Molecular Mechanism for Various Pharmacological Activities of NSAIDS

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
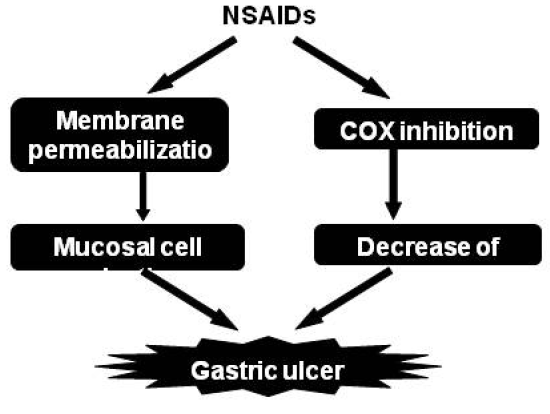
2.1. Molecular mechanisms for NSAID-induced gastric lesions
2.1.1. Direct cytotoxic effect of NSAIDs
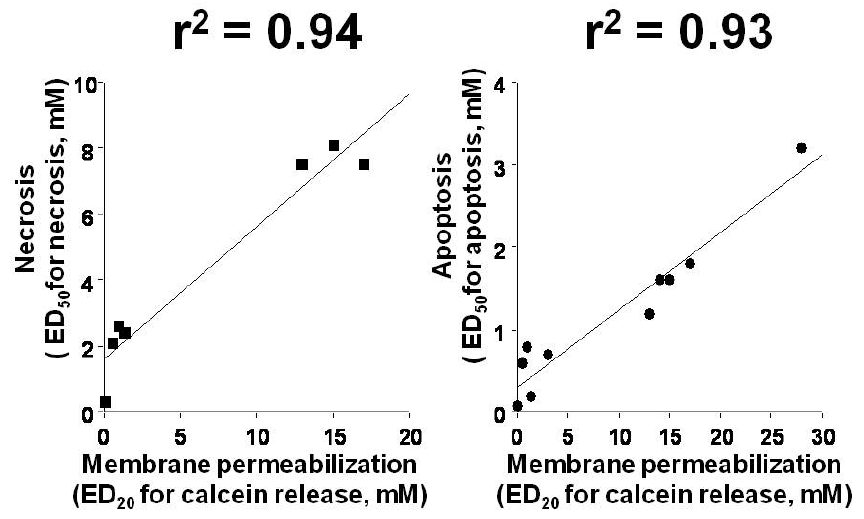
2.1.2. Membrane permeabilization activity of NSAIDs

2.1.3. DNA microarray analysis
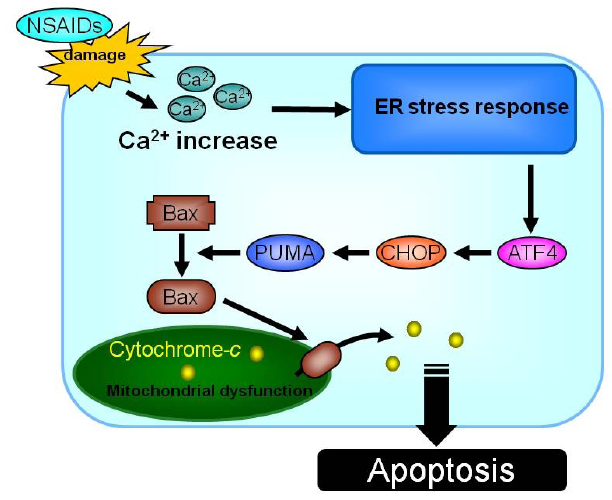
2.1.4. Contribution of the increase in intracellular Ca2+ level and mitochondrial dysfunction to NSAID-induced apoptosis
2.1.5. Contribution of mucosal cell death to NSAID-induced gastric lesions
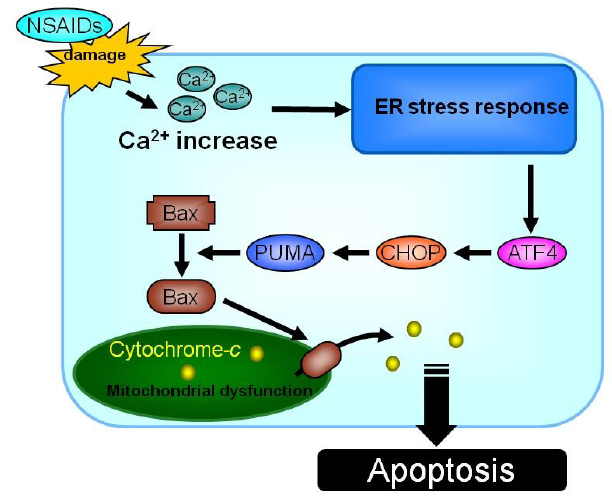
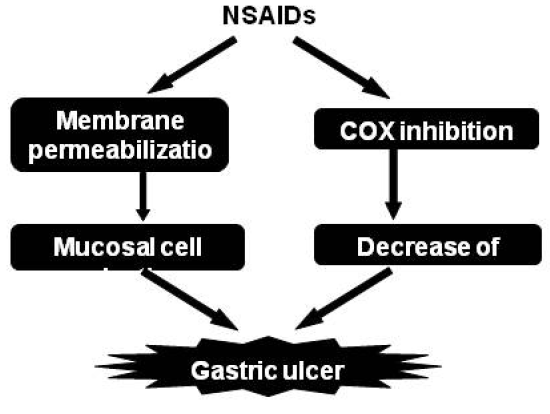
2.1.6. Strategy for development of NSAIDs with lower gastrointestinal side effect
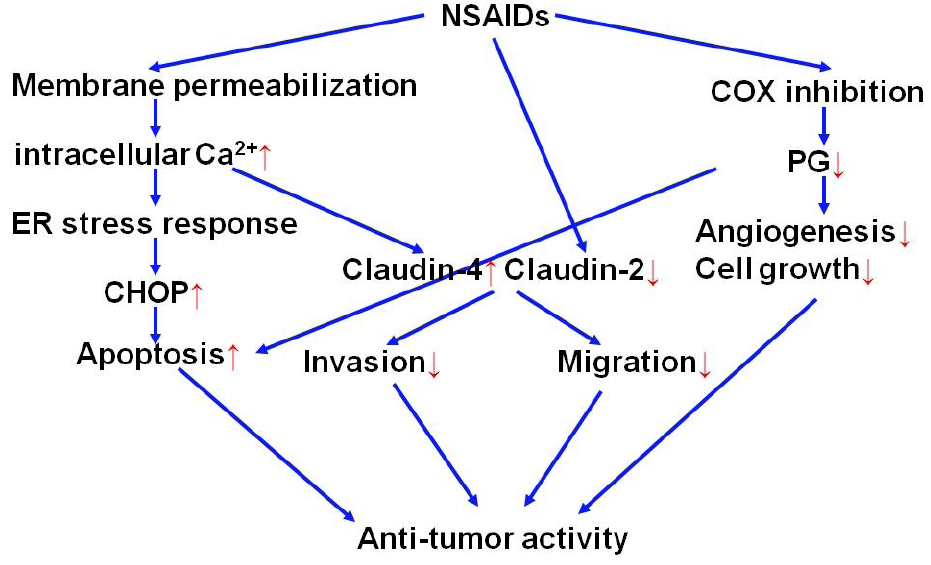
2.2. Molecular mechanisms for anti-tumor activity of NSAIDs
2.2.2. Roles of ER chaperones in anti-tumor activity of NSAIDs
2.2.3. Roles of S100P in anti-tumor activity of NSAIDs
2.2.4. Strategy for development of NSAIDs with potent anti-tumor activity
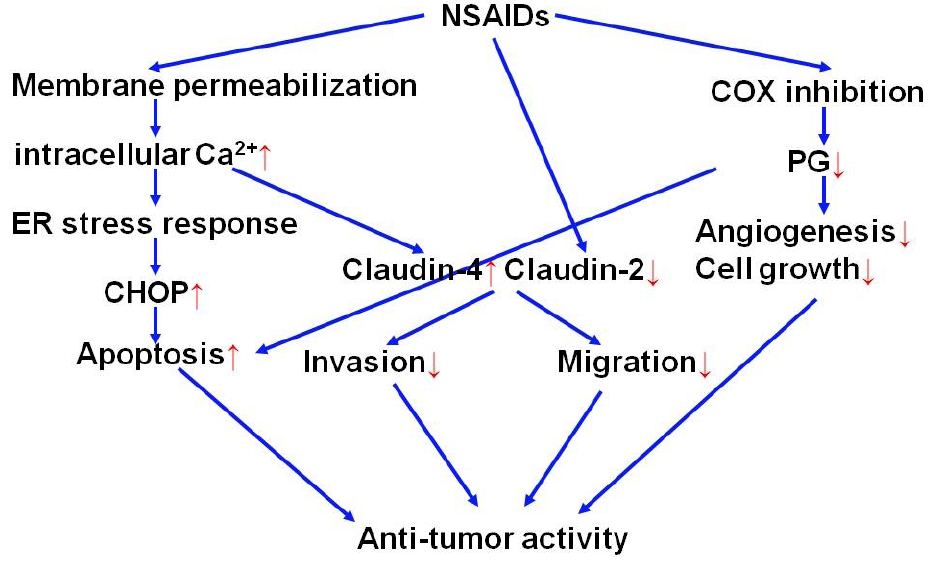
2.3. Molecular mechanisms for anti-AD activity of NSAIDs
2.3.1. Roles of COX-inhibition and PG-decrease in anti-AD activity of NSAIDs
2.3.2. Role of ER chaperones in anti-AD activity of NSAIDs
2.3.3. Strategy for development of NSAIDs with potent anti-AD activity
3. Conclusions
References
- Smalley, W.E.; Ray, W.A.; Daugherty, J.R.; Griffin, M.R. Nonsteroidal anti-inflammatory drugs and the incidence of hospitalizations for peptic ulcer disease in elderly persons. Am. J. Epidemiol. 1995, 141, 539–545. [Google Scholar]
- Hawkey, C.J. Nonsteroidal anti-inflammatory drug gastropathy. Gastroenterology 2000, 119, 521–535. [Google Scholar]
- Singh, G. Recent considerations in nonsteroidal anti-inflammatory drug gastropathy. Am. J. Med. 1998, 105, 31S–38S. [Google Scholar]
- Hoshino, T.; Tsutsumi, S.; Tomisato, W.; Hwang, H.J.; Tsuchiya, T.; Mizushima, T. Prostaglandin E2 protects gastric mucosal cells from apoptosis via EP2 and EP4 receptor activation. J. Biol. Chem. 2003, 278, 12752–12758. [Google Scholar]
- Miller, T.A. Protective effects of prostaglandins against gastric mucosal damage: current knowledge and proposed mechanisms. Am. J. Physiol. 1983, 245, G601–G623. [Google Scholar]
- Hoshino, T.; Takano, T.; Tsutsumi, S.; Tomisato, W.; Tsuchiya, T.; Mizushima, T. Effects of prostaglandin E2 on gastric irritant-induced apoptosis. Dig. Dis. Sci. 2002, 47, 2370–2379. [Google Scholar]
- Tsutsumi, S.; Haruna, R.; Tomisato, W.; Takano, T.; Hoshino, T.; Tsuchiya, T.; Mizushima, T. Effects of prostaglandins on spontaneous apoptosis in gastric mucosal cells. Dig. Dis. Sci. 2002, 47, 84–89. [Google Scholar]
- Vane, J. Towards a better aspirin. Nature 1994, 367, 215–216. [Google Scholar]
- Smith, C.J.; Zhang, Y.; Koboldt, C.M.; Muhammad, J.; Zweifel, B.S.; Shaffer, A.; Talley, J.J.; Masferrer, J.L.; Seibert, K.; Isakson, P.C. Pharmacological analysis of cyclooxygenase-1 in inflammation. Proc. Natl. Acad. Sci. USA 1998, 95, 13313–13318. [Google Scholar]
- Chan, C.C.; Boyce, S.; Brideau, C.; Charleson, S.; Cromlish, W.; Ethier, D.; Evans, J.; Ford, H.A.; Forrest, M.J.; Gauthier, J.Y.; Gordon, R.; Gresser, M.; Guay, J.; Kargman, S.; Kennedy, B.; Leblanc, Y.; Leger, S.; Mancini, J.; O'Neill, G.P.; Ouellet, M.; Patrick, D.; Percival, M.D.; Perrier, H.; Prasit, P.; Rodger, I.; et al. Rofecoxib [Vioxx, MK-0966; 4-(4'-methylsulfonylphenyl)-3-phenyl-2-(5H)-furanone]: a potent and orally active cyclooxygenase-2 inhibitor. Pharmacological and biochemical profiles. J. Pharmacol. Exp. Ther. 1999, 290, 551–560. [Google Scholar] [PubMed]
