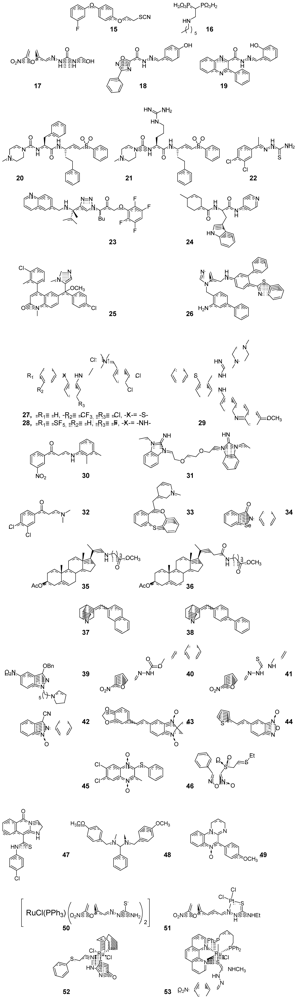
Synthetic Medicinal Chemistry in Chagas’ Disease: Compounds at The Final Stage of “Hit-To-Lead” Phase

Abstract
: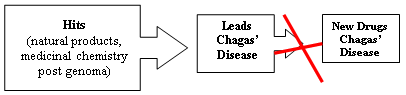
1. Introduction
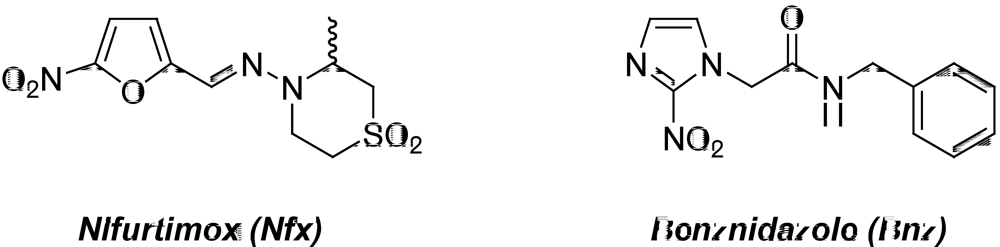
2. Medicinal Chemistry in Chagas’ Disease
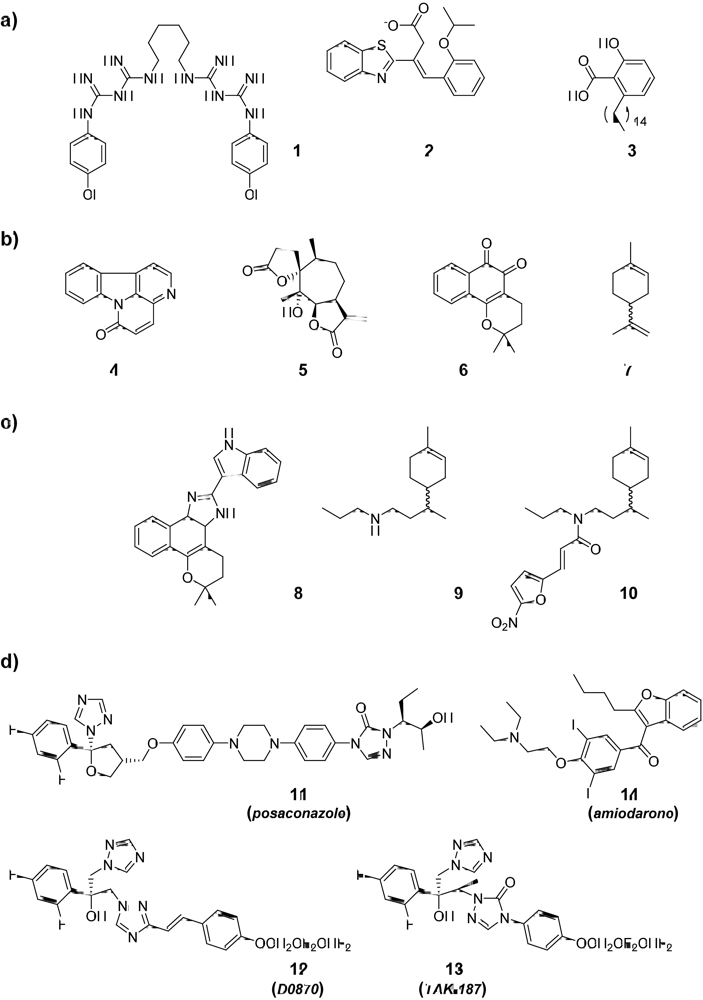
2.1. Compounds from the “Active-To-Hit” Phase

3. Compounds at the Final Stage of “Hit-to-Lead” Phase
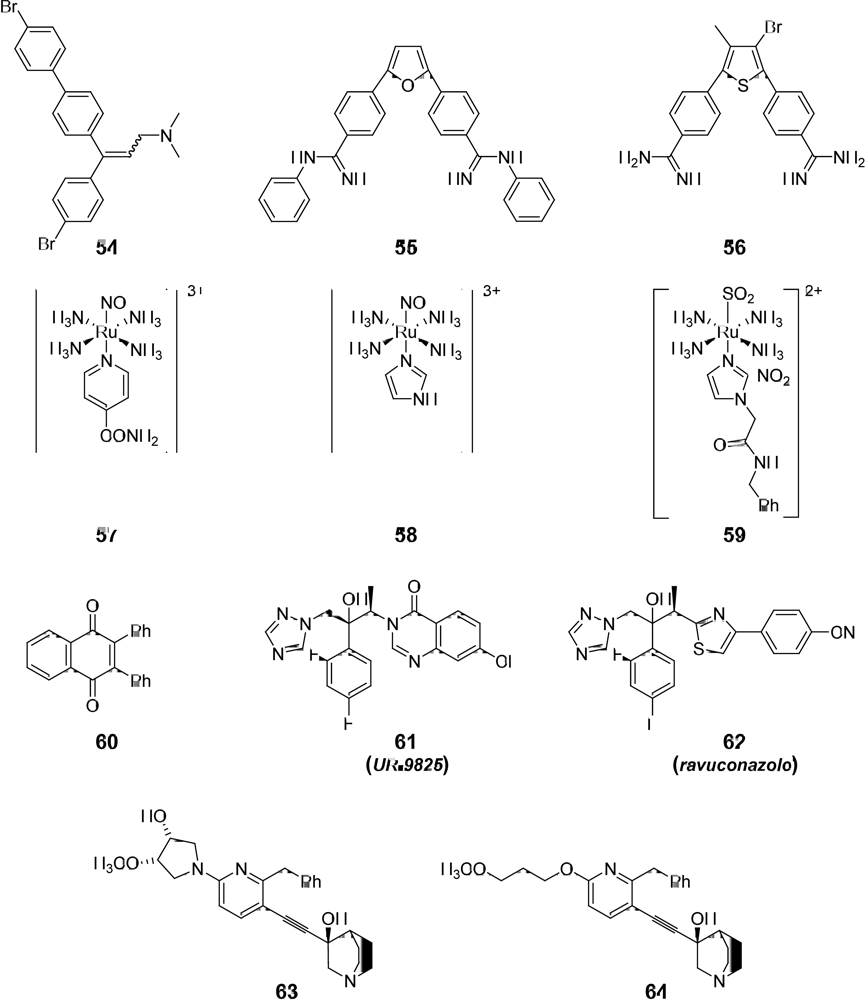
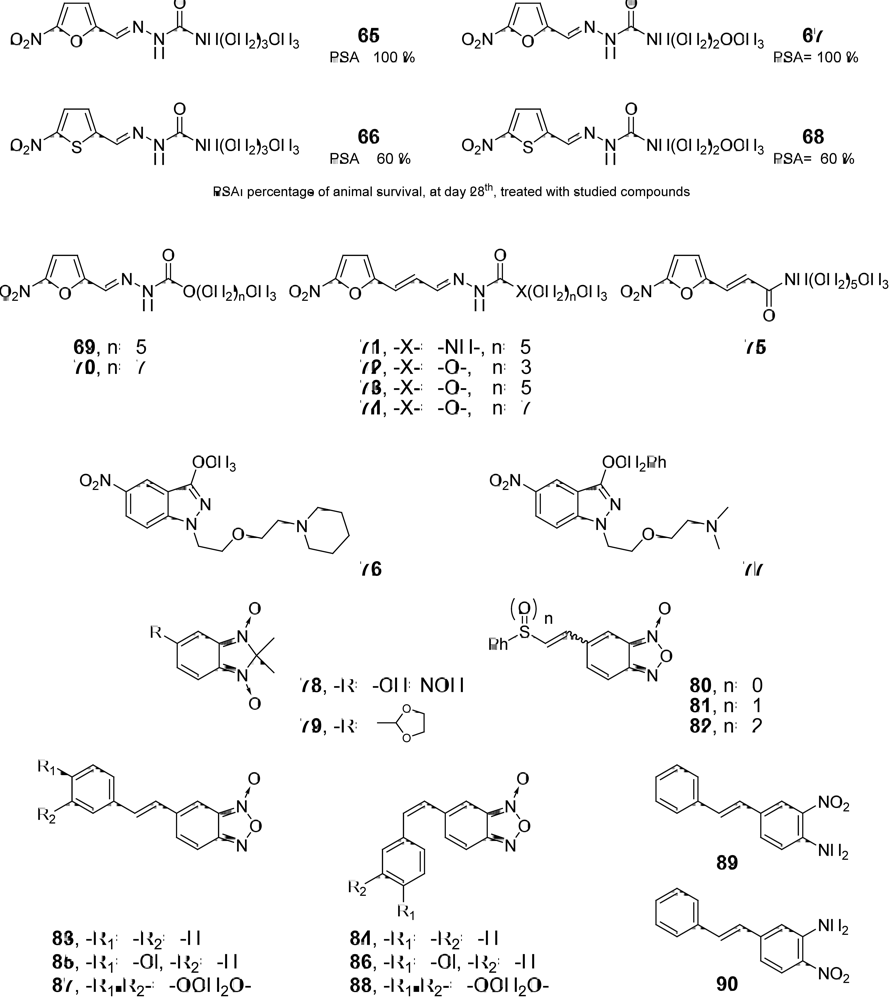
3.1. Drug-like Properties of the anti-T. cruzi compounds at the Ending Stage of “Hit-to-Lead” Phase

{kind=link}
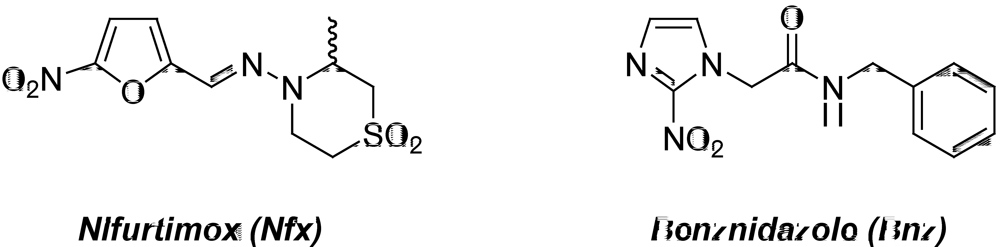{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd. | Meets Lipinski’s rule | Meets Veber’s rule | Drug-likeness1 | Drug-score1 | Toxic effects1 | Alerts for mn2 | Carcinogenic - mutagenic effects2 |
|---|---|---|---|---|---|---|---|
| 4 | y (0)3 | y (0)3 | -0.984 | 0.545 | n6 | I7 | A8 |
| 5 | y (0) | y (0) | -10.47 | 0.22 | y (m,i) | I | C |
| 11 | n (2) | n (1) | 4.51 | 0.05 | y (m,t,i,r) | I | C |
| 12 | y (1) | n (1) | -9.42 | 0.16 | y (i,r) | I | C |
| 13 | y (1) | y (0) | -4.34 | 0.13 | y (i,r) | I | C |
| 20 | y (1) | n (1) | 0.48 | 0.29 | y (i) | I | C |
| 21 | n (3) | n (2) | 1.21 | 0.41 | y (i) | I | C |
| 26 | n (2) | y (0) | -2.68 | 0.05 | y (m,t,i) | I | A,F |
| 5410 | y (1) | y (0) | -3.08 | 0.14 | n | II | C |
| 55 | y (1) | y (0) | 0.38 | 0.28 | n | II | C,D |
| 56 | y (1) | y (0) | -2.61 | 0.29 | n | II | C |
| 57 | y (1) | n (1) | -9 | - | - | I | C |
| 58 | y (1) | n (1) | - | - | - | I | C |
| 59 | n (2) | n (1) | - | - | - | I | C |
| 60 | y (1) | y (0) | -1.38 | 0.23 | y (r) | I | A |
| 61 | y (0) | y (0) | 3.41 | 0.51 | y (r) | I | C |
| 62 | y (0) | y (0) | -7.2 | 0.08 | y (m,t,i,r) | I | C |
| 63 | y (0) | y (0) | 2.76 | 0.64 | n | I | C |
| 64 | y (0) | y (0) | 1.43 | 0.53 | n | I | C |
| 65 | y (0) | y (0) | 1.01 | 0.55 | y (m) | I | A |
| 66 | y (0)3 | y (0)3 | -2.614 | 0.335 | y (m)6 | I7 | A8 |
| 1.57 | 0.81 | n | |||||
| 67 | y (0) | y (0) | I | A | |||
| 68 | y (0) | y (0) | -2.09 | 0.49 | n | I | A |
| 69 | y (0) | y (0) | -15.56 | 0.19 | y (i) | I | A |
| 70 | y (0) | n (1) | -23.57 | 0.21 | y (i) | I | A |
| 71 | y (0) | y (0) | -17.57 | 0.11 | y (m,i) | I | A |
| 72 | y (0) | y (0) | -8.91 | 0.17 | y (m,i) | I | A |
| 73 | y (0) | y (0) | -17.57 | 0.11 | y (m,i) | I | A |
| 74 | y (0) | n (1) | -25.58 | 0.11 | y (m,i) | I | A |
| 75 | y (0) | y (0) | -13.95 | 0.23 | y (m) | I | A |
| 76 | y (0) | y (0) | -5.98 | 0.43 | n | I | A |
| 77 | y (0) | y (0) | -9.05 | 0.40 | n | I | A |
| 78 | y (0) | y (0) | -2.43 | 0.53 | n | I | C |
| 79 | y (0) | y (0) | -8.82 | 0.49 | n | I | C |
| 8010 | y (0) | y (0) | -3.64 | 0.24 | n | I | A |
| 8110 | y (0) | y (0) | -5.44 | 0.32 | n | I | A |
| 8210 | y (0) | y (0) | -12.28 | 0.25 | y (i) | I | A |
| 83 | y (0) | y (0) | -3.53 | 0.28 | n | I | A |
| 84 | y (0) | y (0) | -3.53 | 0.28 | n | I | A |
| 85 | y (1) | y (0) | -0.95 | 0.22 | y (m) | I | A/B |
| 86 | y (1) | y (0) | -0.95 | 0.22 | y (m) | I | A/B |
| 87 | y (0) | y (0) | -1.48 | 0.27 | n | I | A |
| 88 | y (0) | y (0) | -1.48 | 0.27 | n | I | A |
| Nfx | y (0) | y (0) | 0.65 | 0.16 | n (m,t,r) | I | A |
| Bnz | y (0) | y (0) | -3.32 | 0.18 | n (m,r) | I | A |
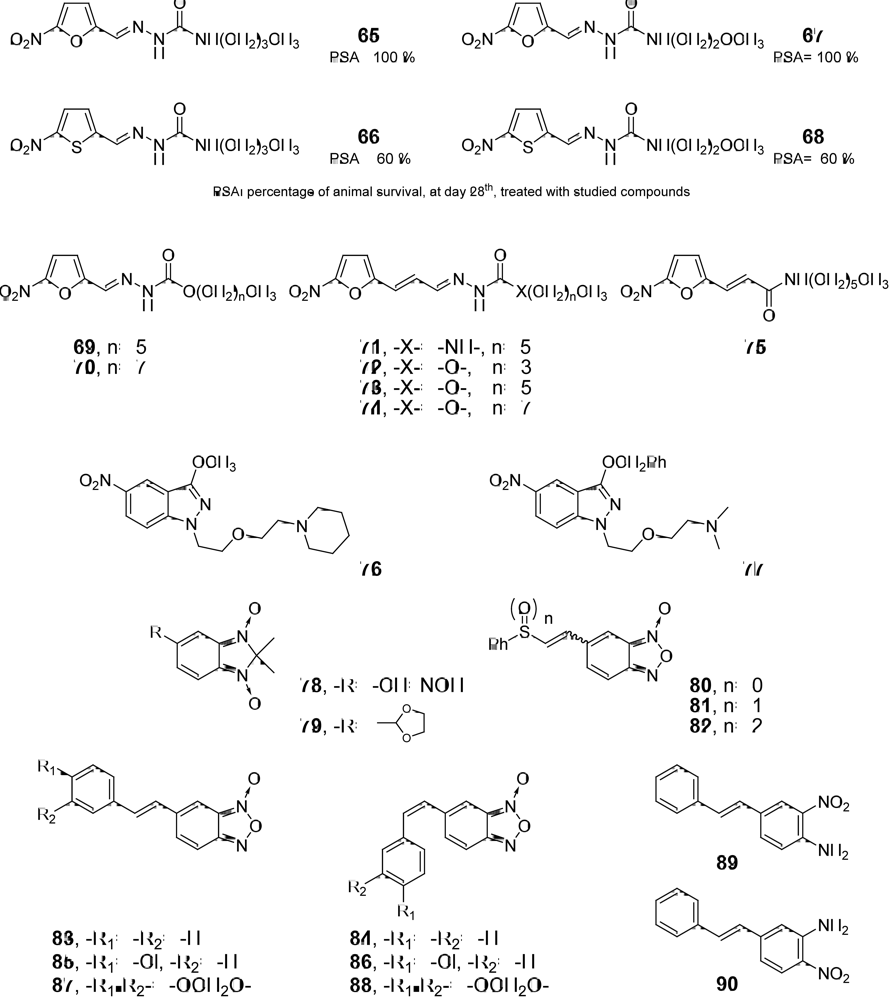
3.2. Scale-Up Information of the anti-T. cruzi Compounds at the Ending Stage of “Hit-to-Lead” Phase
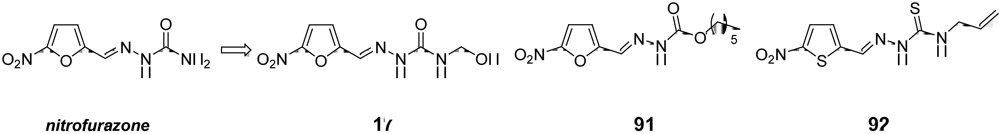
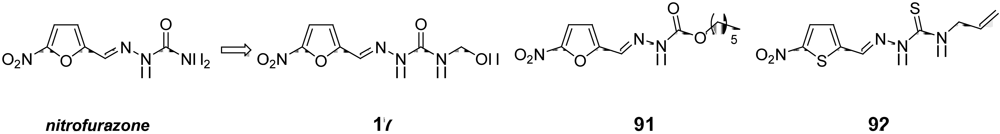
3.3. Candidates to be Submitted at the “Hit-to-Lead” Phase in the Short to Medium-Term
4. Conclusions
Acknowledgments
References and Notes
- Chagas, C. Nova entidade morbida do homen. Resumo geral dos estudos etiológicos e clínicos. Mem. Inst. Oswaldo Cruz 1911, 3, 219–275. [Google Scholar]
- Tekiel, V.; Alba-Soto, C.D.; González Cappa, S.M.; Postan, M.; Sánchez, D.O. Identification of novel vaccine candidates for Chagas’ disease by immunization with sequential fractions of a trypomastigote cDNA expression library. Vaccine 2009, 27, 1323–1332. [Google Scholar] [CrossRef]
- Cerecetto, H.; González, M. Chemotherapy of Chagas' disease: Status and new developments. Curr. Top. Med. Chem. 2002, 2, 1187–213. [Google Scholar] [CrossRef]
- Castro, J.A.; Montalto de Mecca, M.; Bartel, L.C. Toxic side effects of drugs used to treat Chagas’ disease (American Trypanosomiasis). Hum. Exp. Toxicol. 2006, 25, 471–479. [Google Scholar] [CrossRef]
- Jannin, J.; Villa, L. An overview of Chagas disease treatment. Mem. Inst. Oswaldo Cruz 2007, 102, 95–97. [Google Scholar] [CrossRef]
- Marin-Neto, J.A.; Rassi, A.; Morillo, C.A.; Avezum, A.; Connolly, S.J.; Sosa-Estani, S.; Rosas, F.; Yusuf, S. Rationale and design of a randomized placebo-controlled trial assessing the effects of etiologic treatment in Chagas' cardiomyopathy: The BENznidazole Evaluation For Interrupting Trypanosomiasis (BENEFIT). Am. Heart J. 2008, 156, 37–43. [Google Scholar] [CrossRef]
- Priotto, G.; Kasparian, S.; Mutombo, W.; Ngouama, D.; Ghorashian, S.; Arnold, U.; Ghabri, S.; Baudin, E.; Buard, V.; Kazadi-Kyanza, S.; Ilunga, M.; Mutangala, W.; Pohlig, G.; Schmid, C.; Karunakara, U.; Torreele, E.; Kande, V. Nifurtimox-eflornithine combination therapy for second-stage African Trypanosoma brucei gambiense trypanosomiasis: A multicentre, randomised, phase III, non-inferiority trial. Lancet 2009, 374, 56–64. [Google Scholar] [CrossRef]
- Murta, S.M.; Gazzinelli, R.T.; Brener, Z.; Romanha, A.J. Molecular characterization of susceptible and naturally resistant strains of Trypanosoma cruzi to benznidazole and nifurtimox. Mol. Biochem. Parasitol. 1998, 93, 203–214. [Google Scholar] [CrossRef]
- Nagel, R. Genotoxicity studies with two antichagasic drugs. Mutat. Res. 1987, 191, 17–20. [Google Scholar] [CrossRef]
- de Castro, C.R.; Montalto de Mecca, M.; Fanelli, S.L.; de Ferreyra, E.C.; Díaz, E.G.; Castro, J.A. Benznidazole-induced ultrastructural and biochemical alterations in rat esophagus. Toxicology 2003, 191, 189–198. [Google Scholar] [CrossRef]
