Beta-Adrenergic Agonists

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Adrenergic Control of ASM Tone
2.1. Circulating Catecholamines
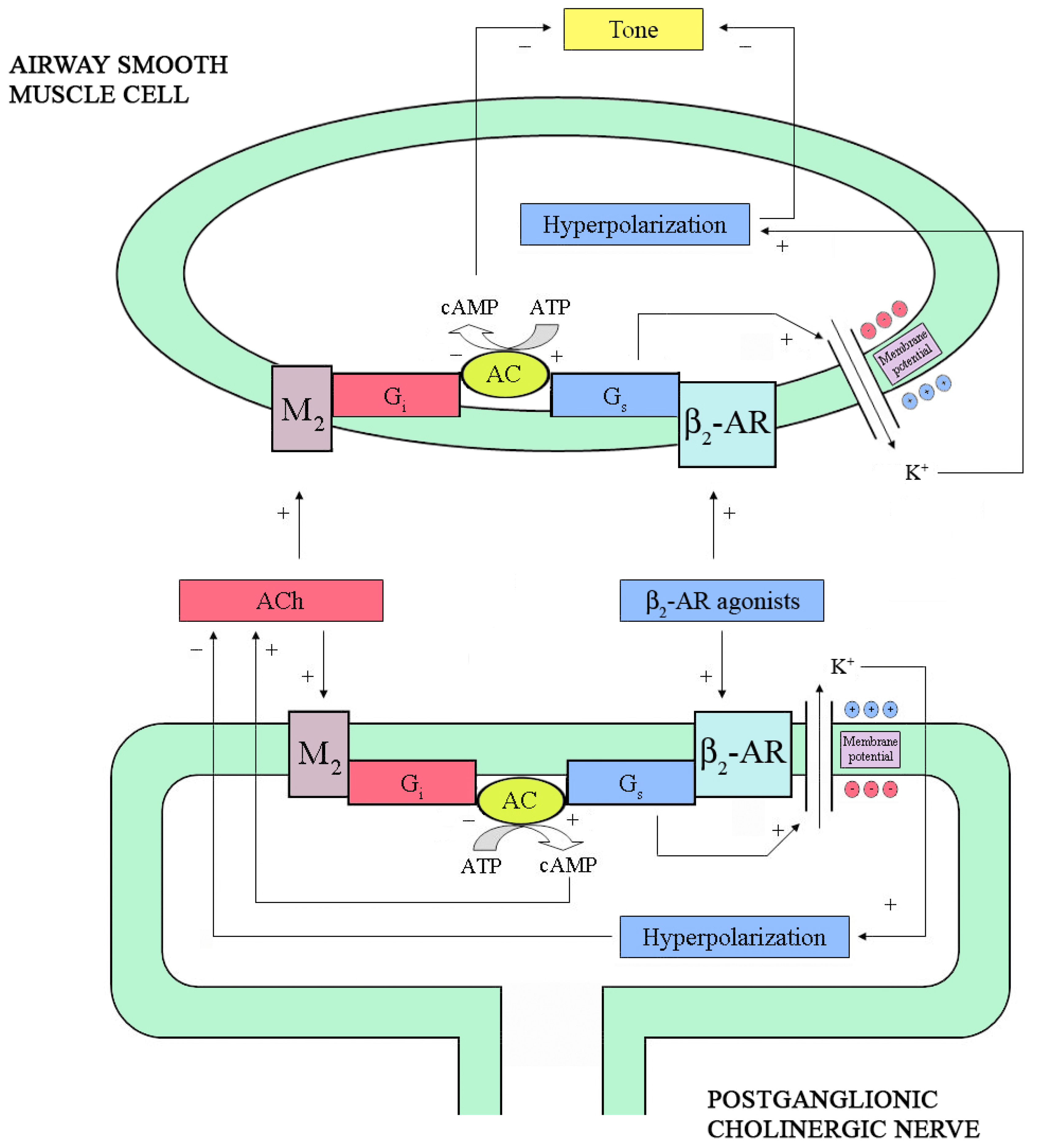
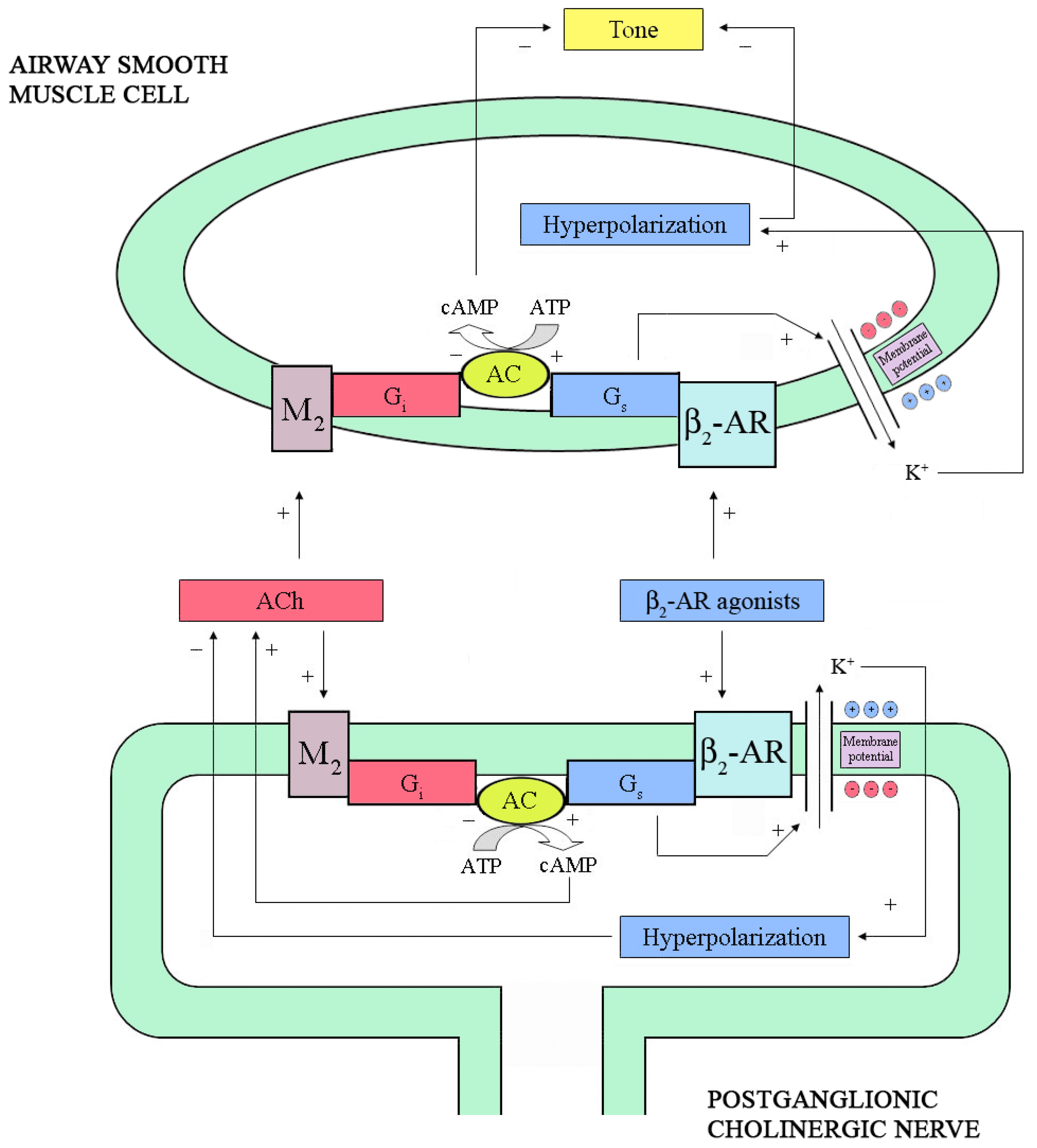
2.2. Neural Interactions
3. Adrenergic Receptors
4. G-Protein-Coupled Receptor Signaling
4.1. ASM Relaxation
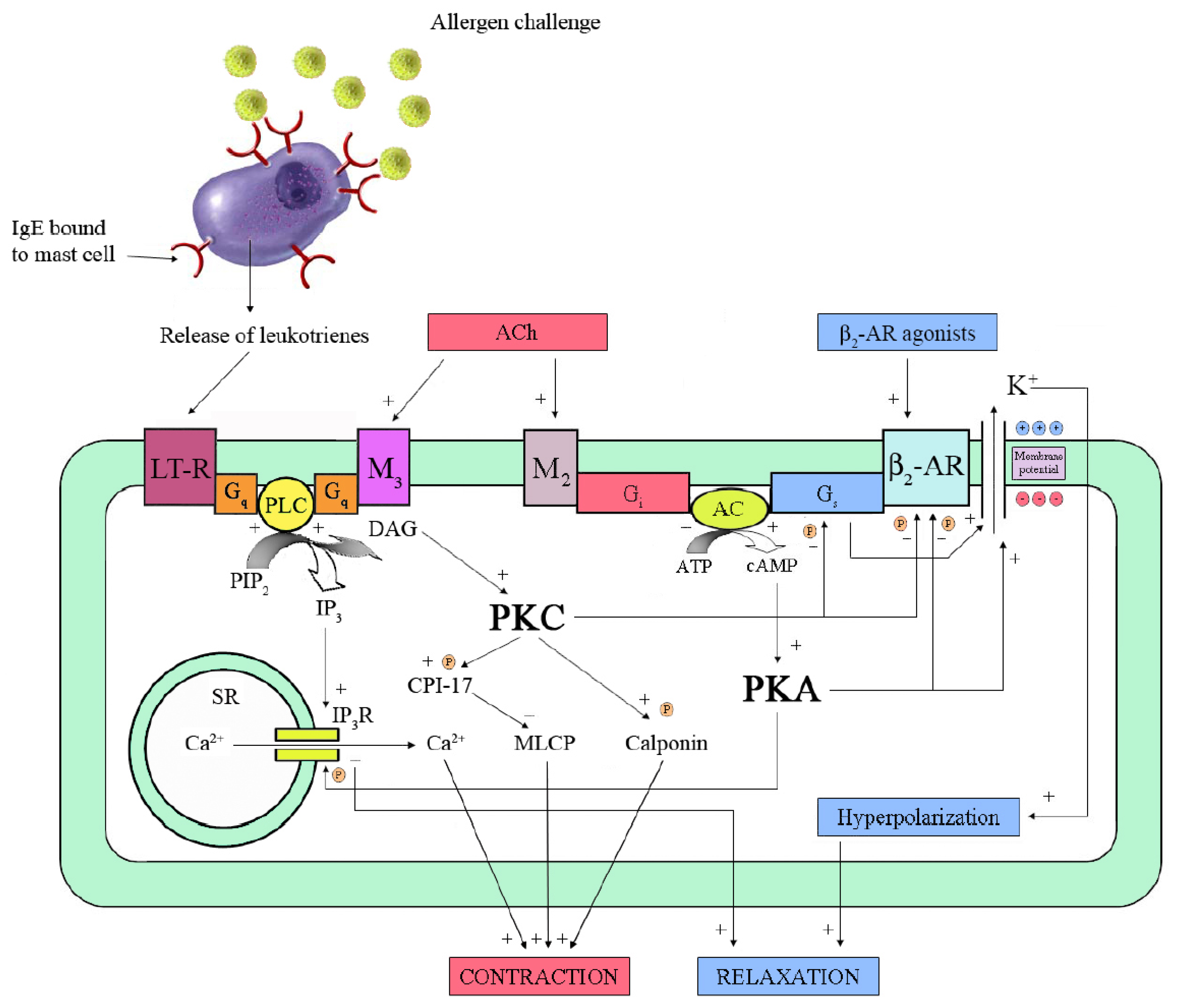
4.2. Anti-Inflammatory Effects
5. β2-AR Dysfunction
5.1. Desensitization by Phosphorylation
 : phosphorylation sites.
: phosphorylation sites.
: phosphorylation sites.
: phosphorylation sites.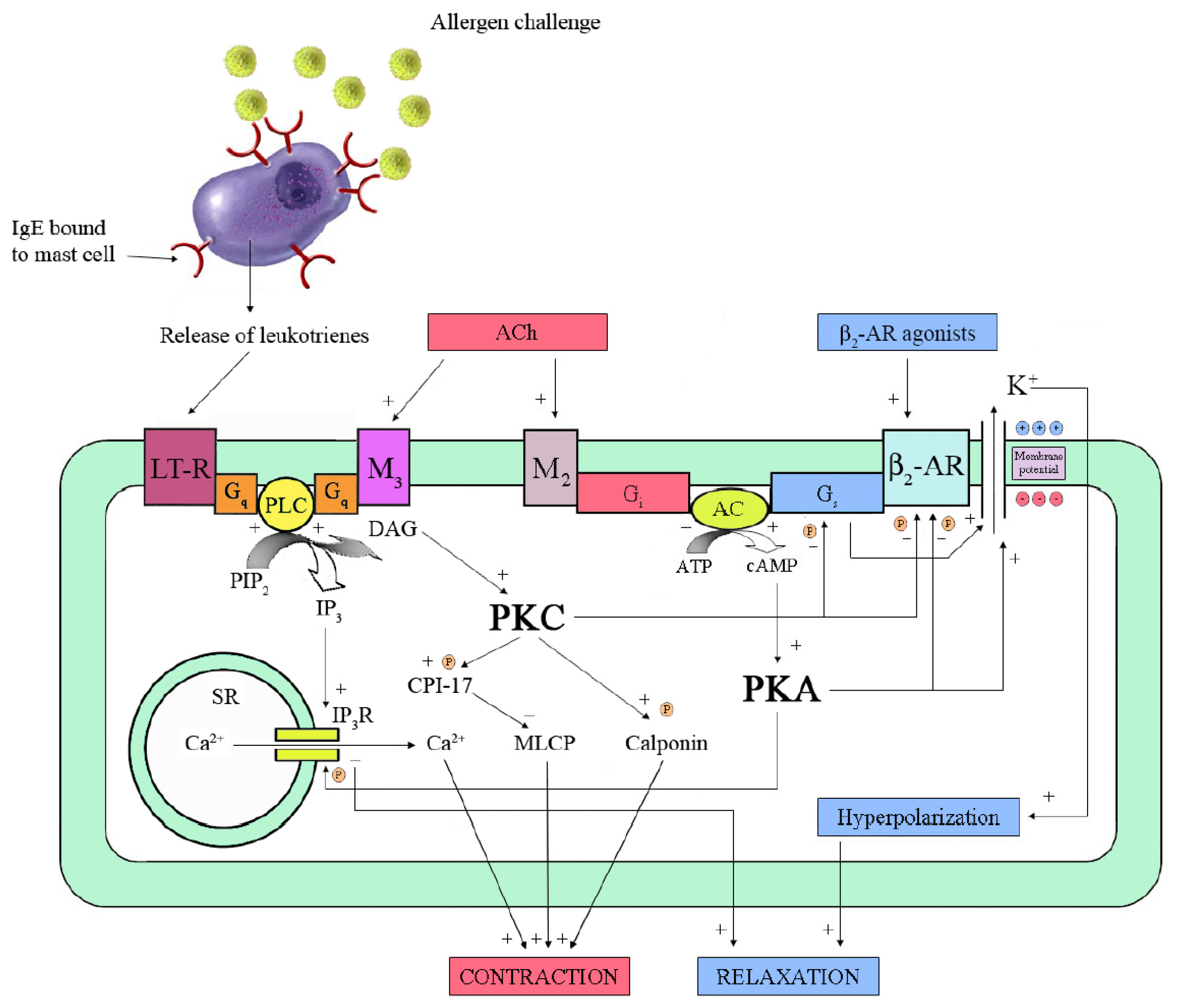
5.2. Desensitization by Sequestration
5.3. Desensitization by Down-Regulation
5.4. The β-AR Paradox
5.5. Reversal of Desensitization
6. Genetic Polymorphisms of the β2-AR
7. Role of β2-AR Agonists in the Treatment of Obstructive Lung Diseases
7.1. Symptom-Relief
7.1.1. Asthma
7.1.2. COPD
7.2. Disease-Control
8. The Great β2-AR Agonists Controversy
8.1. Short-Acting β2-AR Agonists
8.2. Long-Acting β2-AR Agonists
9. Future Prospects and Conclusions
References
- Chen, K.K.; Schmidt, C.F. Ephedrine and related substances. Medicine (Baltimore) 1930, 9, 1–117. [Google Scholar] [CrossRef]
- Ahlquist, R.P. A study of adrenotropic receptors. Am. J. Physiol. 1948, 153, 586–600. [Google Scholar]
- Cazzola, M.; Matera, M.G. Emerging inhaled bronchodilators: an update. Eur. Respir. J. 2009, 34, 757–769. [Google Scholar]
- Rabe, K.F.; Hurd, S.; Anzueto, A.; Barnes, P.J.; Buist, S.A.; Calverley, P.; Fukuchi, Y.; Jenkins, C.; Rodriguez-Roisin, R.; van Weel, C.; et al. Global Initiative for Chronic Obstructive Lung Disease. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. GOLD executive summary. Am. J. Respir. Crit. Care Med. 2007, 176, 532–555. [Google Scholar] [PubMed]
- National Asthma Education and Prevention Program. Expert Panel Report 3 (EPR-3): Guidelines for the Diagnosis and Management of Asthma - summary report 2007. J. Allergy Clin. Immunol. 2007, 120 Suppl., S94–S138. [CrossRef] [PubMed]
- Bateman, E.D.; Hurd, S.S.; Barnes, P.J.; Bousquet, J.; Drazen, J.M.; FitzGerald, M.; Gibson, P.; Ohta, K.; O'Byrne, P.; Pedersen, S.E.; et al. Global strategy for asthma management and prevention: GINA executive summary. Eur. Respir. J. 2008, 31, 143–178. [Google Scholar] [CrossRef] [PubMed]
- Doidge, J.M.; Satchell, D.G. Adrenergic and non-adrenergic inhibitory nerves in mammalian airways. J. Auton. Nerv. Syst. 1982, 5, 83–99. [Google Scholar]
- Barnes, P.J. Neural control of human airways in health and disease. Am. Rev. Respir. Dis. 1986, 134, 1289–1314. [Google Scholar]
- Belvisi, M.G. Overview of the innervation of the lung. Curr. Opin. Pharmacol. 2002, 2, 211–215. [Google Scholar]
- Groneberg, D.A.; Harrison, S.; Dinh, Q.T.; Geppetti, P.; Fischer, A. Tachykinins in the respiratory tract. Curr. Drug Targets 2006, 7, 1005–1010. [Google Scholar]
- Barnes, P.J. Neural control of airway function: new perspectives. Mol. Aspects Med. 1990, 11, 351–423. [Google Scholar]
- Barnes, P.J. Modulation of neurotransmission in airways. Physiol. Rev. 1992, 72, 699–729. [Google Scholar]
- Lees, G.M. A hitch-hiker’s guide to the galaxy of adrenoceptors. Br. J. Med. 1981, 283, 173–178. [Google Scholar]
- Hoffman, B.B.; Lefkowitz, R.J. Catecholamines, sympathomimetic drugs, and adrenergic receptor antagonists. In Goodman & Gilman’sthe Pharmacological Basis of Therapeutics, 9th; Hardman, J.G., Limbird, L.E., Molinoff, P.B., Ruddon, R.W., Goodman, L.S., Gilman, A., Eds.; McGraw-Hill: New York, NY, USA, 1996; pp. 199–248. [Google Scholar]
- Barnes, P.J. Endogenous catecholamines and asthma. J. Allergy Clin. Immunol. 1986, 77, 791–795. [Google Scholar]
- Nadel, J.A.; Barnes, P.J.; Holtzman, M.J. Autonomic factors in hyperreactivity of airway smooth muscle. In Handbook of Physiology. The Respiratory System; Macklem, P.T., Mead, J., Eds.; American Physiological Society: Bethesda, MD, USA, 1986; Volume 3, pp. 693–702, Part 2. [Google Scholar]
- Rhoden, K.J.; Meldrum, L.A.; Barnes, P.J. Inhibition of cholinergic neurotransmission in human airways by β2-adrenoceptors. J. Appl. Physiol. 1988, 65, 700–705. [Google Scholar]
- Bai, T.R.; Lam, R.; Prasad, F.Y. Effects of adrenergic agonists and adenosine on cholinergic neurotransmission in human tracheal smooth muscle. Pulmon. Pharmacol. 1989, 1, 193–199. [Google Scholar]
- Barnes, P.J. Muscarinic receptor subtype in airways. Life Sci. 1993, 52, 521–528. [Google Scholar]
- Lands, A.M.; Arnold, A.; McAuliff, J.P.; Luduena, F.P.; Brown, T.G., Jr. Differentiation of receptor systems activated by sympathomimetic amines. Nature 1967, 214, 597–598. [Google Scholar]
- Carstairs, J.R.; Nimmo, A.J.; Barnes, P.J. Autoradiographic visualization of beta-adrenoceptor subtype in human lung. Am. Rev. Respir. Dis. 1985, 132, 541–547. [Google Scholar]
- Barnes, P.J.; Nadel, J.A.; Skoogh, B.E.; Roberts, J.M. Characterization of beta adrenoceptor subtypes in canine airway smooth muscle by radioligand binding and physiological responses. J. Pharmacol. Exp. Ther. 1983, 225, 456–461. [Google Scholar]
- Hamid, Q.A.; Mak, J.C.; Sheppard, M.N.; Corrin, B.; Venter, J.C.; Barnes, P.J. Localization of beta2-adrenoceptor messenger RNA in human and rat lung using in situ hybridization: correlation with receptor autoradiography. Eur. J. Pharmacol. 1991, 206, 133–138. [Google Scholar]
