Sci. USA 1995, 92, 258–262. [Google Scholar]
- Wyllie, F.S.; Lemoine, N.R.; Barton, C.M.; Dawson, T.; Bond, J.; Wynford-Thomas, D. Direct growth stimulation of normal human epithelial cells by mutant p53. Mol. Carcinog. 1993, 7, 83–88. [Google Scholar]
- Ouellette, M.M.; McDaniel, L.D.; Wright, W.E.; Shay, J.W.; Schultz, R.A. The establishment of telomerase-immortalized cell lines representing human chromosome instability syndromes. Hum. Mol. Genet. 2000, 9, 403–411. [Google Scholar]
- Choi, D.; Whittier, P.S.; Oshima, J.; Funk, W.D. Telomerase expression prevents replicative senescence but does not fully reset mRNA expression patterns in Werner syndrome cell strains. FASEB J. 2001, 15, 1014–1020. [Google Scholar]
- Davis, T.; Kipling, D. Werner Syndrome as an example of inflamm-aging: possible therapeutic opportunities for a progeroid syndrome? Rejuvenation Res. 2006, 9, 402–407. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, Y.; Higashikawa, T.; Tatsumi, M. A retarded rate of DNA replication and normal level of DNA repair in Werner's syndrome fibroblasts in culture. J. Cell. Physiol. 1977, 92, 365–374. [Google Scholar]
- Takeuchi, F.; Hanaoka, F.; Goto, M.; Akaoka, I.; Hori, T.; Yamada, M.; Miyamoto, T. Altered frequency of initiation sites of DNA replication in Werner's syndrome cells. Hum. Genet. 1982, 60, 365–368. [Google Scholar]
- Poot, M.; Hoehn, H.; Runger, T.M.; Martin, G.M. Impaired S-phase transit of Werner syndrome cells expressed in lymphoblastoid cell lines. Exp. Cell Res. 1992, 202, 267–273. [Google Scholar]
- Huot, J.; Houle, F.; Marceau, F.; Landry, J. Oxidative stress-induced actin reorganization mediated by the p38 mitogen-activated protein kinase/heat shock protein 27 pathway in vascular endothelial cells. Circ. Res. 1997, 80, 383–392. [Google Scholar]
- Guay, J.; Lambert, H.; Gingras-Breton, G.; Lavoie, J.N.; Huot, J.; Landry, J. Regulation of actin filament dynamics by p38 map kinase-mediated phosphorylation of heat shock protein 27. J. Cell Sci. 1997, 110 (Pt 3), 357–368. [Google Scholar] [PubMed]
- Wang, W.; Chen, J.X.; Liao, R.; Deng, Q.; Zhou, J.J.; Huang, S.; Sun, P. Sequential activation of the MEK-extracellular signal-regulated kinase and MKK3/6-p38 mitogen-activated protein kinase pathways mediates oncogenic ras-induced premature senescence. Mol. Cell. Biol. 2002, 22, 3389–3403. [Google Scholar]
- Deng, Q.; Liao, R.; Wu, B.L.; Sun, P. High intensity ras signaling induces premature senescence by activating p38 pathway in primary human fibroblasts. J. Biol. Chem. 2004, 279, 1050–1059. [Google Scholar]
- Pichierri, P.; Franchitto, A. Werner syndrome protein, the MRE11 complex and ATR: menage-a-trois in guarding genome stability during DNA replication? Bioessays 2004, 26, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Lopez, A.M.; Jackson, D.A.; Iborra, F.; Cox, L.S. Asymmetry of DNA replication fork progression in Werner's syndrome. Aging Cell 2002, 1, 30–39. [Google Scholar]
- Beddy, D.J.; Watson, W.R.; Fitzpatrick, J.M.; O'Connell, P.R. Critical involvement of stress-activated mitogen-activated protein kinases in the regulation of intracellular adhesion molecule-1 in serosal fibroblasts isolated from patients with Crohn's disease. J. Am. Coll. Surg. 2004, 199, 234–242. [Google Scholar]
