Cell-Penetrating Peptides for Antiviral Drug Development


Abstract
:1. Nature and Scope of the Challenges Presented by Viral Infections
2. The Advent of Cell-Penetrating Peptides

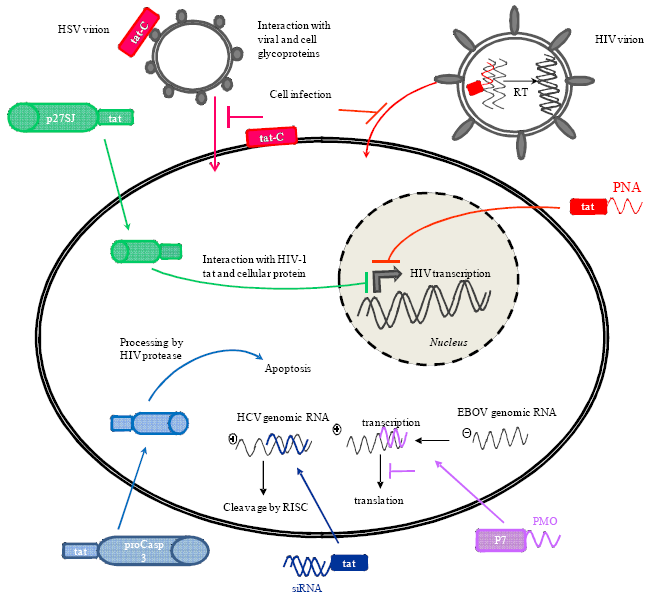{kind=link}
| Antiviral cargo | Targeted virus | Conjugated CPP | Experimental systems | Limitations – CPP composition requirements |
|---|---|---|---|---|
| PMO | RNA viruses | |||
| West Nile virus [66,67] | (RXR)4XB | Cell culture | Dose-dependent toxicity in cell culture and mice | |
| Mouse | ||||
| Japanese encephalitis virus [67] | (RXR)4XB | Cell culture | ||
| St. Louis encephalitis virus [67] | (RXR)4XB | Cell culture | ||
| Dengue virus [68,69,70] | (RXR)4XB | Cell culture | ||
| R5F2R4C | Mouse | |||
| R9F2C | ||||
| SARS coronavirus [71] | R9F2C | Cell culture | ||
| R5F2R4C | ||||
| Mouse hepatitis virus [59,63] | R9F2C (RXR)4XB | Cell culture Mouse | PPMO toxicity in mice when treatment given after MHV challenge | |
| PPMO with higher number of arginine residues exhibit greater antiviral activity in cell culture | ||||
| PPMO with insertions of 6-aminohexanoic acid offer greater protection in mouse | ||||
| Equine arteritis virus [72] | R9F2C | Cell culture | ||
| Porcine reproductive and respiratory syndrome virus [73] | R5F2R4C | Cell culture | ||
| Poliovirus 1 [75] | R9F2C | Cell culture | in vitro toxicity when longer periods of treatment | |
| (RXR)4XB | Mouse | |||
| Human rhinovirus 14 | R9F2C | Cell culture | ||
| [75] | (RXR)4XB | |||
| Coxsackievirus B2 | R9F2C | Cell culture | ||
| [75] | (RXR)4XB | |||
| Coxsackievirus B3 | (RXR)4XB | Cell culture | ||
| [76] | Mouse | |||
| Foot-and-mouth disease virus [74] | R9F2C | Cell culture | ||
| Sindbis virus [77] | R9F2C | Cell culture | ||
| Venezuelan equine encephalitis virus [77] | (RXR)4XB | Cell culture | ||
| Mouse | ||||
| Ebola virus [64,78] | (RXR)4XB | Cell culture Mouse | PPMO with insertions of 6-aminohexanoic acid and higher number of arginine-6-aminohexanoic repeats offer greater protection in mouse | |
| R9F2C | ||||
| (RX)n=2-8B | ||||
| (RB)8B | ||||
| Respiratory syncytial virus [79] | (RXR)4XB | Cell culture | Endosomal entrapment | |
| Mouse | ||||
| Measles virus [80] | (RXR)4XB | Cell culture | ||
| Influenza A virus [81,82,83] | (RXR)4XB | Cell culture | Higher doses of PPMO induced abnormal infiltration of mouse lungs by immune system cells | |
| R5F2R4C | Mouse | |||
| DNA viruses | ||||
| Kaposi’s sarcoma-associated herpesvirus [84,85] | R5F2R4C | Cell culture | ||
| (RXR)4XB | ||||
| Herpesvirus type 1 | (RXR)4XB | Cell culture | ||
| [86] | Mouse | |||
| PNA | HIV-1 [91,92,93,94,95,99,100,101] | Disulfide-linked: | Cell culture Some preliminary mouse studies for tat and penetratin conjugates | Endosomal entrapment requiring lysosomotropic agents Nature of CPP and of CPP-PNA linkage had an effect on conjugate activity |
