NSAIDs, Mitochondria and Calcium Signaling: Special Focus on Aspirin/Salicylates

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
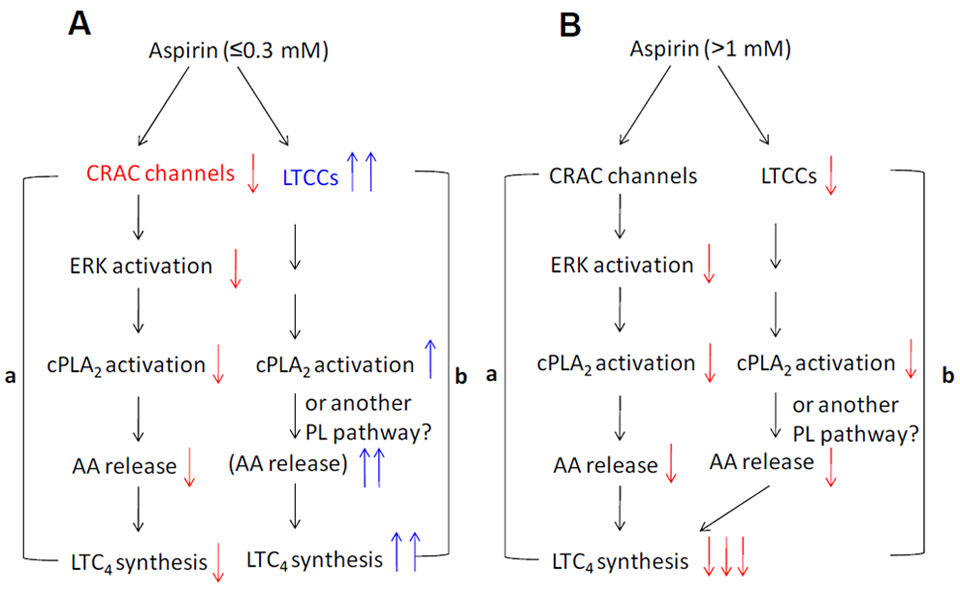
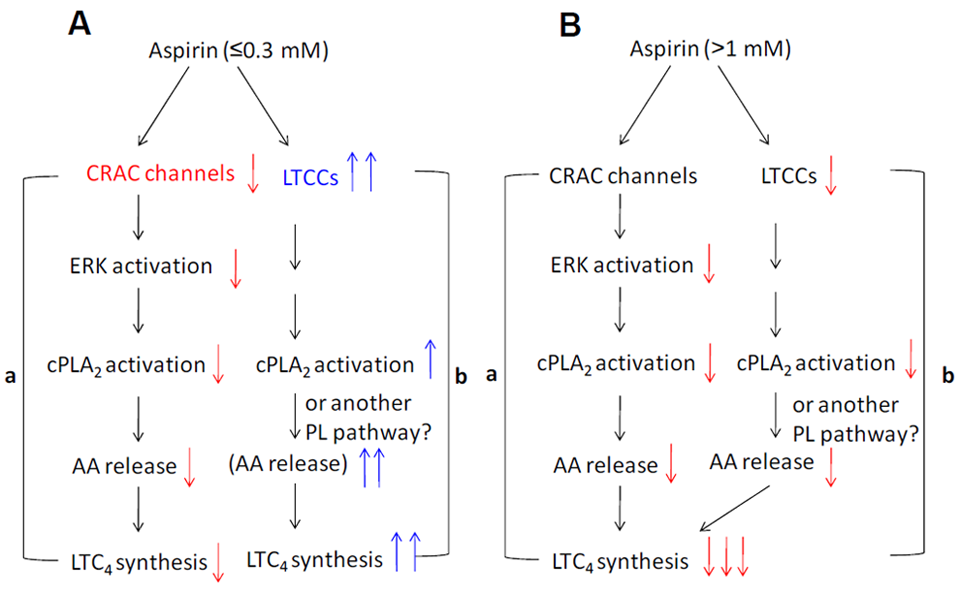
2. COX-Independent Modulation of Mast Cell Activation by NSAIDs
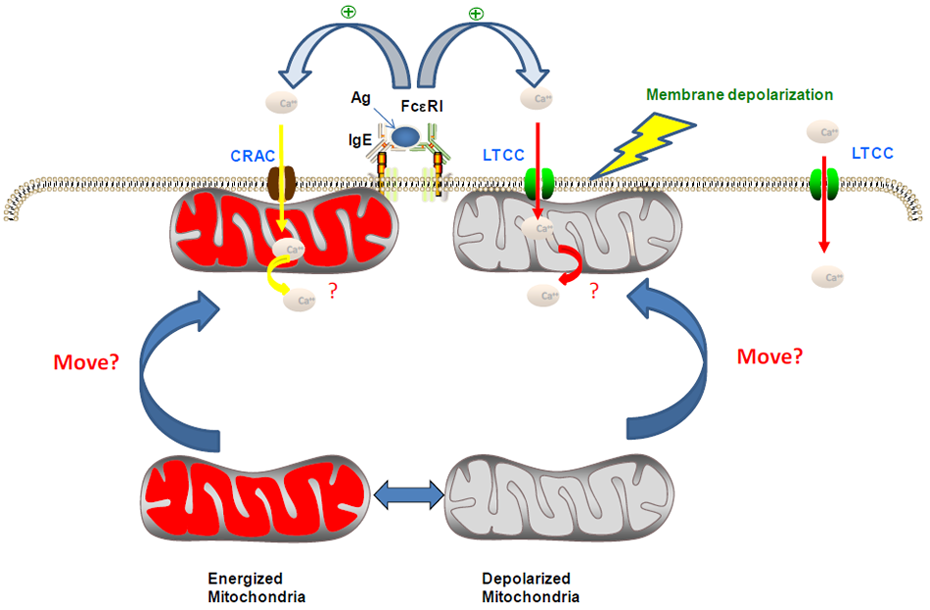
3. Modulation of Ca2+ Channel Activities by NSAIDs
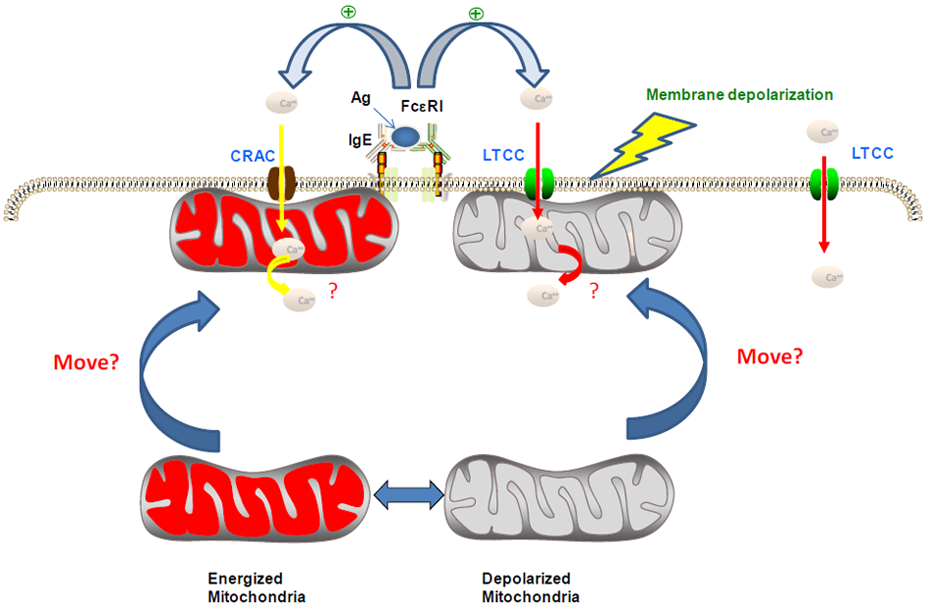
4. Roles of ROS, Ca2+ and Mitochondria in the Chemopreventive Effects of NSAIDs
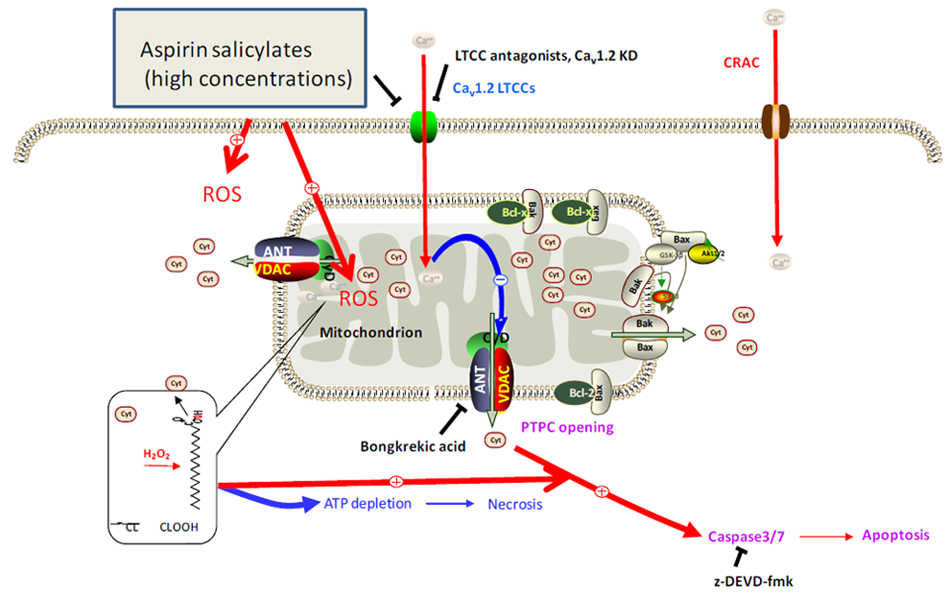
5. Conclusions and Perspectives
Acknowledgments
References
- Vane, J.R. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat. New Biol. 1971, 231, 232–235. [Google Scholar]
- Wu, K.K. Aspirin and other cyclooxygenase inhibitors: New therapeutic insights. Semin. Vasc. Med. 2003, 3, 107–112. [Google Scholar]
- Tegeder, I.; Pfeilschifter, J.; Geisslinger, G. Cyclooxygenase-independent actions of cyclooxygenase inhibitors. FASEB J. 2001, 15, 2057–2072. [Google Scholar]
- Chiabrando, C.; Castelli, M.G.; Cozzi, E.; Fanelli, R.; Campoleoni, A.; Balotta, C.; Latini, R.; Garattini, S. Antiinflammatory action of salicylates: Aspirin is not a prodrug for salicylate against rat carrageenin pleurisy. Eur. J. Pharmacol. 1989, 159, 257–264. [Google Scholar]
- April, P.; Abeles, M.; Baraf, H.; Cohen, S.; Curran, N.; Doucette, M.; Ekholm, B.; Goldlust, B.; Knee, C.M.; Lee, E.; et al. Does the acetyl group of aspirin contribute to the antiinflammatory efficacy of salicylic acid in the treatment of rheumatoid arthritis? Semin. Arthritis Rheum. 1990, 19, 20–28. [Google Scholar] [PubMed]
- Preston, S.J.; Arnold, M.H.; Beller, E.M.; Brooks, P.M.; Buchanan, W.W. Comparative analgesic and anti-inflammatory properties of sodium salicylate and acetylsalicylic acid (aspirin) in rheumatoid arthritis. Br. J. Clin. Pharmacol. 1989, 27, 607–611. [Google Scholar]
- Tegeder, I.; Niederberger, E.; Israr, E.; Gühring, H.; Brune, K.; Euchenhofer, C.; Grösch, S.; Geisslinger, G. Inhibition of NF-kappaB and AP-1 activation by R- and S-flurbiprofen. FASEB J. 2001, 15, 2–4. [Google Scholar]
- Kopp, E.; Ghosh, S. Inhibition of NF-kappa B by sodium salicylate and aspirin. Science 1994, 265, 956–959. [Google Scholar]
- Román, J.; de Arriba, A.F.; Barrón, S.; Michelena, P.; Giral, M.; Merlos, M.; Bailón, E.; Comalada, M.; Gálvez, J.; Zarzuelo, A.; Ramis, I. UR-1505, a new salicylate, blocks T cell activation through nuclear factor of activated T cells. Mol. Pharmacol. 2007, 72, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Mortaz, E.; Redegeld, F.A.; Nijkamp, F.P.; Engels, F. Dual effects of acetylsalicylic acid on mast cell degranulation, expression of cyclooxygenase-2 and release of pro-inflammatory cytokines. Biochem. Pharmacol. 2005, 69, 1049–1057. [Google Scholar]
- Togo, K.; Suzuki, Y.; Yoshimaru, T.; Inoue, T.; Terui, T.; Ochiai, T.; Ra, C. Aspirin and salicylates modulate IgE-mediated leukotriene secretion in mast cells through a dihydropyridine receptor-mediated Ca2+ influx. Clin. Immunol. 2009, 131, 145–156. [Google Scholar]
- Suzuki, Y.; Yoshimaru, T.; Inoue, T.; Terui, T.; Ochiai, T.; Ra, C. Analysis of the mechanism for the development of allergic skin inflammation and the application for its treatment: Aspirin modulation of IgE-dependent mast cell activation: Role of aspirin-induced exacerbation of immediate allergy. J. Pharmacol. Sci. 2009, 110, 237–244. [Google Scholar]
