Prospects for the Use of ATR Inhibitors to Treat Cancer

Abstract
:1. Introduction
2. The PIKK Family

3. Structure of ATR/ATRIP
4. Comparison of ATR and ATM
5. The ATR and ATM Checkpoint Pathways
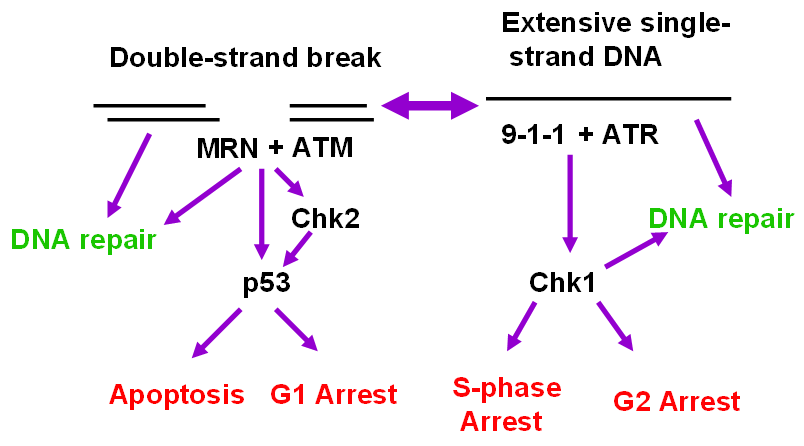
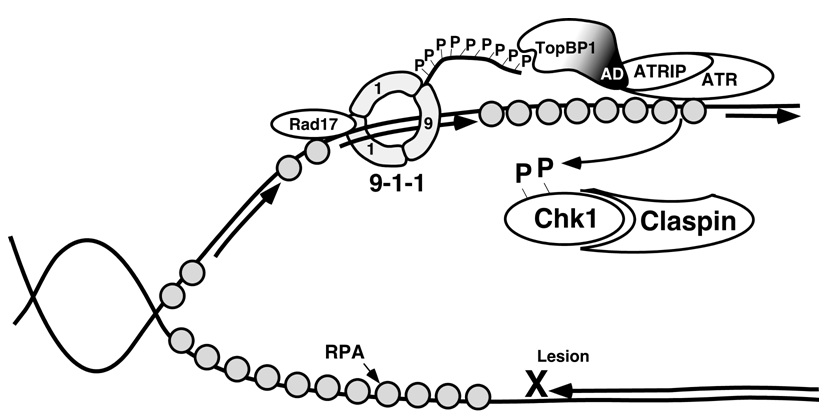
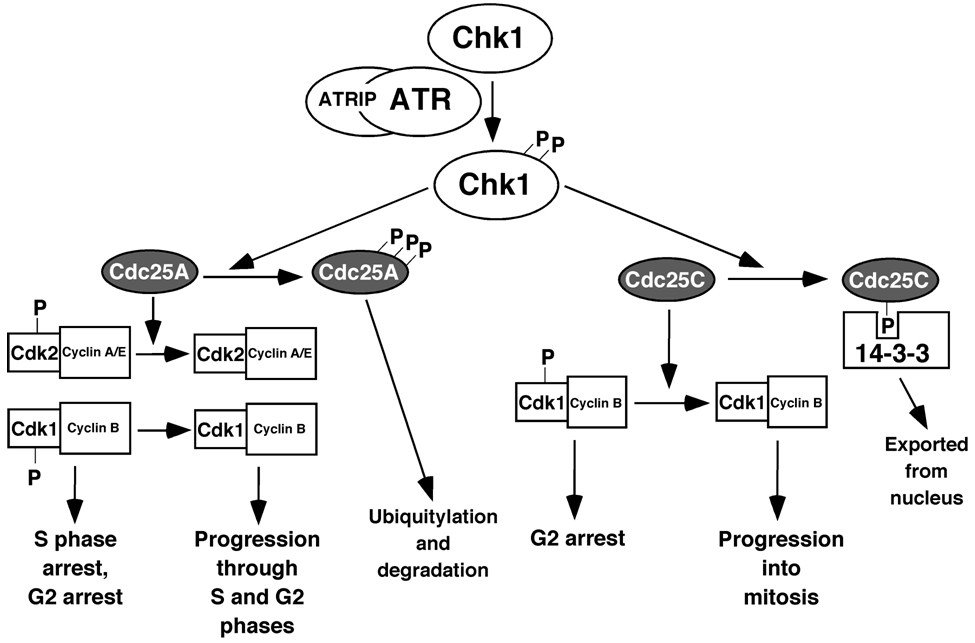
6. Stimuli that Activate the ATR-Chk1 Pathway
6.1. DNA damage
6.2. Fragile sites and repetitive DNA sequences
6.3. Shortened telomeres
7. ATR Substrates
8. Rationale for Inhibiting ATR in Cancer
9. Inhibition of ATR in Cancer Therapy
Known inhibitors of ATR
10. Sensitization of Cancer Cells to Chemotherapeutic Drugs by ATR Inhibition
10.1. Antimetabolites
10.2. Platinating agents
10.3. Alkylating agents
10.4. Topoisomerase poisons
10.5. A view based on ATR mutant cells
11. Potential Problems with ATR Inhibition
12. Clinical Trials Using Chk1 Inhibitors

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor | Combination | Tumor types | Phase | Status | Trial identifier |
|---|---|---|---|---|---|
| UCN-01 | Irinotecan | Solid | I | Completed | NCT00047242 |
| Solid or triple | |||||
| negative breast | I | Recruiting | NCT00031681 | ||
| Topotecan | Ovarian | I | Completed | NCT00072267 | |
| II | Completed | ||||
| Small cell lung | II | Active | NCT00098956 | ||
| Gemcitabine | Pancreatic | I | Completed | NCT00039403 | |
| Cisplatin (2)* | Solid | I | Completed | NCT00012194, | |
| NCT00006464 | |||||
| Carboplatin | Solid | I | Completed | NCT00036777 | |
| Fluorouracil | Pancreatic | II | Completed | NCT00045747 | |
| Solid | I | Completed | NCT00004059 | ||
| Fluorouracil and Leucovorin | Solid | I | Completed | NCT00042861 | |
| Prednisone | Solid or lymphoma | I | Completed | NCT00045500 | |
| Perifosine | Leukemia | I | Recruiting | NCT00301938 | |
| Fludarabine | Lymphoma or leukemia | I | Completed | NCT00019838 | |
| Lymphoma or leukemia | II | Active | |||
| Cytarabine | Leukemia | I | Active | NCT00004263 | |
| AZD7762 | Gemcitabine (2)* | Solid | I | Recruiting | NCT00413686, |
| NCT00937664 | |||||
| Irinotecan | Solid | I | Recruiting | NCT00473616 | |
| XL844 | Gemcitabine | Solid | I | Ongoing | NCT00475917 |
| PF-0477736 | Gemcitabine | Solid | I | Recruiting | NCT00437203 |
| CBP501 | Cisplatin | Solid | I | Active | NCT00551512 |
| Cisplatin and Pemetrexed | Solid | I | Recruiting | NCT00942825 | |
| Malignant pleural | |||||
| Mesothelioma | II | Active | NCT00700336 | ||
| SCH 900776 | Gemcitabine | Solid or lymphoma | I | Recruiting | NCT00779584 |
| Cytarabine | Acute leukemia | I | Recruiting | NCT00907517 |
13. Prospects for Development of ATR Inhibitors
13.1. Issues in the design of small molecule ATR inhibitors
13.2. Approaches for the development of ATR inhibitors
14. Conclusions
Abbreviations
| 5-FU | 5-fluorouracil |
| ATM | ataxia telengiectasia mutated |
| ATR | ataxia telengiectasia and Rad3-related |
| ATRIP | ATR-interacting protein |
| BLM | Bloom helicase |
| BRCA1 | breast cancer (suppressor) 1 |
| Chk1 | checkpoint kinase 1 |
| Chk2 | checkpoint kinase 2 |
| DSB | double-strand break |
| HR | homologous recombination |
| mTOR | mammalian target of rapamycin |
| NHEJ | non-homologous end joining |
| PIKK | phosphoinositide 3-kinase related kinase |
| PRD | PIK regulatory domain |
| RFC | replication factor C |
| RPA | replication protein A |
| ssDNA | single-stranded DNA |
| TOPBP1 | topoisomerase binding protein 1 |
| WRN | Werner’s syndrome protein |
References
- Zhou, B.B.; Elledge, S.J. The DNA damage response: putting checkpoints in perspective. Nature 2000, 408, 433–439. [Google Scholar]
