Targeting the Tumour: Cell Penetrating Peptides for Molecular Imaging and Radiotherapy

{kind=link}
Abstract
:1. Introduction
2. Uptake Mechanisms and Repercussions for Applications
3. Applications of CPPs for in Vivo Molecular Imaging (in Oncology)
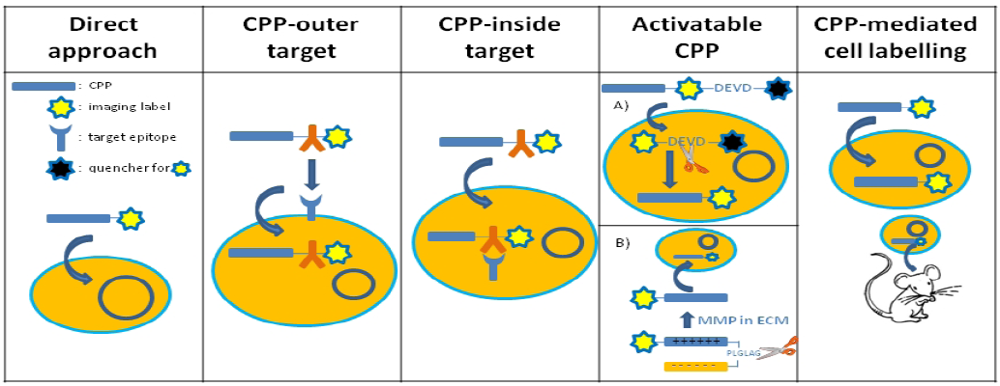
3.1. The Direct Approach
3.2. CPP Constructs with an Intracellular or Intranuclear Target
3.3. CPP Constructs with an Extracellular Target
3.4. Activatable CPP Constructs
3.5. Use of CPP to Track Prelabelled Cells
4. Applications of CPPs for Molecular Radiotherapy (In Oncology)
5. Future Applications: Pitfalls and Considerations
References
- Foerg, C.; Merkle, H.P. On the biomedical promise of cell penetrating peptides: Limits versus prospects. J. Pharm. Sci. 2008, 97, 144–162. [Google Scholar] [CrossRef] [PubMed]
- Juliano, R.; Alam, M.R.; Dixit, V.; Kang, H. Mechanisms and strategies for effective delivery of antisense and siRNA oligonucleotides. Nucleic Acids Res. 2008, 36, 4158–4171. [Google Scholar]
- Kersemans, V.; Kersemans, K.; Cornelissen, B. Cell penetrating peptides for in vivo molecular imaging applications. Curr. Pharm. Des. 2008, 14, 2415–2447. [Google Scholar] [CrossRef] [PubMed]
- Torchilin, V.P. Cell penetrating peptide-modified pharmaceutical nanocarriers for intracellular drug and gene delivery. Biopolymers 2008, 90, 604–610. [Google Scholar]
- Vives, E.; Schmidt, J.; Pelegrin, A. Cell-penetrating and cell-targeting peptides in drug delivery. Biochim. Biophys. Acta 2008, 1786, 126–138. [Google Scholar] [PubMed]
- El-Sayed, A.; Futaki, S.; Harashima, H. Delivery of macromolecules using arginine-rich cell-penetrating peptides: Ways to overcome endosomal entrapment. AAPS J. 2009, 11, 13–22. [Google Scholar]
- Fonseca, S.B.; Pereira, M.P.; Kelley, S.O. Recent advances in the use of cell-penetrating peptides for medical and biological applications. Adv. Drug Deliv. Rev. 2009, 61, 953–964. [Google Scholar]
- Heitz, F.; Morris, M.C.; Divita, G. Twenty years of cell-penetrating peptides: From molecular mechanisms to therapeutics. Br. J. Pharmacol. 2009, 157, 195–206. [Google Scholar]
- Pujals, S.; Giralt, E. Proline-rich, amphipathic cell-penetrating peptides. Adv. Drug Deliv. Rev. 2008, 60, 473–484. [Google Scholar]
- Pujals, S.; Fernandez-Carneado, J.; Ludevid, M.D.; Giralt, E. D-SAP: A new, noncytotoxic, and fully protease resistant cell-penetrating peptide. Chem. Med. Chem. 2008, 3, 296–301. [Google Scholar]
- Schmidt, N.; Mishra, A.; Lai, G.H.; Wong, G.C. Arginine-rich cell-penetrating peptides. FEBS Lett. 2009, in press.. [Google Scholar]
- Johnson, L.N.; Cashman, S.M.; Kumar-Singh, R. Cell-penetrating peptide for enhanced delivery of nucleic acids and drugs to ocular tissues including retina and cornea. Mol. Ther. 2008, 16, 107–114. [Google Scholar]
- Sheng, J.; Oyler, G.; Zhou, B.; Janda, K.; Shoemaker, C.B. Identification and characterization of a novel cell-penetrating peptide. Biochem. Biophys. Res. Commun. 2009, 382, 236–240. [Google Scholar]
- Johnson, L.N.; Cashman, S.M.; Read, S.P.; Kumar-Singh, R. Cell penetrating peptide POD mediates delivery of recombinant proteins to retina, cornea and skin. Vision Res. 2009. [Google Scholar] [CrossRef]
- Duchardt, F.; Ruttekolk, I.R.; Verdurmen, W.P.; Lortat-Jacob, H.; Burck, J.; Hufnagel, H.; Fischer, R.; van den Heuvel, M.; Lowik, D.W.; Vuister, G.W.; Ulrich, A.; de Waard, M.; Brock, R. A Cell-penetrating Peptide Derived from Human Lactoferrin with Conformation-dependent Uptake Efficiency. J. Biol. Chem. 2009, 284, 36099–36108. [Google Scholar]
- Schwarze, S.R.; Ho, A.; Vocero-Akbani, A.; Dowdy, S.F. In vivo protein transduction: Delivery of a biologically active protein into the mouse. Science 1999, 285, 1569–1572. [Google Scholar] [PubMed]