| tat | ||||
| penetratin | ||||
| transportan | ||||
| transportan 21 | ||||
| transportan 22 | ||||
| R6-penetratin | ||||
| Stably-linked: | ||||
| tat | ||||
| transportan | ||||
| transportan 21 | ||||
| Japanese encephalitis virus [96] | tat | Cell culture | ||
| siRNA | Hepatitis C virus [102] | tat | Cell culture | |
| HIV-1 [15] | nonamer arginine (9R) | Mouse | ||
| non-covalent binding | ||||
| Proteins | HIV-1 [103,104,105] | tat | Cell culture | |
| Human papillomavirus type 18 [106] | 9R | Cell culture | Nature of CPP directly impacted the level of antiviral activity | |
| PTD4 | ||||
3. Delivery of Antisense Agents
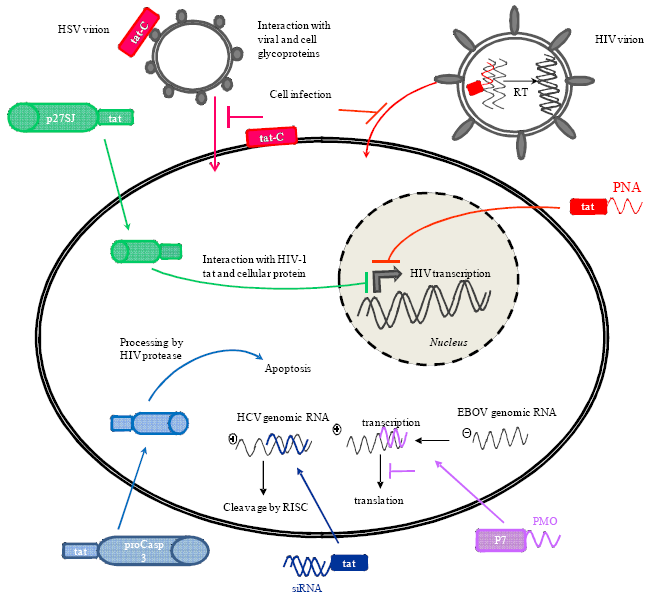
3.1. Delivery of Phosphorodiamidate Morpholino Oligomers (PMOs)
3.1.1. Nature of Peptides Conjugated to PMOs
3.1.2. PPMOs against RNA Viruses
3.1.3. PPMOs against DNA Viruses
3.1.4. Enhancement of PMO Antisense Activity
3.1.5. PPMO Toxicity
3.2. Delivery of Peptide-Nucleic Acids (PNAs)
3.2.2. Preclinical Studies of CPP-PNA Conjugates in Mice
3.3. Delivery of Small Interfering RNAs (siRNAs)
6. Conclusions
References
- Haagmans, B.L.; Andeweg, A.C.; Osterhaus, A. The Application of Genomics to Emerging Zoonotic Viral Diseases. PLos Pathog. 2009, 5, 5. [Google Scholar]
- De Clercq, E. Antivirals and antiviral strategies. Nat. Rev. Microbiol. 2004, 2, 704–720. [Google Scholar]
- Rerks-Ngarm, S.; Pitisuttithum, P.; Nitayaphan, S.; Kaewkungwal, J.; Chiu, J.; Paris, R.; Premsri, N.; Namwat, C.; de Souza, M.; Adams, E.; Benenson, M.; Gurunathan, S.; Tartaglia, J.; McNeil, J.G.; Francis, D.P.; Stablein, D.; Birx, D.L.; Chunsuttiwat, S.; Khamboonruang, C.; Thongcharoen, P.; Robb, M.L.; Michael, N.L.; Kunasol, P.; Kim, J.H.; Investigators, M.-T. Vaccination with ALVAC and AIDSVAX to Prevent HIV-1 Infection in Thailand. N. Engl. J. Med. 2009, 361, 2209–2220. [Google Scholar]
- Wedemeyer, H.; Schuller, E.; Schlaphoff, V.; Stauber, R.E.; Wiegand, J.; Schiefke, I.; Firbas, C.; Jilma, B.; Thursz, M.; Zeuzem, S.; Hofmann, W.P.; Hinrichsen, H.; Tauber, E.; Manns, M.P.; Klade, C.S. Therapeutic vaccine IC41 as late add-on to standard treatment in patients with chronic hepatitis C. Vaccine 2009, 27, 5142–5151. [Google Scholar]
- De Clercq, E. The design of drugs for HIV and HCV. Nat. Rev. Drug Discov. 2007, 6, 1001–1018. [Google Scholar]
- Dykxhoorn, D.M.; Lieberman, J. Silencing viral infection. PLOS Med. 2006, 3, 1000–1004. [Google Scholar]
- Blackburn, G.M.; Gait, M.J.; Loakes, D.; Williams, D.M. Nucleosides and Nucleotides. In Nucleic Acids in Chemistry and Biology, 3rd ed; Royal Society of Chemistry: Cambridge, UK, 2006; pp. 77–142. [Google Scholar]
- Bharti, A.C.; Shukla, S.; Mahata, S.; Hedau, S.; Das, B.C. Anti-human papillomavirus therapeutics: Facts & future. Indian J. Med. Res. 2009, 130, 296–310. [Google Scholar]