- Chabaud-Riou, M.; Firestein, G.S. Expression and activation of mitogen-activated protein kinase kinases-3 and -6 in rheumatoid arthritis. Am. J. Pathol. 2004, 164, 177–184. [Google Scholar]
- Furuzawa-Carballeda, J.; Alcocer-Varela, J. Interleukin-8, interleukin-10, intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 expression levels are higher in synovial tissue from patients with rheumatoid arthritis than in osteoarthritis. Scand. J. Immunol. 1999, 50, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Holtmann, M.H.; Neurath, M.F. Differential TNF-signaling in chronic inflammatory disorders. Curr. Mol. Med. 2004, 4, 439–444. [Google Scholar]
- Hollenbach, E.; Neumann, M.; Vieth, M.; Roessner, A.; Malfertheiner, P.; Naumann, M. Inhibition of p38 MAP kinase- and RICK/NF-κB-signaling suppresses inflammatory bowel disease. FASEB J. 2004, 18, 1550–1552. [Google Scholar]
- Hollenbach, E.; Vieth, M.; Roessner, A.; Neumann, M.; Malfertheiner, P.; Naumann, M. Inhibition of RICK/nuclear factor-κB and p38 signaling attenuates the inflammatory response in a murine model of Crohn disease. J. Biol. Chem. 2005, 280, 14981–14988. [Google Scholar]
- Dinarello, C.A. Inflammatory cytokines: interleukin-1 and tumor necrosis factor as effector molecules in autoimmune diseases. Curr. Opin. Immunol. 1991, 3, 941–948. [Google Scholar]
- Force, T.; Kuida, K.; Namchuk, M.; Parang, K.; Kyriakis, J.M. Inhibitors of protein kinase signaling pathways: emerging therapies for cardiovascular disease. Circulation 2004, 109, 1196–1205. [Google Scholar]
- Yokote, K.; Hara, K.; Mori, S.; Kadowaki, T.; Saito, Y.; Goto, M. Dysadipocytokinemia in werner syndrome and its recovery by treatment with pioglitazone. Diabetes Care 2004, 27, 2562–2563. [Google Scholar]
- Murano, S.; Nakazawa, A.; Saito, I.; Masuda, M.; Morisaki, N.; Akikusa, B.; Tsuboyama, T.; Saito, Y. Increased blood plasminogen activator inhibitor-1 and intercellular adhesion molecule-1 as possible risk factors of atherosclerosis in Werner syndrome. Gerontology 1997, 43 (Suppl. 1), 43–52. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Bonafe, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N.Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [PubMed]
- Giacomoni, P.U. Ageing, science and the cosmetics industry. The micro-inflammatory model serves as a basis for developing effective anti-ageing products for the skin. EMBO Rep. 2005, 6 (S1), S45–S48. [Google Scholar] [CrossRef]
- Vasto, S.; Candore, G.; Balistreri, C.R.; Caruso, M.; Colonna-Romano, G.; Grimaldi, M.P.; Listi, F.; Nuzzo, D.; Lio, D.; Caruso, C. Inflammatory networks in ageing, age-related diseases and longevity. Mech. Ageing Dev. 2007, 128, 83–91. [Google Scholar]
- Kyriakis, J.M.; Avruch, J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol. Rev. 2001, 81, 807–869. [Google Scholar] [PubMed]
- Turjanski, A.G.; Vaque, J.P.; Gutkind, J.S. MAP kinases and the control of nuclear events. Oncogene 2007, 26, 3240–3253. [Google Scholar]