- Dietz, G.P.; Bahr, M. Delivery of bioactive molecules into the cell: The Trojan horse approach. Mol. Cell. Neurosci. 2004, 27, 85–131. [Google Scholar]
- Snyder, E.L.; Dowdy, S.F. Recent advances in the use of protein transduction domains for the delivery of peptides, proteins and nucleic acids in vivo. Exp. Opin Drug Deliv 2005, 2, 43–51. [Google Scholar] [CrossRef]
- Gros, E.; Deshayes, S.; Morris, M.C.; Aldrian-Herrada, G.; Depollier, J.; Heitz, F.; Divita, G. A non-covalent peptide-based strategy for protein and peptide nucleic acid transduction. Biochim. Biophys. Acta 2006, 1758, 384–393. [Google Scholar] [CrossRef] [PubMed]
- Myou, S.; Leff, A.R.; Myo, S.; Boetticher, E.; Meliton, A.Y.; Lambertino, A.T.; Liu, J.; Xu, C.; Munoz, N.M.; Zhu, X. Activation of group IV cytosolic phospholipase A2 in human eosinophils by phosphoinositide 3-kinase through a mitogen-activated protein kinase-independent pathway. J. Immunol. 2003, 171, 4399–4405. [Google Scholar]
- Cao, G.; Pei, W.; Ge, H.; Liang, Q.; Luo, Y.; Sharp, F.R.; Lu, A.; Ran, R.; Graham, S.H.; Chen, J. In Vivo Delivery of a Bcl-xL Fusion Protein Containing the TAT Protein Transduction Domain Protects against Ischemic Brain Injury and Neuronal Apoptosis. J. Neurosci. 2002, 22, 5423–5431. [Google Scholar] [PubMed]
- Chen, L.; Harrison, S.D. Cell-penetrating peptides in drug development: Enabling intracellular targets. Biochem. Soc. Trans. 2007, 35, 821–825. [Google Scholar]
- Szeto, H.H.; Schiller, P.W.; Zhao, K.; Luo, G. Fluorescent dyes alter intracellular targeting and function of cell-penetrating tetrapeptides. FASEB J. 2005, 19, 118–120. [Google Scholar]
- Duchardt, F.; Fotin-Mleczek, M.; Schwarz, H.; Fischer, R.; Brock, R. A comprehensive model for the cellular uptake of cationic cell-penetrating peptides. Traffic 2007, 8, 848–866. [Google Scholar]
- Ziegler, A.; Seelig, J. Interaction of the protein transduction domain of HIV-1 TAT with heparan sulfate: Binding mechanism and thermodynamic parameters. Biophys. J. 2004, 86, 254–263. [Google Scholar]
- Richard, J.P.; Melikov, K.; Vives, E.; Ramos, C.; Verbeure, B.; Gait, M.J.; Chernomordik, L.V.; Lebleu, B. Cell-penetrating peptides. A reevaluation of the mechanism of cellular uptake. J. Biol. Chem. 2003, 278, 585–590. [Google Scholar] [PubMed]
- Fittipaldi, A.; Ferrari, A.; Zoppe, M.; Arcangeli, C.; Pellegrini, V.; Beltram, F.; Giacca, M. Cell membrane lipid rafts mediate caveolar endocytosis of HIV-1 Tat fusion proteins. J. Biol. Chem. 2003, 278, 34141–34149. [Google Scholar]
- Kaplan, I.M.; Wadia, J.S.; Dowdy, S.F. Cationic TAT peptide transduction domain enters cells by macropinocytosis. J Control. Release 2005, 102, 247–253. [Google Scholar]
- Nakase, I.; Niwa, M.; Takeuchi, T.; Sonomura, K.; Kawabata, N.; Koike, Y.; Takehashi, M.; Tanaka, S.; Ueda, K.; Simpson, J.C.; Jones, A.T.; Sugiura, Y.; Futaki, S. Cellular uptake of arginine-rich peptides: Roles for macropinocytosis and actin rearrangement. Mol. Ther. 2004, 10, 1011–1022. [Google Scholar]
- Richard, J.P.; Melikov, K.; Brooks, H.; Prevot, P.; Lebleu, B.; Chernomordik, L.V. Cellular uptake of unconjugated TAT peptide involves clathrin-dependent endocytosis and heparan sulfate receptors. J. Biol. Chem. 2005, 280, 15300–15306. [Google Scholar]
- Vandenbroucke, R.E.; De Smedt, S.C.; Demeester, J.; Sanders, N.N. Cellular entry pathway and gene transfer capacity of TAT-modified lipoplexes. Biochim. Biophys. Acta 2007, 1768, 571–579. [Google Scholar] [PubMed]
- Fischer, R.; Kohler, K.; Fotin-Mleczek, M.; Brock, R. A stepwise dissection of the intracellular fate of cationic cell-penetrating peptides. J. Biol. Chem. 2004, 279, 12625–12635. [Google Scholar]
- Dupont, E.; Prochiantz, A.; Joliot, A. Identification of a signal peptide for unconventional secretion. J. Biol. Chem. 2007, 282, 8994–9000. [Google Scholar]