- Chander, G.; Sulkowski, M.S.; Jenckes, M.W.; Torbenson, M.S.; Herlong, H.F.; Bass, E.B.; Gebo, K.A. Treatment of chronic hepatitis C: A systematic review. Hepatology 2002, 36, 135–144. [Google Scholar]
- McKeegan, K.S.; Borges-Walmsley, M.I.; Walmsley, A.R. Microbial and viral drug resistance mechanisms. Trends Microbiol. 2002, 10, S8–S14. [Google Scholar]
- Zhu, J.M.; Trang, P.; Kim, K.; Zhou, T.H.; Deng, H.Y.; Liu, F.Y. Effective inhibition of Rta expression and lytic replication of Kaposi's sarcoma-associated herpesvirus by human RNase P. Proc. Natl. Acad. Sci. USA 2004, 101, 9073–9078. [Google Scholar]
- Duzgunes, N.; Simoes, S.; Slepushkin, V.; Pretzer, E.; Flasher, D.; Salem, II; Steffan, G.; Konopka, K.; De Lima, M.C.P. Delivery of antiviral agents in liposomes. Liposomes, Part E. Methods Enzymol. 2005, 391, 351–373. [Google Scholar] [CrossRef] [PubMed]
- Clayton, R.; Ohagen, A.; Nicol, F.; Del Vecchio, A.M.; Jonckers, T.H.M.; Goethals, O.; Van Loock, M.; Michiels, L.; Grigsby, J.; Xu, Z.; Zhang, Y.P.; Gutshall, L.L.; Cunningham, M.; Jiang, H.; Bola, S.; Sarisky, R.T.; Hertogs, K. Sustained and specific in vitro inhibition of HIV-1 replication by a protease inhibitor encapsulated in gp120-targeted liposomes. Antiviral Res. 2009, 84, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.H.; Xin, P.; Morrey, J.D.; Clawson, G.A. Self-processing ribozyme cassette: Utility against human papillomavirus 11 E6/E7 mRNA and hepatitis B virus. Mol. Ther. 2004, 9, 596–606. [Google Scholar]
- Kumar, P.; Ban, H.S.; Kim, S.S.; Wu, H.Q.; Pearson, T.; Greiner, D.L.; Laouar, A.; Yao, J.H.; Haridas, V.; Habiro, K.; Yang, Y.G.; Jeong, J.H.; Lee, K.Y.; Kim, Y.H.; Kim, S.W.; Peipp, M.; Fey, G.H.; Manjunath, N.; Shultz, L.D.; Lee, S.K.; Shankar, P. T cell-specific siRNA delivery suppresses HIV-1 infection in humanized mice. Cell 2008, 134, 577–586. [Google Scholar]
- Yu, X.K.; Trang, P.; Shah, S.; Atanasov, I.; Kim, Y.H.; Bai, Y.; Zhou, Z.H.; Liu, F.Y. Dissecting human cytomegalovirus gene function and capsid maturation by ribozyme targeting and electron cryomicroscopy. Proc. Natl. Acad. Sci. USA 2005, 102, 7103–7108. [Google Scholar]
- Shiver, J.W.; Emini, E.A. Recent advances in the development of HIV-1 vaccines using replication-incompetent adenovirus vectors. Annu. Rev. Med. 2004, 55, 355–372. [Google Scholar]
- Fattal, E.; Couvreur, P.; Dubernet, C. "Smart" delivery of antisense oligonucleotides by anionic pH-sensitive liposomes. Adv. Drug Delivery Rev. 2004, 56, 931–946. [Google Scholar] [CrossRef]
- Sekaly, R.P. The failed HIV Merck vaccine study: A step back or a launching point for future vaccine development? J. Exp. Med. 2008, 205, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Vives, E.; Brodin, P.; Lebleu, B. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J. Biol. Chem. 1997, 272, 16010–16017. [Google Scholar]