- Davis, R.J. Signal transduction by the JNK group of MAP kinases. Cell 2000, 103, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Rossomando, A.J.; Payne, D.M.; Weber, M.J.; Sturgill, T.W. Evidence that pp42, a major tyrosine kinase target protein, is a mitogen-activated serine/threonine kinase. Proc. Natl. Acad. Sci. USA 1989, 86, 6940–6943. [Google Scholar]
- Cowley, S.; Paterson, H.; Kemp, P.; Marshall, C.J. Activation of MAP kinase kinase is necessary and sufficient for PC12 differentiation and for transformation of NIH3T3 cells. Cell 1994, 77, 841–852. [Google Scholar]
- Mansour, S.J.; Matten, W.T.; Hermann, A.S.; Candia, J.M.; Rong, S.; Fukasawa, K.; Vande Woude, G.F.; Ahn, N.G. Transformation of mammalian cells by constitutively active MAP kinase kinase. Science 1994, 265, 966–970. [Google Scholar]
- Lee, J.C.; Laydon, J.T.; McDonnell, P.C.; Gallagher, T.F.; Kumar, S.; Green, D.; McNulty, D.; Blumenthal, M.J.; Heys, J.R.; Landvatter, S.W.; Strickler, J.E.; McLaughlin, M.M; Siemens, I.R.; Fisher, S.M.; Livi, G.P.; White, J.R.; Adams, J.L.; Young, P.R. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature 1994, 372, 739–746. [Google Scholar] [PubMed]
- Schieven, G.L. The biology of p38 kinase: a central role in inflammation. Curr. Top. Med. Chem. 2005, 5, 921–928. [Google Scholar]
- Freshney, N.W.; Rawlinson, L.; Guesdon, F.; Jones, E.; Cowley, S.; Hsuan, J.; Saklatvala, J. Interleukin-1 activates a novel protein kinase cascade that results in the phosphorylation of Hsp27. Cell 1994, 78, 1039–1049. [Google Scholar]
- Kumar, S.; Boehm, J.; Lee, J.C. p38 MAP kinases: key signalling molecules as therapeutic targets for inflammatory diseases. Nat. Rev. Drug Discovery 2003, 2, 717–726. [Google Scholar]
- Kim, G.Y.; Mercer, S.E.; Ewton, D.Z.; Yan, Z.; Jin, K.; Friedman, E. The stress-activated protein kinases p38α and JNK1 stabilize p21Cip1 by phosphorylation. J. Biol. Chem. 2002, 277, 29792–29802. [Google Scholar]
- Pagano, G.; Zatterale, A.; Degan, P.; d'Ischia, M.; Kelly, F.J.; Pallardo, F.V.; Kodama, S. Multiple involvement of oxidative stress in werner syndrome phenotype. Biogerontology 2005, 6, 233–243. [Google Scholar]
- Pagano, G.; Zatterale, A.; Degan, P.; d'Ischia, M.; Kelly, F.J.; Pallardo, F.V.; Calzone, R.; Castello, G.; Dunster, C.; Giudice, A.; Kilinc, Y.; Lloret, A.; Manini, P.; Masella, R.; Vuttariello, E.; Warnau, M. In vivo prooxidant state in Werner syndrome (WS): results from three WS patients and two WS heterozygotes. Free Radical Res. 2005, 39, 529–533. [Google Scholar] [CrossRef]
- Davis, T.; Bagley, M.C.; Dix, M.C.; Murziani, P.G.; Rokicki, M.J.; Widdowson, C.S.; Zayed, J.M.; Bachler, M.A.; Kipling, D. Synthesis and in vivo activity of MK2 and MK2 substrate-selective p38αMAPK inhibitors in Werner syndrome cells. Bioorg. Med. Chem. Lett. 2007, 17, 6832–6835. [Google Scholar] [PubMed]
- Davis, T.; Baird, D.M.; Haughton, M.F.; Jones, C.J.; Kipling, D. Prevention of accelerated cell aging in Werner syndrome using a p38 mitogen-activated protein kinase inhibitor. J. Gerontol. A Biol. Sci. Med. Sci. 2005, 60, 1386–1393. [Google Scholar]