- Kastan, M.B.; Bartek, J. Cell-cycle checkpoints and cancer. Nature 2004, 432, 316–323. [Google Scholar]
- Keith, C.T.; Schreiber, S.L. PIK-Related Kinases: DNA Repair, Recombination, and Cell Cycle Checkpoints. Science 1995, 270, 50–51. [Google Scholar]
- Engelman, J.A.; Luo, J.; Cantley, L.C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 2006, 7, 606–619. [Google Scholar]
- Bosotti, R.; Isacchi, A.; Sonnhammer, E.L. FAT: a novel domain in PIK-related kinases. Trends Biochem. Sci. 2000, 25, 225–227. [Google Scholar]
- Sibanda, B.L.; Chirgadze, D.Y.; Blundell, T.L. Crystal structure of DNA-PKcs reveals a large open-ring cradle comprised of HEAT repeats. Nature 2010, 463, 118–121. [Google Scholar]
- Llorca, O.; Rivera-Calzada, A.; Grantham, J.; Willison, K.R. Electron microscopy and 3D reconstructions reveal that human ATM kinase uses an arm-like domain to clamp around double-stranded DNA. Oncogene 2003, 22, 3867–3874. [Google Scholar]
- Chen, X.; Zhao, R.; Glick, G.G.; Cortez, D. Function of the ATR N-terminal domain revealed by an ATM/ATR chimera. Exp. Cell. Res. 2007, 313, 1667–1674. [Google Scholar]
- Cimprich, K.; Cortez, D. ATR: an essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 2008, 9, 616–627. [Google Scholar]
- Zou, L.; Elledge, S.J. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003, 300, 1542–1548. [Google Scholar]
- Ball, H.L.; Ehrhardt, M.R.; Mordes, D.A.; Glick, G.G.; Chazin, W.J.; Cortez, D. Function of a conserved checkpoint recruitment domain in ATRIP proteins. Mol. Cell Biol. 2007, 27, 3367–3377. [Google Scholar]
- Mordes, D.A.; Glick, G.G.; Zhao, R.; Cortez, D. TopBP1 activates ATR through ATRIP and a PIKK regulatory domain. Genes. Dev. 2008, 22, 1478–1489. [Google Scholar]
- Itakura, E.; Umeda, K.; Sekoguchia, E.; Takata, H.; Ohsumib, M.; Matsuura, A. ATR-dependent phosphorylation of ATRIP in response to genotoxic stress. Biochem. Biophys. Res. Commun. 2004, 323, 1197–1202. [Google Scholar]
- Myers, J.S.; Zhao, R.; Xu, X.; Ham, A.-JL.; Cortez, D. Cyclin-dependent kinase 2 dependent phosphorylation of ATRIP regulates the G2-M checkpoint response to DNA damage. Cancer Res. 2007, 67, 6685–6690. [Google Scholar] [CrossRef] [PubMed]
- Venere, M.; Snyder, A.; Zgheib, O.; Halazonetis, T.D. Phosphorylation of ATR-interacting protein on Ser239 mediates an interaction with breast-ovarian cancer susceptibility 1 and checkpoint function. Cancer Res. 2007, 67, 6100–6105. [Google Scholar]
- Abraham, R.T. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes. Dev. 2001, 15, 2177–2196. [Google Scholar]
- Kim, S.T.; Lim, D.S.; Canman, C.E.; Kastan, M.B. Substrate specificities and identification of putative substrates of ATM kinase family members. J. Biol. Chem. 1999, 274, 37538–37543. [Google Scholar]
- Abraham, R.T. Cell Cycle Checkpoint Signaling Through the ATM and ATR Kinases. Genes. Dev. 2001, 15, 2177–2196. [Google Scholar]
- Oakley, G.G.; Loberg, L.I.; Yao, J.; Risinger, M.A.; Yunker, R.L.; Zernik-Kobak, M.; Khanna, K.K.; Lavin, M.F.; Carty, M.P.; Dixon, K. UV-induced hyperphosphorylation of replication protein a depends on DNA replication and expression of ATM protein. Mol. Biol. Cell 2001, 12, 1199–1213. [Google Scholar]
- Cortez, D.; Wang, Y.; Qin, J.; Elledge, S.J. Requirement of ATM-dependent phosphorylation of brca1 in the DNA damage response to double-strand breaks. Science 1999, 286, 1162–1166. [Google Scholar]
- Brown, E.J.; Baltimore, D. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes. Dev. 2000, 14, 397–402. [Google Scholar]
- O'Connell, M.; J.Cimprich, K.A. G2 damage checkpoints: what is the turn-on? J. Cell Sci. 2005, 118, 1–6. [Google Scholar]