- Ligumsky, M.; Golanska, E.M.; Hansen, D.G.; Kauffman, G.J. Aspirin can inhibit gastric mucosal cyclo-oxygenase without causing lesions in rat. Gastroenterology 1983, 84, 756–761. [Google Scholar]
- Mukherjee, D.; Nissen, S.E.; Topol, E.J. Risk of cardiovascular events associated with selective COX-2 inhibitors. JAMA 2001, 286, 954–959. [Google Scholar]
- Mukherjee, D. Selective cyclooxygenase-2 (COX-2) inhibitors and potential risk of cardiovascular events. Biochem. Pharmacol. 2002, 63, 817–821. [Google Scholar]
- Farrow, D.C.; Vaughan, T.L.; Hansten, P.D.; Stanford, J.L.; Risch, H.A.; Gammon, M.D.; Chow, W.H.; Dubrow, R.; Ahsan, H.; Mayne, S.T.; Schoenberg, J.B.; West, A.B.; Rotterdam, H.; Fraumeni, J.F., Jr.; Blot, W.J. Use of aspirin and other nonsteroidal anti-inflammatory drugs and risk of esophageal and gastric cancer. Cancer Epidemiol. Biomarkers Prev. 1998, 7, 97–102. [Google Scholar]
- Sorensen, H.T.; Friis, S.; Norgard, B.; Mellemkjaer, L.; Blot, W.J.; McLaughlin, J.K.; Ekbom, A.; Baron, J.A. Risk of cancer in a large cohort of nonaspirin NSAID users: a population-based study. Br. J. Cancer 2003, 88, 1687–1692. [Google Scholar]
- Gupta, R.A.; Dubois, R.N. Colorectal cancer prevention and treatment by inhibition of cyclooxygenase-2. Nat Rev Cancer 2001, 1, 11–21. [Google Scholar]
- Kismet, K.; Akay, M.T.; Abbasoglu, O.; Ercan, A. Celecoxib: a potent cyclooxygenase-2 inhibitor in cancer prevention. Cancer Detect. Prev. 2004, 28, 127–142. [Google Scholar]
- Tsujii, M.; Kawano, S.; Tsuji, S.; Sawaoka, H.; Hori, M.; DuBois, R.N. Cyclooxygenase regulates angiogenesis induced by colon cancer cells. Cell 1998, 93, 705–716. [Google Scholar]
- Rolland, P.H.; Martin, P.M.; Jacquemier, J.; Rolland, A.M.; Toga, M. Prostaglandin in human breast cancer: Evidence suggesting that an elevated prostaglandin production is a marker of high metastatic potential for neoplastic cells. J. Natl. Cancer Inst. 1980, 64, 1061–1070. [Google Scholar]
- Eberhart, C.E.; Coffey, R.J.; Radhika, A.; Giardiello, F.M.; Ferrenbach, S.; DuBois, R.N. Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology 1994, 107, 1183–1188. [Google Scholar]
- Ristimaki, A.; Honkanen, N.; Jankala, H.; Sipponen, P.; Harkonen, M. Expression of cyclooxygenase-2 in human gastric carcinoma. Cancer Res. 1997, 57, 1276–1280. [Google Scholar]
- Piazza, G.A.; Alberts, D.S.; Hixson, L.J.; Paranka, N.S.; Li, H.; Finn, T.; Bogert, C.; Guillen, J.M.; Brendel, K.; Gross, P.H.; Sperl, G.; Ritchie, J.; Burt, R.W.; Ellsworth, L.; Ahnen, D.J.; Pamukcu, R. Sulindac sulfone inhibits azoxymethane-induced colon carcinogenesis in rats without reducing prostaglandin levels. Cancer Res. 1997, 57, 2909–2915. [Google Scholar]
- Reddy, B.S.; Kawamori, T.; Lubet, R.A.; Steele, V.E.; Kelloff, G.J.; Rao, C.V. Chemopreventive efficacy of sulindac sulfone against colon cancer depends on time of administration during carcinogenic process. Cancer Res. 1999, 59, 3387–3391. [Google Scholar]
- Zhang, X.; Morham, S.G.; Langenbach, R.; Young, D.A. Malignant transformation and antineoplastic actions of nonsteroidal antiinflammatory drugs (NSAIDs) on cyclooxygenase-null embryo fibroblasts. J. Exp. Med. 1999, 190, 451–459. [Google Scholar]