- Tamaoki, J.; Yamauchi, F.; Chiyotani, A.; Yamawaki, I.; Takeuchi, S.; Konno, K. Atypical β-adrenoceptor (β3-adrenoceptor) mediated relaxation of canine isolated bronchial smooth muscle. J. Appl. Physiol. 1993, 74, 297–302. [Google Scholar]
- Martin, C.A.; Naline, E.; Bakdach, H.; Advenier, C. Beta 3-adrenoceptor agonists, BRL 37344 and SR 58611A, do not induce relaxation of human, sheep and guinea-pig airway smooth muscle in vitro. Eur. Respir. J. 1994, 7, 1610–1615. [Google Scholar] [CrossRef] [PubMed]
- Emorine, L.J.; Marullo, S.; Briend-Sutren, M.M.; Patey, G.; Tate, K.; Delavier-Klutchko, C.; Strosberg, A.D. Molecular characterization of the human beta 3-adrenergic receptor. Science 1989, 245, 1118–1121. [Google Scholar]
- Goldie, R.G.; Paterson, J.W.; Spina, D.; Wale, J.L. Classification of β2-adrenoceptors in human isolated bronchus. Br. J. Pharmacol. 1984, 81, 611–615. [Google Scholar]
- Nials, A.T.; Coleman, R.A.; Johnson, M.; Magnussen, H.; Rabe, K.F.; Vardey, C.J. Effects of β-adrenoceptor agonists in human bronchial smooth muscle. Br. J. Pharmacol. 1993, 110, 1112–1116. [Google Scholar] [PubMed]
- Löfdahl, C.G.; Svedmyr, N. Effects of prenalterol in asthmatic patients. Eur. J. Clin. Pharmacol. 1982, 23, 297–302. [Google Scholar]
- Torphy, T.J.; Rinard, G.A.; Rietow, M.G.; Mayer, S.E. Functional antagonism in canine tracheal smooth muscle: inhibition by methacholine of the mechanical and biochemical responses to isoproterenol. J. Pharmacol. Exp. Ther. 1983, 227, 694–699. [Google Scholar]
- Torphy, T.J.; Zheng, C.; Peterson, S.M.; Fiscus, R.R.; Rinard, G.A.; Mayer, S.E. Inhibitory effect of methacholine on drug-induced relaxation, cyclic AMP accumulation, and cyclic AMP-dependent protein kinase activation in canine tracheal smooth muscle. J. Pharmacol. Exp. Ther. 1985, 232, 409–417. [Google Scholar]
- Gross, N.J.; Skorodin, M.S. Anticholinergic, antimuscarinic bronchodilators. Am. Rev. Respir. Dis. 1984, 129, 856–870. [Google Scholar]
- Gilman, A.G. G proteins: Transducers of receptor-generated signals. Annu. Rev. Biochem. 1987, 56, 615–649. [Google Scholar]
- Dohlman, H.G.; Thorner, J.; Caron, M.G.; Lefkowitz, R.J. Model systems for the study of seven-transmembrane-segment receptors. Annu. Rev. Biochem. 1991, 60, 653–688. [Google Scholar]
- Neves, S.R.; Ram, P.T.; Iyengar, R. G protein pathways. Science 2002, 296, 1636–1639. [Google Scholar]
- Dohlman, H.G. A scaffold makes the switch. Sci. Signal. 2008, 1. pe46.. [Google Scholar]
- Liggett, S.B.; Lefkowitz, R.J. Adrenergic receptor-coupled adenylyl cyclase systems: regulation of receptor function by phosphorylation, sequestration and downregulation. In Regulation of Cellular Signal Transduction Pathways by Desensitization and Amplification; Sibley, D., Houslay, M., Eds.; John Wiley & Sons: London, UK, 1993; pp. 71–97. [Google Scholar]
- Giembycz, M.A.; Raeburn, D. Putative substrates for cyclic nucleotide-dependent protein kinases and the control of airway smooth muscle tone. J. Auton. Pharmacol. 1991, 11, 365–398. [Google Scholar]
- Gerthoffer, U.T. Calcium dependence of myosin phosphorylation and airway smooth muscle contraction and relaxation. Am. J. Physiol. 1986, 250, C597–C604. [Google Scholar]
- Hall, I.P.; Hill, S.J. Beta-adrenoceptor stimulation inhibits histamine-stimulated inositol phospholipids hydrolysis in bovine tracheal smooth muscle. Br. J. Pharmacol. 1988, 95, 1204–1212. [Google Scholar]
- Twort, C.H.; van Breemen, C. Human airway smooth muscle in cell culture: control of the intracellular calcium store. Pulm. Pharmacol. 1989, 2, 45–53. [Google Scholar]
- Gunst, S.J.; Stropp, J.Q. Effect of Na-K adenosinetriphosphatase activity on relaxation of canine tracheal smooth muscle. J. Appl. Physiol. 1988, 64, 635–641. [Google Scholar]
- Jones, T.R.; Charette, L.; Garcia, M.L.; Kaczorowski, G.J. Selective inhibition of relaxation of guinea-pig trachea by charybdotoxin, a potent Ca(++)-activated K+ channel inhibitor. J. Pharmacol. Exp. Ther. 1990, 255, 697–706. [Google Scholar]
- Jones, T.R.; Charette, L.; Garcia, M.L.; Kaczorowski, G.J. Interaction of iberiotoxin with beta-adrenoceptor agonists and sodium nitroprusside on guinea pig trachea. J. Appl. Physiol. 1993, 74, 1879–1884. [Google Scholar]
- Tanaka, Y.; Yamashita, Y.; Yamaki, F.; Horinouchi, T.; Shigenobu, K.; Koike, K. MaxiK channel mediates beta2-adrenoceptor-activated relaxation to isoprenaline through cAMP-dependent and -independent mechanisms in guinea-pig tracheal smooth muscle. J. Smooth Muscle Res. 2003, 39, 205–219. [Google Scholar]
- Koike, K.; Yamashita, Y.; Horinouchi, T.; Yamaki, F.; Tanaka, Y. cAMP-independent mechanism is significantly involved in beta2-adrenoceptor-mediated tracheal relaxation. Eur. J. Pharmacol. 2004, 492, 65–70. [Google Scholar]
- Miura, M.; Belvisi, M.G.; Stretton, C.D.; Yacoub, M.H.; Barnes, P.J. Role of potassium channels in bronchodilator responses in human airways. Am. Rev. Respir. Dis. 1992, 146, 132–136. [Google Scholar]
- Kume, H.; Graziano, M.P.; Kotlikoff, M.I. Stimulatory and inhibitory regulation of calcium-activated potassium channels by guanine nucleotide-binding proteins. Proc. Natl. Acad. Sci. USA 1992, 89, 11051–11055. [Google Scholar]