- Gupta, R.A.; DuBois, R.N. Aspirin, NSAIDS, and colon cancer prevention: mechanisms? Gastroenterology 1998, 114, 1095–1098. [Google Scholar] [PubMed]
- Thun, M.J.; Henley, S.J.; Patrono, C. Nonsteroidal anti-inflammatory drugs as anticancer agents: Mechanistic, pharmacologic, and clinical issues. J. Natl. Cancer Inst. 2002, 94, 252–266. [Google Scholar] [PubMed]
- Ulrich, C.M.; Bigler, J.; Potter, J.D. Non-steroidal anti-inflammatory drugs for cancer prevention: promise, perils and pharmacogenetics. Nat. Rev. Cancer 2006, 6, 130–140. [Google Scholar]
- Akre, K.; Ekström, A.M.; Signorello, L.B.; Hansson, L.E.; Nyrén, O. Aspirin and risk for gastric cancer: A population-based case-control study in Sweden. Br. J. Cancer 2001, 84, 965–968. [Google Scholar]
- Schreinemachers, D.M.; Everson, R.B. Aspirin use and lung, colon, and breast cancer incidence in a prospective study. Epidemiology 1994, 5, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, E.J.; Thun, M.J.; Bain, E.B.; Rodriguez, C.; Henley, S.J.; Calle, E.E. A large cohort study of long-term daily use of adult-strength aspirin and cancer incidence. J. Natl. Cancer Inst. 2007, 99, 608–615. [Google Scholar]
- Schildkraut, J.M.; Moorman, P.G.; Halabi, S.; Calingaert, B.; Marks, J.R.; Berchuck, A. Analgesic drug use and risk of ovarian cancer. Epidemiology 2006, 17, 104–107. [Google Scholar]
- Sheng, H.; Shao, J.; Morrow, J.D.; Beauchamp, R.D.; DuBois, R.N. Modulation of apoptosis and Bcl-2 expression by prostaglandin E2 in human colon cancer cells. Cancer Res. 1998, 58, 362–366. [Google Scholar]
- Tsujii, M.; DuBois, R.N. Alterations in cellular adhesion and apoptosis in epithelial cells overexpressing prostaglandin endoperoxide synthase 2. Cell 1995, 83, 493–501. [Google Scholar]
- Masferrer, J.L.; Leahy, K.M.; Koki, A.T.; Zweifel, B.S.; Settle, S.L.; Woerner, B.M.; Edwards, D.A.; Flickinger, A.G.; Moore, R.J.; Seibert, K. Antiangiogenic and antitumor activities of cyclooxygenase-2 inhibitors. Cancer Res. 2000, 60, 1306–1311. [Google Scholar]
- Hanif, R.; Pittas, A.; Feng, Y.; Koutsos, M.I.; Qiao, L.; Staiano-Coico, L.; Shiff, S.I.; Rigas, B. Effects of nonsteroidal anti-inflammatory drugs on proliferation and on induction of apoptosis in colon cancer cells by a prostaglandin-independent pathway. Biochem. Pharmacol. 1999, 52, 235–245. [Google Scholar]
- Lai, M.Y.; Huang, J.A.; Liang, Z.H.; Jiang, H.X.; Tang, G.D. Mechanisms underlying aspirin-mediated growth inhibition and apoptosis induction of cyclooxygenase-2 negative colon cancer cell line SW480. World J. Gastroenterol. 2008, 14, 4227–4233. [Google Scholar]
- Zhang, X.; Morham, S.G.; Langenbach, R.; Young, D.A. Malignant transformation and antineoplastic actions of nonsteroidal antiinflammatory drugs (NSAIDs) on cyclooxygenase-null embryo fibroblasts. J. Exp. Med. 1999, 190, 451–459. [Google Scholar]
- Jana, N.R. NSAIDs and apoptosis. Cell Mol. Life Sci. 2008, 65, 1295–1301. [Google Scholar]
- Sung, J.; Russell, R.I.; Nyeomans, Chan F.K.; Chen, S.; Fock, K.; Goh, K.L.; Kullavanijaya, P.; Kimura, K.; Lau, C.; Louw, J.; Sollano, J.; Triadiafalopulos, G.; Xiao, S,; Brooks, P. Non-steroidal anti-inflammatory drug toxicity in the upper gastrointestinal tract. J. Gastroenterol. Hepatol. 2000, 15 Suppl., G58–G68. [Google Scholar] [CrossRef] [PubMed]