- Hanif, R.; Pittas, A.; Feng, Y.; Koutsos, M.I.; Qiao, L.; Staiano-Coico, L.; Shiff, S.I.; Rigas, B. Effects of nonsteroidal anti-inflammatory drugs on proliferation and on induction of apoptosis in colon cancer cells by a prostaglandin-independent pathway. Biochem. Pharmacol. 1996, 52, 237–245. [Google Scholar]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar]
- Mattson, M.P. Pathways towards and away from Alzheimer's disease. Nature 2004, 430, 631–639. [Google Scholar]
- in t' Veld, B.A.; Ruitenberg, A.; Hofman, A.; Launer, L.J.; van Duijn, C.M.; Stijnen, T.; Breteler, M.M.; Stricker, B.H. Nonsteroidal antiinflammatory drugs and the risk of Alzheimer's disease. N. Engl. J. Med. 2001, 345, 1515–1521. [Google Scholar]
- Lim, G.P.; Yang, F.; Chu, T.; Chen, P.; Beech, W.; Teter, B.; Tran, T.; Ubeda, O.; Ashe, K.H.; Frautschy, S.A.; Cole, G.M. Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer's disease. J. Neurosci. 2000, 20, 5709–5714. [Google Scholar]
- Eriksen, J.L.; Sagi, S.A.; Smith, T.E.; Weggen, S.; Das, P.; McLendon, D.C.; Ozols, V.V.; Jessing, K.W.; Zavitz, K.H.; Koo, E.H.; Golde, T.E. NSAIDs and enantiomers of flurbiprofen target gamma-secretase and lower Abeta 42 in vivo. J. Clin. Invest. 2003, 112, 440–449. [Google Scholar] [PubMed]
- Yan, Q.; Zhang, J.; Liu, H.; Babu-Khan, S.; Vassar, R.; Biere, A.L.; Citron, M.; Landreth, G. Anti-inflammatory drug therapy alters beta-amyloid processing and deposition in an animal model of Alzheimer's disease. J. Neurosci. 2003, 23, 7504–7509. [Google Scholar]
- Weggen, S.; Eriksen, J.L.; Das, P.; Sagi, S.A.; Wang, R.; Pietrzik, C.U.; Findlay, K.A.; Smith, T.E.; Murphy, M.P.; Bulter, T.; Kang, D.E.; Marquez-Sterling, N.; Golde, T.E.; Koo, E.H. A subset of NSAIDs lower amyloidogenic Abeta42 independently of cyclooxygenase activity. Nature 2001, 414, 212–216. [Google Scholar]
- Weggen, S.; Eriksen, J.L.; Sagi, S.A.; Pietrzik, C.U.; Ozols, V.; Fauq, A.; Golde, T.E.; Koo, E.H. Evidence that nonsteroidal anti-inflammatory drugs decrease amyloid beta 42 production by direct modulation of gamma-secretase activity. J. Biol. Chem. 2003, 278, 31831–31837. [Google Scholar]
- Kitamura, Y.; Shimohama, S.; Koike, H.; Kakimura, J.; Matsuoka, Y.; Nomura, Y.; Gebicke-Haerter, P.J.; Taniguchi, T. Increased expression of cyclooxygenases and peroxisome proliferator-activated receptor-gamma in Alzheimer's disease brains. Biochem. Biophys. Res. Commun. 1999, 254, 582–586. [Google Scholar]
- Yasojima, K.; Schwab, C.; McGeer, E.G.; McGeer, P.L. Distribution of cyclooxygenase-1 and cyclooxygenase-2 mRNAs and proteins in human brain and peripheral organs. Brain Res. 1999, 830, 226–236. [Google Scholar]
- Montine, T.J.; Sidell, K.R.; Crews, B.C.; Markesbery, W.R.; Marnett, L.J.; Roberts, L.J., 2nd.; Morrow, J.D. Elevated CSF prostaglandin E2 levels in patients with probable AD. Neurology 1999, 53, 1495–1498. [Google Scholar] [PubMed]
- Ho, L.; Purohit, D.; Haroutunian, V.; Luterman, J.D.; Willis, F.; Naslund, J.; Buxbaum, J.D.; Mohs, R.C.; Aisen, P.S.; Pasinetti, G.M. Neuronal cyclooxygenase 2 expression in the hippocampal formation as a function of the clinical progression of Alzheimer disease. Arch. Neurol. 2001, 58, 487–492. [Google Scholar]
- Andreasson, K.I.; Savonenko, A.; Vidensky, S.; Goellner, J.J.; Zhang, Y.; Shaffer, A.; Kaufmann, W.E.; Worley, P.F.; Isakson, P.; Markowska, A.L. Age-dependent cognitive deficits and neuronal apoptosis in cyclooxygenase-2 transgenic mice. J. Neurosci. 2001, 21, 8198–8209. [Google Scholar]