- Bartel, L.C.; Montalto de Mecca, M.; Fanelli, S.L.; Rodriguez de Castro, C.; Diaz, E.G.; Castro, J.A. Early nifurtimox-induced biochemical and ultrastructural alterations in rat heart. Hum. Exp. Toxicology 2007, 26, 781–788. [Google Scholar] [CrossRef]
- El-Sayed, N.M.; Myler, P.J.; Bartholomeu, D.C.; Nilsson, D.; Aggarwal, G.; Tran, A.N.; Ghedin, E.; Worthey, E.A.; Delcher, A.L.; Blandin, G.; et al. Comparative genomics of trypanosomatid parasitic protozoa. Science 2005, 309, 404–409. [Google Scholar] [CrossRef] [PubMed]
- Burleigh, B.A. Probing Trypanosoma cruzi biology with DNA microarrays. Parasitology 2004, 128, S3–S10. [Google Scholar] [PubMed]
- Heby, O.; Persson, L.; Rentala, M. Targeting the polyamine biosynthetic enzymes: A promising approach to therapy of African sleeping sickness, Chagas' disease, and leishmaniasis. Amino Acids 2007, 33, 359–366. [Google Scholar] [CrossRef]
- Datta, A.K.; Datta, R.; Sen, B. Antiparasitic chemotherapy: Tinkering with the purine salvage pathway. Adv. Exp. Med. Biol. 2008, 625, 116–132. [Google Scholar] [PubMed]
- Niemirowicz, G.; Fernández, D.; Solà, M.; Cazzulo, J.J.; Avilés, F.X.; Gomis-Rüth, F.X. The molecular analysis of Trypanosoma cruzi metallocarboxypeptidase 1 provides insight into fold and substrate specificity. Mol. Microbiol. 2008, 70, 853–866. [Google Scholar] [PubMed]
- Alvarez, V.E.; Kosec, G.; Sant’Anna, C.; Turk, V.; Cazzulo, J.J.; Turk, B. Blocking autophagy to prevent parasite differentiation: A possible new strategy for fighting parasitic infections? Autophagy 2008, 4, 361–363. [Google Scholar] [PubMed]
- Meiering, S.; Inhoff, O.; Mies, J.; Vincek, A.; Garcia, G.; Kramer, B.; Dormeyer, M.; Krauth-Siegel, R.L. Inhibitors of Trypanosoma cruzi trypanothione reductase revealed by virtual screening and parallel synthesis. J. Med. Chem. 2005, 48, 4793–4802. [Google Scholar] [CrossRef]
- Neres, J.; Brewer, M.L.; Ratier, L.; Botti, H.; Buschiazzo, A.; Edwards, P.N.; Mortenson, P.N.; Charlton, M.H.; Alzari, P.M.; Frasch, A.C.; Bryce, R.A.; Douglas, K.T. Discovery of novel inhibitors of Trypanosoma cruzi trans-sialidase from in silico screening. Bioorg. Med. Chem. Lett. 2009, 19, 589–596. [Google Scholar] [CrossRef]
- Freitas, R.F.; Prokopczyk, I.M.; Zottis, A.; Oliva, G.; Andricopulo, A.D.; Trevisan, M.T.; Vilegas, W.; Silva, M.G.; Montanari, C.A. Discovery of novel Trypanosoma cruzi glyceraldehyde-3-phosphate dehydrogenase inhibitors. Bioorg. Med. Chem. 2009, 17, 2476–2482. [Google Scholar] [CrossRef]
- Menezes, I.R.; Lopes, J.C.; Montanari, C.A.; Oliva, G.; Pavão, F.; Castilho, M.S.; Vieira, P.C.; Pupo, M.T. 3D QSAR studies on binding affinities of coumarin natural products for glycosomal GAPDH of Trypanosoma cruzi. J. Comput. Aided Mol. Des. 2003, 17, 277–290. [Google Scholar] [CrossRef]
- Guido, R.V.; Trossini, G.H.; Castilho, M.S.; Oliva, G.; Ferreira, E.I.; Andricopulo, A.D. Structure-activity relationships for a class of selective inhibitors of the major cysteine protease from Trypanosoma cruzi. J. Enzyme Inhib. Med. Chem. 2008, 23, 964–973. [Google Scholar] [CrossRef]
- Trossini, G.H.; Guido, R.V.; Oliva, G.; Ferreira, E.I.; Andricopulo, A.D. Quantitative structure-activity relationships for a series of inhibitors of cruzain from Trypanosoma cruzi: Molecular modeling, CoMFA and CoMSIA studies. J. Mol. Graph. Model 2009, 28, 3–11. [Google Scholar] [CrossRef]
- Saeidnia, S.; Gohari, A.R.; Ito, M.; Kiuchi, F.; Honda, G. Bioactive constituents from Dracocephalum subcapitatum (O. Kuntze) Lipsky. Z. Naturforsch C. 2005, 60, 22–24. [Google Scholar]
- Ferreira, M.E.; Nakayama, H.; de Arias, A.R.; Schinini, A.; de Bilbao, N.V.; Serna, E.; Lagoutte, D.; Soriano-Agatón, F.; Poupon, E.; Hocquemiller, R.; Fournet, A. Effects of canthin-6-one alkaloids from Zanthoxylum chiloperone on Trypanosoma cruzi-infected mice. J. Ethnopharmacol. 2007, 109, 258–263. [Google Scholar] [CrossRef]
- Sülsen, V.P.; Cazorla, S.I.; Frank, F.M.; Redko, F.C.; Anesini, C.A.; Coussio, J.D.; Malchiodi, E.L.; Martino, V.S.; Muschietti, L.V. Trypanocidal and leishmanicidal activities of flavonoids from Argentine medicinal plants. Am. J. Trop. Med. Hyg. 2007, 77, 654–659. [Google Scholar] [PubMed]
- Sülsen, V.P.; Frank, F.M.; Cazorla, S.I.; Anesini, C.A.; Malchiodi, E.L.; Freixa, B.; Vila, R.; Muschietti, L.V.; Martino, V.S. Trypanocidal and leishmanicidal activities of sesquiterpene lactones from Ambrosia tenuifolia Sprengel (Asteraceae). Antimicrob. Agents Chemother. 2008, 52, 2415–2419. [Google Scholar] [CrossRef]
- Aponte, J.C.; Vaisberg, A.J.; Rojas, R.; Sauvain, M.; Lewis, W.H.; Lamas, G.; Gilman, R.H.; Hammond, G.B. A multipronged approach to the study of Peruvian ethnomedicinal plants: A legacy of the ICBG-Peru project. J. Nat. Prod. 2009, 72, 524–526. [Google Scholar] [CrossRef]
- de Castro, S.L.; Pinto, M.C.F.R.; Pinto, A.V. Screening of natural and synthetic drugs against Trypanosoma cruzi. 1 - Establishing a structure/activity relationship. Microbios 1994, 78, 83–90. [Google Scholar] [PubMed]
- Pinto, A.V.; Neves Pinto, C.; Pinto, M.C.F.R.; Santa Rita, R.M.; Pezzella, C.; de Castro, S.L. Trypanocidal activity of synthetic heterocyclic derivatives from active quinones from Tabebuia sp. Arzneim-Forsch 1997, 47, 74–79. [Google Scholar]
- Moura, K.C.G.; Emery, F.S.; Neves-Pinto, C.; Pinto, M.C.F.R.; Dantas, A.P.; Salomão, K.; de Castro, S.L.; Pinto, A.V. Synthesis and trypanocidal activity of naphthoquinones isolated from Tabebuia and heterocyclic derivatives: A review from an interdisciplinary study. J. Braz. Chem. Soc. 2001, 12, 325–338. [Google Scholar]
- Graebin, C.S.; Madeira, M.F.; Yokoyama-Yasunaka, J.K.U.; Miguel, D.C.; Uliana, S.R.B.; Benitez, D.; Cerecetto, H.; González, M.; Gomes da Rosa, R.; Eifler-Lima, V.L. Synthesis and in vitro activity of limonene derivatives against Leishmania and Trypanosoma. Eur. J. Med. Chem. 2010, 45, 1524–1528. [Google Scholar] [CrossRef]
