The Pharmacogenomics of Anti-Hypertensive Therapy

{kind=link}
Abstract
:1. Hypertension—Scale of the Problem
2. Antihypertensive Therapy—Control and Resistance
3. Variability in Antihypertensive Drug Response
4. Genetic and Non-Genetic Contribution to Drug Response
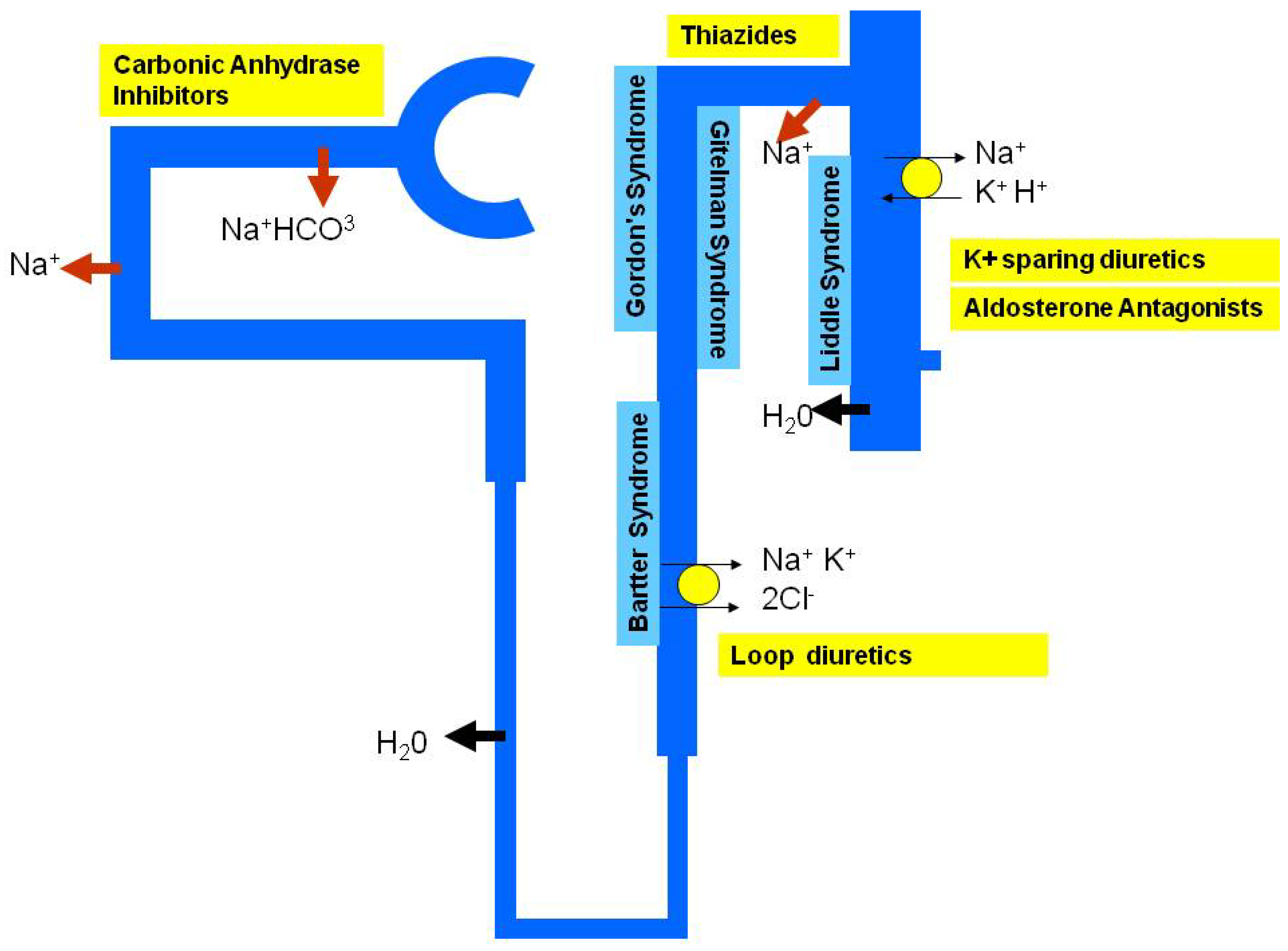
5. Monogenic Hypertension and Personalised Treatment
6. Essential Hypertension and Personalised Treatment
7. Designing a Pharmacogenetic Study of Antihypertensive Treatment
8. Conclusions
References
- Lewington, S.; Clarke, R.; Qizilbash, N.; Peto, R.; Collins, R. Age-specific relevance of usual blood pressure to vascular mortality: A meta-analysis of individual data for one million adults in 61 prospective studies. Lancet 2002, 360(9349), 1903–1913. [Google Scholar] [PubMed]
- Kearney, P.M.; Whelton, M.; Reynolds, K.; Muntner, P.; Whelton, P.K.; He, J. Global burden of hypertension: analysis of worldwide data. Lancet 2005, 365(9455), 217–223. [Google Scholar] [PubMed]
- Ezzati, M.; Lopez, A.D.; Rodgers, A.; Vander, H.S.; Murray, C.J. Selected major risk factors and global and regional burden of disease. Lancet 2002, 360(9343), 1347–1360. [Google Scholar] [PubMed]
- Hajjar, I.; Kotchen, T.A. Trends in prevalence, awareness, treatment, and control of hypertension in the United States, 1988-2000. JAMA 2003, 290(2), 199–206. [Google Scholar] [CrossRef] [PubMed]
- Lloyd-Jones, D.M.; Evans, J.C.; Larson, M.G.; O'Donnell, C.J.; Roccella, E.J.; Levy, D. Differential control of systolic and diastolic blood pressure : Factors associated with lack of blood pressure control in the community. Hypertension 2000, 36(4), 594–599. [Google Scholar] [PubMed]
- Thoenes, M.; Neuberger, H.R.; Volpe, M.; Khan, B.V.; Kirch, W.; Bohm, M. Antihypertensive drug therapy and blood pressure control in men and women: An international perspective. J. Hum. Hypertens. 2010, 24(5), 336–344. [Google Scholar] [CrossRef] [PubMed]
- Cushman, W.C.; Ford, C.E.; Cutler, J.A.; Margolis, K.L.; Davis, B.R.; Grimm, R.H.; Black, H.R.; Hamilton, B.P.; Holland, J.; Nwachuku, C.; Papademetriou, V.; Probstfield, J.; Wright, J.T., Jr.; Alderman, M.H.; Weiss, R.J.; Piller, L.; Bettencourt, J.; Walsh, S.M. Success and predictors of blood pressure control in diverse North American settings: The antihypertensive and lipid-lowering treatment to prevent heart attack trial (ALLHAT). J. Clin. Hypertens. 2002, 4(6), 393–404. [Google Scholar] [CrossRef]
- Canzanello, V.J.; Baranco-Pryor, E.; Rahbari-Oskoui, F.; Schwartz, G.L.; Boerwinkle, E.; Turner, S.T.; Chapman, A.B. Predictors of blood pressure response to the angiotensin receptor blocker candesartan in essential hypertension. Am. J. Hypertens. 2008, 21(1), 61–66. [Google Scholar] [CrossRef] [PubMed]