- Mayor, S.; Pagano, R.E. Pathways of clathrin-independent endocytosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 603–612. [Google Scholar]
- Polyakov, V.; Sharma, V.; Dahlheimer, J.L.; Pica, C.M.; Luker, G.D.; Piwnica-Worms, D. Novel Tat-peptide chelates for direct transduction of technetium-99m and rhenium into human cells for imaging and radiotherapy. Bioconjug. Chem. 2000, 11, 762–771. [Google Scholar]
- Bullok, K.E.; Dyszlewski, M.; Prior, J.L.; Pica, C.M.; Sharma, V.; Piwnica-Worms, D. Characterization of novel histidine-tagged Tat-peptide complexes dual-labeled with (99m)Tc-tricarbonyl and fluorescein for scintigraphy and fluorescence microscopy. Bioconjug. Chem. 2002, 13, 1226–1237. [Google Scholar]
- Barnett, E.M.; Elangovan, B.; Bullok, K.E.; Piwnica-Worms, D. Selective cell uptake of modified Tat peptide-fluorophore conjugates in rat retina in ex vivo and in vivo models. Invest. Ophthalmol. Vis. Sci. 2006, 47, 2589–2595. [Google Scholar] [PubMed]
- Bullok, K.; Piwnica-Worms, D. Synthesis and characterization of a small, membrane-permeant, caspase-activatable far-red fluorescent peptide for imaging apoptosis. J. Med. Chem. 2005, 48, 5404–5407. [Google Scholar] [CrossRef] [PubMed]
- Barnett, E.M.; Zhang, X.; Maxwell, D.; Chang, Q.; Piwnica-Worms, D. Single-cell imaging of retinal ganglion cell apoptosis with a cell-penetrating, activatable peptide probe in an in vivo glaucoma model. Proc. Natl. Acad. Sci. USA 2009, 106, 9391–9396. [Google Scholar]
- Maxwell, D.; Chang, Q.; Zhang, X.; Barnett, E.M.; Piwnica-Worms, D. An improved cell-penetrating, caspase-activatable, near-infrared fluorescent peptide for apoptosis imaging. Bioconjug. Chem. 2009, 20, 702–709. [Google Scholar] [PubMed]
- Chen, B.; Liu, Q.; Zhang, Y.; Xu, L.; Fang, X. Transmembrane delivery of the cell-penetrating peptide conjugated semiconductor quantum dots. Langmuir 2008, 24, 11866–11871. [Google Scholar]
- Medintz, I.L.; Pons, T.; Delehanty, J.B.; Susumu, K.; Brunel, F.M.; Dawson, P.E.; Mattoussi, H. Intracellular delivery of quantum dot-protein cargos mediated by cell penetrating peptides. Bioconjug. Chem. 2008, 19, 1785–1795. [Google Scholar]
- Michalet, X.; Pinaud, F.F.; Bentolila, L.A.; Tsay, J.M.; Doose, S.; Li, J.J.; Sundaresan, G.; Wu, A.M.; Gambhir, S.S.; Weiss, S. Quantum dots for live cells, in vivo imaging, and diagnostics. Science 2005, 307, 538–544. [Google Scholar] [PubMed]
- Bauer, C.; Bauder-Wuest, U.; Mier, W.; Haberkorn, U.; Eisenhut, M. 131I-labeled peptides as caspase substrates for apoptosis imaging. J. Nucl. Med. 2005, 46, 1066–1074. [Google Scholar]
- Lahorte, C.M.; Vanderheyden, J.L.; Steinmetz, N.; Van de Wiele, C.; Dierckx, R.A.; Slegers, G. Apoptosis-detecting radioligands: Current state of the art and future perspectives. Eur. J. Nucl. Med. Mol. Imaging 2004, 31, 887–919. [Google Scholar]
- Hu, M.; Chen, P.; Wang, J.; Chan, C.; Scollard, D.A.; Reilly, R.M. Site-specific conjugation of HIV-1 tat peptides to IgG: A potential route to construct radioimmunoconjugates for targeting intracellular and nuclear epitopes in cancer. Eur. J. Nucl. Med. Mol. Imaging 2006, 33, 301–310. [Google Scholar]
- Hu, M.; Chen, P.; Wang, J.; Scollard, D.A.; Vallis, K.A.; Reilly, R.M. 123I-labeled HIV-1 tat peptide radioimmunoconjugates are imported into the nucleus of human breast cancer cells and functionally interact in vitro and in vivo with the cyclin-dependent kinase inhibitor, p21(WAF-1/Cip-1). Eur. J. Nucl. Med. Mol. Imaging 2007, 34, 368–377. [Google Scholar] [PubMed]
- Hu, M.; Wang, J.; Chen, P.; Reilly, R.M. HIV-1 Tat peptide immunoconjugates differentially sensitize breast cancer cells to selected antiproliferative agents that induce the cyclin-dependent kinase inhibitor p21WAF-1/CIP-1. Bioconjug. Chem. 2006, 17, 1280–1287. [Google Scholar]