- Álvarez, G.; Gerpe, A.; Benitez, D.; Garibotto, F.; Zacchino, S.; Graebin, C.S.; Gomes da Rosa, R.; Eifler-Lima, V.L.; González, M.; Cerecetto, H. New limonene-hybrid derivatives with anti-T. cruzi activity. Lett. Drug Des. Dis. 2010, 7. in press. [Google Scholar]
- Faúndez, M.; López-Muñoz, R.; Torres, G.; Morello, A.; Ferreira, J.; Kemmerling, U.; Orellana, M.; Maya, J.D. Buthionine sulfoximine has anti-Trypanosoma cruzi activity in a murine model of acute Chagas' disease and enhances the efficacy of nifurtimox. Antimicrob. Agents Chemother. 2008, 52, 1837–1839. [Google Scholar] [CrossRef]
- López-Muñoz, R.; Faúndez, M.; Klein, S.; Escanilla, S.; Torres, G.; Lee-Liu, D.; Ferreira, J.; Kemmerling, U.; Orellana, M.; Morello, A.; Ferreira, A.; Maya, J.D. Trypanosoma cruzi: In vitro effect of aspirin with nifurtimox and benznidazole. Exp. Parasitol. 2010, 124, 167–171. [Google Scholar] [CrossRef]
- Soeiro, M.N.C.; Dantas, A.P.; Daliry, A.; da Silva, C.F.; Batista, D.G.J.; de Souza, E.M.; Oliveira, G.M.; Salomão, K.; Batista, M.M.; Pacheco, M.G.O.; Bernardino da Silva, P.; Santa-Rita, R.M.; Menna Barreto, R.F.S.; Boykin, D.W.; de Castro, S.L. Experimental chemotherapy for Chagas disease: 15 years of research contributions from in vivo and in vitro studies. Mem. Inst. Oswaldo Cruz 2009, 104, 301–310. [Google Scholar] [CrossRef]
- Menna Barreto, R.F.; Salomão, K.; Dantas, A.P.; Santa-Rita, R.M.; Soares, M.J.; Barbosa, H.S.; de Castro, S.L. Different cell death pathways induced by drugs in Trypanosoma cruzi: An ultrastructural study. Micron 2009, 40, 157–168. [Google Scholar] [CrossRef]
- Urbina, J.A.; Lira, R.; Visbal, G.; Bartrolí, J. In vitro antiproliferative effects and mechanism of action of the new triazole derivative UR-9825 against the protozoan parasite Trypanosoma (Schizotrypanum)cruzi. Antimicrob. Agents Chemother. 2000, 44, 2498–2502. [Google Scholar] [CrossRef]
- Urbina, J.A. Ergosterol biosynthesis and drug development for Chagas disease. Mem. Inst. Oswaldo Cruz 2009, 104, 311–318. [Google Scholar] [CrossRef]
- Courchesne, W.E. Characterization of a novel, broad-based fungicidal activity for the antiarrhythmic drug amiodarone. J. Pharmacol. Exp. Ther. 2002, 300, 195–199. [Google Scholar] [CrossRef]
- Benaim, G.; Sanders, J.M.; Garcia-Marchan, Y.; Colina, C.; Lira, R.; Caldera, A.R.; Payares, G.; Sanoja, C.; Burgos, J.M.; Leon-Rossell, A.; Concepcion, J.L.; Schijman, A.G.; Levin, M.; Oldfield, E.; Urbina, J.A. Amiodarone has intrinsic anti-Trypanosoma cruzi activity and acts synergistically with Posaconazole. J. Med. Chem. 2006, 49, 892–899. [Google Scholar] [CrossRef]
- Available online: http://apps.who.int/tdr/svc/grants or http://www.dndi.org/ (accessed on 24 March 2010).
- Elhalem, E.; Bailey, B.N.; Docampo, R.; Ujváry, I.; Szajnman, S.H.; Rodriguez, J.B. Design, synthesis, and biological evaluation of aryloxyethyl thiocyanate derivatives against Trypanosoma cruzi. J. Med. Chem. 2002, 45, 3984–3999. [Google Scholar] [CrossRef]
- Szajnman, S.H.; Yan, W.; Bailey, B.N.; Docampo, R.; Elhalem, E.; Rodriguez, J.B. Design and synthesis of aryloxyethyl thiocyanate derivatives as potent inhibitors of Trypanosoma cruzi proliferation. J. Med. Chem. 2000, 43, 1826–1840. [Google Scholar] [CrossRef]
- Urbina, J.A.; Concepcion, J.L.; Montalvetti, A.; Rodriguez, J.B.; Docampo, R. Mechanism of action of 4-phenoxyphenoxyethyl thiocyanate (WC-9) against Trypanosoma cruzi, the causative agent of Chagas' disease. Antimicrob. Agents Chemother. 2003, 47, 2047–2050. [Google Scholar] [CrossRef]
- Liñares, G.G.; Gismondi, S.; Codesido, N.O.; Moreno, S.N.; Docampo, R.; Rodriguez, J.B. Fluorine-containing aryloxyethyl thiocyanate derivatives are potent inhibitors of Trypanosomacruzi and Toxoplasma gondii proliferation. Bioorg. Med. Chem. Lett. 2007, 17, 5068–5071. [Google Scholar] [CrossRef]
- Szajnman, S.H.; Ravaschino, E.L.; Docampo, R.; Rodriguez, J.B. Synthesis and biological evaluation of 1-amino-1,1-bisphosphonates derived from fatty acids against Trypanosoma cruzi targeting farnesyl pyrophosphate synthase. Bioorg. Med. Chem. Lett. 2005, 15, 4685–4690. [Google Scholar] [CrossRef]
- Szajnman, S.H.; García Liñares, G.E.; Li, Z.H.; Jiang, C.; Galizzi, M.; Bontempi, E.J.; Ferella, M.; Moreno, S.N.; Docampo, R.; Rodriguez, J.B. Synthesis and biological evaluation of 2-alkylaminoethyl-1,1-bisphosphonic acids against Trypanosoma cruzi and Toxoplasma gondii targeting farnesyl diphosphate synthase. Bioorg. Med. Chem. 2008, 16, 3283–3290. [Google Scholar] [CrossRef]
- Lima Leite, A.C.; Souza de Lima, R.; de M. Moreira, D.R.; de O. Cardoso, M.V.; Gouveia de Brito, A.C.; Farias dos Santos, L.M.; Zaldini Hernandes, M.; Costa Kiperstok, A.; Santana de Lima, R.; Soares, M.B.P. Synthesis, docking, and in vitro activity of thiosemicarbazones, aminoacyl-thiosemicarbazides and acyl-thiazolidones against Trypanosoma cruzi. Bioorg. Med. Chem. 2006, 14, 3749–3757. [Google Scholar] [CrossRef]
- Leite, A.C.L.; Moreira, D.R.M.; Cardoso, M.V.O.; Hernandes, M.Z.; Pereira, V.R.A.; Silva, R.O.; Kiperstok, A.C.; Lima, M.S.; Soares, M.B.P. Synthesis, cruzain docking, and in vitro studies of aryl-4-oxothiazolylhydrazones against Trypanosoma cruzi. Chem. Med. Chem. 2007, 2, 1339–1345. [Google Scholar]
- Trossini, G.H.; Malvezzi, A.; T-do Amaral, A.; Rangel-Yagui, C.O.; Izidoro, M.A.; Cezari, M.H.; Juliano, L.; Chin, C.M.; Menezes, C.M.; Ferreira, E.I. Cruzain inhibition by hydroxymethylnitrofurazone and nitrofurazone: Investigation of a new target in Trypanosoma cruzi. J. Enzyme Inhib. Med. Chem. 2010, 25, 62–67. [Google Scholar] [CrossRef]
- dos Santos Filho, J.M.; Lima Leite, A.C.; de Oliveira, B.G.; Magalhães Moreira, D.G.; Lima, M.S.; Botelho Pereira Soares, M.; Leite, L.F.C.C. Design, synthesis and cruzain docking of 3-(4-substituted-aryl)-1,2,4-oxadiazole-N-acylhydrazones as anti-Trypanosoma cruzi agents. Bioorg. Med. Chem. 2009, 17, 6682–6691. [Google Scholar] [CrossRef]