- Davis, T.; Bachler, M.A.; Wyllie, F.S.; Bagley, M.C.; Kipling, D. Evaluating the role of p38 MAP kinase in the growth of Werner syndrome fibroblasts. Ann. N.Y. Acad. Sci. 2010, 1197. in press. [Google Scholar]
- Goldstein, D.M.; Gabriel, T. Pathway to the clinic: inhibition of P38 MAP kinase. A review of ten chemotypes selected for development. Curr. Top. Med. Chem. 2005, 5, 1017–1029. [Google Scholar] [CrossRef] [PubMed]
- Choy, E.H.; Panayi, G.S. Cytokine pathways and joint inflammation in rheumatoid arthritis. N. Engl. J. Med. 2001, 344, 907–916. [Google Scholar]
- Gallagher, T.F.; Fier-Thompson, S.M.; Garigpati, R.S.; Sorenson, M.E.; Smietana, J.M.; Lee, D.; Bender, P.E.; Lee, J.C.; Laydon, J.T.; Griswold, D.E.; Chabot-Fletcher, M.C.; Breton, J.J.; Adams, J.L. 2,4,5- triarylimidazole inhibitors of IL-1 biosynthesis. Bioorg. Med. Chem. Lett. 1995, 5, 1103–1206. [Google Scholar]
- Wilson, K.P.; Fitzgibbon, M.J.; Caron, P.R.; Griffith, J.P.; Chen, W.; McCaffrey, P.G.; Chambers, S.P.; Su, M.S. Crystal structure of p38 mitogen-activated protein kinase. J. Biol. Chem. 1996, 271, 27696–27700. [Google Scholar]
- Zhu, X.; Kim, J.L.; Newcomb, J.R.; Rose, P.E.; Stover, D.R.; Toledo, L.M.; Zhao, H.; Morgenstern, K.A. Structural analysis of the lymphocyte-specific kinase Lck in complex with non-selective and Src family selective kinase inhibitors. Structure 1999, 7, 651–661. [Google Scholar]
- Madhusudan, P.A.; Xuong, N.H.; Taylor, S.S. Crystal structure of a transition state mimic of the catalytic subunit of cAMP-dependent protein kinase. Nat. Struct. Biol. 2002, 9, 273–277. [Google Scholar]
- Tong, L.; Pav, S.; White, D.M.; Rogers, S.; Crane, K.M.; Cywin, C.L.; Brown, M.L.; Pargellis, C.A. A highly specific inhibitor of human p38 MAP kinase binds in the ATP pocket. Nat. Struct. Biol. 1997, 4, 311–316. [Google Scholar]
- Wilson, K.P.; McCaffrey, P.G.; Hsiao, K.; Pazhanisamy, S.; Galullo, V.; Bemis, G.W.; Fitzgibbon, M.J.; Caron, P.R.; Murcko, M.A.; Su, M.S. The structural basis for the specificity of pyridinylimidazole inhibitors of p38 MAP kinase. Chem. Biol. 1997, 4, 423–431. [Google Scholar]
- Wang, Z.; Canagarajah, B.J.; Boehm, J.C.; Kassisa, S.; Cobb, M.H.; Young, P.R.; Abdel-Meguid, S.; Adams, J.L.; Goldsmith, E.J. Structural basis of inhibitor selectivity in MAP kinases. Structure 1998, 6, 1117–1128. [Google Scholar]
- Diller, D.J.; Lin, T.H.; Metzger, A. The discovery of novel chemotypes of p38 kinase inhibitors. Curr. Top. Med. Chem. 2005, 5, 953–965. [Google Scholar]
- Davis, T.; Haughton, M.F.; Jones, C.J.; Kipling, D. Prevention of accelerated cell ageing in the Werner syndrome. Ann. N.Y. Acad. Sci. 2006, 1067, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Davis, T.; Kipling, D. Assessing the role of stress signalling via p38 MAP kinase in the premature senescence of Ataxia Telangiectasia and Werner syndrome fibroblasts. Biogerontology 2009, 10, 253–266. [Google Scholar]
- Chang, S.; Multani, A.S.; Cabrera, N.G.; Naylor, M.L.; Laud, P.; Lombard, D.; Pathak, S.; Guarente, L.; DePinho, R.A. Essential role of limiting telomeres in the pathogenesis of Werner syndrome. Nat. Genet. 2004, 36, 877–882. [Google Scholar]