- Cornelissen, B.; Kersemans, V.; McLarty, K.; Tran, L.; Vallis, K.A.; Reilly, R.M. In vivo monitoring of intranuclear p27(kip1) protein expression in breast cancer cells during trastuzumab (Herceptin) therapy. Nucl. Med. Biol. 2009, 36, 811–819. [Google Scholar] [CrossRef] [PubMed]
- Banath, J.P.; Olive, P.L. Expression of phosphorylated histone H2AX as a surrogate of cell killing by drugs that create DNA double-strand breaks. Cancer Res. 2003, 63, 4347–4350. [Google Scholar]
- Cornelissen, B.; Kersemans, V.; Sleeth, K.; Darbar, S.; Smart, S.; Vallis, K. Imaging of DNA double strand breaks in vivo using fluorophore-labelled TAT-immunoconjugates. J. Nucl. Med. 2009, 50, 1015. [Google Scholar]
- Gallazzi, F.; Wang, Y.; Jia, F.; Shenoy, N.; Landon, L.A.; Hannink, M.; Lever, S.Z.; Lewis, M.R. Synthesis of radiometal-labeled and fluorescent cell-permeating peptide-PNA conjugates for targeting the bcl-2 proto-oncogene. Bioconjug. Chem. 2003, 14, 1083–1095. [Google Scholar]
- Zhang, Y.M.; Tung, C.H.; He, J.; Liu, N.; Yanachkov, I.; Liu, G.; Rusckowski, M.; Vanderheyden, J.L. Construction of a novel chimera consisting of a chelator-containing Tat peptide conjugated to a morpholino antisense oligomer for technetium-99m labeling and accelerating cellular kinetics. Nucl. Med. Biol. 2006, 33, 263–269. [Google Scholar]
- Heckl, S.; Pipkorn, R.; Waldeck, W.; Spring, H.; Jenne, J.; von der Lieth, C.W.; Corban-Wilhelm, H.; Debus, J.; Braun, K. Intracellular visualization of prostate cancer using magnetic resonance imaging. Cancer Res. 2003, 63, 4766–4772. [Google Scholar]
- Wang, Y.; Nakamura, K.; Liu, X.; Kitamura, N.; Kubo, A.; Hnatowich, D.J. Simplified preparation via streptavidin of antisense oligomers/carriers nanoparticles showing improved cellular delivery in culture. Bioconjug. Chem. 2007, 18, 1338–1343. [Google Scholar] [CrossRef] [PubMed]
- Su, W.; Mishra, R.; Pfeuffer, J.; Wiesmuller, K.H.; Ugurbil, K.; Engelmann, J. Synthesis and cellular uptake of a MR contrast agent coupled to an antisense peptide nucleic acid--cell- penetrating peptide conjugate. Contrast Media Mol. Imaging 2007, 2, 42–49. [Google Scholar]
- Jain, M.; Chauhan, S.C.; Singh, A.P.; Venkatraman, G.; Colcher, D.; Batra, S.K. Penetratin improves tumor retention of single-chain antibodies: A novel step toward optimization of radioimmunotherapy of solid tumors. Cancer Res. 2005, 65, 7840–7846. [Google Scholar]
- Costantini, D.L.; Chan, C.; Cai, Z.; Vallis, K.A.; Reilly, R.M. (111)In-labeled trastuzumab (Herceptin) modified with nuclear localization sequences (NLS): An Auger electron-emitting radiotherapeutic agent for HER2/neu-amplified breast cancer. J. Nucl. Med. 2007, 48, 1357–1368. [Google Scholar] [PubMed]
- Costantini, D.; McLarty, K.; Lee, H.; Done, S.; Vallis, K.; Reilly, R. The pharmacokinetics, normal tissue toxicity and anti-tumor effects of [111]In-NLS-trastuzumab in mice bearing HER2-overexpressing breast cancer xenografts. J. Nucl. Med. 2009, 50, 571. [Google Scholar]
- Myrberg, H.; Zhang, L.; Mae, M.; Langel, U. Design of a tumor-homing cell-penetrating peptide. Bioconjug. Chem. 2008, 19, 70–75. [Google Scholar]
- Mae, M.; Myrberg, H.; El-Andaloussi, S.; Langel, U. Design of a tumour homing cell-penetrating peptide for drug delivery. Int. J. Pept. Res. Ther. 2009, 15, 1–15. [Google Scholar]
- Jiang, T.; Olson, E.S.; Nguyen, Q.T.; Roy, M.; Jennings, P.A.; Tsien, R.Y. Tumor imaging by means of proteolytic activation of cell-penetrating peptides. Proc. Natl. Acad. Sci. USA 2004, 101, 17867–71782. [Google Scholar]
- Todd, A.A.; Olson, E.S.; Jiang, T.; Wong, E.H.; Nguyen, Q.; Scadeng, M.; Ellies, L.; Tsien, R.Y. Tumor imaging and therapy with activatable cell penetrating peptides. In First AACR International Conference on Molecular Diagnostics in Cancer Therapeutic Development, 12–15 September 2006.