- Rokutan, K.; Yamada, M.; Torigoe, J.; Saito, T. Transforming growth factor-beta inhibits proliferation and maturation of cultured guinea pig gastric pit cells. Am. J. Physiol. 1998, 275, G526–G533. [Google Scholar] [PubMed]
- Tomisato, W.; Tsutsumi, S.; Rokutan, K.; Tsuchiya, T.; Mizushima, T. NSAIDs induce both necrosis and apoptosis in guinea pig gastric mucosal cells in primary culture. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, G1092–G1100. [Google Scholar]
- Aburaya, M.; Tanaka, K.; Hoshino, T.; Tsutsumi, S.; Suzuki, K.; Makise, M.; Akagi, R.; Mizushima, T. Heme oxygenase-1 protects gastric mucosal cells against non-steroidal anti-inflammatory drugs. J. Biol. Chem. 2006, 281, 33422–33432. [Google Scholar]
- Tomisato, W.; Tsutsumi, S.; Hoshino, T.; Hwang, H.J.; Mio, M.; Tsuchiya, T.; Mizushima, T. Role of direct cytotoxic effects of NSAIDs in the induction of gastric lesions. Biochem. Pharmacol. 2004, 67, 575–585. [Google Scholar]
- Tomisato, W.; Tanaka, K.; Katsu, T.; Kakuta, H.; Sasaki, K.; Tsutsumi, S.; Hoshino, T.; Aburaya, M.; Li, D.; Tsuchiya, T.; Suzuki, K.; Yokomizo, K.; Mizushima, T. Membrane permeabilization by non-steroidal anti-inflammatory drugs. Biochem. Biophys. Res. Commun. 2004, 323, 1032–1039. [Google Scholar]
- Tanaka, K.; Tomisato, W.; Hoshino, T.; Ishihara, T.; Namba, T.; Aburaya, M.; Katsu, T.; Suzuki, K.; Tsutsumi, S.; Mizushima, T. Involvement of intracellular Ca2+ levels in nonsteroidal anti-inflammatory drug-induced apoptosis. J. Biol. Chem. 2005, 280, 31059–31067. [Google Scholar]
- Mima, S.; Tsutsumi, S.; Ushijima, H.; Takeda, M.; Fukuda, I.; Yokomizo, K.; Suzuki, K.; Sano, K.; Nakanishi, T.; Tomisato, W.; Tsuchiya, T.; Mizushima, T. Induction of claudin-4 by nonsteroidal anti-inflammatory drugs and its contribution to their chemopreventive effect. Cancer Res. 2005, 65, 1868–1876. [Google Scholar]
- Tsutsumi, S.; Gotoh, T.; Tomisato, W.; Mima, S.; Hoshino, T.; Hwang, H.J.; Takenaka, H.; Tsuchiya, T.; Mori, M.; Mizushima, T. Endoplasmic reticulum stress response is involved in nonsteroidal anti-inflammatory drug-induced apoptosis. Cell Death Differ. 2004, 11, 1009–1016. [Google Scholar]
- Namba, T.; Hoshino, T.; Tanaka, K.; Tsutsumi, S.; Ishihara, T.; Mima, S.; Suzuki, K.; Ogawa, S.; Mizushima, T. Up-regulation of 150-kDa oxygen-regulated protein by celecoxib in human gastric carcinoma cells. Mol. Pharmacol 2007, 71, 860–870. [Google Scholar]
- Yu, J.; Zhang, L.; Hwang, P.M.; Kinzler, K.W.; Vogelstein, B. PUMA induces the rapid apoptosis of colorectal cancer cells. Mol. Cell 2001, 7, 673–682. [Google Scholar]
- Nakano, K.; Vousden, K.H. PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell 2001, 7, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Letai, A.; Bassik, M.C.; Walensky, L.D.; Sorcinelli, M.D.; Weiler, S.; Korsmeyer, S.J. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2002, 2, 183–192. [Google Scholar]
- Li, J.; Lee, B.; Lee, A.S. Endoplasmic reticulum stress-induced apoptosis: multiple pathways and activation of p53-up-regulated modulator of apoptosis (PUMA) and NOXA by p53. J. Biol. Chem. 2006, 281, 7260–7270. [Google Scholar]
- Reimertz, C.; Kogel, D.; Rami, A.; Chittenden, T.; Prehn, J.H. Gene expression during ER stress-induced apoptosis in neurons: induction of the BH3-only protein Bbc3/PUMA and activation of the mitochondrial apoptosis pathway. J Cell Biol 2003, 162, 587–597. [Google Scholar]