- Grattan, C.E. Aspirin sensitivity and urticaria. Clin. Exp. Dermatol. 2003, 28, 123–127. [Google Scholar]
- Ying, S.; Corrigan, C.J.; Lee, T.H. Mechanisms of aspirin-sensitive asthma. Allergol. Int. 2004, 53, 111–119. [Google Scholar]
- Morita, E.; Kunie, K.; Matsuo, H. Food-dependent exercise-induced anaphylaxis. J. Dermatol. Sci. 2007, 47, 109–117. [Google Scholar]
- Kinet, J.P. The high-affinity IgE receptor (FcεRI): From physiology to pathology. Annu. Rev. Immunol. 1999, 17, 931–972. [Google Scholar]
- Mortaz, E.; Engels, F.; Nijkamp, F.P.; Redegeld, F.A. New insights on the possible role of mast cells in aspirin-induced asthma. Curr. Mol. Pharmacol. 2009, 2, 182–189. [Google Scholar]
- Bae, J.S.; Kim, S.H.; Ye, Y.M.; Yoon, H.J.; Suh, C.H.; Nahm, D.H.; Park, H.S. Significant association of FcεRIα promoter polymorphisms with aspirin-intolerant chronic urticaria. J. Allergy Clin. Immunol. 2007, 119, 449–456. [Google Scholar] [PubMed]
- Sullivan, S.; Dahlén, B.; Dahlén, S.E.; Kumlin, M. Increased urinary excretion of the prostaglandin D2 metabolite 9α, 11β-prostaglandin F2 after aspirin challenge supports mast cell activation in aspirin-induced airway obstruction. J. Allergy Clin. Immunol. 1996, 98, 421–432. [Google Scholar]
- Mita, H.; Endoh, S.; Kudoh, M.; Kawagishi, Y.; Kobayashi, M.; Taniguchi, M.; Akiyama, K. Possible involvement of mast-cell activation in aspirin provocation of aspirin-induced asthma. Allergy 2001, 56, 1061–1067. [Google Scholar]
- Wang, X.S.; Wu, A.Y.; Leung, P.S.; Lau, H.Y. PGE2 suppresses excessive anti-IgE induced cysteinyl leucotrienes production in mast cells of patients with aspirin exacerbated respiratory disease. Allergy 2007, 62, 620–627. [Google Scholar]
- Stevenson, D.D.; Hankammer, M.A.; Mathison, D.A.; Christiansen, S.C.; Simon, R.A. Aspirin desensitization treatment of aspirin-sensitive patients with rhinosinusitis-asthma: Long-term outcomes. J. Allergy Clin. Immunol. 1996, 98, 751–758. [Google Scholar]
- Leslie, C.C. Properties and regulation of cytosolic phospholipase A2. J. Biol. Chem. 1997, 272, 16709–16712. [Google Scholar]
- Gijón, M.A.; Leslie, C.C. Regulation of arachidonic acid release and cytosolic phospholipase A2 activation. J. Leukoc. Biol. 1999, 65, 330–336. [Google Scholar]
- Nemenoff, R.A.; Winitz, S.; Qian, N.X.; Van Putten, V.; Johnson, G.L.; Heasley, L.E. Phosphorylation and activation of a high molecular weight form of phospohlipase A2 by p42 microtubule-associated protein 2 kinase and protein kinase C. J. Biol. Chem. 1993, 268, 1960–1964. [Google Scholar]
- Hefner, Y.; Borsch-Haubold, A.G.; Murakami, M.; Wilde, J.I.; Pasquet, S.; Schieltz, D.; Ghomashchi, F.; Yates, J.R., III.; Armstrong, C.G.; Paterson, A.; Cohen, P.; Fukunaga, R.; Hunter, T.; Kudo, I.; Watson, S.P.; Gelb, M.H. Serine 727 phosphorylation and activation of cytosolic phospholipase A2 by MNK1-related protein kinases. J. Biol. Chem. 2000, 275, 37542–37551. [Google Scholar] [PubMed]
- Parekh, A.B.; Putney, J.W., Jr. Store-operated calcium channels. Physiol. Rev. 2005, 85, 757–810. [Google Scholar]