- Conant, K.; St Hillaire, C.; Nagase, H.; Visse, R.; Gary, D.; Haughey, N.; Anderson, C.; Turchan, J.; Nath, A. Matrix metalloproteinase 1 interacts with neuronal integrins and stimulates dephosphorylation of Akt. J. Biol. Chem. 2004, 279, 8056–8062. [Google Scholar]
- Galis, Z.S.; Johnson, C.; Godin, D.; Magid, R.; Shipley, J.M.; Senior, R.M.; Ivan, E. Targeted disruption of the matrix metalloproteinase-9 gene impairs smooth muscle cell migration and geometrical arterial remodeling. Circ. Res. 2002, 91, 852–859. [Google Scholar]
- Lee, M.H.; Verma, V.; Maskos, K.; Nath, D.; Knauper, V.; Dodds, P.; Amour, A.; Murphy, G. Engineering N-terminal domain of tissue inhibitor of metalloproteinase (TIMP)-3 to be a better inhibitor against tumour necrosis factor-alpha-converting enzyme. Biochem. J. 2002, 364, 227–234. [Google Scholar]
- Lockhart, A.C.; Braun, R.D.; Yu, D.; Ross, J.R.; Dewhirst, M.W.; Humphrey, J.S.; Thompson, S.; Williams, K.M.; Klitzman, B.; Yuan, F.; Grichnik, J.M.; Proia, A.D.; Conway, D.A.; Hurwitz, H.I. Reduction of wound angiogenesis in patients treated with BMS-275291, a broad spectrum matrix metalloproteinase inhibitor. Clin. Cancer Res. 2003, 9, 586–593. [Google Scholar]
- Stefanidakis, M.; Koivunen, E. Cell-surface association between matrix metalloproteinases and integrins: Role of the complexes in leukocyte migration and cancer progression. Blood 2006, 108, 1441–1450. [Google Scholar]
- Bremer, C.; Tung, C.H.; Weissleder, R. Molecular imaging of MMP expression and therapeutic MMP inhibition. Acad. Radiol. 2002, 9, 314–315. [Google Scholar]
- Kopka, K.; Breyholz, H.J.; Wagner, S.; Law, M.P.; Riemann, B.; Schroer, S.; Trub, M.; Guilbert, B.; Levkau, B.; Schober, O.; Schafers, M. Synthesis and preliminary biological evaluation of new radioiodinated MMP inhibitors for imaging MMP activity in vivo. Nucl. Med. Biol. 2004, 31, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Oltenfreiter, R.; Staelens, L.; Hillaert, U.; Heremans, A.; Noel, A.; Frankenne, F.; Slegers, G. Synthesis, radiosynthesis, in vitro and preliminary in vivo evaluation of biphenyl carboxylic and hydroxamic matrix metalloproteinase (MMP) inhibitors as potential tumor imaging agents. Appl. Radiat. Isot. 2005, 62, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.H.; Fei, X.; DeGrado, T.R.; Wang, J.Q.; Stone, K.L.; Martinez, T.D.; Gay, D.J.; Baity, W.L.; Mock, B.H.; Glick-Wilson, B.E.; Sullivan, M.L.; Miller, K.D.; Sledge, G.W.; Hutchins, G.D. Synthesis, biodistribution and micro-PET imaging of a potential cancer biomarker carbon-11 labeled MMP inhibitor (2R)-2-[[4-(6-fluorohex-1-ynyl)phenyl]sulfonylamino]-3-methylbutyric acid [11C]methyl ester. Nucl. Med. Biol. 2003, 30, 753–760. [Google Scholar]
- Zheng, Q.H.; Fei, X.; Liu, X.; Wang, J.Q.; Bin Sun, H.; Mock, B.H.; Lee Stone, K.; Martinez, T.D.; Miller, K.D.; Sledge, G.W.; Hutchins, G.D. Synthesis and preliminary biological evaluation of MMP inhibitor radiotracers [11C]methyl-halo-CGS 27023A analogs, new potential PET breast cancer imaging agents. Nucl. Med. Biol. 2002, 29, 761–770. [Google Scholar]
- Watkins, C.L.; Brennan, P.; Fegan, C.; Takayama, K.; Nakase, I.; Futaki, S.; Jones, A.T. Cellular uptake, distribution and cytotoxicity of the hydrophobic cell penetrating peptide sequence PFVYLI linked to the proapoptotic domain peptide PAD. J. Control. Release 2009, 140, 237–244. [Google Scholar]
- Bullok, K.E.; Maxwell, D.; Kesarwala, A.H.; Gammon, S.; Prior, J.L.; Snow, M.; Stanley, S.; Piwnica-Worms, D. Biochemical and in vivo characterization of a small, membrane-permeant, caspase-activatable far-red fluorescent peptide for imaging apoptosis. Biochemistry 2007, 46, 4055–4065. [Google Scholar] [PubMed]