- Romeiro, N.C.; Aguirre, G.; Hernández, P.; González, M.; Cerecetto, H.; Aldana, I.; Pérez-Silanes, S.; Monge, A.; Barreiro, E.J.; Lima, L.M. Synthesis, trypanocidal activity and docking studies of novel quinoxaline-N-acylhydrazones, designed as cruzain inhibitors candidates. Bioorg. Med. Chem. 2009, 17, 641–652. [Google Scholar] [CrossRef]
- McKerrow, J.H.; Doyle, P.S.; Engel, J.C.; Podust, L.M.; Robertson, S.A.; Ferreira, R.; Saxton, T.; Arkin, M.; Kerr, I.D.; Brinen, L.S.; Craik, C.S. Two approaches to discovering and developing new drugs for Chagas disease. Mem. Inst. Oswaldo Cruz 2009, 104, 263–269. [Google Scholar] [CrossRef]
- Roush, W.R.; Gwaltney, S.L.; Cheng, J.; Scheidt, K.A.; McKerrow, J.H.; Hansell, E. Vinyl sulfonate esters and vinyl sulfonamides: Potent, irreversible inhibitors of cysteine proteases. J. Am. Chem. Soc. 1998, 120, 10994–10995. [Google Scholar] [CrossRef]
- Roush, W.R.; Cheng, J.; Knapp-Reed, B.; Alvarez-Hernandez, A.; McKerrow, J.H.; Hansell, E.; Engel, J.C. Potent second generation vinyl sulfonamide inhibitors of the trypanosomal cysteine protease cruzain. Bioorg. Med. Chem. Lett. 2001, 11, 2759–2762. [Google Scholar] [CrossRef]
- Meléndez-López, S.G.; Herdman, S.; Hirata, K.; Choi, M.H.; Choe, Y.; Craik, C.; Caffrey, C.R.; Hansel, E.; Chávez-Munguía, B.; Chen, Y.T.; Roush, W.R.; McKerrow, J.; Eckmann, L.; Guo, J.; Stanley, S.L.; Reed, S.L. Use of recombinant Entamoeba histolytica cysteine proteinase to identify a potent inhibitor of amebic invasion in a human colonic model. Eukaryot. Cell 2007, 6, 1130–1136. [Google Scholar] [CrossRef]
- Fujii, N.; Mallari, J.P.; Hansell, E.J.; Mackey, Z.; Doyle, P.; Zhou, Y.M.; Gut, J.; Rosenthal, P.J.; McKerrow, J.H.; Guy, R.K. Discovery of potent thiosemicarbazone inhibitors of rhodesain and cruzain. Bioorg. Med. Chem. Lett. 2005, 15, 121–123. [Google Scholar] [CrossRef]
- Brak, K.; Doyle, P.S.; McKerrow, J.H.; Ellman, J.A. Identification of a new class of nonpeptidic inhibitors of cruzain. J Am Chem Soc. 2008, 130, 6404–6410. [Google Scholar] [CrossRef]
- Podust, L.M.; von Kries, J.P.; Nasser Eddine, A.; Kim, Y.; Yermalitskaya, L.V.; Kuehne, R.; Ouellet, H.; Warrier, T.; Alteköster, M.; Lee, J.S.; Rademann, J.; Oschkinat, H.; Kaufmann, S.H.E.; Waterman, M.R. Small molecule scaffolds for CYP51 inhibitors identified by high-throughput screening and defined by X-ray crystallography. Antimicrob. Agents Chemother. 2007, 51, 3915–3923. [Google Scholar] [CrossRef]
- Chen, C.K.; Doyle, P.S.; Yermalitskaya, L.V.; Mackey, Z.B.; Ang, K.K.H.; McKerrow, J.H.; Podust, L.M. Trypanosoma cruzi CYP51 inhibitor derived from a Mycobacterium tuberculosis screen Hit. PLoS Negl. Trop. Dis. 2009, 3, e372. [Google Scholar] [CrossRef]
- Hucke, O.; Gelb, M.H.; Verlinde, C.L.M.J.; Buckner, F.S. The protein farnesyltransferase inhibitor Tipifarnib as a new lead for the development of drugs against Chagas disease. J. Med. Chem. 2005, 48, 5415–5418. [Google Scholar] [CrossRef]
- Kraus, J.M.; Verlinde, C.L.; Karimi, M.; Lepesheva, G.I.; Gelb, M.H.; Buckner, F.S. Rational modification of a candidate cancer drug for use against Chagas disease. J. Med. Chem. 2009, 52, 1639–1647, Erratum in: J. Med. Chem. 2009, 52, 4549. [Google Scholar] [CrossRef] [PubMed]
- Chennamaneni, N.K.; Arif, J.; Buckner, F.S.; Gelb, M.H. Isoquinoline-based analogs of the cancer drug clinical candidate tipifarnib as anti-Trypanosoma cruzi agents. Bioorg. Med. Chem. Lett. 2009, 19, 6582–6584. [Google Scholar] [CrossRef]
- Suryadevara, P.K.; Olepu, S.; Lockman, J.W.; Ohkanda, J.; Karimi, M.; Verlinde, C.L.; Kraus, J.M.; Schoepe, J.; Van Voorhis, W.C.; Hamilton, A.D.; Buckner, F.S.; Gelb, M.H. Structurally simple inhibitors of lanosterol 14alpha-demethylase are efficacious in a rodent model of acute Chagas disease. J. Med. Chem. 2009, 52, 3703–3715. [Google Scholar] [CrossRef]
- Krauth-Siegel, R.L.; Comini, M.A. Redox control in trypanosomatids, parasitic protozoa with trypanothione-based thiol metabolism. Biochim. Biophys. Acta 2008, 1780, 1236–1248. [Google Scholar] [CrossRef] [PubMed]
- Stump, B.; Eberle, C.; Kaiser, M.; Brun, R.; Krauth-Siegel, R.L.; Diederich, F. Diaryl sulfide-based inhibitors of trypanothione reductase: Inhibition potency, revised binding mode and antiprotozoal activities. Org. Biomol. Chem. 2008, 6, 3935–3947. [Google Scholar] [CrossRef]
- Stump, B.; Eberle, C.; Schweizer, W.B.; Marcel Kaiser, M.; Brun, R.; Krauth-Siegel, R.L.; Lentz, D.; Diederich, F. Pentafluorosulfanyl as a novel building block for enzyme inhibitors: Trypanothione reductase inhibition and antiprotozoal activities of diarylamines. Chem. Bio. Chem. 2009, 10, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Eberle, C.; Burkhard, J.A.; Stump, B.; Kaiser, M.; Brun, R.; Krauth-Siegel, R.S.; Diederich, F. Synthesis, inhibition potency, binding mode, and antiprotozoal activities of fluorescent inhibitors of trypanothione reductase based on mepacrine-conjugated diaryl sulfide scaffolds. Chem. Med. Chem. 2009, 4, 2034–2044. [Google Scholar]
- Martyn, D.C.; Jones, D.C.; Fairlamb, A.H.; Clardy, J. High-throughput screening affords novel and selective trypanothione reductase inhibitors with anti-trypanosomal activity. Bioorg. Med. Chem. Lett. 2007, 17, 1280–1283. [Google Scholar] [CrossRef]
- Holloway, G.A.; Baell, J.B.; Fairlamb, A.H.; Novello, P.M.; Parisot, J.P.; Richardson, J.; Watson, K.G.; Street, I.P. Discovery of 2-iminobenzimidazoles as a new class of trypanothione reductase inhibitor by high-throughput screening. Bioorg. Med. Chem. Lett. 2007, 17, 1422–1427. [Google Scholar] [CrossRef]
- Perez-Pineiro, R.; Burgos, A.; Jones, D.C.; Andrew, L.C.; Rodriguez, H.; Suarez, M.; Fairlamb, A.H.; Wishart, D.S. Development of a novel virtual screening cascade protocol to identify potential trypanothione reductase inhibitors. J. Med. Chem. 2009, 52, 1670–1680. [Google Scholar] [CrossRef]