- Blaufox, M.D.; Lee, H.B.; Davis, B.; Oberman, A.; Wassertheil-Smoller, S.; Langford, H. Renin predicts diastolic blood pressure response to nonpharmacologic and pharmacologic therapy. JAMA 1992, 267(9), 1221–1225. [Google Scholar] [PubMed]
- Calhoun, D.A.; Jones, D.; Textor, S.; Goff, D.C.; Murphy, T.P.; Toto, R.D.; White, A.; Cushman, W.C.; White, W.; Sica, D.; Ferdinand, K.; Giles, T.D.; Falkner, B.; Carey, R.M. Resistant hypertension: diagnosis, evaluation, and treatment: A scientific statement from the American Heart Association Professional Education Committee of the Council for High Blood Pressure Research. Circulation 2008, 117(25), e510–e526. [Google Scholar] [PubMed]
- Chapman, A.B.; Schwartz, G.L.; Boerwinkle, E.; Turner, S.T. Predictors of antihypertensive response to a standard dose of hydrochlorothiazide for essential hypertension. Kidney Int. 2002, 61(3), 1047–1055. [Google Scholar] [CrossRef] [PubMed]
- Brewster, L.M.; van Montfrans, G.A.; Kleijnen, J. Systematic review: Antihypertensive drug therapy in black patients. Ann. Intern. Med. 2004, 141(8), 614–627. [Google Scholar]
- Johnson, J.A. Ethnic differences in cardiovascular drug response: Potential contribution of pharmacogenetics. Circulation 2008, 118(13), 1383–1393. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.J.; Cruickshank, J.K.; Dominiczak, A.F.; MacGregor, G.A.; Poulter, N.R.; Russell, G.I.; Thom, S.; Williams, B. Better blood pressure control: How to combine drugs. J. Hum. Hypertens. 2003, 17(2), 81–86. [Google Scholar] [PubMed]
- Blumenfeld, J.D.; Laragh, J.H. Renin system analysis: A rational method for the diagnosis and treatment of the individual patient with hypertension. Am. J. Hypertens. 1998, 11(7), 894–896. [Google Scholar] [CrossRef] [PubMed]
- Laragh, J.H. Renin profiling for diagnosis, risk assessment, and treatment of hypertension. Kidney Int. 1993, 44(5), 1163–1175. [Google Scholar] [CrossRef] [PubMed]
- Buhler, F.R.; Bolli, P.; Kiowski, W.; Erne, P.; Hulthen, U.L.; Block, L.H. Renin profiling to select antihypertensive baseline drugs. Renin inhibitors for high-renin and calcium entry blockers for low-renin patients. Am. J. Med. 1984, 77(2A), 36–42. [Google Scholar] [PubMed]
- Laragh, J.H.; Resnick, L.M. Recognizing and treating two types of long-term vasoconstriction in hypertension. Kidney Int. 1988, 25, S162–S174. [Google Scholar]
- Williams, B.; Poulter, N.R.; Brown, M.J.; Davis, M.; McInnes, G.T.; Potter, J.F.; Sever, P.S.; Thom, S.M. British Hypertension Society guidelines for hypertension management 2004 (BHS-IV): summary. BMJ 2004, 328(7440), 634–640. [Google Scholar] [PubMed]
- Padmanabhan, S.; Delles, C.; Dominiczak, A.F. Genetic factors in hypertension. Arch. Med. Sci. 2009, 5(2A), S212–S219. [Google Scholar]