- Yeh, H.Y.; Yates, M.V.; Mulchandani, A.; Chen, W. Visualizing the dynamics of viral replication in living cells via Tat peptide delivery of nuclease-resistant molecular beacons. Proc. Natl. Acad. Sci. USA 2008, 105, 17522–17525. [Google Scholar]
- Dmitriev, R.I.; Zhdanov, A.V.; Ponomarev, G.V.; Yashunski, D.V.; Papkovsky, D.B. Intracellular oxygen-sensitive phosphorescent probes based on cell-penetrating peptides. Anal. Biochem. 2009, 398, 24–33. [Google Scholar]
- Liu, M.; Guo, Y.M.; Wu, Q.F.; Yang, J.L.; Wang, P.; Wang, S.C.; Guo, X.J.; Qiang, Y.Q.; Duan, X.Y. Paramagnetic particles carried by cell-penetrating peptide tracking of bone marrow mesenchymal stem cells, a research in vitro. Biochem. Biophys. Res. Commun. 2006, 347, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Guo, Y.M.; Yang, J.L.; Wang, P.; Zhao, L.Y.; Shen, N.; Wang, S.C.; Guo, X.J.; Wu, Q.F. Application of cell penetrating peptide in magnetic resonance imaging of bone marrow mesenchymal stem cells. Acta Biochim. Biophys. Sin. (Shanghai) 2006, 38, 865–873. [Google Scholar] [PubMed]
- Guo, Y.M.; Liu, M.; Yang, J.L.; Guo, X.J.; Wang, S.C.; Duan, X.Y.; Wang, P. Intercellular imaging by a polyarginine derived cell penetrating peptide labeled magnetic resonance contrast agent, diethylenetriamine pentaacetic acid gadolinium. Chin. Med. J. (Engl) 2007, 120, 50–55. [Google Scholar] [PubMed]
- Bhorade, R.; Weissleder, R.; Nakakoshi, T.; Moore, A.; Tung, C.H. Macrocyclic chelators with paramagnetic cations are internalized into mammalian cells via a HIV-tat derived membrane translocation peptide. Bioconjug. Chem. 2000, 11, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Josephson, L.; Tung, C.H.; Moore, A.; Weissleder, R. High-efficiency intracellular magnetic labeling with novel superparamagnetic-Tat peptide conjugates. Bioconjug. Chem. 1999, 10, 186–191. [Google Scholar]
- Nitin, N.; LaConte, L.E.; Zurkiya, O.; Hu, X.; Bao, G. Functionalization and peptide-based delivery of magnetic nanoparticles as an intracellular MRI contrast agent. J. Biol. Inorg. Chem. 2004, 9, 706–712. [Google Scholar]
- Stephenson, K.A.; Banerjee, S.R.; Sogbein, O.O.; Levadala, M.K.; McFarlane, N.; Boreham, D.R.; Maresca, K.P.; Babich, J.W.; Zubieta, J.; Valliant, J.F. A new strategy for the preparation of peptide-targeted technetium and rhenium radiopharmaceuticals. The automated solid-phase synthesis, characterization, labeling, and screening of a peptide-ligand library targeted at the formyl peptide receptor. Bioconjug. Chem. 2005, 16, 1189–1195. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, K.A.; Zubieta, J.; Banerjee, S.R.; Levadala, M.K.; Taggart, L.; Ryan, L.; McFarlane, N.; Boreham, D.R.; Maresca, K.P.; Babich, J.W.; Valliant, J.F. A new strategy for the preparation of peptide-targeted radiopharmaceuticals based on an fmoc-lysine-derived single amino acid chelate (SAAC). automated solid-phase synthesis, NMR characterization, and in vitro screening of fMLF(SAAC)G and fMLF[(SAAC-Re(CO)3)+]G. Bioconjug. Chem. 2004, 15, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Bartholoma, M.; Valliant, J.; Maresca, K.P.; Babich, J.; Zubieta, J. Single amino acid chelates (SAAC): A strategy for the design of technetium and rhenium radiopharmaceuticals. Chem. Commun. (Camb.) 2009, 5, 493–512. [Google Scholar] [PubMed]
- Chen, P.; Wang, J.; Hope, K.; Jin, L.; Dick, J.; Cameron, R.; Brandwein, J.; Minden, M.; Reilly, R.M. Nuclear localizing sequences promote nuclear translocation and enhance the radiotoxicity of the anti-CD33 monoclonal antibody HuM195 labeled with 111In in human myeloid leukemia cells. J. Nucl. Med. 2006, 47, 827–836. [Google Scholar]