- Roos-Mattjus, P.; Vroman, B.T.; Burtelow, M.A.; Rauen, M.; Eapen, A.K.; Karnitz, L.M. Genotoxin-Induced Rad9-Hus1-Rad1 (9-1-1) Chromatin Association is an Early Checkpoint Signaling Event. J. Biol. Chem. 2002, 277, 43809–43812. [Google Scholar]
- Zou, L.; Cortez, D.; Elledge, S.J. Regulation of ATR substrate selection by Rad17-dependent loading of Rad9 complexes onto chromatin. Genes Dev. 2002, 16, 198–208. [Google Scholar]
- Ellison, V.; Stillman, B. Biochemical characterization of DNA damage checkpoint complexes: clamp loader and clamp complexes with specificity for 5' recessed DNA. PLoS Biol. 2003, 1, E33. [Google Scholar]
- Delacroix, S.; Wagner, J.M.; Kobayashi, M.; Yamamoto, K.; Karnitz, L.M. The Rad9-Hus1-Rad1 (9-1-1) clamp activates checkpoint signaling via TopBP1. Genes Dev. 2007, 21, 1472–1477. [Google Scholar]
- Kumagai, A.; Lee, J.; Yoo, H.Y.; Dunphy, W.G. TopBP1 activates the ATR-ATRIP complex. Cell 2006, 124, 943–955. [Google Scholar]
- Mordes, D.A.; Cortez, D. Activation of ATR and related PIKKs. Cell Cycle 2008, 7, 2809–2812. [Google Scholar]
- Liu, Q.; Guntuku, S.; Cui, X.; Matsuoka, S.; Cortez, D.; Tamai, K.; Luo, G.; Carattini-Rivera, S.; DeMayo, F.; Bradley, A.; Donehower, L.A.; Elledge, S.J. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000, 14, 1448–1459. [Google Scholar]
- Kumagai, A.; Dunphy, W.G. Claspin, a novel protein required for the activation of Chk1 during a DNA replication checkpoint response in Xenopus egg extracts. Mol. Cell 2000, 6, 839–849. [Google Scholar]
- Mickle, K.L.; Oliva, A.; Huberman, J.A.; Leatherwood, J. Checkpoint effects and telomere amplification during DNA re-replication in fission yeast. BMC Mol. Biol. 2007, 8, 119. [Google Scholar]
- Loegering, D.; Arlander, S.J.; Hackbarth, J.; Vroman, B.T.; Roos-Mattjus, P.; Hopkins, K.M.; Lieberman, H.B.; Karnitz, L.M.; Kaufmann, S.H. Rad9 protects cells from topoisomerase poison-induced cell death. J. Biol. Chem. 2004, 279, 18641–18647. [Google Scholar]
- Flatten, K.; Dai, N.T.; Vroman, B.T.; Loegering, D.; Erlichman, C.; Karnitz, L.M.; Kaufmann, S.H. The role of checkpoint kinase 1 in sensitivity to topoisomerase I poisons. J. Biol. Chem. 2005, 280, 14349–14355. [Google Scholar]
- Kemp, M.G.; Akan, Z.; Yilmaz, S.; Grillo, M.; Smith-Roe, S.L.; Kang, T.H.; Cordeiro-Stone, M.; Kaufmann, W.K.; Abraham, R.T.; Sancar, A.; Unsal-Kacmaz, K. Tipin-RPA interaction mediates Chk1 phosphorylation by ATR in response to genotoxic stress. J. Biol. Chem. 2010. [Google Scholar]
- Smith, K.D.; Fu, M.A.; Brown, E.J. Tim-Tipin dysfunction creates an indispensible reliance on the ATR-Chk1 pathway for continued DNA synthesis. J. Cell Biol. 2009, 187, 15–23. [Google Scholar]
- Sorensen, C.S.; Syljuasen, R.G.; Falck, J.; Schroeder, T.; Rönnstrand, L.; Khanna, K.K.; Zhou, B.; Bartek, J.; Lukas, J. Chk1 regulates the S phase checkpoint by coupling the phsiological turnover and ionizing radiation-induced accelerated proteolysis of Cdc25A. Cancer Cell 2003, 3, 247–258. [Google Scholar]
- Sanchez, Y.; Wong, C.; Thoma, R.S.; Richman, R.; Wu, Z.; Piwnica-Worms, H.; Elledge, S.J. Conservation of the Chk1 checkpoint pathway in mammals: linkage of DNA damage to Cdk regulation through Cdc25. Science 1997, 277, 1497–1501. [Google Scholar]
- Peng, C.Y.; Graves, P.R.; Thoma, R.S.; Wu, Z.; Shaw, A.S.; Piwnica-Worms, H. Mitotic and G2 checkpoint control: regulation of 14-3-3 protein binding by phosphorylation of Cdc25C on serine-216. Science 1997, 277, 1501–1505. [Google Scholar]
- Bartek, J.; Lukas, C.; Lukas, J. Checking on DNA damage in S phase. Nat. Rev. Mol. Cell Biol. 2004, 5, 792–804. [Google Scholar]