- Lifton, R.P.; Dluhy, R.G.; Powers, M.; Rich, G.M.; Gutkin, M.; Fallo, F.; Gill, J.R., Jr.; Feld, L.; Ganguly, A.; Laidlaw, J.C. Hereditary hypertension caused by chimaeric gene duplications and ectopic expression of aldosterone synthase. Nat. Genet. 1992, 2(1), 66–74. [Google Scholar] [CrossRef] [PubMed]
- Geller, D.S.; Farhi, A.; Pinkerton, N.; Fradley, M.; Moritz, M.; Spitzer, A.; Meinke, G.; Tsai, F.T.; Sigler, P.B.; Lifton, R.P. Activating mineralocorticoid receptor mutation in hypertension exacerbated by pregnancy. Science 2000, 289(5476), 119–123. [Google Scholar] [PubMed]
- Shimkets, R.A.; Warnock, D.G.; Bositis, C.M.; Nelson-Williams, C.; Hansson, J.H.; Schambelan, M.; Gill, J.R., Jr.; Ulick, S.; Milora, R.V.; Findling, J.W. Liddle's syndrome: Heritable human hypertension caused by mutations in the beta subunit of the epithelial sodium channel. Cell 1994, 79(3), 407–414. [Google Scholar] [CrossRef] [PubMed]
- Hansson, J.H.; Nelson-Williams, C.; Suzuki, H.; Schild, L.; Shimkets, R.; Lu, Y.; Canessa, C.; Iwasaki, T.; Rossier, B.; Lifton, R.P. Hypertension caused by a truncated epithelial sodium channel gamma subunit: Genetic heterogeneity of Liddle syndrome. Nat. Genet. 1995, 11(1), 76–82. [Google Scholar] [CrossRef] [PubMed]
- Wilson, F.H.; Disse-Nicodeme, S.; Choate, K.A.; Ishikawa, K.; Nelson-Williams, C.; Desitter, I.; Gunel, M.; Milford, D.V.; Lipkin, G.W.; Achard, J.M.; Feely, M.P.; Dussol, B.; Berland, Y.; Unwin, R.J.; Mayan, H.; Simon, D.B.; Farfel, Z.; Jeunemaitre, X.; Lifton, R.P. Human hypertension caused by mutations in WNK kinases. Science 2001, 293(5532), 1107–1112. [Google Scholar] [PubMed]
- Carretero, O.A.; Oparil, S. Essential hypertension. Part I: Definition and etiology. 2000, 101(3), 329–335. [Google Scholar] [PubMed]
- Luft, F.C. Twins in cardiovascular genetic research. Hypertension 2001, 37(2), 350–356. [Google Scholar] [PubMed]
- Mongeau, J.G.; Biron, P.; Sing, C.F. The influence of genetics and household environment upon the variability of normal blood pressure: The Montreal Adoption Survey. Clin. Exp. Hypertens. 1986, 8(4&5), 653–660. [Google Scholar] [CrossRef]
- Cowley, A.W., Jr. The genetic dissection of essential hypertension. Nat. Rev. Genet. 2006, 7(11), 829–840. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.C.; Hunt, S.C.; Kardia, S.; Turner, S.T.; Chakravarti, A.; Schork, N.; Olshen, R.; Curb, D.; Jaquish, C.; Boerwinkle, E.; Rao, D.C. An investigation of genome-wide associations of hypertension with microsatellite markers in the family blood pressure program (FBPP). Hum. Genet. 2007, 121(5), 577–590. [Google Scholar] [CrossRef] [PubMed]