- Bain, J.; Plater, L.; Elliott, M.; Shpiro, N.; Hastie, C.J.; McLauchlan, H.; Klevernic, I.; Arthur, J.S.; Alessi, D.R.; Cohen, P. The selectivity of protein kinase inhibitors: a further update. Biochem. J. 2007, 408, 297–315. [Google Scholar]
- Godl, K.; Wissing, J.; Kurtenbach, A.; Habenberger, P.; Blencke, S.; Gutbrod, H.; Salassidis, K.; Stein-Gerlach, M.; Missio, A.; Cotten, M.; Daub, H. An efficient proteomics method to identify the cellular targets of protein kinase inhibitors. Proc. Natl. Acad. Sci. USA 2003, 100, 15434–15439. [Google Scholar]
- Regan, J.; Breitfelder, S.; Cirillo, P.; Gilmore, T.; Graham, A.G.; Hickey, E.; Klaus, B.; Madwed, J.; Moriak, M.; Moss, N.; Pargellis, C.; Pav, S.; Proto, A.; Swinamer, A.; Tong, L.; Torcellini, C. Pyrazole urea-based inhibitors of p38 MAP kinase: from lead compound to clinical candidate. J. Med. Chem. 2002, 45, 2994–3008. [Google Scholar]
- Brown, K.K.; Heitmeyer, S.A.; Hookfin, E.B.; Hsieh, L.; Buchalova, M.; Taiwo, Y.O.; Janusz, M.J. P38 MAP kinase inhibitors as potential therapeutics for the treatment of joint degeneration and pain associated with osteoarthritis. J. Inflamm. (Lond.) 2008, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Haddad, J.J. VX-745. Vertex Pharmaceuticals. Curr. Opin. Investig. Drugs 2001, 2, 1070–1076. [Google Scholar] [PubMed]
- Weidsman, M.; Furst, D.; Schiff, M. A double-blind, placebo-controlled trial of VX-745, an oral p38 mitogen activated protein kinase (MAPK) inhibitor, in patients with rheumatoid arthritis (RA). In Presented at: The 2002 annual European Congress of Rheumatology, Stockholm, Sweden, 12–15 June 2002.
- Goldstein, D.M.; Kuglstatter, A.; Lou, Y.; Soth, M.J. Selective p38α inhibitors clinically evaluated for the treatment of chronic inflammatory disorders. J. Med. Chem. 2010, 53, 2345–2353. [Google Scholar]
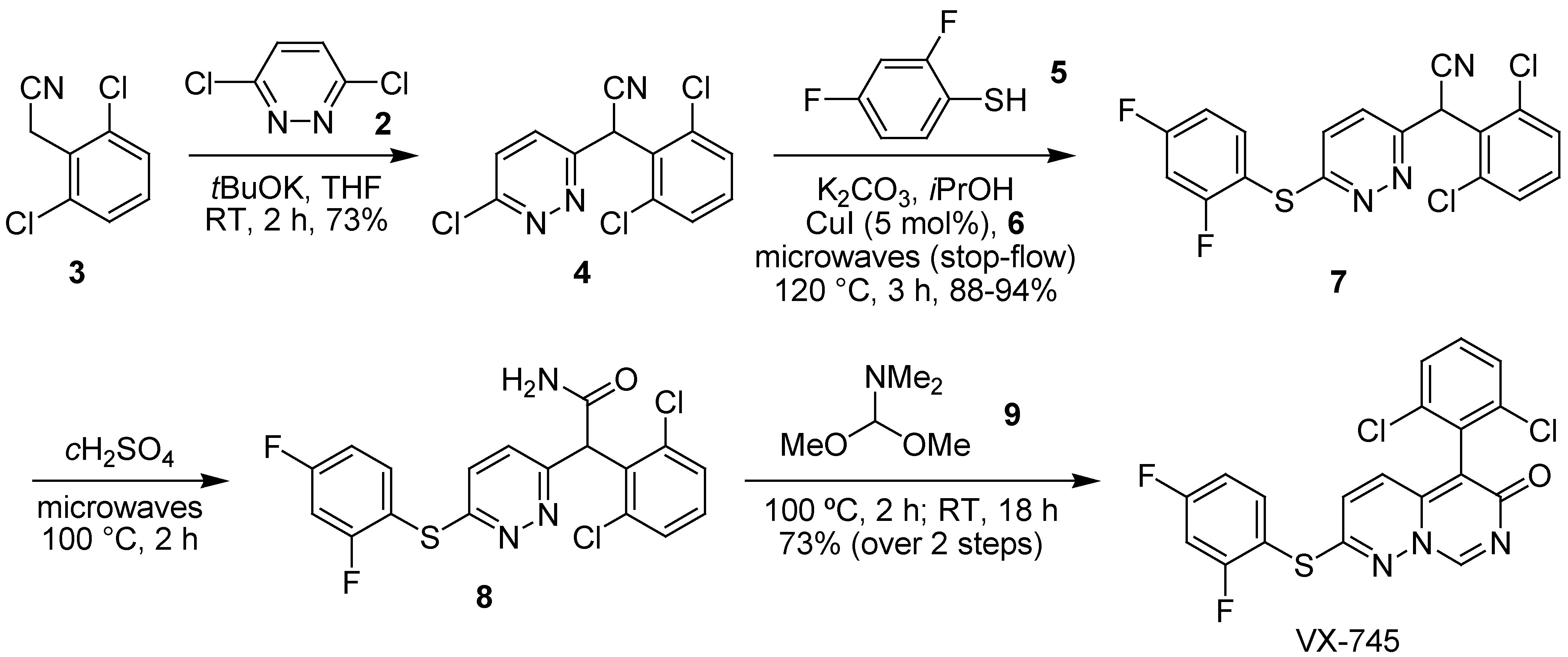
- Bagley, M.C.; Davis, T.; Dix, M.C.; Rokicki, M.J.; Kipling, D. Rapid synthesis of VX-745: p38 MAP kinase inhibition in Werner syndrome cells. Bioorg. Med. Chem. Lett. 2007, 17, 5107–5110. [Google Scholar]
- Bagley, M.C.; Dix, M.C.; Fusillo, V. Rapid Ullmann-type synthesis of aryl sulfides using a copper(I) catalyst and ligand under microwave irradiation. Tetrahedron Lett. 2009, 50, 3661–3664. [Google Scholar]
- Bagley, M.C.; Davis, T.; Dix, M.C.; Fusillo, V.; Pigeaux, M.; Rokicki, M.J.; Kipling, D. Microwave-assisted Ullmann C-S bond formation: synthesis of the p38α MAPK clinical candidate VX-745. J. Org. Chem. 2009, 74, 8336–8342. [Google Scholar]
- Bagley, M.C.; Davis, T.; Dix, M.C.; Fusillo, V.; Pigeaux, M.; Rokicki, M.J.; Kipling, D. Gramme-scale synthesis of the p38α MAPK inhibitor VX-745 for pre-clinical studies into Werner syndrome. Future Med. Chem. 2010, 2. in press. [Google Scholar]
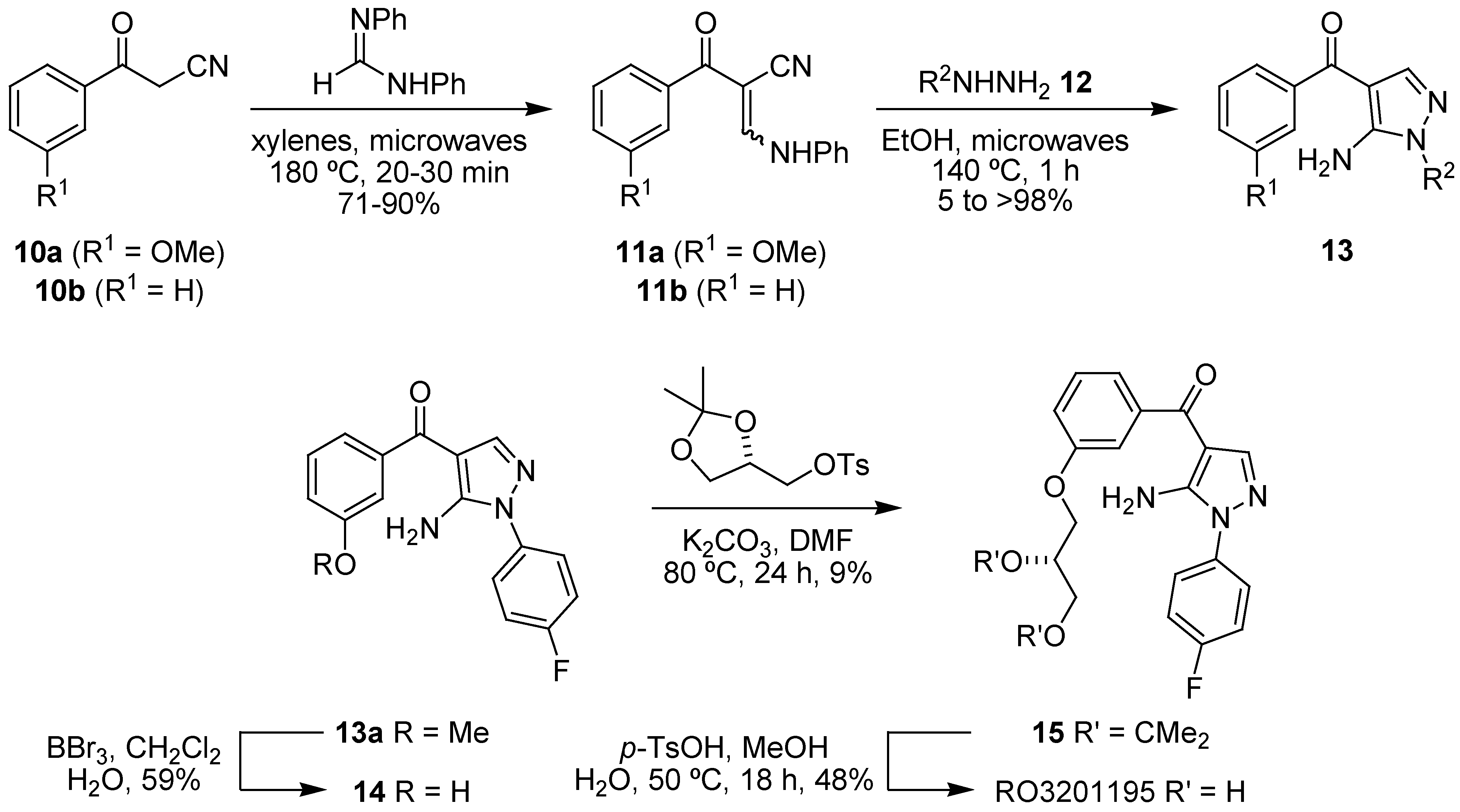
- Goldstein, D.M.; Alfredson, T.; Bertrand, J.; Browner, M.F.; Clifford, K.; Dalrymple, S.A.; Dunn, J.; Freire-Moar, J.; Harris, S.; Labadie, S.S.; La Fargue, J.; Lapierre, J.M.; Larrabee, S.; Li, F.; Papp, E.; McWeeney, D.; Ramesha, C.; Roberts, R.; Rotstein, D.; San Pablo, B.; Sjogren, E.B.; So, O.Y.; Talamas, F.X.; Tao, W.; Trejo, A.; Villasenor, A.; Welch, M.; Welch, T.; Weller, P.; Whiteley, P.E.; Young, K.; Zipfel, S. Discovery of S-[5-amino-1-(4-fluorophenyl)-1H-pyrazol-4-yl]-[3-(2,3-dihydroxypropoxy)phenyl]methanone (RO3201195), an orally bioavailable and highly selective inhibitor of p38 MAP kinase. J. Med. Chem. 2006, 49, 1562–1575. [Google Scholar]