- Shirahige, K.; Hori, Y.; Shiraishi, K.; Yamashita, M.; Takahashi, K.; Obuse, C.; Tsurimoto, T.; Yoshikawa, H. Regulation of DNA-replication origins during cell-cycle progression. Nature 1998, 395, 618–621. [Google Scholar]
- Feijoo, C.; Hall-Jackson, C.; Wu, R.; Jenkins, D.; Leitch, J.; Gilbert, D.M.; Smythe, C. Activation of mammalian Chk1 during DNA replication arrest: a role for Chk1 in the intra-S phase checkpoint monitoring replication origin firing. J. Cell Biol. 2001, 154, 913–923. [Google Scholar]
- Heffernan, T.P.; Simpson, D.A.; Frank, A.R.; Heinloth, A.N.; Paules, R.S.; Cordeiro-Stone, M.; Kaufmann, W.K. An ATR- and Chk1-dependent S checkpoint inhibits replicon initiation following UVC-induced DNA damage. Mol. Cell Biol. 2002, 22, 8552–8561. [Google Scholar]
- Unsal-Kacmaz, K.; Chastain, P.D.; Qu, P.; Minoo, P.; Cordeiro-Stone, M.; Sancar, A.; Kaufmann, W.K. The human Tim/Tipin complex coordinates an Intra-S checkpoint response to UV that slows replication fork displacement. Mol. Cell Biol. 2007, 27, 3131–3142. [Google Scholar]
- Liu, E.; Lee, A.Y.; Chiba, T.; Olson, E.; Sun, P.; Wu, X. The ATR-mediated S phase checkpoint prevents rereplication in mammalian cells when licensing control is disrupted. J. Cell Biol. 2007, 179, 643–657. [Google Scholar]
- Kastan, M.B.; Lim, D.S. The many substrates and functions of ATM. Nat. Rev. Mol. Cell. Biol. 2000, 1, 179–186. [Google Scholar]
- Vousden, K.H.; Lu, X. Live or let die: the cell's response to p53. Nat. Rev. Cancer 2002, 2, 594–604. [Google Scholar]
- Lowe, S.W.; Cepro, E.; Evan, G. Intrinsic tumour suppression. Nature 2004. [Google Scholar]
- Cliby, W.A.; Roberts, C.J.; Cimprich, K.A.; Stringer, C.M.; Lamb, J.R.; Schreiber, S.L.; Friend, S.H. Overexpression of a kinase-inactive ATR protein causes sensitivity to DNA-damaging agents and defects in cell cycle checkpoints. Embo. J. 1998, 17, 159–169. [Google Scholar]
- Ewald, B.; Sampath, D.; Plunkett, W. ATM and the Mre11-Rad50-Nbs1 complex respond to nucleoside analogue-induced stalled replication forks and contribute to drug resistance. Cancer Res. 2008, 68, 7947–7955. [Google Scholar]
- Zhao, H.; Piwnica-Worms, H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol. Cell Biol. 2001, 21, 4129–4139. [Google Scholar]
- Pichiorri, F.; Ishii, H.; Okumura, H.; Trapasso, F.; Wang, Y.; Huebner, K. Molecular parameters of genome instability: roles of fragile genes at common fragile sites. J. Cell Biochem. 2008, 104, 1525–1533. [Google Scholar]
- Casper, A.M.; Nghiem, P.; Arlt, M.F.; Glover, T.W. ATR regulates fragile site stability. Cell 2002, 111, 779–789. [Google Scholar]
- Durkin, S.G.; Arlt, M.F.; Howlett, N.G.; Glover, T.W. Depletion of CHK1, but not CHK2, induces chromosomal instability and breaks at common fragile sites. Oncogene 2006, 25, 4381–4388. [Google Scholar]
- Entezam, A.; Usdin, K. ATR protects the genome against CGG.CCG-repeat expansion in Fragile X premutation mice. Nucleic Acids Res. 2008, 36, 1050–1056. [Google Scholar] [CrossRef] [PubMed]
- Cha, R.S.; Kleckner, N. ATR homolog Mec1 promotes fork progression, thus averting breaks in replication slow zones. Science 2002, 297, 602–606. [Google Scholar]
- Lenzmeier, B.A.; Freudenreich, C.H. Trinucleotide repeat instability: A hairpin curve at the crossroads of replication, recombination, and repair. Cytogenet. Genome Res. 2003, 100, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, M.; Gustafson, T.L.; Majors, E.R.; Freudenreich, C.H. Expanded CAG repeats activate the DNA damage checkpoint pathway. Mol. Cell 2004, 15, 287–293. [Google Scholar]