- Caulfield, M.; Munroe, P.; Pembroke, J.; Samani, N.; Dominiczak, A.; Brown, M.; Benjamin, N.; Webster, J.; Ratcliffe, P.; O'Shea, S.; Papp, J.; Taylor, E.; Dobson, R.; Knight, J.; Newhouse, S.; Hooper, J.; Lee, W.; Brain, N.; Clayton, D.; Lathrop, G.M.; Farrall, M.; Connell, J. Genome-wide mapping of human loci for essential hypertension. Lancet 2003, 361(9375), 2118–2123. [Google Scholar] [PubMed]
- Padmanabhan, S.; Wallace, C.; Munroe, P.B.; Dobson, R.; Brown, M.; Samani, N.; Clayton, D.; Farrall, M.; Webster, J.; Lathrop, M.; Caulfield, M.; Dominiczak, A.F.; Connell, J.M. Chromosome 2p shows significant linkage to antihypertensive response in the British Genetics of Hypertension Study. Hypertension 2006, 47(3), 603–608. [Google Scholar] [CrossRef] [PubMed]
- Barkley, R.A.; Chakravarti, A.; Cooper, R.S.; Ellison, R.C.; Hunt, S.C.; Province, M.A.; Turner, S.T.; Weder, A.B.; Boerwinkle, E. Positional identification of hypertension susceptibility genes on chromosome 2. Hypertension 2004, 43(2), 477–482. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, S.; Melander, O.; Hastie, C.; Menni, C.; Delles, C.; Connell, J.M.; Dominiczak, A.F. Hypertension and genome-wide association studies: Combining high fidelity phenotyping and hypercontrols. J. Hypertens. 2008, 26(7), 1275–1281. [Google Scholar] [CrossRef] [PubMed]
- Arnett, D.K.; Davis, B.R.; Ford, C.E.; Boerwinkle, E.; Leiendecker-Foster, C.; Miller, M.B.; Black, H.; Eckfeldt, J.H. Pharmacogenetic association of the angiotensin-converting enzyme insertion/deletion polymorphism on blood pressure and cardiovascular risk in relation to antihypertensive treatment: The Genetics of Hypertension-Associated Treatment (GenHAT) study. Circulation 2005, 111(25), 3374–3383. [Google Scholar] [PubMed]
- Brunner, M.; Cooper-DeHoff, R.M.; Gong, Y.; Karnes, J.H.; Langaee, T.Y.; Pepine, C.J.; Johnson, J.A. Factors influencing blood pressure response to trandolapril add-on therapy in patients taking verapamil SR (from the International Verapamil SR/Trandolapril [INVEST] Study). Am. J. Cardiol. 2007, 99(11), 1549–1554. [Google Scholar] [CrossRef] [PubMed]
- Turner, S.T.; Schwartz, G.L.; Chapman, A.B.; Boerwinkle, E. WNK1 kinase polymorphism and blood pressure response to a thiazide diuretic. Hypertension 2005, 46(4), 758–765. [Google Scholar] [CrossRef] [PubMed]
- Arnett, D.K.; Claas, S.A.; Lynch, A.I. Has pharmacogenetics brought us closer to 'personalized medicine' for initial drug treatment of hypertension? Curr. Opin. Cardiol. 2009, 24(4), 333–339. [Google Scholar] [CrossRef] [PubMed]
- Pacanowski, M.A.; Gong, Y.; Cooper-DeHoff, R.M.; Schork, N.J.; Shriver, M.D.; Langaee, T.Y.; Pepine, C.J.; Johnson, J.A. beta-adrenergic receptor gene polymorphisms and beta-blocker treatment outcomes in hypertension. Clin. Pharmacol. Ther. 2008, 84(6), 715–721. [Google Scholar] [CrossRef] [PubMed]