- Belvisi, M.G.; Patel, H.J.; Takahashi, T.; Barnes, P.J.; Giembycz, M.A. Paradoxical facilitation of acetylcholine release from parasympathetic nerves innervating guinea-pig trachea by isoprenaline. Br. J. Pharmacol. 1996, 117, 1413–1420. [Google Scholar]
- De Haas, J.R.; Terpstra, J.S.; Van der Zwaag, M.; Kockelbergh, P.G.; Roffel, A.D.F; Zaagsma, J. Facilitatory β2-adrenoceptors on cholinergic and adrenergic nerve endings of the guinea pig trachea. Am. J. Physiol. Lung Cell. Mol. Physiol. 1999, 276, L420–L425. [Google Scholar]
- Zhang, X.Y.; Olszewski, M.A.; Robinson, N.E. β2-Adrenoceptor activation augments acetylcholine release from tracheal parasympathetic nerves. Am. J. Physiol. Lung Cell. Mol. Physiol. 1995, 268, L950–L956. [Google Scholar]
- Wessler, I.; Reinheimer, T.; Brunn, G.; Anderson, G.P.; Maclagan, J.; Rackè, K. β2-adrenoceptors mediate inhibition of [3H]-acetylcholine release from the isolated rat and guinea-pig trachea: role of the airway mucosa and prostaglandins. Br. J. Pharmacol. 1994, 113, 1221–1230. [Google Scholar] [PubMed]
- Seamon, K.B.; Padgett, W.; Daly, J.W. Forskolin: Unique diterpene activator of adenylate cyclase in membranes and in intact cells. Proc. Natl. Acad. Sci. USA 1981, 78, 3363–3367. [Google Scholar]
- Patel, H.J.; Giembycz, M.A.; Keeling, J.E.; Barnes, P.J.; Belvisi, M.G. Inhibition of cholinergic neurotransmission in guinea pig trachea by NS1619, a putative activator of large-conductance, calcium-activated potassium channels. J. Pharmacol. Exp. Ther. 1998, 286, 952–958. [Google Scholar] [PubMed]
- Brichetto, L.; Song, P.; Crimi, E.; Rehder, K.; Brusasco, V. Modulation of cholinergic responsiveness through the β-adrenoceptor signal transmission pathway in bovine trachealis. J. Appl. Physiol. 2003, 95, 735–741. [Google Scholar]
- Freund-Michel, V.C.; Birrell, M.A.; Giembycz, M.A.; Hele, D.J.; Haj-Yahia, S.; Belvisi, M.G. Beta2-agonists block tussive responses in guinea pigs via an atypical cAMP-dependent pathway. Eur. Respir. J. 2010, 35, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Chong, C.F.; Chen, C.C.; Ma, H.P.; Wu, Y.C.; Chen, Y.C.; Wang, T.L. Comparison of lidocaine and bronchodilator inhalation treatments for cough suppression in patients with chronic obstructive pulmonary disease. Emerg. Med. J. 2005, 22, 429–432. [Google Scholar]
- Mulrennan, S.; Wright, C.; Thompson, R.; Goustas, P.; Morice, A. Effect of salbutamol on smoking related cough. Pulm. Pharmacol. Ther. 2004, 17, 127–131. [Google Scholar]
- Chang, A.B.; Phelan, P.D.; Carlin, J.B.; Sawyer, S.M.; Robertson, C.F. A randomised, placebo controlled trial of inhaled salbutamol and beclomethasone for recurrent cough. Arch. Dis. Child. 1998, 79, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.A.; Adamson, D.L.; Choudry, N.B.; Fuller, R.W. The effect of altering airway tone on the sensitivity of the cough reflex in normal volunteers. Eur. Respir. J. 1991, 4, 1078–1079. [Google Scholar]
- Brusasco, V.; Crimi, E.; Gherson, G.; Nardelli, R.; Oldani, V.; Francucci, B.; Della Cioppa, S.; Senn, S.; Fabbri, L. Actions other than smooth muscle relaxation may play a role in the protective effects of formoterol on the allergen-induced late asthmatic reaction. Pulm. Pharmacol. Ther. 2002, 15, 399–406. [Google Scholar]
- Johnson, S.R.; Knox, A.J. Synthetic functions of airway smooth muscle in asthma. Trends Pharmacol. Sci. 1997, 18, 288–292. [Google Scholar]
- Baroffio, M.; Crimi, E.; Brusasco, V. Airway smooth muscle as a model for new investigative drugs in asthma. Ther. Adv. Respir. Dis. 2008, 2, 129–139. [Google Scholar]
- Baroffio, M.; Barisione, G.; Crimi, E.; Brusasco, V. Noninflammatory mechanisms of airway hyper-responsiveness in bronchial asthma: an overview. Ther. Adv. Respir. Dis. 2009, 3, 163–174. [Google Scholar]
- Black, J.L.; Roth, M. Intrinsic asthma: is it intrinsic to the smooth muscle? Clin. Exp. Allergy 2009, 39, 962–965. [Google Scholar] [CrossRef] [PubMed]
- Kaur, M.; Holden, N.S.; Wilson, S.M.; Sukkar, M.B.; Chung, K.F.; Barnes, P.J.; Newton, R.; Giembycz, M.A. Effect of beta2-adrenoceptor agonists and other cAMP-elevating agents on inflammatory gene expression in human ASM cells: a role for protein kinase A. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 295, L505–L514. [Google Scholar]
- Lefkowitz, R.J.; Hausdorff, W.P.; Caron, M.G. Role of phosphorylation in desensitization of the beta-adrenoceptor. Trends Pharmacol. Sci. 1990, 11, 190–194. [Google Scholar]
- Bai, T.R.; Mak, J.C.W.; Barnes, P.J. A comparison of β-adrenergic receptors and in vitro relaxant responses to isoproterenol in asthmatic airway smooth muscle. Am. J. Respir. Cell. Mol. Biol. 1992, 6, 647–651. [Google Scholar] [PubMed]
- Brusasco, V.; Crimi, E.; Baroffio, M. Allergic airway inflammation and beta-adrenoceptor dysfunction. Cell. Biochem. Biophys. 2006, 44, 129–138. [Google Scholar]
- Szentivanyi, A. Beta adrenergic theory of atopic abnormality in bronchial asthma. J. Allergy 1968, 42, 203–232. [Google Scholar]
- Trian, T.; Ge, Q.; Moir, L.M.; Burgess, J.K.; Kuo, C.; King, N.J.; Reddel, H.K.; Black, J.L.; Oliver, B.G.; McParland, B.E. Rhinovirus-induced exacerbations of asthma - How is the beta2-adrenoceptor implicated? Am. J. Respir. Cell. Mol. Biol. 2009. Epub ahead of print.. [Google Scholar]