- Kotturi, M.F.; Hunt, S.V.; Jefferies, W.A. Roles of CRAC and Cav-like channels in T cells: More than one gatekeeper? Trends Pharmacol. Sci. 2006, 27, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Inoue, T.; Ra, C. L-type Ca2+ channels: A new player in the regulation of Ca2+ signaling, cell activation and cell survival in immune cells. Mol. Immunol. 2010, 47, 640–648. [Google Scholar]
- Bodi, I.; Mikala, G.; Koch, S.E.; Akhter, S.A.; Schwartz, A. The L-type calcium channel in the heart: the beat goes on. J. Clin. Invest. 2005, 115, 3306–3317. [Google Scholar]
- Jiang, Y.; Ruta, V.; Chen, J.; Lee, A.; MacKinnon, R. The principle of gating charge movement in a voltage-dependent K+ channel. Nature 2003, 423, 42–48. [Google Scholar]
- Kotturi, M.F.; Carlow, D.A.; Lee, J.C.; Ziltener, H.J.; Jefferies, W.A. Identification and functional characterization of voltage-dependent calcium channels in T lymphocytes. J. Biol. Chem. 2003, 278, 46949–46960. [Google Scholar]
- Stokes, L.; Gordon, J.; Grafton, G. Non-voltage-gated L-type Ca2+ channels in human T cells. J. Biol. Chem. 2004, 279, 19566–19573. [Google Scholar]
- Brereton, H.M.; Harland, M.L.; Froscio, M.; Petronijevic, T.; Barritt, G.J. Novel variants of voltage-operated calcium channel alpha 1-subunit transcripts in a rat liver-derived cell line: Deletion in the IVS4 voltage sensing region. Cell Calcium 1997, 22, 39–52. [Google Scholar]
- Yoshimaru, T.; Suzuki, Y.; Inoue, T.; Ra, C. L-type Ca2+ channels in mast cells: activation by membrane depolarization and distinct roles in regulating mediator release from store-operated Ca2+ channels. Mol. Immunol. 2009, 46, 1267–1277. [Google Scholar]
- Suzuki, Y.; Yoshimaru, T.; Inoue, T.; Nunomura, S.; Ra, C. The high-affinity immunoglobulin E receptor (FcεRI) regulates mitochondrial calcium uptake and a dihydropyridine receptor-mediated calcium influx in mast cells: Role of the FcεRIβ chain immunoreceptor tyrosine-based activation motif. Biochem. Pharmacol. 2008, 75, 1492–1503. [Google Scholar]
- Chang, W.C.; Parekh, A.B. Close functional coupling between Ca2+ release-activated Ca2+ channels, arachidonic acid release, and leukotriene C4 secretion. J. Biol. Chem. 2004, 279, 29994–29999. [Google Scholar] [PubMed]
- Chang, W.C.; Nelson, C.; Parekh, A.B. Ca2+ influx through CRAC channels activates cytosolic phospholipase A2, leukotriene C4 secretion, and expression of c-fos through ERK-dependent and -independent pathways in mast cells. FASEB J. 2006, 20, 2381–2383. [Google Scholar] [CrossRef] [PubMed]
- Feske, S. Calcium signalling in lymphocyte activation and disease. Nat. Rev. Immunol. 2007, 7, 690–702. [Google Scholar]
- Vig, M.; Kinet, J.P. Calcium signaling in immune cells. Nat. Immunol. 2009, 10, 21–27. [Google Scholar]
- Vig, M.; DeHaven, W.I.; Bird, G.S.; Billingsley, J.M.; Wang, H.; Rao, P.E.; Hutchings, A.B.; Jouvin, M.H.; Putney, J.W., Jr.; Kinet, J.P. Defective mast cell effector functions in mice lacking the CRACM1 pore subunit of store-operated calcium release-activated calcium channels. Nat. Immunol. 2008, 9, 89–96. [Google Scholar]
- Makowska, A.; Zablocki, K.; Duszyński, J. The role of mitochondria in the regulation of calcium influx into Jurkat cells. Eur. J. Biochem. 2000, 267, 877–884. [Google Scholar]
- Szczeklik, A.; Stevenson, D.D. Aspirin-induced asthma: advances in pathogenesis, diagnosis, and management. J. Allergy Clin. Immunol. 2003, 111, 913–921. [Google Scholar] [CrossRef] [PubMed]