- Lemaitre, R.N.; Heckbert, S.R.; Sotoodehnia, N.; Bis, J.C.; Smith, N.L.; Marciante, K.D.; Hindorff, L.A.; Lange, L.A.; Lumley, T.S.; Rice, K.M.; Wiggins, K.L.; Psaty, B.M. beta1- and beta2-adrenergic receptor gene variation, beta-blocker use and risk of myocardial infarction and stroke. Am. J. Hypertens. 2008, 21(3), 290–296. [Google Scholar] [PubMed]
- Johnson, J.A.; Zineh, I.; Puckett, B.J.; McGorray, S.P.; Yarandi, H.N.; Pauly, D.F. Beta 1-adrenergic receptor polymorphisms and antihypertensive response to metoprolol. Clin. Pharmacol. Ther. 2003, 74(1), 44–52. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, Z.Q.; Yu, B.N.; Xu, F.H.; Mo, W.; Zhou, G.; Liu, Y.Z.; Li, Q.; Zhou, H.H. beta1-Adrenergic receptor polymorphisms influence the response to metoprolol monotherapy in patients with essential hypertension. Clin. Pharmacol. Ther. 2006, 80(1), 23–32. [Google Scholar] [CrossRef] [PubMed]
- Psaty, B.M.; Smith, N.L.; Heckbert, S.R.; Vos, H.L.; Lemaitre, R.N.; Reiner, A.P.; Siscovick, D.S.; Bis, J.; Lumley, T.; Longstreth, W.T., Jr.; Rosendaal, F.R. Diuretic therapy, the alpha-adducin gene variant, and the risk of myocardial infarction or stroke in persons with treated hypertension. JAMA 2002, 287(13), 1680–1689. [Google Scholar] [PubMed]
- Davis, B.R.; Arnett, D.K.; Boerwinkle, E.; Ford, C.E.; Leiendecker-Foster, C.; Miller, M.B.; Black, H.; Eckfeldt, J.H. Antihypertensive therapy, the alpha-adducin polymorphism, and cardiovascular disease in high-risk hypertensive persons: The Genetics of Hypertension-Associated Treatment Study. Pharmacogenomics J. 2007, 7(2), 112–122. [Google Scholar] [CrossRef] [PubMed]
- Gerhard, T.; Gong, Y.; Beitelshees, A.L.; Mao, X.; Lobmeyer, M.T.; Cooper-DeHoff, R.M.; Langaee, T.Y.; Schork, N.J.; Shriver, M.D.; Pepine, C.J.; Johnson, J.A. Alpha-adducin polymorphism associated with increased risk of adverse cardiovascular outcomes: Results from GENEtic Substudy of the INternational VErapamil SR-trandolapril STudy (INVEST-GENES). Am. Heart J. 2008, 156(2), 397–404. [Google Scholar] [CrossRef] [PubMed]
- Manunta, P.; Lavery, G.; Lanzani, C.; Braund, P.S.; Simonini, M.; Bodycote, C.; Zagato, L.; Delli, C.S.; Tantardini, C.; Brioni, E.; Bianchi, G.; Samani, N.J. Physiological interaction between alpha-adducin and WNK1-NEDD4L pathways on sodium-related blood pressure regulation. Hypertension 2008, 52(2), 366–372. [Google Scholar] [CrossRef] [PubMed]
- De Buyzere, M. Selective genetic advantages for users of thiazide diuretics. Is there a case for the 460Trp variant of alpha-adducin? J. Hypertens. 2009, 27(1), 24–27. [Google Scholar] [CrossRef] [PubMed]
- Kelley-Hedgepeth, A.; Peter, I.; Kip, K.; Montefusco, M.; Kogan, S.; Cox, D.; Ordovas, J.; Levy, D.; Reis, S.; Mendelsohn, M.; Housman, D.; Huggins, G. The protective effect of KCNMB1 E65K against hypertension is restricted to blood pressure treatment with beta-blockade. J. Hum. Hypertens. 2008, 22(7), 512–515. [Google Scholar] [CrossRef] [PubMed]