- Kersemans, V.; Cornelissen, B.; Minden, M.D.; Brandwein, J.; Reilly, R.M. Drug-resistant AML cells and primary AML specimens are killed by 111In-anti-CD33 monoclonal antibodies modified with nuclear localizing peptide sequences. J. Nucl. Med. 2008, 49, 1546–1554. [Google Scholar]
- Cornelissen, B.; Darbar, S.; Sleeth, K.; Kersemans, V.; Vallis, K. Amplification of IR-induced DNA damage by Auger electron treatment with TAT-radioimmunoconjugates. J. Nucl. Med. 2009, 50, 638. [Google Scholar]
- Jain, M.; Venkatraman, G.; Batra, S.K. Cell-penetrating peptides and antibodies: A new direction for optimizing radioimmunotherapy. Eur. J. Nucl. Med. Mol. Imaging 2007, 34, 973–977. [Google Scholar]
- Anderson, D.C.; Nichols, E.; Manger, R.; Woodle, D.; Barry, M.; Fritzberg, A.R. Tumor cell retention of antibody Fab fragments is enhanced by an attached HIV TAT protein-derived peptide. Biochem. Biophys. Res. Commun. 1993, 194, 876–884. [Google Scholar]
- Niesner, U.; Halin, C.; Lozzi, L.; Gunthert, M.; Neri, P.; Wunderli-Allenspach, H.; Zardi, L.; Neri, D. Quantitation of the tumor-targeting properties of antibody fragments conjugated to cell-permeating HIV-1 TAT peptides. Bioconjug. Chem. 2002, 13, 729–736. [Google Scholar]
- Kameyama, S.; Horie, M.; Kikuchi, T.; Omura, T.; Takeuchi, T.; Nakase, I.; Sugiura, Y.; Futaki, S. Effects of cell-permeating peptide binding on the distribution of 125I-labeled Fab fragment in rats. Bioconjug. Chem. 2006, 17, 597–602. [Google Scholar]
- Nagahara, H.; Vocero-Akbani, A.M.; Snyder, E.L.; Ho, A.; Latham, D.G.; Lissy, N.A.; Becker-Hapak, M.; Ezhevsky, S.A.; Dowdy, S.F. Transduction of full-length TAT fusion proteins into mammalian cells: TAT-p27Kip1 induces cell migration. Nat. Med. 1998, 4, 1449–1452. [Google Scholar]
- Moschos, S.A.; Jones, S.W.; Perry, M.M.; Williams, A.E.; Erjefalt, J.S.; Turner, J.J.; Barnes, P.J.; Sproat, B.S.; Gait, M.J.; Lindsay, M.A. Lung delivery studies using siRNA conjugated to TAT(48-60) and penetratin reveal peptide induced reduction in gene expression and induction of innate immunity. Bioconjug. Chem. 2007, 18, 1450–1459. [Google Scholar]
- Moschos, S.A.; Williams, A.E.; Lindsay, M.A. Cell-penetrating-peptide-mediated siRNA lung delivery. Biochem. Soc. Trans. 2007, 35, 807–810. [Google Scholar]
- Schorderet, D.F.; Manzi, V.; Canola, K.; Bonny, C.; Arsenijevic, Y.; Munier, F.L.; Maurer, F. D-TAT transporter as an ocular peptide delivery system. Clin. Exp. Ophthalmol. 2005, 33, 628–635. [Google Scholar]
- Sethuraman, V.A.; Bae, Y.H. TAT peptide-based micelle system for potential active targeting of anti-cancer agents to acidic solid tumors. J. Control. Release 2007, 118, 216–224. [Google Scholar]
- Stubbs, M.; McSheehy, P.M.; Griffiths, J.R.; Bashford, C.L. Causes and consequences of tumour acidity and implications for treatment. Mol. Med. Today 2000, 6, 15–19. [Google Scholar]
- Kale, A.A.; Torchilin, V.P. Enhanced transfection of tumor cells in vivo using "Smart" pH-sensitive TAT-modified pegylated liposomes. J. Drug Target 2007, 15, 538–545. [Google Scholar] [CrossRef] [PubMed]
- Bidwell, G.L., 3rd ed.; Davis, A.N.; Fokt, I.; Priebe, W.; Raucher, D. A thermally targeted elastin-like polypeptide-doxorubicin conjugate overcomes drug resistance. Invest New Drugs 2007, 25, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Bidwell, G.L., 3rd ed.; Fokt, I.; Priebe, W.; Raucher, D. Development of elastin-like polypeptide for thermally targeted delivery of doxorubicin. Biochem. Pharmacol. 2007, 73, 620–631. [Google Scholar] [CrossRef] [PubMed]