- Ishihara, T.; Hoshino, T.; Namba, T.; Tanaka, K.; Mizushima, T. Involvement of up-regulation of PUMA in non-steroidal anti-inflammatory drug-induced apoptosis. Biochem. Biophys. Res. Commun. 2007, 356, 711–717. [Google Scholar]
- Belton, O.; Byrne, D.; Kearney, D.; Leahy, A.; Fitzgerald, D.J. Cyclooxygenase-1 and -2-dependent prostacyclin formation in patients with atherosclerosis. Circulation 2000, 102, 840–845. [Google Scholar]
- Ray, W.A.; Griffin, M.R.; Stein, C.M. Cardiovascular toxicity of valdecoxib. N. Engl. J. Med. 2004, 351, 2767. [Google Scholar]
- Arai, Y.; Tanaka, K.; Ushijima, H.; Tomisato, W.; Tsutsumi, S.; Aburaya, M.; Hoshino, T.; Yokomizo, K.; Suzuki, K.; Katsu, T.; Tsuchiya, T.; Mizushima, T. Low direct cytotoxicity of nabumetone on gastric mucosal cells. Dig. Dis. Sci. 2005, 50, 1641–1646. [Google Scholar]
- Tomisato, W.; Tanaka, K.; Tsutsumi, S.; Hoshino, T.; Yokomizo, K.; Suzuki, K.; Katsu, T.; Mizushima, T. Low direct cytotoxicity and cytoprotective effects of nitric oxide releasing indomethacin. Dig. Dis. Sci. 2005, 50, 1927–1937. [Google Scholar]
- Hirakawa, T.; Rokutan, K.; Nikawa, T.; Kishi, K. Geranylgeranylacetone induces heat shock proteins in cultured guinea pig gastric mucosal cells and rat gastric mucosa. Gastroenterology 1996, 111, 345–357. [Google Scholar]
- Katsuno, M.; Sang, C.; Adachi, H.; Minamiyama, M.; Waza, M.; Tanaka, F.; Doyu, M.; Sobue, G. Pharmacological induction of heat-shock proteins alleviates polyglutamine-mediated motor neuron disease. Proc Natl Acad Sci U S A 2005, 102, 16801–16806. [Google Scholar]
- Mathew, A.; Morimoto, R.I. Role of the heat-shock response in the life and death of proteins. Ann. NY Acad. Sci. 1998, 851, 99–111. [Google Scholar]
- Morimoto, R.I. Regulation of the heat shock transcriptional response: cross talk between a family of heat shock factors, molecular chaperones, and negative regulators. Genes Dev. 1998, 12, 3788–3796. [Google Scholar] [CrossRef] [PubMed]
- Tomisato, W.; Tsutsumi, S.; Tsuchiya, T.; Mizushima, T. Geranylgeranylacetone protects guinea pig gastric mucosal cells from gastric stressor-induced necrosis by induction of heat-shock proteins. Biol. Pharm. Bull. 2001, 24, 887–891. [Google Scholar]
- Tomisato, W.; Takahashi, N.; Komoto, C.; Rokutan, K.; Tsuchiya, T.; Mizushima, T. Geranylgeranylacetone protects cultured guinea pig gastric mucosal cells from indomethacin. Dig. Dis. Sci. 2000, 45, 1674–1679. [Google Scholar]
- Suemasu, S.; Tanaka, K.; Namba, T.; Ishihara, T.; Katsu, T.; Fujimoto, M.; Adachi, H.; Sobue, G.; Takeuchi, K.; Nakai, A.; Mizushima, T. A role for HSP70 in protecting against indomethacin-induced gastric lesions. J. Biol. Chem. 2009, 284, 19705–19715. [Google Scholar]
- Tanaka, K.; Tsutsumi, S.; Arai, Y.; Hoshino, T.; Suzuki, K.; Takaki, E.; Ito, T.; Takeuchi, K.; Nakai, A.; Mizushima, T. Genetic evidence for a protective role of heat shock factor 1 against irritant-induced gastric lesions. Mol. Pharmacol. 2007, 71, 985–993. [Google Scholar]
- Asano, T.; Tanaka, K.; Yamakawa, N.; Adachi, H.; Sobue, G.; Goto, H.; Takeuchi, K.; Mizushima, T. HSP70 confers protection against indomethacin-induced lesions of the small intestine. J. Pharmacol. Exp. Ther. 2009, 330, 458–467. [Google Scholar]
- Tanaka, K.; Namba, T.; Arai, Y.; Fujimoto, M.; Adachi, H.; Sobue, G.; Takeuchi, K.; Nakai, A.; Mizushima, T. Genetic Evidence for a Protective Role for Heat Shock Factor 1 and Heat Shock Protein 70 against Colitis. J. Biol. Chem. 2007, 282, 23240–23252. [Google Scholar]