- Khadaroo, B.; Teixeira, M.T.; Luciano, P.; Eckert-Boulet, N.; Germann, S.M.; Simon, M.N.; Gallina, I.; Abdallah, P.; Gilson, E.; Géli, V.; Lisby, M. The DNA damage response at eroded telomeres and tethering to the nuclear pore complex. Nat. Cell Biol. 2009, 11, 980–987. [Google Scholar]
- d'Adda di Fagagna, F.; Reaper, P.M.; Clay-Farrace, L.; Fiegler, H.; Carr, P.; von Zglinicki, T.; Saretzki, G.; Carter, N.P.; Jackson, S.P. A DNA damage checkpoint response in telomere-initiated senescence. Nature 2003, 426, 194–198. [Google Scholar]
- Ritchie, K.B.; Mallory, J.C.; Petes, T.D. Interactions of TLC1 (which encodes the RNA subunit of telomerase), TEL1, and MEC1 in regulating telomere length in the yeast Saccharomyces cerevisiae. Mol. Cell Biol. 1999, 19, 6065–6075. [Google Scholar] [PubMed]
- Francia, S.; Weiss, R.S.; Hande, M.P.; Freire, R.; di Fagagna, F.A. Telomere and telomerase modulation by the mammalian Rad9/Rad1/Hus1 DNA-damage-checkpoint complex. Curr. Biol. 2006, 16, 1551–1558. [Google Scholar]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R., III; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; Shiloh, Y.; Gygi, S.P.; Elledge, S.J. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [PubMed]
- Shechter, D.; Costanzo, V.; Gautier, J. ATR and ATM regulate the timing of DNA replication origin firing. Nat. Cell Biol. 2004, 6, 648–655. [Google Scholar]
- Maya-Mendoza, A.; Petermann, E.; Gillespie, D.A.F.; Caldecott, K.W.; Jackson, D.A. Chk1 regulates the density of active replication origins during the vertebrate S phase. Embo. J. 2007, 26, 2719–2731. [Google Scholar]
- Brush, G.S.; Morrow, D.M.; Hieter, P.; Kelly, T.J. The ATM homologue MEC1 is required for phosphorylation of replication protein A in yeast. Proc. Natl. Acad. Sci. USA 1996, 93, 15075–15080. [Google Scholar]
- Liu, J.S.; Kuo, S.R.; Melendy, T. Phosphorylation of replication protein A by S-phase checkpoint kinases. DNA Repair (Amst) 2006, 5, 369–380. [Google Scholar] [PubMed]
- Cortez, D.; Glick, G.; Elledge, S.J. Minichromosome maintenance proteins are direct targets of the ATM and ATR checkpoint kinases. Proc. Natl. Acad. Sci. USA 2004, 101, 10078–10083. [Google Scholar]
- Yoo, H.Y.; Shevchenko, A.; Shevchenko, A.; Dunphy, W.G. Mcm2 is a direct substrate of ATM and ATR during DNA damage and DNA replication checkpoint responses. J. Biol. Chem. 2004, 279, 53353–53364. [Google Scholar]
- Trenz, K.; Errico, A.; Costanzo, V. Plx1 is required for chromosomal DNA replication under stressful conditions. Embo J. 2008, 27, 876–885. [Google Scholar]
- Chen, J. Ataxia telangiectasia-related protein is involved in the phosphorylation of BRCA1 following deoxyribonucleic acid damage. Cancer Res. 2000, 60, 5037–5039. [Google Scholar]
- Pichierri, P.; Rosselli, F.; Franchitto, A. Werner's syndrome protein is phosphorylated in an ATR/ATM-dependent manner following replication arrest and DNA damage induced during the S phase of the cell cycle. Oncogene 2003, 22, 1491–1500. [Google Scholar]
- Tripathi, V.; Kaur, S.; Sengupta, S. Phosphorylation-dependent interactions of BLM and 53BP1 are required for their anti-recombinogenic roles during homologous recombination. Carcinogenesis 2008, 29, 52–61. [Google Scholar]
- Khakhar, R.R.; Cobb, J.A.; Bjergbaek, L.; Hickson, I.D.; Gasser, S.M. RecQ helicases: multiple roles in genome maintenance. Trends Cell Biol. 2003, 13, 493–501. [Google Scholar]
- Bachrati, C.Z.; Borts, R.H.; Hickson, I.D. Mobile D-loops are a preferred substrate for the Bloom's syndrome helicase. Nucleic Acids Res. 2006, 34, 2269–2279. [Google Scholar]
- Opresko, P.L.; Sowd, G.; Wang, H. The Werner syndrome helicase/exonuclease processes mobile D-loops through branch migration and degradation. PLoS One 2009, 4, e4825. [Google Scholar]