- Arm, J.P.; Austen, K.F. Leukotriene receptors and aspirin sensitivity. N. Engl. J. Med. 2002, 347, 1524–1526. [Google Scholar]
- Juergens, U.R.; Christiansen, S.C.; Stevenson, D.D.; Zuraw, B.L. Inhibition of monocyte leukotriene B4 production after aspirin desensitization. J. Allergy Clin. Immunol. 1995, 96, 148–156. [Google Scholar]
- Hail, N., Jr.; Lotan, R. Cancer chemoprevention and mitochondria: Targeting apoptosis in transformed cells via the disruption of mitochondrial bioenergetics/redox state. Mol. Nutr. Food Res. 2009, 53, 49–67. [Google Scholar]
- Scatena, R.; Bottoni, P.; Botta, G.; Martorana, G.E.; Giardina, B. The role of mitochondria in pharmacotoxicology: A reevaluation of an old, newly emerging topic. Am. J. Cell Physiol. 2007, 293, C12–C21. [Google Scholar]
- Sun, S.Y.; Hail, N., Jr.; Lotan, R. Apoptosis as a novel target for cancer chemoprevention. J. Natl. Cancer Inst. 2002, 96, 662–672. [Google Scholar]
- Yin, M.J.; Yamamoto, Y.; Gaynor, R.B. The anti-inflammatory agents aspirin and salicylate inhibit the activity of IκB kinase β. Nature 1998, 396, 77–80. [Google Scholar]
- Hsu, A.L.; Ching, T.T.; Wang, D.S.; Song, X.; Rangnekar, V.M.; Chen, C.S. The cyclooxygenase-2 inhibitor celecoxib induces apoptosis by blocking Akt activation in human prostate cancer cells independently of Bcl-2. J. Biol. Chem. 2000, 275, 11397–11403. [Google Scholar]
- Lincová, E.; Hampl, A.; Pernicová, Z.; Starsíchová, A.; Krcmár, P.; Machala, M.; Kozubík, A.; Soucek, K. Multiple defects in negative regulation of the PKB/Akt pathway sensitise human cancer cells to the antiproliferative effect of non-steroidal anti-inflammatory drugs. Biochem. Pharmacol. 2009, 78, 561–572. [Google Scholar]
- Zhou, X.M.; Wong, B.C.; Fan, X.M.; Zhang, H.B.; Lin, M.C.; Kung, H.F.; Fan, D.M.; Lam, S.K. Non-steroidal anti-inflammatory drugs induce apoptosis in gastric cancer cells through up-regulation of bax and bak. Carcinogenesis 2001, 22, 1393–1397. [Google Scholar]
- Gu, Q.; Wang, J.D.; Xia, H.H.; Lin, M.C.; He, H.; Zou, B.; Tu, S.P.; Yang, Y.; Liu, X.G.; Lam, S.K.; Wong, W.M.; Chan, A.O.; Yuen, M.F.; Kung, H.F.; Wong, B.C. Activation of the caspase-8/Bid and Bax pathways in aspirin-induced apoptosis in gastric cancer. Carcinogenesis 2005, 26, 541–546. [Google Scholar]
- Ho, C.C.; Yang, X.W.; Lee, T.L.; Liao, P.H.; Yang, S.H.; Tsai, C.H.; Chou, M.Y. Activation of the caspase-8/Bid and Bax pathways in aspirin-induced apoptosis in gastric cancer. Eur. J. Clin. Invest. 2003, 33, 875–882. [Google Scholar]
- Zimmermann, K.C.; Waterhouse, N.J.; Goldstein, J.C.; Schuler, M.; Green, D.R. Aspirin induces apoptosis through release of cytochrome c from mitochondria. Neoplasia 2000, 2, 505–513. [Google Scholar]
- Piqué, M.; Barragán, M.; Dalmau, M.; Bellosillo, B.; Pons, G.; Gil, J. Aspirin induces apoptosis through mitochondrial cytochrome c release. FEBS Lett. 2000, 480, 193–196. [Google Scholar]
- Bellosillo, B.; Piqué, M.; Barragán, M.; Castaño, E.; Villamor, N.; Colomer, D.; Montserrat, E.; Pons, G.; Gil, J. Aspirin and salicylate induce apoptosis and activation of caspases in B-cell chronic lymphocytic leukemia cells. Blood 1998, 92, 1406–1414. [Google Scholar]