- Beitelshees, A.L.; Gong, Y.; Wang, D.; Schork, N.J.; Cooper-DeHoff, R.M.; Langaee, T.Y.; Shriver, M.D.; Sadee, W.; Knot, H.J.; Pepine, C.J.; Johnson, J.A. KCNMB1 genotype influences response to verapamil SR and adverse outcomes in the INternational VErapamil SR/Trandolapril STudy (INVEST). Pharmacogenet. Genomics 2007, 17(9), 719–729. [Google Scholar] [CrossRef] [PubMed]
- Turner, S.T.; Bailey, K.R.; Fridley, B.L.; Chapman, A.B.; Schwartz, G.L.; Chai, H.S.; Sicotte, H.; Kocher, J.P.; Rodin, A.S.; Boerwinkle, E. Genomic association analysis suggests chromosome 12 locus influencing antihypertensive response to thiazide diuretic. Hypertension 2008, 52(2), 359–365. [Google Scholar] [CrossRef] [PubMed]
- Kurland, L.; Lind, L.; Melhus, H. Using genotyping to predict responses to anti-hypertensive treatment. Trends Pharmacol. Sci. 2005, 26(9), 443–447. [Google Scholar] [PubMed]
- Link, E.; Parish, S.; Armitage, J.; Bowman, L.; Heath, S.; Matsuda, F.; Gut, I.; Lathrop, M.; Collins, R. SLCO1B1 variants and statin-induced myopathy—A genomewide study. N. Engl. J. Med. 2008, 359(8), 789–799. [Google Scholar] [PubMed]
- Eichler, E.E.; Flint, J.; Gibson, G.; Kong, A.; Leal, S.M.; Moore, J.H.; Nadeau, J.H. Missing heritability and strategies for finding the underlying causes of complex disease. Nat. Rev. Genet 2010, 11(6), 446–450. [Google Scholar] [CrossRef] [PubMed]
- Cantor, R.M.; Lange, K.; Sinsheimer, J.S. Prioritizing GWAS results: A review of statistical methods and recommendations for their application. Am. J. Hum. Genet 2010, 86(1), 6–22. [Google Scholar] [CrossRef] [PubMed]
- Chanock, S.J.; Manolio, T.; Boehnke, M.; Boerwinkle, E.; Hunter, D.J.; Thomas, G.; Hirschhorn, J.N.; Abecasis, G.; Altshuler, D.; Bailey-Wilson, J.E.; Brooks, L.D.; Cardon, L.R.; Daly, M.; Donnelly, P.; Fraumeni, J.F., Jr.; Freimer, N.B.; Gerhard, D.S.; Gunter, C.; Guttmacher, A.E.; Guyer, M.S.; Harris, E.L.; Hoh, J.; Hoover, R.; Kong, C.A.; Merikangas, K.R.; Morton, C.C.; Palmer, L.J.; Phimister, E.G.; Rice, J.P.; Roberts, J.; Rotimi, C.; Tucker, M.A.; Vogan, K.J.; Wacholder, S.; Wijsman, E.M.; Winn, D.M.; Collins, F.S. Replicating genotype-phenotype associations. Nature 2007, 447(7145), 655–660. [Google Scholar] [PubMed]
- Padmanabhan, S.; Menni, C.; Lee, W.K.; Laing, S.; Brambilla, P.; Sega, R.; Perego, R.; Grassi, G.; Cesana, G.; Delles, C.; Mancia, G.; Dominiczak, A.F. The effects of sex and method of blood pressure measurement on genetic associations with blood pressure in the PAMELA study. J. Hypertens. 2010, 28(3), 465–477. [Google Scholar] [PubMed]