- Ho, G.P.; Margossian, S.; Taniguchi, T.; D'Andrea, A.D. Phosphorylation of FANCD2 on two novel sites is required for mitomycin C resistance. Mol. Cell Biol. 2006, 26, 7005–7015. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, U.K.; Senderowicz, A.M.; Ferbeyre, G. RNA silencing of checkpoint regulators sensitizes p53-defective prostate cancer cells to chemotherapy while sparing normal cells. Cancer Res. 2005, 65, 2872–2881. [Google Scholar]
- Pan, Y.; Ren, K.H.; He, H.W.; Shao, R.G. Knockdown of Chk1 sensitizes human colon carcinoma HCT116 cells in a p53-dependent manner to lidamycin through abrogation of a G2/M checkpoint and induction of apoptosis. Cancer Biol. Ther. 2009, 8, 1559–1566. [Google Scholar]
- Tao, Y.; Leteur, C.; Zhang, P.; Castedo, M.; Pierré, A.; Golsteyn, R.M.; Bourhis, J.; Kroemer, G.; Deutsch, E. Radiosensitization by Chir-124, a selective CHK1 inhibitor: Effects of p53 and cell cycle checkpoints. Cell Cycle 2009, 8, 1196–1205. [Google Scholar]
- Powell, S.N.; DeFrank, J.S.; Connell, P.; Eogan, M.; Preffer, F.; Dombkowski, D.; Tang, W.; Friend, S. Differential sensitivity of p53(-) and p53(+) cells to caffeine-induced radiosensitization and override of G2 delay. Cancer Res. 1995, 55, 1643–1648. [Google Scholar]
- Strunz, A.M.; Peschke, P.; Waldeck, W.; Ehemann, V.; Kissel, M.; Debus, J. Preferential radiosensitization in p53-mutated human tumour cell lines by pentoxifylline-mediated disruption of the G2/M checkpoint control. Int. J. Radiat. Biol. 2002, 78, 721–732. [Google Scholar]
- Rodriguez-Bravo, V.; Guaita-Esteruelas, S.; Salvador, N.; Bachs, O.; Agell, N. Different S/M checkpoint responses of tumor and non tumor cell lines to DNA replication inhibition. Cancer Res. 2007, 67, 11648–11656. [Google Scholar]
- Sarkaria, J.N.; Busby, E.C.; Tibbetts, R.S.; Roos, P.; Taya, Y.; Karnitz, L.M.; Abraham, R.T. Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine. Cancer Res. 1999, 59, 4375–4382. [Google Scholar]
- Abraham, R.T. The ATM-related kinase, hSMG-1, bridges genome and RNA surveillance pathways. DNA Repair (Amst) 2004, 3, 919–925. [Google Scholar] [CrossRef]
- Nishida, H.; Tatewaki, N.; Nakajima, Y.; Magara, T.; Ko, K.M.; Hamamori, Y.; Konishi, T. Inhibition of ATR protein kinase activity by schisandrin B in DNA damage response. Nucleic Acids Res. 2009, 37, 5678–5689. [Google Scholar]
- Nghiem, P.; Park, P.K.; Kim, Y.; Vaziri, C.; Schreiber, S.L. ATR inhibition selectively sensitizes G1 checkpoint-deficient cells to lethal premature chromatin condensation. Proc. Natl. Acad. Sci. USA 2001, 98, 9092–9097. [Google Scholar]
- Wilsker, D.; Bunz, F. Loss of ataxia telangiectasia mutated- and Rad3-related function potentiates the effects of chemotherapeutic drugs on cancer cell survival. Mol. Cancer Ther. 2007, 6, 1406–1413. [Google Scholar]
- Plunkett, W.; Huang, P.; Xu, Y.Z.; Heinemann, V.; Grunewald, R.; Gandhi, V. Gemcitabine: metabolism, mechanisms of action, and self-potentiation. Semin. Oncol. 1995, 22 (Suppl. 11), 3–10. [Google Scholar]
- Karnitz, L.M.; Flatten, K.S.; Wagner, J.M.; Loegering, D.; Hackbarth, J.S.; Arlander, S.J.H.; Vroman, B.T.; Thomas, M.B.; Baek, Y.-U.; Hopkins, K.M.; Lieberman, H.B.; Chen, J.; Cliby, W.A.; Kaufmann, S.H. Gemcitabine-induced activation of checkpoint signaling pathways that affect tumor cell survival. Mol. Pharmacol. 2005, 68, 1636–1644. [Google Scholar]
- Parsels, L.A.; Morgan, M.A.; Tanska, D.M.; Parsels, J.D.; Palmer, B.D.; Booth, R.J.; Denny, W.A.; Canman, C.E.; Kraker, A.J.; Lawrence, T.S.; Maybaum, J. Gemcitabine sensitization by checkpoint kinase 1 inhibition correlates with inhibition of a Rad51 DNA damage response in pancreatic cancer cells. Mol. Cancer Ther. 2009, 8, 45–54. [Google Scholar]