- Anderson, J.M.; Van Itallie, C.M. Tight junctions and the molecular basis for regulation of paracellular permeability. Am. J. Physiol. 1995, 269, G467–G475. [Google Scholar]
- Soler, A.P.; Miller, R.D.; Laughlin, K.V.; Carp, N.Z.; Klurfeld, D.M.; Mullin, J.M. Increased tight junctional permeability is associated with the development of colon cancer. Carcinogenesis 1999, 20, 1425–1431. [Google Scholar]
- Li, D.; Mrsny, R.J. Oncogenic Raf-1 disrupts epithelial tight junctions via downregulation of occludin. J. Cell Biol. 2000, 148, 791–800. [Google Scholar]
- Hoevel, T.; Macek, R.; Swisshelm, K.; Kubbies, M. Reexpression of the TJ protein CLDN1 induces apoptosis in breast tumor spheroids. Int. J. Cancer 2004, 108, 374–383. [Google Scholar]
- Michl, P.; Barth, C.; Buchholz, M.; Lerch, M.M.; Rolke, M.; Holzmann, K.H.; Menke, A.; Fensterer, H.; Giehl, K.; Lohr, M.; Leder, G.; Iwamura, T.; Adler, G.; Gress, T.M. Claudin-4 expression decreases invasiveness and metastatic potential of pancreatic cancer. Cancer Res. 2003, 63, 6265–6271. [Google Scholar]
- Mima, S.; Takehara, M.; Takada, H.; Nishimura, T.; Hoshino, T.; Mizushima, T. NSAIDs suppress the expression of claudin-2 to promote invasion activity of cancer cells. Carcinogenesis 2008, 29, 1994–2000. [Google Scholar]
- Lee, A.S. The glucose-regulated proteins: stress induction and clinical applications. Trends Biochem. Sci. 2001, 26, 504–510. [Google Scholar]
- Reddy, R.K.; Mao, C.; Baumeister, P.; Austin, R.C.; Kaufman, R.J.; Lee, A.S. Endoplasmic reticulum chaperone protein GRP78 protects cells from apoptosis induced by topoisomerase inhibitors: role of ATP binding site in suppression of caspase-7 activation. J. Biol. Chem. 2003, 278, 20915–20924. [Google Scholar]
- Morris, J.A.; Dorner, A.J.; Edwards, C.A.; Hendershot, L.M.; Kaufman, R.J. Immunoglobulin binding protein (BiP) function is required to protect cells from endoplasmic reticulum stress but is not required for the secretion of selective proteins. J. Biol. Chem. 1997, 272, 4327–4334. [Google Scholar]
- Tsukamoto, Y.; Kuwabara, K.; Hirota, S.; Kawano, K.; Yoshikawa, K.; Ozawa, K.; Kobayashi, T.; Yanagi, H.; Stern, D.M.; Tohyama, M.; Kitamura, Y.; Ogawa, S. Expression of the 150-kd oxygen-regulated protein in human breast cancer. Lab. Invest. 1998, 78, 699–706. [Google Scholar]
- Miyagi, T.; Hori, O.; Koshida, K.; Egawa, M.; Kato, H.; Kitagawa, Y.; Ozawa, K.; Ogawa, S.; Namiki, M. Antitumor effect of reduction of 150-kDa oxygen-regulated protein expression on human prostate cancer cells. Int. J. Urol. 2002, 9, 577–585. [Google Scholar]
- Wang, W.H.; Huang, J.Q.; Zheng, G.F.; Lam, S.K.; Karlberg, J.; Wong, B.C. Non-steroidal anti-inflammatory drug use and the risk of gastric cancer: a systematic review and meta-analysis. J. Natl. Cancer Inst. 2003, 95, 1784–1791. [Google Scholar]
- Tsutsumi, S.; Namba, T.; Tanaka, K.I.; Arai, Y.; Ishihara, T.; Aburaya, M.; Mima, S.; Hoshino, T.; Mizushima, T. Celecoxib upregulates endoplasmic reticulum chaperones that inhibit celecoxib-induced apoptosis in human gastric cells. Oncogene 2006, 25, 1018–1029. [Google Scholar]
- Becker, T.; Gerke, V.; Kube, E.; Weber, K. S100P, a novel Ca(2+)-binding protein from human placenta. cDNA cloning, recombinant protein expression and Ca2+ binding properties. Eur. J. Biochem. 1992, 207, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Shyu, R.Y.; Huang, S.L.; Jiang, S.Y. Retinoic acid increases expression of the calcium-binding protein S100P in human gastric cancer cells. J. Biomed. Sci. 2003, 10, 313–319. [Google Scholar]