- Redlak, M.J.; Power, J.J.; Miller, T.A. Role of mitochondria in aspirin-induced apoptosis in human gastric epithelial cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 289, G731–G738. [Google Scholar]
- Spitz, G.A.; Furtado, C.M.; Sola-Penna, M.; Zancan, P. Acetylsalicylic acid and salicylic acid decrease tumor cell viability and glucose metabolism modulating 6-phosphofructo-1-kinase structure and activity. Biochem. Pharmacol. 2009, 77, 46–53. [Google Scholar]
- Biban, C.; Tassani, V.; Toninello, A.; Siliprandi, D.; Siliprandi, N. The alterations in the energy linked properties induced in rat liver mitochondria by acetylsalicylate are prevented by cyclosporin A or Mg2+. Biochem. Pharmacol. 1995, 50, 497–500. [Google Scholar]
- Danial, N.N.; Korsmeyer, S.J. Cell death: Critical control points. Cell 2004, 116, 205–219. [Google Scholar]
- Yan, N.; Shi, Y. Mechanisms of apoptosis through structural biology. Annu. Rev. Cell Dev. Biol. 2005, 21, 35–56. [Google Scholar]
- Lemasters, J.J.; Theruvath, T.P.; Zhong, Z.; Nieminen, A.L. Mitochondrial calcium and the permeability transition in cell death. Biochim. Biophys. Acta 2009, 1787, 1395–1401. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Brennerb, C. The adenine nucleotide translocase: A central component of the mitochondrial permeability transition pore and key player in cell death. Curr. Med. Chem. 2003, 10, 1507–1525. [Google Scholar]
- Zhivotovsky, B.; Galluzzi, L.; Kepp, O.; Kroemer, G. Adenine nucleotide translocase: A component of the phylogenetically conserved cell death machinery. Cell Death Differ. 2009, 16, 1419–1425. [Google Scholar]
- Hail, N., Jr. Mitochondria: A novel target for the chemoprevention of cancer. Apoptosis 2005, 10, 687–705. [Google Scholar]
- Brenner, C.; Grimm, S. The permeability transition pore complex in cancer cell death. Oncogene 2006, 25, 4744–4756. [Google Scholar]
- Skulachev, V.P. Why are mitochondria involved in apoptosis? Permeability transition pores and apoptosis as selective mechanisms to eliminate superoxide-producing mitochondria and cell. FEBS Lett. 1996, 397, 7–10. [Google Scholar]
- Orrenius, S.; Gogvadze, V.; Zhivotovsky, B. Mitochondrial oxidative stress: implications for cell death. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 143–183. [Google Scholar]
- Ralph, S.J.; Rodríguez-Enríquez, S.; Neuzil, J.; Moreno-Sánchez, R. Bioenergetic pathways in tumor mitochondria as targets for cancer therapy and the importance of the ROS-induced apoptotic trigger. Mol. Aspects Med. 2010, 31, 29–59. [Google Scholar]
- Ott, M.; Zhivotovsky, B.; Orrenius, S. Role of cardiolipin in cytochrome c release from mitochondria. Cell Death Differ. 2007, 14, 1243–1247. [Google Scholar]
- Kroemer, G.; Petit, P.; Zamzami, N.; Vayssière, J.L.; Mignotte, B. The biochemistry of programmed cell death. FASEB J. 1995, 9, 1277–1287. [Google Scholar]
- Kizaki, H.; Tadakuma, T.; Odaka, C.; Muramatsu, J.; Ishimura, Y. Activation of a suicide process of thymocytes through DNA fragmentation by calcium ionophores and phorbol esters. J. Immunol. 1989, 143, 1790–1794. [Google Scholar]
- Tadakuma, T.; Kizaki, H.; Odaka, C.; Kubota, R.; Ishimura, Y.; Yagita, H.; Okumura, K. CD4+CD8+ thymocytes are susceptible to DNA fragmentation induced by phorbol ester, calcium ionophore and anti-CD3 antibody. Eur. J. Immunol. 1990, 20, 779–784. [Google Scholar]