- Matthews, D.J.; Yakes, F.M.; Chen, J.; Tadano, M.; Bornheim, L.; Clary, D.O.; Tai, A.; Wagner, J.M.; Miller, N.; Kim, Y.D.; Robertson, S.; Murray, L.; Karnitz, L.M. Pharmacological abrogation of S-phase checkpoint enhances the anti-tumor activity of gemcitabine in vivo. Cell Cycle 2007, 6, 104–110. [Google Scholar] [CrossRef]
- Mesa, R.A.; Loegering, D.; Powell, H.L.; Flatten, K.; Arlander, S.J.H.; Dai, N.T.; Heldebrant, M.P.; Vroman, B.T.; Smith, B.D.; Karp, J.E.; Eyck, C.J.T.; Erlichman, C.; Kaufmann, S.H.; Karnitz, L.M. Heat shock protein 90 inhibition sensitizes acute myelogenous leukemia cells to cytarabine. Blood 2005, 106, 318–327. [Google Scholar]
- Jardim, M.J.; Wang, Q.; Furumai, R.; Wakeman, T.; Goodman, B.K.; Wang, X.-F. Reduced ATR or Chk1 expression leads to chromosome instability and chemosensitization of mismatch repair-deficient colorectal cancer cells. Mol. Biol. Cell 2009, 20, 3801–3809. [Google Scholar]
- Ganzinelli, M.; Carrassa, L.; Crippa, F.; Tavecchio, M.; Broggini, M.; Damia, G. Checkpoint kinase 1 down-regulation by an inducible small interfering RNA expression system sensitized in vivo tumors to treatment with 5-fluorouracil. Clin. Cancer Res. 2008, 14, 5131–5141. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.M.; Karnitz, L.M. Cisplatin-induced DNA damage activates replication checkpoint signaling components that differentially affect tumor cell survival. Mol. Pharmacol. 2009, 76, 208–214. [Google Scholar]
- Lewis, K.A.; Lilly, K.K.; Reynolds, E.A.; Sullivan, W.P.; Kaufmann, S.H.; Cliby, W.A. Ataxia telangiectasia and rad3-related kinase contributes to cell cycle arrest and survival after cisplatin but not oxaliplatin. Mol. Cancer Ther. 2009, 8, 855–863. [Google Scholar]
- Nieborowska-Skorska, M.; Stoklosa, T.; Datta, M.; Czechowska, A.; Rink, L.; Slupianek, A.; Koptyra, M.; Seferynska, I.; Krszyna, K.; Blasiak, J.; Skorski, T. ATR-Chk1 axis protects BCR/ABL leukemia cells from the lethal effect of DNA double-strand breaks. Cell Cycle 2006, 5, 994–1000. [Google Scholar] [PubMed]
- Caporali, S.; Levati, L.; Starace, G.; Ragone, G.; Bonmassar, E.; Alvino, E.; D'Atri, S. AKT is activated in an ataxia-telangiectasia and Rad3-related-dependent manner in response to temozolomide and confers protection against drug-induced cell growth inhibition. Mol. Pharmacol. 2008, 74, 173–183. [Google Scholar]
- Caporali, S.; Falcinelli, S.; Starace, G.; Russo, M.T.; Bonmassar, E.; Jiricny, J.; D'Atri, S. DNA damage induced by temozolomide signals to both ATM and ATR: role of the mismatch repair system. Mol. Pharmacol. 2004, 66, 478–491. [Google Scholar]
- Cliby, W.A.; Lewis, K.A.; Lilly, K.K.; Kaufmann, S.H. S phase and G2 arrests induced by topoisomerase I poisons are dependent on ATR kinase function. J. Biol. Chem. 2002, 277, 1599–1606. [Google Scholar]
- Takai, H.; Tominaga, K.; Motoyama, N.; Minamishima, Y.A.; Nagahama, H.; Tsukiyama, T.; Ikeda, K.; Nakayama, K.; Nakanishi, M.; Nakayama, K. Aberrant cell cycle checkpoint function and early embryonic death in Chk1(-/-) mice. Genes Dev. 2000, 14, 1439–1447. [Google Scholar]
- Lara, P.N., Jr.; Mack, P.C.; Synold, T.; Frankel, P.; Longmate, J.; Gumerlock, P.H.; Doroshow, J.H.; Gandara, D.R. The cyclin-dependent kinase inhibitor UCN-01 plus cisplatin in advanced solid tumors: a California cancer consortium phase I pharmacokinetic and molecular correlative trial. Clin. Cancer Res. 2005, 11, 4444–4450. [Google Scholar]
- Edelman, M.J.; Bauer, K.S., Jr.; Wu, S.; Smith, R.; Bisacia, S.; Dancey, J. Phase I and pharmacokinetic study of 7-hydroxystaurosporine and carboplatin in advanced solid tumors. Clin. Cancer Res. 2007, 13, 2667–2674. [Google Scholar]