- Jones, D.C.; Ariza, A.; Chow, W.H.A.; Oza, S.L.; Fairlamb, A.H. Comparative structural, kinetic and inhibitor studies of Trypanosoma brucei trypanothione reductase with T. cruzi. Mol. Biochem. Parasitol. 2010, 169, 12–19. [Google Scholar] [CrossRef]
- Khabnadideh, S.; Pez, D.; Musso, A.; Brun, R.; Ruiz Pérez, L.M.; González-Pacanowska, D.; Gilbert, I.H. Design, synthesis and evaluation of 2,4-diaminoquinazolines as inhibitors of trypanosomal and leishmanial dihydrofolate reductase. Bioorg. Med. Chem. 2005, 13, 2637–2649. [Google Scholar] [CrossRef]
- Orenes Lorente, S.; Jimenez Jimenez, C.; Gros, l.; Yardley, V.; de Luca-Fradley, k.; Croft, S.L.; Urbina, J.A.; Ruiz-Perez, L.M.; Gonzalez Pacanowska, D.; Gilbert, I.H. Preparation of transition-state analogues of sterol 24-methyl transferase as potential anti-parasitics. Bioorg. Med. Chem. 2005, 13, 435–5453. [Google Scholar] [CrossRef]
- Braga, M.V.; Magaraci, F.; Lorente, S.O.; Gilbert, I.; de Souza, W. Effects of inhibitors of Delta24(25)-sterol methyl transferase on the ultrastructure of epimastigotes of Trypanosoma cruzi. Microsc. Microanal. 2005, 11, 506–515. [Google Scholar] [CrossRef]
- Gros, L.; Orenes Lorente, S.; Jimenez Jimenez, C.; Yardley, V.; Rattray, L.; Wharton, H.; Little, S.; Croft, S.L.; Ruiz-Perez, L.M.; Gonzalez-Pacanowska, D.; Gilbert, I.H. Evaluation of Azasterols as Anti-Parasitics. J. Med. Chem. 2006, 49, 6094–6103. [Google Scholar] [CrossRef]
- Gigante, F.; Kaiser, M.; Brun, R.; Gilbert, I.H. SAR studies on azasterols as potential anti-trypanosomal and anti-leishmanial agents. Bioorg. Med. Chem. 2009, 17, 5950–5961. [Google Scholar] [CrossRef]
- Orenes Lorente, S.; Gómez, R.; Jiménez, C.; Cammerer, S.; Yardley, V.; de Luca-Fradley, K.; Croft, S.L.; Ruiz Perez, L.M.; Urbina, J.; Gonzalez Pacanowska, D.; Gilbert, I.H. Biphenylquinuclidines as inhibitors of squalene synthase and growth of parasitic protozoa. Bioorg. Med. Chem. 2005, 13, 3519–3529. [Google Scholar] [CrossRef]
- Sealey-Cardona, M.; Cammerer, S.; Jones, S.; Ruiz-Pérez, L.M.; Brun, R.; Gilbert, I.H.; Urbina, J.A.; González-Pacanowska, D. Kinetic characterization of squalene synthase from Trypanosoma cruzi: Selective inhibition by quinuclidine derivatives. Antimicrob. Agents Chemother. 2007, 51, 2123–2129. [Google Scholar] [CrossRef]
- Cammerer, S.B.; Jimenez, C.; Jones, S.; Gros, L.; Orenes Lorente, S.; Rodrigues, C.; Rodrigues, J.C.F.; Caldera, A.; Ruiz Perez, L.M.; de Souza, W.; Kaiser, M.; Brun, R.; Urbina, J.A.; Gonzalez Pacanowska, D.; Gilbert, I.H. Quinuclidine derivatives as potential antiparasitics. Antimicrob. Agents Chemother. 2007, 51, 4049–4061. [Google Scholar] [CrossRef]
- Arán, V.J.; Ochoa, C.; Boiani, L.; Buccino, P.; Cerecetto, H.; Gerpe, A.; González, M.; Montero, D.; Nogal, J.J.; Gómez-Barrio, A.; Azqueta, A.; López de Ceráin, A.; Piro, O.E.; Castellano, E.E. Synthesis and biological properties of new 5-nitroindazole derivatives. Bioorg. Med. Chem. 2005, 13, 3197–3207. [Google Scholar] [CrossRef]
- Gerpe, A.; Odreman-Nuñez, I.; Draper, P.; Boiani, L.; Urbina, J.A.; González, M.; Cerecetto, H. Heteroallyl-containing 5-nitrofuranes as new anti-Trypanosoma cruzi agents with a dual mechanism of action. Bioorg. Med. Chem. 2008, 16, 569–577. [Google Scholar] [CrossRef]
- Rodríguez, J.; Arán, V.J.; Boiani, L.; Olea-Azar, C.; Lavaggi, M.L.; González, M.; Cerecetto, H.; Maya, J.D.; Carrasco-Pozo, C.; Cosoy, H.S. New potent 5-nitroindazole derivatives as inhibitors of Trypanosoma cruzi growth: Synthesis, biological evaluation, and mechanism of action studies. Bioorg. Med. Chem. 2009, 17, 8186–8196. [Google Scholar] [CrossRef]
- Gerpe, A.; Alvarez, G.; Benítez, D.; Boiani, L.; Quiroga, M.; Hernández, P.; Sortino, M.; Zacchino, S.; González, M.; Cerecetto , H. 5-Nitrofuranes and 5-nitrothiophenes with anti-Trypanosoma cruzi activity and ability to accumulate squalene. Bioorg. Med. Chem. 2009, 17, 7500–7509. [Google Scholar] [CrossRef]
- Aguirre, G.; Cerecetto, H.; Di Maio, R.; González, M.; Alfaro, M.E.; Jaso, A.; Zarranz, B.; Ortega, M.A.; Aldana, I.; Monge-Vega, A. Quinoxaline N,N'-dioxide derivatives and related compounds as growth inhibitors of Trypanosoma cruzi.Structure-activity relationships. Bioorg. Med. Chem. Lett. 2004, 14, 3835–3839. [Google Scholar] [CrossRef]
- Gerpe, A.; Aguirre, G.; Boiani, L.; Cerecetto, H.; González, M.; Olea-Azar, C.; Rigol, C.; Maya, J.D.; Morello, A.; Piro, O.E.; et al. Indazole N-oxide derivatives as antiprotozoal agents: Synthesis, biological evaluation and mechanism of action studies. Bioorg. Med. Chem. 2006, 14, 3467–3480. [Google Scholar] [CrossRef]
- Boiani, L.; Aguirre, G.; González, M.; Cerecetto, H.; Chidichimo, A.; Cazzulo, J.J.; Bertinaria, M.; Guglielmo, S. Furoxan-, alkylnitrate-derivatives and related compounds as anti-trypanosomatid agents: Mechanism of action studies. Bioorg. Med. Chem. 2008, 16, 7900–7907. [Google Scholar] [CrossRef]
- Ancizu, S.; Moreno, E.; Torres, E.; Burguete, A.; Pérez-Silanes, S.; Benítez, D.; Villar, R.; Solano, B.; Marín, A.; Aldana, I.; Cerecetto, H.; González, M.; Monge, A. Heterocyclic-2-carboxylic acid (3-cyano-1,4-di-N-oxidequinoxalin-2-yl) amide derivatives as hits for the development of neglected disease drugs. Molecules 2009, 14, 2256–2272. [Google Scholar] [CrossRef]
- Boiani, M.; Boiani, L.; Merlino, A.; Hernández, P.; Chidichimo, A.; Cazzulo, J.J.; Cerecetto, H.; González, M. Second generation of 2H-benzimidazole 1,3-dioxide derivatives as anti-trypanosomatid agents: Synthesis, biological evaluation, and mode of action studies. Eur. J. Med. Chem. 2009, 44, 4426–4433. [Google Scholar] [CrossRef]
- Castro, D.; Boiani, L.; Benitez, D.; Hernández, P.; Merlino, A.; Gil, C.; Olea-Azar, C.; González, M.; Cerecetto, H.; Porcal, W. Anti-trypanosomatid benzofuroxans and deoxygenated analogues: Synthesis using polymer-supported triphenylphosphine, biological evaluation and mechanism of action studies. Eur. J. Med. Chem. 2009, 44, 5055–5065. [Google Scholar] [CrossRef]