- Bagley, M.C.; Davis, T.; Dix, M.C.; Murziani, P.G.; Rokicki, M.J.; Kipling, D. Microwave-assisted synthesis of 5-aminopyrazol-4-yl ketones and the p38(MAPK) inhibitor RO3201195 for study in Werner syndrome cells. Bioorg. Med. Chem. Lett. 2008, 18, 3745–3748. [Google Scholar]
- Bagley, M.C.; Davis, T.; Dix, M.C.; Murziani, P.G.; Rokicki, M.J.; Kipling, D. Microwave-assisted synthesis of a pyrazolyl ketone library for evaluation as p38 MAPK inhibitors in Werner syndrome cells. Future Med. Chem. 2010, 2, 203–213. [Google Scholar]
- Pargellis, C.; Tong, L.; Churchill, L.; Cirillo, P.F.; Gilmore, T.; Graham, A.G.; Grob, P.M.; Hickey, E.R.; Moss, N.; Pav, S.; Regan, J. Inhibition of p38 MAP kinase by utilizing a novel allosteric binding site. Nat. Struct. Biol. 2002, 9, 268–272. [Google Scholar]
- Sullivan, J.E.; Holdgate, G.A.; Campbell, D.; Timms, D.; Gerhardt, S.; Breed, J.; Breeze, A.L.; Bermingham, A.; Pauptit, R.A.; Norman, R.A.; Embrey, K.J.; Read, J.; VanScyoc, W.S.; Ward, W.H. Prevention of MKK6-dependent activation by binding to p38α MAP kinase. Biochemistry 2005, 44, 16475–16490. [Google Scholar]
- Gruenbaum, L.M.; Schwartz, R.; Woska, J.R., Jr.; DeLeon, R.P.; Peet, G.W.; Warren, T.C.; Capolino, A.; Mara, L.; Morelock, M.M.; Shrutkowski, A.; Jones, J.W.; Pargellis, C.A. Inhibition of pro-inflammatory cytokine production by the dual p38/JNK2 inhibitor BIRB796 correlates with the inhibition of p38 signaling. Biochem. Pharmacol. 2009, 77, 422–432. [Google Scholar]
- Kuma, Y.; Sabio, G.; Bain, J.; Shpiro, N.; Marquez, R.; Cuenda, A. BIRB796 inhibits all p38 MAPK isoforms in vitro and in vivo. J. Biol. Chem. 2005, 280, 19472–19479. [Google Scholar] [PubMed]
- Bagley, M.C.; Davis, T.; Dix, M.C.; Widdowson, C.S.; Kipling, D. Microwave-assisted synthesis of N-pyrazole ureas and the p38α inhibitor BIRB 796 for study into accelerated cell ageing. Org. Biomol. Chem. 2006, 4, 4158–4164. [Google Scholar] [CrossRef] [PubMed]
- Quinn, J.F.; Bryant, C.E.; Golden, K.C.; Gregg, B.T. Rapid reduction of heteroaromatic nitro groups using catalytic transfer hydrogenation with microwave heating. Tetrahedron Lett. 2010, 51, 786–789. [Google Scholar]
- Almansa, C.; Virgili, M. Pyrazolopyridine derivatives. International Patent Application WO 2004076450, 2004. [Google Scholar]
- Soliva, R.; Gelpi, J.L.; Almansa, C.; Virgili, M.; Orozco, M. Dissection of the recognition properties of p38 MAP kinase. Determination of the binding mode of a new pyridinyl-heterocycle inhibitor family. J. Med. Chem. 2007, 50, 283–293. [Google Scholar] [PubMed]
- Almansa, C. New pyrazolopyridines as p38MAP kinase inhibitors. In Presented at: 2nd RSC-SCI Symposium on kinase inhibitor design, NV Organon, Oss, The Netherlands, 14–15 May 2007.
- Treatment of musculoskeletal and connective tissue diseases. Drug Data Rep. 2008, 310, 715.