- Laporte, J.D.; Moore, P.E.; Panettieri, R.A.; Moeller, W.; Heyder, J.; Shore, S.A. Prostanoids mediate IL-1β-induced β-adrenergic hyporesponsiveness in human airway smooth muscle cells. Am. J. Physiol. 1998, 275, L491–L501. [Google Scholar]
- Hausdorff, W.P.; Caron, M.G.; Lefkowitz, R.J. Turning off the signal: desensitization of beta-adrenergic receptor function. FASEB J. 1990, 4, 2881–2889. [Google Scholar]
- Song, P.; Milanese, M.; Crimi, E.; Bruzzone, S.; Zocchi, E.; Rehder, K.; Brusasco, V. G(s) protein dysfunction in allergen-challenged human isolated passively sensitized bronchi. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 279, L209–L215. [Google Scholar]
- Milanese, M.; Riccio, A.M.; Gamalero, C.; De Giovanni, B.; Brichetto, L.; Baroffio, M.; Crimi, E.; Brusasco, V.; Canonica, G.W. A model of allergen-driven human airway contraction: beta2- pathway dysfunction without cytokine involvement. Ann. Allergy Asthma Immunol. 2005, 94, 273–278. [Google Scholar]
- Song, P.; Milanese, M.; Crimi, E.; Rehder, K.; Brusasco, V. Allergen challenge of passively sensitized human bronchi alters M2- and beta2-receptor function. Am. J. Respir. Crit. Care Med. 1997, 155, 1230–1234. [Google Scholar]
- Song, P.; Crimi, E.; Milanese, M.; Duan, J.; Rehder, K.; Brusasco, V. Anti-inflammatory agents and allergen-induced beta2-receptor dysfunction in isolated human bronchi. Am. J. Respir. Crit. Care Med. 1998, 158, 1809–1814. [Google Scholar]
- Rovati, G.E.; Baroffio, M.; Citro, S.; Brichetto, L.; Ravasi, S.; Milanese, M.; Crimi, E.; Brusasco, V. Cysteinyl-leukotrienes in the regulation of beta2-adrenoceptor function: an in vitro model of asthma. Respir. Res. 2006, 7, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Benovic, J.L.; Mayor, F., Jr.; Staniszewski, C.; Lefkowitz, R.J.; Caron, M.G. Purification and characterization of the beta-adrenergic receptor kinase. J. Biol. Chem. 1987, 262, 9026–9032. [Google Scholar]
- Benovic, J.L.; Kühn, H.; Weyand, I.; Codina, J.; Caron, M.G.; Lefkowitz, R.J. Functional desensitization of the isolated beta-adrenergic receptor by the beta-adrenergic receptor kinase: potential role of an analog of the retinal protein arrestin (48-kDa protein). Proc. Natl. Acad. Sci. USA 1987, 84, 8879–8882. [Google Scholar]
- Lohse, M.J.; Benovic, J.L.; Codina, J.; Caron, M.G.; Lefkowitz, R.J. Beta-Arrestin: A protein that regulates beta-adrenergic receptor function. Science 1990, 248, 1547–1550. [Google Scholar]
- Rajagopal, S.; Kim, J.; Ahn, S.; Craig, S.; Lam, C.M.; Gerard, N.P.; Gerard, C.; Lefkowitz, R.J. Beta-arrestin- but not G protein-mediated signaling by the "decoy" receptor CXCR7. Proc. Natl. Acad. Sci. USA 2010, 107, 628–632. [Google Scholar]
- Nino, G.; Hu, A.; Grunstein, J.S.; Grunstein, M. Mechanism regulating proasthmatic effects of prolonged homologous β2-adrenergic receptor desensitization in airway smooth muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L746–L757. [Google Scholar]
- Wang, W.C.; Mihlbachler, K.A.; Brunnett, A.C.; Liggett, S.B. Targeted transgenesis reveals discrete attenuator functions of GRK and PKA in airway beta2-adrenergic receptor physiologic signaling. Proc. Natl. Acad. Sci. USA 2009, 106, 15007–15012. [Google Scholar]
- Giembycz, M.A. An estimation of beta2-adrenoceptor reserve on human bronchial smooth muscle for some sympathomimetic bronchodilators. Br. J. Pharmacol. 2009, 158, 287–299. [Google Scholar]
- Yu, S.S.; Lefkowitz, R.J.; Hausdorff, W.P. Beta-adrenergic receptor sequestration. A potential mechanism of receptor resensitization. J. Biol. Chem. 1993, 268, 337–341. [Google Scholar] [PubMed]
- Hadcock, J.R.; Wang, H.Y.; Malbon, C.C. Agonist-induced destabilization of beta-adrenergic receptor mRNA. Attenuation of glucocorticoid-induced up-regulation of beta-adrenergic receptors. J. Biol. Chem. 1989, 264, 19928–19933. [Google Scholar] [PubMed]
- Collins, S.; Bouvier, M.; Bolanowski, M.A.; Caron, M.G.; Lefkowitz, R.J. cAMP stimulates transcription of the beta2-adrenergic receptor gene in response to short-term agonist exposure. Proc. Natl. Acad. Sci. USA 1989, 86, 4853–4857. [Google Scholar]
- Huang, L.Y.; Tholanikunnel, B.G.; Vakalopoulou, E.; Malbon, C.C. The M(r) 35,000 beta-adrenergic receptor mRNA-binding protein induced by agonists requires both an AUUUA pentamer and U-rich domains for RNA recognition. J. Biol. Chem. 1993, 268, 25769–25775. [Google Scholar]
- Bouvier, M.; Collins, S.; O'Dowd, B.F.; Campbell, P.T.; de Blasi, A.; Kobilka, B.K.; MacGregor, C.; Irons, G.P.; Caron, M.G.; Lefkowitz, R.J. Two distinct pathways for cAMP-mediated down-regulation of the beta2-adrenergic receptor. Phosphorylation of the receptor and regulation of its mRNA level. J. Biol. Chem. 1989, 264, 16786–16792. [Google Scholar] [PubMed]
- Valiquette, M.; Bonin, H.; Hnatowich, M.; Caron, M.G.; Lefkowitz, R.J.; Bouvier, M. Involvement of tyrosine residues located in the carboxyl tail of the human beta2-adrenergic receptor in agonist-induced down-regulation of the receptor. Proc. Natl. Acad. Sci. USA 1990, 87, 5089–5093. [Google Scholar]
- Kraan, J.; Koëter, G.H.; vd Mark, T.W.; Sluiter, H.J.; de Vries, K. Changes in bronchial hyperreactivity induced by 4 weeks of treatment with antiasthmatic drugs in patients with allergic asthma: a comparison between budesonide and terbutaline. J. Allergy Clin. Immunol. 1985, 76, 628–636. [Google Scholar]