- Lavaggi, M.L.; Aguirre, G.; Boiani, L.; Orelli, L.; García, B.; Cerecetto, H.;. González. Pyrimido[1,2-a]quinoxaline 6-oxide and phenazine 5,10-dioxide derivatives and related compounds as growth inhibitors of Trypanosoma cruzi. Eur. J. Med. Chem. 2008, 43, 1737–1741. [Google Scholar] [CrossRef]
- Caterina, M.C.; Perillo, I.A.; Boiani, L.; Pezaroglo, H.; Cerecetto, H.; González, M.; Salerno, A. Imidazolidines as new anti-Trypanosoma cruzi agents: Biological evaluation and structure-activity relationships. Bioorg. Med. Chem. 2008, 16, 2226–34. [Google Scholar] [CrossRef]
- Bollini, M.; Casal, J.J.; Alvarez, D.E.; Boiani, L.; González, M.; Cerecetto, H.; Bruno, A.M. New potent imidazoisoquinolinone derivatives as anti-Trypanosoma cruzi agents: Biological evaluation and structure-activity relationships. Bioorg. Med. Chem. 2009, 17, 1437–1444. [Google Scholar] [CrossRef]
- Navarro, M.; Cisneros-Fajardo, E.J.; Lehmann, T.; Sánchez-Delgado, R.A.; Atencio, R.; Silva, P.; Lira, R.; Urbina, J.A. Toward a novel metal-based chemotherapy against tropical diseases. 6. Synthesis and characterization of new copper(II) and gold(I) clotrimazole and ketoconazole complexes and evaluation of their activity against Trypanosoma cruzi. Inorg. Chem. 2001, 40, 6879–6884. [Google Scholar] [CrossRef]
- Sánchez-Delgado, R.A.; Lazardi, K.; Rincón, L.; Urbina, J.A. Toward a novel metal-based chemotherapy against tropical diseases. 1. Enhancement of the efficacy of clotrimazole against Trypanosoma cruzi by complexation to ruthenium in RuCl2(clotrimazole)2. J. Med. Chem. 1993, 36, 2041–2043. [Google Scholar] [CrossRef]
- Urquiola, C.; Vieites, M.; Aguirre, G.; Marín, A.; Solano, B.; Arrambide, G.; Noblía, P.; Lavaggi, M.L.; Torre, M.H.; González, M.; Monge, A.; Gambino, D.; Cerecetto, H. Improving anti-trypanosomal activity of 3-aminoquinoxaline-2-carbonitrile N1,N4-dioxide derivatives by complexation with vanadium . Bioorg. Med. Chem. 2006, 14, 5503–5509. [Google Scholar] [CrossRef]
- Vieites, M.; Otero, L.; Santos, D.; Toloza, J.; Figueroa, R.; Norambuena, E.; Olea-Azar, C.; Aguirre, G.; Cerecetto, H.; González, M.; Morello, A.; Maya, J.D.; Garat, B.; Gambino, D. Platinum(II) metal complexes as potential anti-Trypanosoma cruzi agents. J. Inorg. Biochem. 2008, 102, 1033–43. [Google Scholar] [CrossRef]
- Vieites, M.; Smircich, P.; Parajón-Costa, B.; Rodríguez, J.; Galaz, V.; Olea-Azar, C.; Otero, L.; Aguirre, G.; Cerecetto, H.; González, M.; Gómez-Barrio, A.; Garat, B.; Gambino, D. Potent in vitro anti-Trypanosoma cruzi activity of pyridine-2-thiol N-oxide metal complexes having an inhibitory effect on parasite-specific fumarate reductase. J. Biol. Inorg. Chem. 2008, 13, 723–735. [Google Scholar] [CrossRef]
- Vieites, M.; Otero, L.; Santos, D.; Olea-Azar, C.; Norambuena, E.; Aguirre, G.; Cerecetto, H.; González, M.; Kemmerling, U.; Morello, A.; Maya, D.J.; Gambino, D. Platinum-based complexes of bioactive 3-(5-nitrofuryl)acroleine thiosemicarbazones showing anti-Trypanosoma cruzi activity. J. Inorg. Biochem. 2009, 103, 411–418. [Google Scholar] [CrossRef]
- Pagano, M.; Demoro, B.; Toloza, J.; Boiani, L.; González, M.; Cerecetto, H.; Olea-Azar, C.; Norambuena, E.; Gambino, D.; Otero, L. Effect of ruthenium complexation on trypanocidal activity of 5-nitrofuryl containing thiosemicarbazones. Eur. J. Med. Chem. 2009, 44, 4937–4943. [Google Scholar] [CrossRef]
- Donnici, C.L.; Araújo, M.H.; Oliveira, H.S.; Magalhães Moreira, D.R.; Alves Pereira, V.R.; de Assis Souza, M.; Accioly Brelaz de Castro, M.C.; Lima Leite, A.C. Ruthenium complexes endowed with potent anti-Trypanosoma cruzi activity: Synthesis, biological characterization and structure–activity relationships. Bioorg. Med. Chem. 2009, 17, 5038–5043. [Google Scholar] [CrossRef]
- Pérez-Rebolledo, A.; Teixeira, L.R.; Batista, A.A.; Mangrich, A.S.; Aguirre, G.; Cerecetto, H.; González, M.; Hernández, P.; Ferreira, A.M.; Speziali, N.L.; Beraldo, H. 4-Nitroacetophenone-derived thiosemicarbazones and their copper(II) complexes with significant in vitro anti-trypanosomal activity. Eur. J. Med. Chem. 2008, 43, 939–948. [Google Scholar] [CrossRef]
- Otero, L.; Aguirre, G.; Boiani, L.; Denicola, A.; Rigol, C.; Olea-Azar, C.; Maya, J.D.; Morello, A.; González, M.; Gambino, D.; Cerecetto, H. Nitrofurylsemicarbazone rhenium and ruthenium complexes as anti-trypanosomal agents. Eur. J. Med. Chem. 2006, 41, 1231–1239. [Google Scholar] [CrossRef]
- Otero, L.; Vieites, M.; Boiani, L.; Denicola, A.; Rigol, C.; Opazo, L.; Olea-Azar, C.; Maya, J.D.; Morello, A.; Krauth-Siegel, R.L.; et al. Novel antitrypanosomal agents based on palladium nitrofurylthiosemicarbazone complexes: DNA and redox metabolism as potential therapeutic targets. J. Med. Chem. 2006, 49, 3322–3331. [Google Scholar] [CrossRef]
- Otero, L.; Smircich, P.; Vieites, M.; Ciganda, M.; Severino, P.C.; Terenzi, H.; Cerecetto, H.; Gambino, D.; Garat, B. DNA conformational changes and cleavage by ruthenium(II) nitrofurylsemicarbazone complexes. J. Inorg. Biochem. 2007, 101, 74–79. [Google Scholar] [CrossRef]
- Pereira, D.G.; De Castro, S.L.; Duran, N. Activity of N,N-dimethyl-2-propen-1-amine derivatives in mice experimentally infected with Trypanosoma cruzi. Acta Trop. 1998, 69, 205–211. [Google Scholar] [CrossRef]
- Oliveira, D.A.; Pereira, D.G.; Fernandes, A.M.A.P.; De Castro, S.L.; Souza Brito, A.R.M.; De Souza, A.O.; Duran, N. Trypanocidal activity of 2-propen-1-amine derivatives on trypomastigotes culture and in animal model. Parasitol. Res. 2005, 95, 161–166. [Google Scholar] [CrossRef]
- De Souza, E.M.; Melo, G.; Boykin, D.W.; Wilson, W.D.; Hu, Q.; Soeiro, M.N.C. Tripanocidal activity of the phenyl-substituted analogue of furamidine DB569 against Trypanosoma cruzi infection in vivo. J. Antimicrob. Chemoth. 2006, 58, 610–614. [Google Scholar] [CrossRef]
- De Souza, E.M.; Oliveira, G.M.; Soeiro, M.N.C. Electrocardiographic findings in acutely and chronically Trypanosoma cruzi-infected mice treated by a phenyl-substituted analogue of furamidine DB569. Drug Targets Insights 2007, 2, 61–69. [Google Scholar]