- Birkenkamp-Demtroder, K.; Olesen, S.H.; Sorensen, F.B.; Laurberg, S.; Laiho, P.; Aaltonen, L.A.; Orntoft, T.F. Differential gene expression in colon cancer of the caecum versus the sigmoid and rectosigmoid. Gut 2005, 54, 374–384. [Google Scholar]
- Arumugam, T.; Simeone, D.M.; Van Golen, K.; Logsdon, C.D. S100P promotes pancreatic cancer growth, survival, and invasion. Clin. Cancer Res. 2005, 11, 5356–5364. [Google Scholar] [CrossRef] [PubMed]
- Logsdon, C.D.; Simeone, D.M.; Binkley, C.; Arumugam, T.; Greenson, J.K.; Giordano, T.J.; Misek, D.E.; Kuick, R.; Hanash, S. Molecular profiling of pancreatic adenocarcinoma and chronic pancreatitis identifies multiple genes differentially regulated in pancreatic cancer. Cancer Res. 2003, 63, 2649–2657. [Google Scholar]
- Wang, G.; Platt-Higgins, A.; Carroll, J.; de Silva Rudland, S.; Winstanley, J.; Barraclough, R.; Rudland, P.S. Induction of metastasis by S100P in a rat mammary model and its association with poor survival of breast cancer patients. Cancer Res. 2006, 66, 1199–1207. [Google Scholar]
- Averboukh, L.; Liang, P.; Kantoff, P.W.; Pardee, A.B. Regulation of S100P expression by androgen. Prostate 1996, 29, 350–355. [Google Scholar]
- Donato, R. S100: a multigenic family of calcium-modulated proteins of the EF-hand type with intracellular and extracellular functional roles. Int. J. Biochem. Cell Biol. 2001, 33, 637–668. [Google Scholar]
- Arumugam, T.; Simeone, D.M.; Schmidt, A.M.; Logsdon, C.D. S100P stimulates cell proliferation and survival via receptor for activated glycation end products (RAGE). J. Biol. Chem. 2004, 279, 5059–5065. [Google Scholar]
- Fuentes, M.K.; Nigavekar, S.S.; Arumugam, T.; Logsdon, C.D.; Schmidt, A.M.; Park, J.C.; Huang, E.H. RAGE activation by S100P in colon cancer stimulates growth, migration, and cell signaling pathways. Dis. Colon Rectum. 2007, 50, 1230–1240. [Google Scholar] [PubMed]
- Arumugam, T.; Ramachandran, V.; Logsdon, C.D. Effect of cromolyn on S100P interactions with RAGE and pancreatic cancer growth and invasion in mouse models. J. Natl. Cancer Inst. 2006, 98, 1806–1818. [Google Scholar]
- Koltzscher, M.; Neumann, C.; Konig, S.; Gerke, V. Ca2+-dependent binding and activation of dormant ezrin by dimeric S100P. Mol. Biol Cell 2003, 14, 2372–2384. [Google Scholar]
- Filipek, A.; Jastrzebska, B.; Nowotny, M.; Kuznicki, J. CacyBP/SIP, a calcyclin and Siah-1-interacting protein, binds EF-hand proteins of the S100 family. J. Biol. Chem. 2002, 277, 28848–28852. [Google Scholar] [PubMed]
- Hunter, K.W. Ezrin, a key component in tumor metastasis. Trends Mol. Med 2004, 10, 201–204. [Google Scholar]
- Namba, T.; Homan, T.; Nishimura, T.; Mima, S.; Hoshino, T.; Mizushima, T. Up-regulation of S100P expression by non-steroidal anti-inflammatory drugs and its role in anti-tumorigenic effects. J. Biol. Chem. 2009, 284, 4158–4167. [Google Scholar]
- Kaufman, R.J. Orchestrating the unfolded protein response in health and disease. J. Clin. Invest. 2002, 110, 1389–1398. [Google Scholar]
- Ron, D. Translational control in the endoplasmic reticulum stress response. J. Clin. Invest. 2002, 110, 1383–1388. [Google Scholar]
- Yoshida, H.; Okada, T.; Haze, K.; Yanagi, H.; Yura, T.; Negishi, M.; Mori, K. ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol. Cell Biol. 2000, 20, 6755–6767. [Google Scholar]