- Pickering, T.G.; Hall, J.E.; Appel, L.J.; Falkner, B.E.; Graves, J.; Hill, M.N.; Jones, D.W.; Kurtz, T.; Sheps, S.G.; Roccella, E.J. Recommendations for blood pressure measurement in humans and experimental animals: part 1: Blood pressure measurement in humans: A statement for professionals from the Subcommittee of Professional and Public Education of the American Heart Association Council on High Blood Pressure Research. Circulation 2005, 111(5), 697–716. [Google Scholar] [PubMed]
- Trazzi, S.; Mutti, E.; Frattola, A.; Imholz, B.; Parati, G.; Mancia, G. Reproducibility of non-invasive and intra-arterial blood pressure monitoring: implications for studies on antihypertensive treatment. J. Hypertens. 1991, 9(2), 115–119. [Google Scholar] [PubMed]
- Mancia, G.; Parati, G.; Pomidossi, G.; Grassi, G.; Casadei, R.; Zanchetti, A. Alerting reaction and rise in blood pressure during measurement by physician and nurse. Hypertension 1987, 9(2), 209–215. [Google Scholar] [PubMed]
- Andersen, U.O.; Henriksen, J.H.; Jensen, G. Sources of measurement variation in blood pressure in large-scale epidemiological surveys with follow-up. Blood Press 2002, 11(6), 357–365. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, K.; O'Loughlin, J.; Chen, S.; Karp, I.; Paradis, G.; Tremblay, J.; Hamet, P.; Pilote, L. Emergence of sex differences in prevalence of high systolic blood pressure: Analysis of a longitudinal adolescent cohort. Circulation 2006, 114(24), 2663–2670. [Google Scholar] [CrossRef] [PubMed]
- Hardy, R.; Kuh, D.; Whincup, P.H.; Wadsworth, M.E. Age at puberty and adult blood pressure and body size in a British birth cohort study. J. Hypertens. 2006, 24(1), 59–66. [Google Scholar] [CrossRef] [PubMed]
- Seda, O.; Tremblay, J.; Gaudet, D.; Brunelle, P.L.; Gurau, A.; Merlo, E.; Pilote, L.; Orlov, S.N.; Boulva, F.; Petrovich, M.; Kotchen, T.A.; Cowley, A.W., Jr.; Hamet, P. Systematic, genome-wide, sex-specific linkage of cardiovascular traits in French Canadians. Hypertension 2008, 51(4), 1156–1162. [Google Scholar] [PubMed]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an Open Access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Padmanabhan, S.; Paul, L.; Dominczak, A.F. The Pharmacogenomics of Anti-Hypertensive Therapy. Pharmaceuticals 2010, 3, 1779-1791. https://doi.org/10.3390/ph3061779
Padmanabhan S, Paul L, Dominczak AF. The Pharmacogenomics of Anti-Hypertensive Therapy. Pharmaceuticals. 2010; 3(6):1779-1791. https://doi.org/10.3390/ph3061779
Chicago/Turabian StylePadmanabhan, Sandosh, Laura Paul, and Anna F. Dominczak. 2010. "The Pharmacogenomics of Anti-Hypertensive Therapy" Pharmaceuticals 3, no. 6: 1779-1791. https://doi.org/10.3390/ph3061779
APA StylePadmanabhan, S., Paul, L., & Dominczak, A. F. (2010). The Pharmacogenomics of Anti-Hypertensive Therapy. Pharmaceuticals, 3(6), 1779-1791. https://doi.org/10.3390/ph3061779