- Silva, C.F.; Batista, M.M.; Batista, D.G.; de Souza, E.M.; Silva, P.B.; Oliveira, G.M.; Batista, M.M.; Shareef, A.R.; Boykin, D.W.; Soeiro, M.N.C. In vitro and in vivo studies of the trypanocidal activity of a diarylthiophene diamidine against Trypanosoma cruzi. Antimicrob. Agents Chemother. 2008, 52, 3307–3314. [Google Scholar] [CrossRef]
- Silva, J.J.N.; Osakabe, A.L.; Pavanelli, W.R.; Silva, J.S.; Franco, D.W. In vitro and in vivo antiproliferative and trypanocidal activities of ruthenium NO donors. British J. Pharmacol. 2007, 152, 112–121. [Google Scholar] [CrossRef]
- Silva, J.J.N.; Pavanelli, W.R.; Salazar Gutierrez, F.R.; Chagas Alves Lima, F.; Ferreira da Silva, A.B.; Silva, J.S.; Franco, W.R. Complexation of the anti-Trypanosoma cruzi drug Benznidazole improves solubility and efficacy. J. Med. Chem. 2008, 51, 4104–4114. [Google Scholar] [CrossRef]
- Shanmugasundarama, M.; Garcia-Martinez, I.; Li, Q.; Estrada, A.; Martinez, N.E.; Martinez, L.E. Microwave-assisted solidphase Dötz benzannulation reactions: A facile synthesis of 2,3-disubstituted-1,4 naphthoquinones. Tetrahedron Lett. 2005, 46, 7545–7548. [Google Scholar] [CrossRef]
- Ramos, E.I.; Garza, K.M.; Krauth-Siegel, R.L.; Bader, J.; Martinez, L.E.; Maldonado, R.A. 2,3-Diphenyl-1,4-naphthoquinone: A potential chemotherapeutic agent against Trypanosoma cruzi. J. Parasitol. 2009, 95, 461–466. [Google Scholar] [CrossRef]
- Engel, J.C.; Doyle, P.S.; Hsieh, I.; McKerrow, J.H. Cysteine protease inhibitors cure an experimental Trypanosoma cruzi infection. J. Exp. Med. 1998, 188, 725–734. [Google Scholar] [CrossRef]
- Barr, S.C.; Warner, K.L.; Kornreic, B.G.; Piscitelli, J.; Wolfe, A.; Benet, L.; McKerrow, J.H. A cysteine protease inhibitor protects dogs from cardiac damage during infection by Trypanosoma cruzi. Antimicrob. Agents Chemother. 2005, 49, 5160–5161. [Google Scholar] [CrossRef]
- Doyle, P.S.; Zhou, Y.M.; Engel, J.C.; McKerrow, J.H. Cysteine protease inhibitor cures Chagas disease in an immunodeficient-mouse model of infection. Antimicrob. Agents Chemother. 2007, 51, 3932–3939. [Google Scholar] [CrossRef]
- Abdulla, M.H.; Lim, K.C. Sajid; McKerrow, J.H.; Caffrey, C.R. Schistosomiasis Mansoni: Novel chemotherapy using a cysteine protease inhibitor. PLoS Medicine 2007, 4, e14. [Google Scholar] [CrossRef]
- Urbina, J.A.; Docampo, R. Specific chemotherapy of Chagas disease: Controversies and advances. Trends Parasitol. 2003, 19, 495–501. [Google Scholar] [CrossRef]
- Urbina, J.A.; Payares, G.; Molina, J.; Sanoja, C.; Liendo, A.; Lazardi, K.; Piras, M.M.; Piras, R.; Perez, N.; Wincker, P.; Ryley, J.F. Cure of short- and long-term experimental Chagas’ disease using D0870. Science 1996, 273, 969–971. [Google Scholar] [CrossRef] [PubMed]
- Urbina, J.A.; Payares, G.; Sanoja, C.; Lira, R.; Romanha, A.J. In vitro and in vivo activities of ravuconazole on Trypanosoma cruzi, the causative agent of Chagas disease. Intern. J. Antimicrob. Agents 2003, 21, 27–38. [Google Scholar] [CrossRef]
- Urbina, J.A.; Payares, G.; Sanoja, C.; Molina, J.; Lira, R.; Brener, Z.; Romanha, A.J. Parasitological cure of acute and chronic experimental Chagas disease using the long-acting experimental triazole TAK-187. Activity against drug resistant Trypanosoma cruzi strains. Int. J. Antimicrob. Agents 2003, 21, 39–48. [Google Scholar] [CrossRef]
- Corrales, M.; Cardozo, R.; Segura, M.A.; Urbina, J.A.; Basombrio, M.A. Comparative efficacies of TAK-187, a long-lasting ergosterol biosynthesis inhibitor, and benznidazole in preventing cardiac damage in a murine model of Chagas’ disease. Antimicrob. Agents Chemother. 2005, 49, 1556–1560. [Google Scholar] [CrossRef]
- Silva, D.T.; de Meirelles, M.N.; Almeida, D.; Urbina, J.A.; Pereira, M.C. Cytoskeleton reassembly in cardiomyocytes infected by Trypanosoma cruzi is triggered by treatment with ergosterol biosynthesis inhibitors. Int. J. Antimicrob. Agents 2006, 27, 530–537. [Google Scholar] [CrossRef]
- Ferraz, M.L.; Gazzinelli, R.T.; Alves, R.O.; Urbina, J.A.; Romanha, A.J. The anti-Trypanosoma cruzi activity of posaconazole in a murine model of acute Chagas’ disease is less dependent on gamma interferon than that of benznidazole. Antimicrob. Agents Chemother. 2007, 51, 1359–1364. [Google Scholar] [CrossRef]
- Ferraz, M.L.; Gazzinelli, R.T.; Alves, R.O.; Urbina, J.A.; Romanha, A.J. Absence of CD4+ T lymphocytes, CD8+ T lymphocytes, or B lymphocytes has different effects on the efficacy of posaconazole and benznidazole in treatment of experimental acute Trypanosoma cruzi infection. Antimicrob. Agents Chemother. 2009, 53, 174–179. [Google Scholar] [CrossRef]
- Rachel, C.; Sudhakar, P.; Mark, L.; Josephine, L.; Vijay, B. Pharmacokinetics, safety, and tolerability of oral posaconazole administered in single and multiple doses in healthy adults. Antimicrob. Agents Chemother. 2003, 47, 2788–2795. [Google Scholar] [CrossRef]
- Pinazo, M.J.; Espinosa, G.; Gállego, M.; López-Chejade, P.; Urbina, J.A.; Gascon, J. Treatment with posaconazole of a patient with systemic lupus erythematosus and Chagas disease. Am. J. Trop. Med. Hyg. 2010, in press. [Google Scholar]
- Urbina, J.A.; Concepcion, J.L.; Caldera, A.; Payares, G.; Sanoja, C.; Otomo, T.; Hiyoshi, H. In vitro and in vivo activities of E5700 and ER-119884, two novel orally active squalene synthase inhibitors, against Trypanosoma cruzi. Antimicrob. Agents Chemother. 2004, 48, 2379–2387. [Google Scholar] [CrossRef]
- Cerecetto, H.; Di Maio, R.; Gonzalez, M.; Risso, M.; Sagrera, G.; Seoane, G.; Denicola, A.; Peluffo, G.; Quijano, C.; Stoppani, A.O.M.; Paulino, M.; Olea-Azar, C.; Basombrıo, M.A. Synthesis and anti-trypanosomal evaluation of E-isomers of 5-nitro-2-furaldehyde and 5-nitrothiophene-2-carboxaldehyde semicarbazones. Eur. J. Med. Chem. 2000, 35, 343–350. [Google Scholar] [CrossRef]