- Ribeiro, J.M.; Carson, D.A. Ca2+/Mg2+-dependent endonuclease from human spleen: Purification, properties, and role in apoptosis. Biochemistry 1993, 32, 9129–9136. [Google Scholar] [CrossRef] [PubMed]
- Merćep, M.; Noguchi, P.D.; Ashwell, J.D. The cell cycle block and lysis of an activated T cell hybridoma are distinct processes with different Ca2+ requirements and sensitivity to cyclosporine A. J. Immunol. 1989, 142, 4085–4092. [Google Scholar]
- McConkey, D.J.; Hartzell, P.; Amador-Perez, F.J.; Orrenius, S.; Jondal, M. Calcium-dependent killing of immature thymocytes by stimulation via the CD3/T cell receptor complex. J. Immunol. 1989, 143, 1801–1806. [Google Scholar]
- Rodriguez-Tarduchy, G.; Prupti, M.; Lopez-Rivas, A.; Collins, M.K.L. Inhibition of apoptosis by calcium ionophores in IL-3-dependent bone marrow cells is dependent upon production of IL-4. J. Immunol. 1992, 148, 1416–1422. [Google Scholar]
- Lampe, P.A.; Cornbrooks, E.B.; Juhasz, A.; Johnson, E.J.; Franklin, J.L. Suppression of programmed neuronal death by a thapsigargin-induced Ca2+ influx. J. Neurobiol. 1995, 26, 205–212. [Google Scholar]
- Nicotera, P.; Zhivotovsky, B.; Orrenius, S. Nuclear calcium transport and the role of calcium in apoptosis. Cell Calcium 1994, 16, 279–288. [Google Scholar]
- Dowd, D.R. Calcium regulation of apoptosis. Adv. Second Messenger Phosphopotein Res. 1995, 30, 255–280. [Google Scholar]
- Green, D.R.; Reed, J.C. Mitochondria and apoptosis. Science 1998, 281, 1309–1312. [Google Scholar]
- Zhu, L.P.; Yu, X.D.; Ling, S.; Brown, R.A.; Kuo, T.H. Mitochondrial Ca2+ homeostasis in the regulation of apoptotic and necrotic cell deaths. Cell Calcium 2000, 28, 107–117. [Google Scholar]
- Kuo, T.H.; Zhu, L.; Golden, K.; Marsh, J.D.; Bhattacharya, S.K.; Liu, B.F. Altered Ca2+ homeostasis and impaired mitochondrial function in cardiomyopathy. Mol. Cell Biochem. 2002, 272, 187–199. [Google Scholar]
- Vad, N.M.; Yount, G.; Moridani, M.Y. Biochemical mechanism of acetylsalicylic acid (Aspirin) selective toxicity toward melanoma cell lines. Melanoma Res. 2008, 18, 386–399. [Google Scholar]
- Zhao, W.; Mackenzie, G.G.; Murray, O.T.; Zhang, Z.; Rigas, B. Phosphoaspirin (MDC-43), a novel benzyl ester of aspirin, inhibits the growth of human cancer cell lines more potently than aspirin: A redox-dependent effect. Carcinogenesis 2009, 30, 512–519. [Google Scholar] [CrossRef] [PubMed]
- Tesei, A.; Zoli, W.; Fabbri, F.; Leonetti, C.; Rosetti, M.; Bolla, M.; Amadori, D.; Silvestrini, R. NCX 4040, an NO-donating acetylsalicylic acid derivative: Efficacy and mechanisms of action in cancer cells. Nitric Oxide 2008, 19, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Yoshimaru, T.; Inoue, T.; Ra, C. CaV1.2 L-type Ca2+ channel protects mast cells against activation-induced cell death by preventing mitochondrial integrity disruption. Mol. Immunol. 2009, 46, 2370–2380. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.T.; Nagaba, Y.; Cross, H.S.; Wrba, F.; Zhang, L.; Guggino, S.E. The mRNA of L-type calcium channel elevated in colon cancer: Protein distribution in normal and cancerous colon. Am. J. Pathol. 2000, 157, 1549–1562. [Google Scholar]
- Zawadzki, A.; Liu, Q.; Wang, Y.; Melander, A.; Jeppsson, B.; Thorlacius, H. Verapamil inhibits L-type calcium channel mediated apoptosis in human colon cancer cells. Dis. Colon Rectum. 2008, 51, 1696–1702. [Google Scholar]