- Mihara, K.; Almansa, C.; Smeets, R.L.; Loomans, E.E.; Dulos, J.; Vink, P.M.; Rooseboom, M.; Kreutzer, H.; Cavalcanti, F.; Boots, A.M.; Nelissen, R.L. A potent and selective p38 inhibitor protects against bone damage in murine collagen-induced arthritis: a comparison with neutralization of mouse TNFα. Br. J. Pharmacol. 2008, 154, 153–164. [Google Scholar]
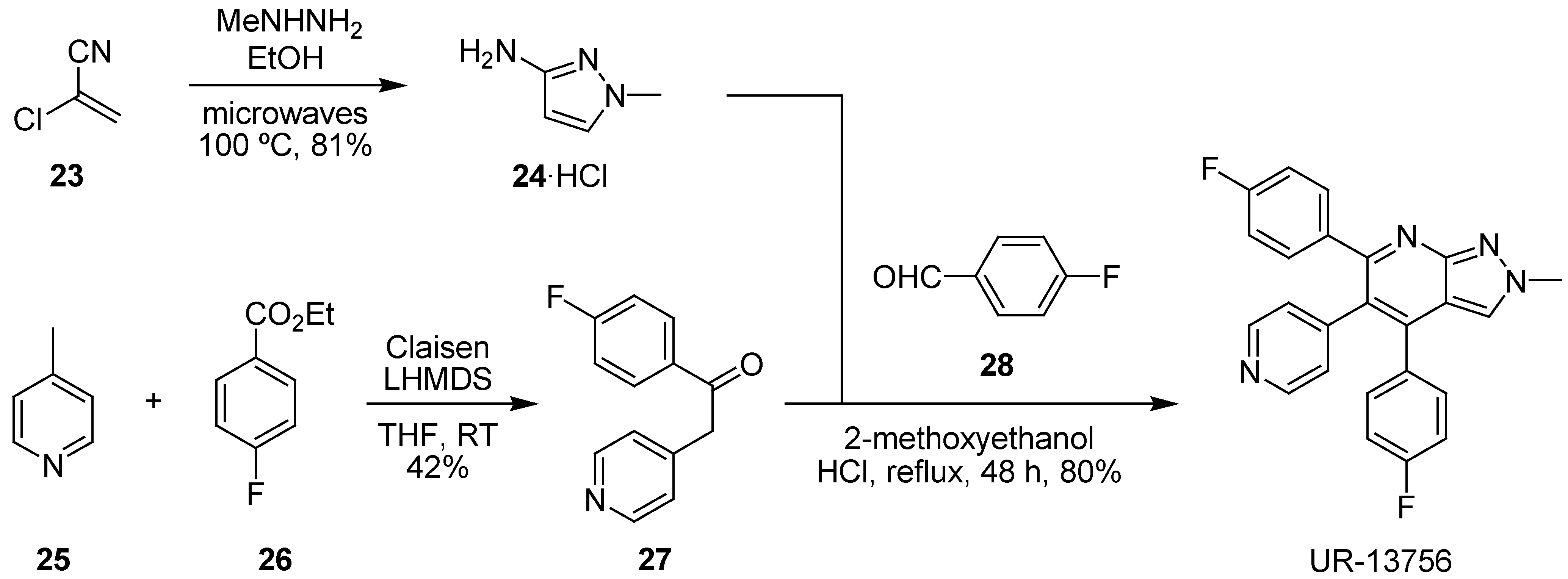
- Bagley, M.C.; Davis, T.; Rokicki, M.J.; Widdowson, C.S.; kipling, D. Synthesis of the highly selective p38 MAPK inhibitor UR-13756 for possible therapeutic use in Werner syndrome. Future Med. Chem. 2010, 2, 193–201. [Google Scholar]
- Almansa, C.; Virgili, M.; Carceller, E.; Grima-Poveda, P. Versatile three component coupling for the synthesis of pyrazolopyridines and other pyrido fused systems. Heterocycles 2008, 75, 1695–1709. [Google Scholar]
- Chang, S. A mouse model of Werner Syndrome: what can it tell us about aging and cancer? Int. J. Biochem. Cell Biol. 2005, 37, 991–999. [Google Scholar] [CrossRef] [PubMed]
- Bowman, M.D.; Schmink, J.R.; McGowan, C.M.; Kormos, C.M.; Leadbeater, N.E. Scale-up of microwave-promoted reactions to the multigram level using sealed-vessel microwave apparatus. Org. Process Res. Dev. 2008, 12, 1078–1088. [Google Scholar] [CrossRef]
- Schmink, J.R.; Kormos, C.M.; Devine, W.G.; Leadbeater, N.E. Exploring the scope for scale-up of organic chemistry using a large batch microwave reactor. Org. Process Res. Dev. 2010, 14, 205–214. [Google Scholar]
- Stadler, A.; Yousefi, B.H.; Dallinger, D.; Walla, P.; Van der Eychen, E.; Kaval, N.; Kappe, C.O. Scalability of microwave-assisted organic synthesis. From single-mode to multimode parallel batch reactors. Org. Process Res. Dev. 2003, 7, 707–716. [Google Scholar] [CrossRef]
- Glasnov, T.N.; Kappe, C.O. Microwave-assisted synthesis under continuous-flow conditions. Macromol. Rapid Commun. 2006, 28, 395–410. [Google Scholar]
- Bagley, M.C.; Jenkins, R.L.; Lubinu, M.C.; Mason, C.; Wood, R. A simple continuous flow microwave reactor. J. Org. Chem. 2005, 70, 7003–7006. [Google Scholar]
- Bagley, M.C.; Fusillo, V.; Jenkins, R.L.; Lubinu, M.C.; Mason, C. Continuous flow processing from microreactors to mesoscale: the Bohlmann-Rahtz cyclodehydration reaction. Org. Biomol. Chem. 2010, 8, 2245–2251. [Google Scholar]
- Shi, Y.; Kotlyarov, A.; Laabeta, K.; Gruber, A.D.; Butt, E.; Marcus, K.; Meyer, H.E.; Friedrich, A.; Volk, H.D.; Gaestel, M. Elimination of protein kinase MK5/PRAK activity by targeted homologous recombination. Mol. Cell. Biol. 2003, 23, 7732–7741. [Google Scholar]
- Manke, I.A.; Nguyen, A.; Lim, D.; Stewart, M.Q.; Elia, A.E.; Yaffe, M.B. MAPKAP kinase-2 is a cell cycle checkpoint kinase that regulates the G2/M transition and S phase progression in response to UV irradiation. Mol. Cell 2005, 17, 37–48. [Google Scholar]
- Dean, J.L.; Sully, G.; Clark, A.R.; Saklatvala, J. The involvement of AU-rich element-binding proteins in p38 mitogen-activated protein kinase pathway-mediated mRNA stabilisation. Cell Signal. 2004, 16, 1113–1121. [Google Scholar]