- Welch, S.; Hirte, H.W.; Carey, M.S.; Hotte, S.J.; Tsao, M.-S.; Brown, S.; Pond, G.R.; Dancey, J.E.; Oza, A.M. UCN-01 in combination with topotecan in patients with advanced recurrent ovarian cancer: a study of the Princess Margaret Hospital Phase II consortium. Gynecol. Oncol. 2007, 106, 305–310. [Google Scholar]
- Jimeno, A.; Rudek, M.A.; Purcell, T.; Laheru, D.A.; Messersmith, W.A.; Dancey, J.; Carducci, M.A.; Baker, S.D.; Hidalgo, M.; Donehower, R.C. Phase I and pharmacokinetic study of UCN-01 in combination with irinotecan in patients with solid tumors. Cancer Chemother. Pharmacol. 2008, 61, 423–433. [Google Scholar]
- Kortmansky, J.; Shah, M.A.; Kaubisch, A.; Weyerbacher, A.; Yi, S.; Tong, W.; Sowers, R.; Gonen, M.; O'Reilly, E.; Kemeny, N.; Ilson, D.I.; Saltz, L.B.; Maki, R.G.; Kelsen, D.P.; Schwartz, G.K. Phase I trial of the cyclin-dependent kinase inhibitor and protein kinase C inhibitor 7-hydroxystaurosporine in combination with Fluorouracil in patients with advanced solid tumors. J. Clin. Oncol. 2005, 23, 1875–1884. [Google Scholar]
- Zhang, J.; Yang, P.L.; Gray, N.S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. [Google Scholar]
- Allen, L.F.; Sebolt-Leopold, J.; Meyer, M.B. CI-1040 (PD184352), a targeted signal transduction inhibitor of MEK (MAPKK). Semin. Oncol. 2003, 30, 105–116. [Google Scholar]
- Cohen, M.S.; Zhang, C.; Shokat, K.M.; Taunton, J. Structural bioinformatics-based design of selective, irreversible kinase inhibitors. Science 2005, 308, 1318–1321. [Google Scholar]
- Rabindran, S.K.; Discafani, C.M.; Rosfjord, E.C.; Baxter, M.; Floyd, M.B.; Golas, J.; Hallett, W.A.; Johnson, B.D.; Nilakantan, R.; Overbeek, E.; Reich, M.F.; Shen, R.; Shi, X.; Tsou, H.-R.; Wang, Y.-F.; Wissner, A. Antitumor activity of HKI-272, an orally active, irreversible inhibitor of the HER-2 tyrosine kinase. Cancer Res. 2004, 64, 3958–3965. [Google Scholar] [PubMed]
- Kobayashi, S.; Ji, H.; Yuza, Y.; Meyerson, M.; Wong, K.-K.; Tenen, D.G.; Halmos, B. An alternative inhibitor overcomes resistance caused by a mutation of the epidermal growth factor receptor. Cancer Res. 2005, 65, 7096–7101. [Google Scholar]
- Willmore, E.; de Caux, S.; Sunter, N.J.; Tilby, M.J.; Jackson, G.H.; Austin, C.A.; Durkacz, B.W. A novel DNA-dependent protein kinase inhibitor, NU7026, potentiates the cytotoxicity of topoisomerase II poisons used in the treatment of leukemia. Blood 2004, 103, 4659–4665. [Google Scholar] [CrossRef] [PubMed]
- Hickson, I.; Zhao, Y.; Richardson, C.J.; Green, S.J.; Martin, N.M.B.; Orr, A.I.; Reaper, P.M.; Jackson, S.P.; Curtin, N.J.; Smith, G.C.M. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 2004, 64, 9152–9159. [Google Scholar]
- Liu, P.; Cheng, H.; Roberts, T.M.; Zhao, J.J. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discov. 2009, 8, 627–644. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wagner, J.M.; Kaufmann, S.H. Prospects for the Use of ATR Inhibitors to Treat Cancer. Pharmaceuticals 2010, 3, 1311-1334. https://doi.org/10.3390/ph3051311
Wagner JM, Kaufmann SH. Prospects for the Use of ATR Inhibitors to Treat Cancer. Pharmaceuticals. 2010; 3(5):1311-1334. https://doi.org/10.3390/ph3051311
Chicago/Turabian StyleWagner, Jill M., and Scott H. Kaufmann. 2010. "Prospects for the Use of ATR Inhibitors to Treat Cancer" Pharmaceuticals 3, no. 5: 1311-1334. https://doi.org/10.3390/ph3051311
APA StyleWagner, J. M., & Kaufmann, S. H. (2010). Prospects for the Use of ATR Inhibitors to Treat Cancer. Pharmaceuticals, 3(5), 1311-1334. https://doi.org/10.3390/ph3051311