- Hoshino, T.; Nakaya, T.; Homan, T.; Tanaka, K.; Sugimoto, Y.; Araki, W.; Narita, M.; Narumiya, S.; Suzuki, T.; Mizushima, T. Involvement of prostaglandin E2 in production of amyloid-beta peptides both in vitro and in vivo. J. Biol. Chem. 2007, 282, 32676–32688. [Google Scholar] [PubMed]
- Coleman, R.A.; Smith, W.L.; Narumiya, S. International Union of Pharmacology classification of prostanoid receptors: properties, distribution, and structure of the receptors and their subtypes. Pharmacol. Rev. 1994, 46, 205–229. [Google Scholar] [PubMed]
- Hoshino, T.; Namba, T.; Takehara, M.; Nakaya, T.; Sugimoto, Y.; Araki, W.; Narumiya, S.; Suzuki, T.; Mizushima, T. Prostaglandin E2 stimulates the production of amyloid-beta peptides through internalization of the EP4 receptor. J. Biol. Chem. 2009, 284, 18493–18502. [Google Scholar]
- Selkoe, D.J. Translating cell biology into therapeutic advances in Alzheimer's disease. Nature 1999, 399, A23–A31. [Google Scholar]
- Mattson, M.P.; Gary, D.S.; Chan, S.L.; Duan, W. Perturbed endoplasmic reticulum function, synaptic apoptosis and the pathogenesis of Alzheimer's disease. Biochem. Soc. Symp. 2001, 151–162. [Google Scholar]
- Yan, S.D.; Fu, J.; Soto, C.; Chen, X.; Zhu, H.; Al-Mohanna, F.; Collison, K.; Zhu, A.; Stern, E.; Saido, T.; Tohyama, M.; Ogawa, S.; Roher, A.; Stern, D. An intracellular protein that binds amyloid-beta peptide and mediates neurotoxicity in Alzheimer's disease. Nature 1997, 389, 689–695. [Google Scholar]
- Kakimura, J.; Kitamura, Y.; Takata, K.; Tsuchiya, D.; Taniguchi, T.; Gebicke-Haerter, P.J.; Smith, M.A.; Perry, G.; Shimohama, S. Possible involvement of ER chaperone Grp78 on reduced formation of amyloid-beta deposits. Ann. NY Acad. Sci. 2002, 977, 327–332. [Google Scholar]
- Hoozemans, J.J.; Veerhuis, R.; Van Haastert, E.S.; Rozemuller, J.M.; Baas, F.; Eikelenboom, P.; Scheper, W. The unfolded protein response is activated in Alzheimer's disease. Acta Neuropathol. (Berl) 2005, 110, 165–172. [Google Scholar] [CrossRef]
- Yoo, B.C.; Kim, S.H.; Cairns, N.; Fountoulakis, M.; Lubec, G. Deranged expression of molecular chaperones in brains of patients with Alzheimer's disease. Biochem. Biophys. Res. Commun. 2001, 280, 249–258. [Google Scholar]
- Hoshino, T.; Nakaya, T.; Araki, W.; Suzuki, K.; Suzuki, T.; Mizushima, T. Endoplasmic reticulum chaperones inhibit the production of amyloid-beta peptides. Biochem. J. 2007, 402, 581–589. [Google Scholar]
- Lleo, A.; Berezovska, O.; Herl, L.; Raju, S.; Deng, A.; Bacskai, B.J.; Frosch, M.P.; Irizarry, M.; Hyman, B.T. Nonsteroidal anti-inflammatory drugs lower Abeta42 and change presenilin 1 conformation. Nat. Med. 2004, 10, 1065–1066. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mizushima, T. Molecular Mechanism for Various Pharmacological Activities of NSAIDS. Pharmaceuticals 2010, 3, 1614-1636. https://doi.org/10.3390/ph3051614
Mizushima T. Molecular Mechanism for Various Pharmacological Activities of NSAIDS. Pharmaceuticals. 2010; 3(5):1614-1636. https://doi.org/10.3390/ph3051614
Chicago/Turabian StyleMizushima, Tohru. 2010. "Molecular Mechanism for Various Pharmacological Activities of NSAIDS" Pharmaceuticals 3, no. 5: 1614-1636. https://doi.org/10.3390/ph3051614
APA StyleMizushima, T. (2010). Molecular Mechanism for Various Pharmacological Activities of NSAIDS. Pharmaceuticals, 3(5), 1614-1636. https://doi.org/10.3390/ph3051614