- Kerrebijn, K.F.; van Essen-Zandvliet, E.E.; Neijens, H.J. Effect of long-term treatment with inhaled corticosteroids and beta-agonists on the bronchial responsiveness in children with asthma. J. Allergy Clin. Immunol. 1987, 79, 653–659. [Google Scholar]
- van Schayck, C.P.; Graafsma, S.J.; Visch, M.B.; Dompeling, E.; van Weel, C.; van Herwaarden, C.L. Increased bronchial hyperresponsiveness after inhaling salbutamol during 1 year is not caused by subsensitization to salbutamol. J. Allergy Clin. Immunol. 1990, 86, 793–800. [Google Scholar]
- Cheung, D.; Timmers, M.C.; Zwinderman, A.H.; Bel, E.H.; Dijkman, J.H.; Sterk, P.J. Long-term effects of a long-acting beta2-adrenoceptor agonist, salmeterol, on airway hyperresponsiveness in patients with mild asthma. N. Engl. J. Med. 1992, 327, 1198–1203. [Google Scholar] [PubMed]
- McGraw, D.W.; Almoosa, K.F.; Paul, R.J.; Kobilka, B.K.; Liggett, S.B. Antithetic regulation by β-adrenergic receptors of Gq receptor signaling via phospholipase C underlies the airway β-agonist paradox. J. Clin. Invest. 2003, 112, 619–626. [Google Scholar] [PubMed]
- Barnes, P.J. Scientific rationale for using a single inhaler for asthma control. Eur. Respir. J. 2007, 29, 587–595. [Google Scholar]
- Callaerts-Vegh, Z.; Evans, K.L.; Dudekula, N.; Cuba, D.; Knoll, B.J.; Callaerts, P.F.; Giles, H.; Shardonofsky, F.R.; Bond, R.A. Effects of acute and chronic administration of beta-adrenoceptor ligands on airway function in a murine model of asthma. Proc. Natl. Acad. Sci. USA 2004, 101, 4948–4953. [Google Scholar]
- Nguyen, L.P.; Omoluabi, O.; Parra, S.; Frieske, J.M.; Clement, C.; Ammar-Aouchiche, Z.; Ho, S.B.; Ehre, C.; Kesimer, M.; Knoll, B.J.; et al. Chronic exposure to beta-blockers attenuates inflammation and mucin content in a murine asthma model. Am. J. Respir. Cell. Mol. Biol. 2008, 38, 256–262. [Google Scholar] [PubMed]
- Hanania, N.A.; Singh, S.; El-Wali, R.; Flashner, M.; Franklin, A.E.; Garner, W.J.; Dickey, B.F.; Parra, S.; Ruoss, S.; Shardonofsky, F.; et al. The safety and effects of the beta-blocker, nadolol, in mild asthma: an open-label pilot study. Pulm. Pharmacol. Ther. 2008, 21, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Lipworth, B.J.; Williamson, P.A. Beta blockers for asthma: a double-edged sword. Lancet 2009, 373, 104–105. [Google Scholar]
- Hanania, N.A.; Dickey, B.F.; Bond, R.A. Clinical implications of the intrinsic efficacy of beta-adrenoceptor drugs in asthma: full, partial and inverse agonism. Curr. Opin. Pulm. Med. 2010, 16, 1–5. [Google Scholar]
- Davies, A.O.; Lefkowitz, R.J. In vitro desensitization of beta-adrenergic receptors in human neutrophils. Attenuation by corticosteroids. J. Clin. Invest. 1983, 71, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Brichetto, L.; Milanese, M.; Song, P.; Patrone, M.; Crimi, E.; Rehder, K.; Brusasco, V. Beclomethasone rapidly ablates allergen-induced beta2-adrenoceptor pathway dysfunction in human isolated bronchi. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 284, L133–L139. [Google Scholar]
- Green, S.A.; Turki, J.; Hall, I.P.; Liggett, S.B. Implications of genetic variability of human beta2-adrenergic receptor structure. Pulm. Pharmacol. 1995, 8, 1–10. [Google Scholar]
- Liggett, S.B. Genetics of beta2-adrenergic receptor variants in asthma. Clin. Exp. Allergy 1995, Suppl. 2, 89–94. [Google Scholar] [CrossRef]
- Kazani, S.; Wechsler, M.E.; Israel, E. The role of pharmacogenomics in improving the management of asthma. J. Allergy Clin. Immunol. 2010, 125, 295–302. [Google Scholar]
- Taylor, D.R.; Drazen, J.M.; Herbison, G.P.; Yandava, C.N.; Hancox, R.J.; Town, G.I. Asthma exacerbations during long term beta-agonist use: influence of beta2-adrenoceptor polymorphism. Thorax 2000, 55, 762–767. [Google Scholar]
- Basu, K.; Palmer, C.N.; Tavendale, R.; Lipworth, B.J.; Mukhopadhyay, S. Beta2-receptor genotype predisposes to exacerbations in steroid-treated asthmatic patients taking frequent albuterol or salmeterol. J. Allergy Clin. Immunol. 2009, 124, 1188–1194. [Google Scholar]
- Wechsler, M.E.; Kunselman, S.J.; Chinchilli, V.M.; Bleecker, E.; Boushey, H.A.; Calhoun, W.J.; Ameredes, B.T.; Castro, M.; Craig, T.J.; Denlinger, L.; et al. Effect of beta2-adrenergic receptor polymorphism on response to long-acting beta2-agonist in asthma (LARGE trial): A genotype-stratified, randomised, placebo-controlled, crossover trial. Lancet 2009, 374, 1754–1764. [Google Scholar] [PubMed]
- Wang, W.C.; Mihlbachler, K.A.; Bleecker, E.R.; Weiss, S.T.; Liggett, S.B. A polymorphism of G-protein coupled receptor kinase 5 alters agonist-promoted desensitization of beta2-adrenergic receptors. Pharmacogenet. Genomics 2008, 18, 729–732. [Google Scholar]
- Hall, I.P.; Wheatley, A.; Wilding, P.; Liggett, S.B. Association of Glu 27 beta2-adrenoceptor polymorphism with lower airway reactivity in asthmatic subjects. Lancet 1995, 345, 1213–1214. [Google Scholar]
- Litonjua, A.A.; Lasky-Su, J.; Schneiter, K.; Tantisira, K.G.; Lazarus, R.; Klanderman, B.; Lima, J.J.; Irvin, C.G.; Peters, S.P.; Hanrahan, J.P.; et al. ARG1 is a novel bronchodilator response gene: screening and replication in four asthma cohorts. Am. J. Respir. Crit. Care Med. 2008, 178, 688–694. [Google Scholar] [CrossRef] [PubMed]