- Duraisamy, S.; Bajpai, M.; Bughani, U.; Dastidar, S.G.; Ray, A.; Chopra, P. MK2: a novel molecular target for anti-inflammatory therapy. Expert Opin. Ther. Targets 2008, 12, 921–936. [Google Scholar]
- Davidson, W.; Frego, L.; Peet, G.W.; Kroe, R.R.; Labadia, M.E.; Lukas, S.M.; Snow, R.J.; Jakes, S.; Grygon, C.A.; Pargellis, C.; Werneburg, B.G. Discovery and characterization of a substrate selective p38α inhibitor. Biochemistry 2004, 43, 11658–11671. [Google Scholar]
- Anderson, D.R.; Hegde, S.; Reinhard, E.; Gomez, L.; Vernier, W.F.; Lee, L.; Liu, S.; Sambandam, A.; Snider, P.A.; Masih, L. Aminocyanopyridine inhibitors of mitogen activated protein kinase-activated protein kinase 2 (MK-2). Bioorg. Med. Chem. Lett. 2005, 15, 1587–1590. [Google Scholar]
- Anderson, D.R.; Meyers, M.J.; Vernier, W.F.; Mahoney, M.W.; Kurumbail, R.G.; Caspers, N.; Poda, G.I.; Schindler, J.F.; Reitz, D.B.; Mourey, R.J. Pyrrolopyridine inhibitors of mitogen-activated protein kinase-activated protein kinase 2 (MK-2). J. Med. Chem. 2007, 50, 2647–2654. [Google Scholar]
- Goldberg, D.R.; Choi, Y.; Cogan, D.; Corson, M.; DeLeon, R.; Gao, A.; Gruenbaum, L.; Hao, M.H.; Joseph, D.; Kashem, M.A.; Miller, C.; Moss, N.; Netherton, M.R.; Pargellis, C.P.; Pelletier, J.; Sellati, R.; Skow, D.; Torcellini, C.; Tseng, Y.C.; Wang, J.; Wasti, R.; Werneburg, B.; Wu, J.P.; Xiong, Z. Pyrazinoindolone inhibitors of MAPKAP-K2. Bioorg. Med. Chem. Lett. 2008, 18, 938–941. [Google Scholar]
- Trujillo, J.I.; Meyers, M.J.; Anderson, D.R.; Hegde, S.; Mahoney, M.W.; Vernier, W.F.; Buchler, I.P.; Wu, K.K.; Yang, S.; Hartmann, S.J.; Reitz, D.B. Novel tetrahydro-β-carboline-1-carboxylic acids as inhibitors of mitogen activated protein kinase-activated protein kinase 2 (MK-2). Bioorg. Med. Chem. Lett. 2007, 17, 4657–4663. [Google Scholar]
- Schlapbach, A.; Huppertz, C. Low-molecular weight MK2 inhibitors: a tough nut to crack. Future Med. Chem. 2009, 1, 1243–1257. [Google Scholar]
- Genovese, M.C. Inhibition of p38: has the fat lady sung? Arthritis Rheum. 2009, 60, 317–320. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an Open Access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bagley, M.C.; Davis, T.; Murziani, P.G.S.; Widdowson, C.S.; Kipling, D. Use of p38 MAPK Inhibitors for the Treatment of Werner Syndrome. Pharmaceuticals 2010, 3, 1842-1872. https://doi.org/10.3390/ph3061842
Bagley MC, Davis T, Murziani PGS, Widdowson CS, Kipling D. Use of p38 MAPK Inhibitors for the Treatment of Werner Syndrome. Pharmaceuticals. 2010; 3(6):1842-1872. https://doi.org/10.3390/ph3061842
Chicago/Turabian StyleBagley, Mark C., Terence Davis, Paola G. S. Murziani, Caroline S. Widdowson, and David Kipling. 2010. "Use of p38 MAPK Inhibitors for the Treatment of Werner Syndrome" Pharmaceuticals 3, no. 6: 1842-1872. https://doi.org/10.3390/ph3061842
APA StyleBagley, M. C., Davis, T., Murziani, P. G. S., Widdowson, C. S., & Kipling, D. (2010). Use of p38 MAPK Inhibitors for the Treatment of Werner Syndrome. Pharmaceuticals, 3(6), 1842-1872. https://doi.org/10.3390/ph3061842