- Gannon, C.J.; Cherukuri, P.; Yakobson, B.I.; Cognet, L.; Kanzius, J.S.; Kittrell, C.; Weisman, R.B.; Pasquali, M.; Schmidt, H.K.; Smalley, R.E.; Curley, S.A. Carbon nanotube-enhanced thermal destruction of cancer cells in a noninvasive radiofrequency field. Cancer 2007, 110, 2654–2665. [Google Scholar]
- Gao, Z.G.; Fain, H.D.; Rapoport, N. Controlled and targeted tumor chemotherapy by micellar-encapsulated drug and ultrasound. J. Control. Release 2005, 102, 203–222. [Google Scholar]
- Beer, A.J.; Haubner, R.; Sarbia, M.; Goebel, M.; Luderschmidt, S.; Grosu, A.L.; Schnell, O.; Niemeyer, M.; Kessler, H.; Wester, H.J.; Weber, W.A.; Schwaiger, M. Positron emission tomography using [18F]Galacto-RGD identifies the level of integrin alpha(v)beta3 expression in man. Clin. Cancer Res. 2006, 12, 3942–3949. [Google Scholar]
- Gehlsen, K.R.; Argraves, W.S.; Pierschbacher, M.D.; Ruoslahti, E. Inhibition of in vitro tumor cell invasion by Arg-Gly-Asp-containing synthetic peptides. J. Cell Biol. 1988, 106, 925–930. [Google Scholar] [CrossRef] [PubMed]
- Begley, R.; Liron, T.; Baryza, J.; Mochly-Rosen, D. Biodistribution of intracellularly acting peptides conjugated reversibly to Tat. Biochem. Biophys. Res. Commun. 2004, 318, 949–954. [Google Scholar]
- Toro, A.; Grunebaum, E. TAT-mediated intracellular delivery of purine nucleoside phosphorylase corrects its deficiency in mice. J. Clin. Invest. 2006, 116, 2717–2726. [Google Scholar]
- Elmquist, A.; Hansen, M.; Langel, U. Structure-activity relationship study of the cell-penetrating peptide pVEC. Biochim. Biophys. Acta 2006, 1758, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Elmquist, A.; Langel, U. In vitro uptake and stability study of pVEC and its all-D analog. Biol. Chem. 2003, 384, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Elmquist, A.; Lindgren, M.; Bartfai, T.; Langel, U. VE-cadherin-derived cell-penetrating peptide, pVEC, with carrier functions. Exp. Cell Res. 2001, 269, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Rennert, R.; Wespe, C.; Beck-Sickinger, A.G.; Neundorf, I. Developing novel hCT derived cell-penetrating peptides with improved metabolic stability. Biochim. Biophys. Acta 2006, 1758, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Horton, K.L.; Stewart, K.M.; Fonseca, S.B.; Guo, Q.; Kelley, S.O. Mitochondria-penetrating peptides. Chem. Biol. 2008, 15, 375–382. [Google Scholar]
- Mahon, K.P.; Potocky, T.B.; Blair, D.; Roy, M.D.; Stewart, K.M.; Chiles, T.C.; Kelley, S.O. Deconvolution of the cellular oxidative stress response with organelle-specific Peptide conjugates. Chem. Biol. 2007, 14, 923–930. [Google Scholar]
- Howl, J.; Nicholl, I.D.; Jones, S. The many futures for cell-penetrating peptides: How soon is now? Biochem. Soc. Trans. 2007, 35, 767–769. [Google Scholar] [CrossRef] [PubMed]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kersemans, V.; Cornelissen, B. Targeting the Tumour: Cell Penetrating Peptides for Molecular Imaging and Radiotherapy. Pharmaceuticals 2010, 3, 600-620. https://doi.org/10.3390/ph3030600
Kersemans V, Cornelissen B. Targeting the Tumour: Cell Penetrating Peptides for Molecular Imaging and Radiotherapy. Pharmaceuticals. 2010; 3(3):600-620. https://doi.org/10.3390/ph3030600
Chicago/Turabian StyleKersemans, Veerle, and Bart Cornelissen. 2010. "Targeting the Tumour: Cell Penetrating Peptides for Molecular Imaging and Radiotherapy" Pharmaceuticals 3, no. 3: 600-620. https://doi.org/10.3390/ph3030600
APA StyleKersemans, V., & Cornelissen, B. (2010). Targeting the Tumour: Cell Penetrating Peptides for Molecular Imaging and Radiotherapy. Pharmaceuticals, 3(3), 600-620. https://doi.org/10.3390/ph3030600