- Nelson, H.S. β-Adrenergic bronchodilators. N. Engl. J. Med. 1995, 333, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Lipworth, B.J.; Struthers, A.D.; McDevitt, D.G. Tachyphylaxis to systemic but not to airway responses during prolonged therapy with high dose inhaled salbutamol in asthmatics. Am. Rev. Respir. Dis. 1989, 140, 586–592. [Google Scholar]
- Drazen, J.M.; Israel, E.; Boushey, H.A.; Chinchilli, V.M.; Fahy, J.V.; Fish, J.E.; Lazarus, S.C.; Lemanske, R.F.; Martin, R.J.; Peters, S.P.; et al. Comparison of regularly scheduled with as-needed use of albuterol in mild asthma. Asthma Clinical Research Network. N. Engl. J. Med. 1996, 335, 841–847. [Google Scholar] [CrossRef] [PubMed]
- Israel, E.; Drazen, J.M.; Liggett, S.B.; Boushey, H.A.; Cherniack, R.M.; Chinchilli, V.M.; Cooper, D.M.; Fahy, J.V.; Fish, J.E.; Ford, J.G.; et al. The effect of polymorphisms of the beta(2)-adrenergic receptor on the response to regular use of albuterol in asthma. Am. J. Respir. Crit. Care Med. 2000, 162, 75–80. [Google Scholar] [PubMed]
- Israel, E.; Chinchilli, V.M.; Ford, J.G.; Boushey, H.A.; Cherniack, R.; Craig, T.J.; Deykin, A.; Fagan, J.K.; Fahy, J.V.; Fish, J.; et al. Use of regularly scheduled albuterol treatment in asthma: genotype-stratified, randomised, placebo-controlled cross-over trial. Lancet 2004, 364, 1505–1512. [Google Scholar] [PubMed]
- Milanese, M.; Saporiti, R.; Bartolini, S.; Pellegrino, R.; Baroffio, M.; Brusasco, V.; Crimi, E. Bronchodilator effects of exercise hyperpnea and albuterol in mild-to-moderate asthma. J. Appl. Physiol. 2009, 107, 494–499. [Google Scholar]
- Hendeles, L.; Colice, G.L.; Meyer, R.J. Withdrawal of albuterol inhalers containing chlorofluorocarbon propellants. N. Engl. J. Med. 2007, 356, 1344–1351. [Google Scholar]
- Ramsdell, J.W.; Colice, G.L.; Ekholm, B.P.; Klinger, N.M. Cumulative dose response study comparing HFA-134a albuterol sulfate and conventional CFC albuterol in patients with asthma. Ann. Allergy Asthma Immunol. 1998, 81, 593–599. [Google Scholar]
- Cates, C.J.; Crilly, J.A.; Rowe, B.H. Holding chambers (spacers) versus nebulizers for beta-agonist treatment of acute asthma. Cochrane Database Syst. Rev. 2006, 2, CD000052–CD000052. [Google Scholar]
- Henderson, W.R., Jr.; Banerjee, E.R.; Chi, E.Y. Differential effects of (S)- and (R)-enantiomers of albuterol in a mouse asthma model. J. Allergy Clin. Immunol. 2005, 116, 332–340. [Google Scholar]
- Berger, W.E.; Milgrom, H.; Skoner, D.P.; Tripp, K.; Parsey, M.V.; Baumgartner, R.A. Xopenex Pediatric Asthma Group. Evaluation of levalbuterol metered dose inhaler in pediatric patients with asthma: a double-blind, randomized, placebo- and active-controlled trial. Curr. Med. Res. Opin. 2006, 22, 1217–1226. [Google Scholar] [CrossRef] [PubMed]
- O'Byrne, P.M.; Bisgaard, H.; Godard, P.P.; Pistolesi, M.; Palmqvist, M.; Zhu, Y.; Ekström, T. Bateman ED Budesonide/formoterol combination therapy as both maintenance and reliever medication in asthma. Am. J. Respir. Crit. Care Med. 2005, 171, 129–136. [Google Scholar] [PubMed]
- Pearlman, D.S.; Chervinsky, P.; LaForce, C.; Seltzer, J.M.; Southern, D.L.; Kemp, J.P.; Dockhorn, R.J.; Grossman, J.; Liddle, R.F.; Yancey, S.W.; et al. A comparison of salmeterol with albuterol in the treatment of mild-to-moderate asthma. N. Engl. J. Med. 1992, 327, 1420–1425. [Google Scholar] [PubMed]
- Nelson, J.A.; Strauss, L.; Skowronski, M.; Ciufo, R.; Novak, R.; McFadden, E.R., Jr. Effect of long-term salmeterol treatment on exercise-induced asthma. N. Engl. J. Med. 1998, 339, 141–146. [Google Scholar]
- Weinberger, M.; Abu-Hasan, M. Life-threatening asthma during treatment with salmeterol. N. Engl. J. Med. 2006, 355, 852–853. [Google Scholar]
- Smyth, E.T.; Pavord, I.D.; Wong, C.S.; Wisniewski, A.F.; Williams, J.; Tattersfield, A.E. Interaction and dose equivalence of salbutamol and salmeterol in patients with asthma. Br. Med. J. 1993, 306, 543–545. [Google Scholar]
- Appleton, S.; Poole, P.; Smith, B.; Veale, A.; Bara, A. Long-acting β2-agonists for chronic obstructive pulmonary disease patients with poorly reversible airflow limitation. Cochrane Database Syst. Rev. 2002, 3, CD001104. [Google Scholar] [PubMed]
- D'Urzo, A.D.; De Salvo, M.C.; Ramirez-Rivera, A.; Almeida, J.; Sichletidis, L.; Rapatz, G.; Kottakis, J. FOR-INT-03 Study Group. In patients with COPD, treatment with a combination of formoterol and ipratropium is more effective than a combination of salbutamol and ipratropium: a 3-week, randomized, double-blind, within-patient, multicenter study. Chest 2001, 119, 1347–1356. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J.; Stockley, R.A. COPD: current therapeutic interventions and future approaches. Eur. Respir. J. 2005, 25, 1084–1106. [Google Scholar]
- Goldkorn, A.; Diotto, P.; Burgess, C.; Weatherall, M.; Holt, S.; Beasley, R.; Siebers, R. The pulmonary and extra-pulmonary effects of high-dose formoterol in COPD: a comparison with salbutamol. Respirology 2004, 9, 102–108. [Google Scholar]
- Sovrani, M.P.; Whale, C.I.; Tattersfield, A.E. A benefit-risk assessment of inhaled long-acting β2-agonists in the management of obstructive pulmonary disease. Drug Safety 2004, 27, 689–715. [Google Scholar]