- Boiani, M.; Boiani, L; Denicola, A; Torres de Ortiz, S.; Serna, E.; Vera de Bilbao, N.; Sanabria, L.; Yaluff, G.; Nakayama, H.; Rojas de Arias, A.; Vega, C.; Rolón, M.; Gómez-Barrios, A; Cerecetto, H.; González, M. 2H-Benzimidazole 1,3-dioxide derivatives: A new family of water-soluble anti-trypanosomatid. J. Med. Chem. 2006, 49, 3215–3220. [Google Scholar] [CrossRef]
- Porcal, W.; Hernandez, P.; Boiani, M.; Aguirre, G.; Boiani, L.; Chidichimo, A.; Cazzulo, J.J.;.;Campillo; Paez, J.A.; Castro, A.; et al. In vivo anti-Chagas vinylthio-, vinylsulfinyl-, and vinylsulfonylbenzofuroxan derivatives. J. Med. Chem. 2007, 50, 6004–6015. [Google Scholar] [CrossRef]
- Boiani, L; Davies, C.; Arredondo, C; Porcal, W.; Merlino, A.; Gerpe, A.; Boiani, M.; Pacheco, J.P.; Basombrio, M.A.; Cerecetto, H.; Gonzalez, M. In vivo studies of 5-arylethenylbenzofuroxans in acute murine models of Chagas disease. Eur. J. Med. Chem. 2008, 43, 2229–2237. [Google Scholar] [CrossRef]
- Boiani, L.; Gerpe, A.; Aran, V.J.; Torres de Ortiz, S.; Serna, E.; Vera de Bilbao, N.; Sanabria, L.; Yaluff, G.; Nakayama, H.; Rojas de Arias, A.; et al. In vitro and in vivo antitrypanosomatid activity of 5-nitroindazoles. Eur. J. Med. Chem. 2009, 44, 1034–1040. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, E.; Murguiondo, M.G.; Arias, M.G.; Arredondo, C.; Pintos, C.; Aguirre, G.; Fernandez, M.; Basmadjian, Y.; Rosa, R.; Pacheco, J.P.; et al. 5-Nitro-2-furyl derivative actives against Trypanosoma cruzi: Preliminary in vivostudies. Eur. J. Med. Chem. 2009, 44, 3909–3914. [Google Scholar] [CrossRef]
- Pérez-Cruz, F.; Jullian, C.; Rodriguez, J.; Arán, V.J.; Olea-Azar, C. Molecular encapsulation of 5-nitroindazole derivatives in 2,6-dimethyl-β-cyclodextrin: Electrochemical and spectroscopic studies. Bioorg. Med. Chem. 2009, 17, 4604–4611. [Google Scholar] [CrossRef]
- Rodríguez, J.; Gerpe, A.; Aguirre, G.; Kemmerling, U.; Piro, O.E.; Arán, V.J.; Maya, J.D.; Olea-Azar, C.; González, M.; Cerecetto, H. Study of 5-nitroindazoles' anti-Trypanosoma cruzi mode of action: Electrochemical behaviour and ESR spectroscopic studies. Eur. J. Med. Chem. 2009, 44, 1545–1553. [Google Scholar] [CrossRef]
- Porcal, W.; Merlino, A.; Boiani, M.; Gerpe, A; Gonzalez, M.; Cerecetto, H. Arylethenylbenzofuroxan derivatives as drugs for Chagas disease: Multigram batch synthesis using a Wittig-Boden process. Org. Proc. Res. Dev. 2008, 12, 156–162. [Google Scholar] [CrossRef]
- Gerpe, A; Merlino, A.; Boiani, M.; Porcal, W.; Fagiolino, P.; Gonzalez, M.; Cerecetto, H. Development of a HPLC method for the determination of antichagasic phenylethenylbenzofuroxans and its major synthetic secondary products in the chemical production processes. J. Pharm. Biomed. Anal. 2008, 47, 88–94. [Google Scholar] [CrossRef]
- Boiani, M; Merlino, A; Gerpe, A; Porcal, W.; Croce, F; Depaula, S; Rodriguez, A; Cerecetto, H; Gonzalez, M. o-Nitroanilines as major metabolic products of anti-Trypanosoma cruzi 5-phenylethenylbenzofuroxans in microsomal and cytosolic fractions of rat hepatocytes and in whole parasitic cells. Xenobiotica 2009, 39, 236–248. [Google Scholar] [CrossRef]
- Cabrera, M.; Lavaggi, M.L.; Hernandez, P; Merlino, A.; Gerpe, A.; Porcal, W.; Boiani, M.; Ferreira, A.; Monge, A.; Lopez de Cerain, A.; Gonzalez, M.; Cerecetto, H. Cytotoxic, mutagenic and genotoxic effects of new anti-T. cruzi 5-phenylethenylbenzofuroxans. Contribution of phase I metabolites on the mutagenicity induction. Toxicol. Lett. 2009, 190, 140–149. [Google Scholar] [CrossRef]
- Martins, F.T.; Ayala, A.P.; Porcal, W.; Cerecetto, H.; González, M.; Ellena, J. Structural relationships in the solid state of the anti-chagas agent (E)-phenylethenylbenzofuroxan. Mol. Divers. 2009. [Google Scholar]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Cramer, G.M.; Ford, R.A.; Hall, R.L. Estimation of toxic hazard – A decision tree approach. J. Cosmet. Toxicol. 1978, 16, 255–276. [Google Scholar] [CrossRef]
- Benigni, R.; Bossa, C.; Netzeva, T.; Rodomonte, A.; Tsakovska, I. Mechanistic QSAR of aromatic amines: New models for discriminating between mutagens and nonmutagens, and validation of models for carcinogens. Environ. Mol. Mutag. 2007, 48, 754–771. [Google Scholar] [CrossRef]
- Chung, M.C.; Gonçalves, M.F.; Colli, W.; Ferreira, E.I.; Miranda, M.T. Synthesis and in vitro evaluation of potential antichagasic dipeptide prodrugs of primaquine. J. Pharm. Sci. 1997, 86, 1127–1131. [Google Scholar] [CrossRef]
- Chung, M.C.; Güido, R.V.; Martinelli, T.F.; Gonçalves, M.F.; Polli, M.C.; Botelho, K.C.; Varanda, E.A.; Colli, W.; Miranda, M.T.; Ferreira, E.I. Synthesis and in vitro evaluation of potential antichagasic hydroxymethylnitrofurazone (NFOH-121): A new nitrofurazone prodrug. Bioorg. Med. Chem. 2003, 11, 4779–4783. [Google Scholar] [CrossRef]
- Aguirre, G.; Cabrera, E.; Cerecetto, H.; Di Maio, R.; González, M.; Seoane, G.; Duffaut, A.; Denicola, A.; Gil, M.J.; Martínez-Merino, V. Design, synthesis and biological evaluation of new potent 5-nitrofuryl derivatives as anti-Trypanosoma cruzi agents. Studies of trypanothione binding site of trypanothione reductase as target for rational design. Eur. J. Med. Chem. 2004, 39, 421–431. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Cerecetto, H.; González, M. Synthetic Medicinal Chemistry in Chagas’ Disease: Compounds at The Final Stage of “Hit-To-Lead” Phase. Pharmaceuticals 2010, 3, 810-838. https://doi.org/10.3390/ph3040810
Cerecetto H, González M. Synthetic Medicinal Chemistry in Chagas’ Disease: Compounds at The Final Stage of “Hit-To-Lead” Phase. Pharmaceuticals. 2010; 3(4):810-838. https://doi.org/10.3390/ph3040810
Chicago/Turabian StyleCerecetto, Hugo, and Mercedes González. 2010. "Synthetic Medicinal Chemistry in Chagas’ Disease: Compounds at The Final Stage of “Hit-To-Lead” Phase" Pharmaceuticals 3, no. 4: 810-838. https://doi.org/10.3390/ph3040810
APA StyleCerecetto, H., & González, M. (2010). Synthetic Medicinal Chemistry in Chagas’ Disease: Compounds at The Final Stage of “Hit-To-Lead” Phase. Pharmaceuticals, 3(4), 810-838. https://doi.org/10.3390/ph3040810