- Baumann, S.; Fas, S.C.; Giaisi, M.; Müller, W.W.; Merling, A.; Gülow, K.; Edler, L.; Krammer, P.H.; Li-Weber, M. Wogonin preferentially kills malignant lymphocytes and suppresses T-cell tumor growth by inducing PLCgamma1- and Ca2+-dependent apoptosis. Blood 2008, 111, 2354–2363. [Google Scholar]
- Inoue, T.; Suzuki, Y.; Yoshimaru, T.; Ra, C. Nitric oxide protects mast cells from activation-induced cell death: The role of the phosphatidylinositol-3-kinase-Akt-endothelial nitric oxide synthase pathway. J. Leukoc. Biol. 2008, 83, 1218–1229. [Google Scholar]
- Suzuki, Y.; Inoue, T.; Ra, C. Endothelial nitric oxide synthase is essential for nitric oxide generation, L-type Ca2+ channel activation and survival in RBL-2H3 mast cells. Biochim. Biophys. Acta 2010, 1803, 372–385. [Google Scholar] [PubMed]
- Martelli, A.M.; Faenza, I.; Billi, A.M.; Manzoli, L.; Evangelisti, C.; Fala, F.; Cocco, L. Intranuclear 3'-phosphoinositide metabolism and Akt signaling: New mechanisms for tumorigenesis and protection against apoptosis? Cell Signal. 2006, 18, 1101–1107. [Google Scholar] [CrossRef] [PubMed]
- Dimmeler, S.; Zeiher, A.M. Nitric oxide-an endothelial cell survival factor. Cell Death Differ. 1999, 6, 964–968. [Google Scholar]
- Choi, B.M.; Pae, H.O.; Jang, S.I.; Kim, Y.M.; Chung, H.T. Nitric oxide as a pro-apoptotic as well as anti-apoptotic modulator. J. Biochem. Mol. Biol. 2002, 35, 116–126. [Google Scholar]
- Parcellier, A.; Tintignac, L.A.; Zhuravleva, E.; Hemmings, B.A. PKB and the mitochondria: AKTing on apoptosis. Cell Signal. 2007, 20, 21–30. [Google Scholar]
- Furuke, K.; Burd, P.R.; Horvath-Arcidiacono, J.A.; Hori, K.; Mostowski, H.; Bloom, E.T. Human NK cells express endothelial nitric oxide synthase, and nitric oxide protects them from activation-induced cell death by regulating expression of TNF-alpha. J. Immunol. 1999, 163, 1473–1480. [Google Scholar]
- Ho, F.M.; Lin, W.W.; Chen, B.C.; Chao, C.M.; Yang, C.R.; Lin, L.Y.; Lai, C.C.; Liu, S. H.; Liau, C.S. High glucose-induced apoptosis in human vascular endothelial cells is mediated through NF-kappaB and c-Jun NH2-terminal kinase pathway and prevented by PI3K/Akt/eNOS pathway. Cell Signal. 2006, 18, 391–399. [Google Scholar]
- Ying, L.; Hofseth, L.J. An emerging role for endothelial nitric oxide synthase in chronic inflammation and cancer. Cancer Res. 2007, 67, 1407–1410. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Suzuki, Y.; Inoue, T.; Ra, C. NSAIDs, Mitochondria and Calcium Signaling: Special Focus on Aspirin/Salicylates. Pharmaceuticals 2010, 3, 1594-1613. https://doi.org/10.3390/ph3051594
Suzuki Y, Inoue T, Ra C. NSAIDs, Mitochondria and Calcium Signaling: Special Focus on Aspirin/Salicylates. Pharmaceuticals. 2010; 3(5):1594-1613. https://doi.org/10.3390/ph3051594
Chicago/Turabian StyleSuzuki, Yoshihiro, Toshio Inoue, and Chisei Ra. 2010. "NSAIDs, Mitochondria and Calcium Signaling: Special Focus on Aspirin/Salicylates" Pharmaceuticals 3, no. 5: 1594-1613. https://doi.org/10.3390/ph3051594
APA StyleSuzuki, Y., Inoue, T., & Ra, C. (2010). NSAIDs, Mitochondria and Calcium Signaling: Special Focus on Aspirin/Salicylates. Pharmaceuticals, 3(5), 1594-1613. https://doi.org/10.3390/ph3051594