- Lazarus, S.C.; Boushey, H.A.; Fahy, J.V.; Chinchilli, V.M.; Lemanske, R.F., Jr.; Sorkness, C.A.; Kraft, M.; Fish, J.E.; Peters, S.P.; Craig, T.; et al. Long-acting beta2-agonist monotherapy vs continued therapy with inhaled corticosteroids in patientswith persistent asthma: a randomized controlled trial. JAMA 2001, 285, 2583–2593. [Google Scholar]
- Gibson, P.G.; Powell, H.; Ducharme, F.M. Differential effects of maintenance long-acting beta-agonist and inhaled corticosteroid on asthma control and asthma exacerbations. J. Allergy Clin. Immunol. 2007, 119, 344–350. [Google Scholar]
- Woolcock, A.; Lundback, B.; Ringdal, N.; Jacques, L.A. Comparison of addition of salmeterol to inhaled steroids with doubling of the dose of inhaled steroids. Am. J. Respir. Crit. Care Med. 1996, 153, 1481–1488. [Google Scholar]
- Pauwels, R.A.; Löfdahl, C.G.; Postma, D.S.; Tattersfield, A.E.; O'Byrne, P.; Barnes, P.J.; Ullman, A. Effect of inhaled formoterol and budesonide on exacerbations of asthma. N. Engl. J. Med. 1997, 337, 1405–1411, [Erratum, N. Engl. J. Med.1998, 338, 139.]. [Google Scholar] [CrossRef] [PubMed]
- Giembycz, M.A.; Kaur, M.; Leigh, R.; Newton, R. A Holy Grail of asthma management: toward understanding how long-acting β2-adrenoceptor agonists enhance the clinical efficacy of inhaled corticosteroids. Br. J. Pharmacol. 2008, 153, 1090–1104. [Google Scholar]
- Papi, A.; Canonica, G.W.; Maestrelli, P.; Paggiaro, P.L.; Olivieri, D.; Pozzi, E.; Crimi, N.; Vignola, A.M.; Morelli, P.; Nicolini, G.; et al. Rescue use of beclomethasone and albuterol in a single inhaler for mild asthma. N. Engl. J. Med. 2007, 356, 2040–2052. [Google Scholar] [CrossRef] [PubMed]
- Barger, G.; Dale, H.H. Chemical structure and sympathomimetic action of amines. J. Physiol. (Lond.) 1910, 41, 19–59. [Google Scholar] [PubMed]
- Benson, R.L.; Perlman, F. Clinical effects of epinephrine by inhalation. J. Allergy 1948, 19, 129–140. [Google Scholar]
- Davies, D.S. Metabolism of isoprenaline and other bronchodilator drugs in man and dog. Bull. Physiopathol. Respir. 1972, 8, 679–682. [Google Scholar]
- Speizer, F.E.; Doll, R.; Heaf, P. Observations on recent increase in mortality from asthma. Br. Med. J. 1968, 1, 335–339. [Google Scholar]
- Fraser, P.; Doll, R. Geographical variations in the epidemic of asthma deaths. Br. J. Prev. Soc. Med. 1971, 25, 34–36. [Google Scholar] [Green Version]
- Jackson, R.T. ; Beaglehole, R.; Rea, H.H; Sutherland, D.C. Mortality from asthma : a new epidemic in New Zealand. Br. Med. J. 1982, 285, 771–774. [Google Scholar] [Green Version]
- Speizer, F.E.; Doll, R.; Heaf, P.; Strang, L.B. Investigation into use of drugs preceding death from asthma. Br. Med. J. 1968, 1, 339–343. [Google Scholar] [Green Version]
- Conolly, M.E.; Davies, D.S.; Dollery, C.T.; George, C.F. Resistance to beta-adrenoceptor stimulants (a possible explanation for the rise in ashtma deaths). Br. J. Pharmacol. 1971, 43, 389–402. [Google Scholar] [Green Version]
- Wilson, J.D.; Sutherland, D.C.; Thomas, A.C. Has the change to beta-agonists combined with oral theophylline increased cases of fatal asthma? Lancet 1981, 1, 1235–1237. [Google Scholar] [PubMed][Green Version]
- Grant, I.W.B. Asthma in New Zealand. Br. Med. J. 1983, 286, 374–377. [Google Scholar] [Green Version]
- Crane, J.; Flatt, A.; Jackson, R.; Ball, M.; Pearce, N.; Burgess, C.; Kwong, T.; Beasley, R. Prescribed fenoterol and death from asthma in New Zealand, 1981-83; Case-control study. Lancet 1989, 1, 917–922. [Google Scholar] [PubMed][Green Version]
- Pearce, N.; Grainger, J.; Atkinson, M.; Crane, J.; Burgess, C.; Culling, C.; Windom, H.; Beasley, R. Case-control study of prescribed fenoterol and death from asthma in New Zealand, 1977-81. Thorax 1990, 45, 170–175. [Google Scholar] [Green Version]
- Spitzer, W.O.; Suissa, S.; Ernst, P.; Horwitz, R.I.; Habbick, B.; Cockcroft, D.; Boivin, J.F.; McNutt, M.; Buist, A.S.; Rebuck, A.S. The use of beta-agonists and the risk of death and near death from asthma. N. Engl. J. Med. 1992, 326, 501–506. [Google Scholar] [Green Version]
- Taylor, D.R.; Sears, M.R.; Herbison, G.P.; Flannery, E.M.; Print, C.G.; Lake, D.C.; Yates, D.M.; Lucas, M.K.; Li, Q. Regular inhaled beta agonist in asthma: effect on exacerbations and lung function. Thorax 1993, 48, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Dennis, S.M.; Sharp, S.J.; Vickers, M.R.; Frost, C.D.; Crompton, G.K.; Barnes, P.J.; Lee, T.H. Regular inhaled salbutamol and asthma control: the TRUST randomized trial. Lancet 2000, 355, 1675–1679. [Google Scholar]
- The British Guidelines on Asthma Management 1995 Review and Position Statement. Thorax 1997, 52 Suppl. 1, S1–S20. [CrossRef]
- Ullman, A.; Svedmyr, N. Salmeterol, a new long-acting inhaled beta2-adrenoceptor agonist: comparison with salbutamol in adult asthmatic patients. Thorax 1988, 43, 674–678. [Google Scholar]
- Faulds, D.; Hollingshead, L.M.; Goa, K.L. Formoterol. A review of its pharmacologic properties and therapeutic efficacy in reversible obstructive airways disease. Drugs 1991, 42, 115–137. [Google Scholar] [CrossRef] [PubMed]
- Grove, A.; Lipworth, B.J. Bronchodilator subsensitivity to salbutamol after twice daily salmeterol in asthmatic patients. Lancet 1995, 346, 201–206. [Google Scholar]
- Newnham, D.M.; McDevitt, D.G.; Lipworth, B.J. Bronchodilator subsensitivity after chronic dosing with eformoterol in patients with asthma. Am. J. Med. 1994, 97, 29–37. [Google Scholar]
- Yates, D.H.; Kharitonov, S.A.; Barnes, P.J. An inhaled glucocorticoid does not prevent tolerance to the bronchoprotective effect of a long-acting β2-agonist. Am. J. Respir. Crit. Care Med. 1996, 154, 1603–1607. [Google Scholar]
- Arvidsson, P.; Larsson, S.; Löfdahl, C-G.; Melander, B.; Svedmyr, N.; Wåhlander, L. Inhaled formoterol during one year in asthma: a comparison with salbutamol. Eur. Respir. J. 1991, 4, 1168–1173. [Google Scholar] [PubMed]
- Castle, W.; Fuller, R.; Hall, J.; Palmer, J. Serevent nationwide surveillance study: comparison of salmeterol with salbutamol in asthmatic patients who require regular bronchodilator treatment. Br. Med. J. 1993, 306, 1034–1037. [Google Scholar]
- Nelson, H.S.; Weiss, S.T.; Bleecker, E.R.; Yancey, S.W.; Dorinsky, P.M. The Salmeterol Multicenter Asthma Research Trial. A comparison of usual pharmacotherapy for asthma or usual pharmacotherapy plus salmeterol. Chest 2006, 129, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Anderson, H.R.; Ayres, J.G.; Sturdy, P.M.; Bland, J.M.; Butland, B.K.; Peckitt, C.; Taylor, J.C.; Victor, C.R. Bronchodilator treatment and deaths from asthma: case-control study. Br. Med. J. 2005, 330, 117–123. [Google Scholar]
- Maringe, C.; Rickard, K.; DiSantostefano, R.; Davis, K.; Kiri, V. Concomitant use of long-acting β-agonists with inhaled corticosteroids among asthma patients in the UK primary care. Eur. Respir. J. 2007, 30 Suppl. 51, 3698. [Google Scholar]
- Food and Drug Administration Center for Drug Evaluation and Research. Summary Minutes of the Pulmonary-Allergy Drugs Advisory Committee. Available online: www.fda.gov/ohrms/dockets/ac/05/minutes/2005-4148M1_Final.pdf Last accessed: July 2008. Last updated: June 13, 2005..
- Mann, M.; Chowdhury, B.; Sullivan, E.; Nicklas, R; Anthracite, R.; Meyer, R.J. Serious asthma exacerbations in asthmatics treated with high-dose formoterol. Chest 2003, 124, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, J.; LaForce, C.; Friedman, B.; Sokol, W.; Till, D.; Della Cioppa, G.; van As, A. Formoterol, 24 µg bid, and serious asthma exacerbations: similar rates compared with formoterol, 12 µg bid, with and without extra doses taken on demand, and placebo. Chest 2006, 129, 27–38. [Google Scholar] [PubMed]
- Sears, M.R.; Ottosson, A.; Radner, F.; Suissa, S. .Long-acting β-agonists: a review of formoterol safety data from asthma clinical trials. Eur. Respir. J. 2009, 33, 21–32. [Google Scholar]
- Battram, C.; Charlton, S.J.; Cuenoud, B.; Dowling, M.R.; Fairhurst, R.A.; Farr, D.; Fozard, J.R.; Leighton-Davies, J.R.; Lewis, C.A.; McEvoy, L.; et al. In vitro and in vivo pharmacological characterization of 5-[(R)-2-(5,6-diethyl-indan-2-ylamino)-1-hydroxy-ethyl]-8-hydroxy-1H-quinolin-2-one (indacaterol), a novel inhaled β2 adrenoceptor agonist with a 24-h duration of action. J. Pharmacol. Exp. Ther. 2006, 317, 762–770. [Google Scholar] [CrossRef] [PubMed]
- Sturton, R.G.; Trifilieff, A.; Nicholson, A.G.; Barnes, P.J. Pharmacological characterization of indacaterol, a novel once daily inhaled β2-adrenoceptor agonist, on small airways in human and rat precision-cut lung slices. J. Pharmacol. Exp. Ther. 2008, 324, 270–275. [Google Scholar] [PubMed]
- Beeh, K.M.; Derom, E.; Kanniess, F.; Cameron, R.; Higgins, M.; van As, A. Indacaterol, a novel inhaled β2-agonist, provides sustained 24 h bronchodilation in asthma. Eur. Respir. J. 2007, 29, 871–878. [Google Scholar] [CrossRef] [PubMed]
- Pauwels, R.A.; Löfdahl, C.G.; Postma, D.S.; Tattersfield, A.E.; O’Byrne, P.; Barnes, P.J.; Ullman, A. Effect of inhaled formoterol and budesonide on exacerbations of asthma. Formoterol and Corticosteroids Establishing Therapy (FACET) International Study Group. N. Engl. J. Med. 1997, 337, 1405–1411. [Google Scholar] [PubMed]
- Price, D.; Dutchman, D.; Mawson, A.; Bodalia, B.; Duggan, S.; Todd, P. Early asthma control and maintenance with eformoterol following reduction of inhaled corticosteroid dose. Thorax 2002, 57, 791–798. [Google Scholar]
- Currie, G.P.; Lee, D.K.; Srivastava, P. Long-acting bronchodilator or leukotriene modifier as add-on therapy to inhaled corticosteroids in persistent asthma? Chest 2005, 128, 2954–2962. [Google Scholar] [PubMed]
- FDA Public Health Advisory. Serevent Diskus (salmeterol xinafoate inhalation powder), Advair Diskus (fluticasone propionate & salmeterol inhalation powder), Foradil Aerolizer (formoterol fumarate inhalation powder). Date last updated: May 15, 2006.. Available online: www.fda.gov/cder/drug/advisory/LABA.htm October 30, 2008.
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Barisione, G.; Baroffio, M.; Crimi, E.; Brusasco, V. Beta-Adrenergic Agonists. Pharmaceuticals 2010, 3, 1016-1044. https://doi.org/10.3390/ph3041016
Barisione G, Baroffio M, Crimi E, Brusasco V. Beta-Adrenergic Agonists. Pharmaceuticals. 2010; 3(4):1016-1044. https://doi.org/10.3390/ph3041016
Chicago/Turabian StyleBarisione, Giovanni, Michele Baroffio, Emanuele Crimi, and Vito Brusasco. 2010. "Beta-Adrenergic Agonists" Pharmaceuticals 3, no. 4: 1016-1044. https://doi.org/10.3390/ph3041016
APA StyleBarisione, G., Baroffio, M., Crimi, E., & Brusasco, V. (2010). Beta-Adrenergic Agonists. Pharmaceuticals, 3(4), 1016-1044. https://doi.org/10.3390/ph3041016