- Frankel, A.D.; Pabo, C.O. Cellular uptake of the Tat protein from human immunodeficiency virus. Cell 1988, 55, 1189–1193. [Google Scholar]
- Derossi, D.; Joliot, A.H.; Chassaing, G.; Prochiantz, A. The 3rd helix of the antennapedia homeodomain translocates through biological membranes. J. Biol. Chem. 1994, 269, 10444–10450. [Google Scholar]
- Elliott, G.; Ohare, P. Intercellular trafficking and protein delivery by a herpesvirus structural protein. Cell 1997, 88, 223–233. [Google Scholar]
- Phelan, A.; Elliott, G.; O'Hare, P. Intercellular delivery of functional p53 by the herpesvirus protein VP22. Nat. Biotechnol. 1998, 16, 440–443. [Google Scholar]
- Arruda, S.; Bomfim, G.; Knights, R.; Huimabyron, T.; Riley, L.W. Cloning of an Mycobacterium-tuberculosis DNA fragment associated with entry and survival inside cells. Science 1993, 261, 1454–1457. [Google Scholar] [PubMed]
- Lu, S.W.; Tager, L.A.; Chitale, S.; Riley, L.W. A cell-penetrating peptide derived from mammalian cell uptake protein of Mycobacterium tuberculosis. Anal. Biochem. 2006, 353, 7–14. [Google Scholar]
- Pooga, M.; Hallbrink, M.; Zorko, M.; Langel, U. Cell penetration by transportan. FASEB J. 1998, 12, 67–77. [Google Scholar]
- Oehlke, J.; Scheller, A.; Wiesner, B.; Krause, E.; Beyermann, M.; Klauschenz, E.; Melzig, M.; Bienert, M. Cellular uptake of an alpha-helical amphipathic model peptide with the potential to deliver polar compounds into the cell interior non-endocytically. Biochim. Biophys. Acta Biomembr. 1998, 1414, 127–139. [Google Scholar]
- Rothbard, J.B.; Garlington, S.; Lin, Q.; Kirschberg, T.; Kreider, E.; McGrane, P.L.; Wender, P.A.; Khavari, P.A. Conjugation of arginine oligomers to cyclosporin A facilitates topical delivery and inhibition of inflammation. Nat. Med. 2000, 6, 1253–1257. [Google Scholar]
- Järver, P.; Langel, K.; El-Andaloussi, S.; Langel, U. Applications of cell-penetrating peptides in regulation of gene expression. Biochem. Soc. Trans. 2007, 35, 770–774. [Google Scholar]
- Henriques, S.T.; Melo, M.N.; Castanho, M. Cell-penetrating peptides and antimicrobial peptides: How different are they? Biochem. J. 2006, 399, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Gupta, B.; Levchenko, T.S.; Torchilin, V.P. Intracellular delivery of large molecules and small particles by cell-penetrating proteins and peptides. Adv. Drug Delivery Rev. 2005, 57, 637–651. [Google Scholar]
- Fawell, S.; Seery, J.; Daikh, Y.; Moore, C.; Chen, L.L.; Pepinsky, B.; Barsoum, J. Tat-mediated delivery of heterologous proteins into cells. Proc. Natl. Acad. Sci. USA 1994, 91, 664–668. [Google Scholar]
- Morris, M.C.; Vidal, P.; Chaloin, L.; Heitz, F.; Divita, G. A new peptide vector for efficient delivery of oligonucleotides into mammalian cells. Nucleic Acids Res. 1997, 25, 2730–2736. [Google Scholar]
- Troy, C.M.; Derossi, D.; Prochiantz, A.; Greene, L.A.; Shelanski, M.L. Downregulation of Cu/Zn superoxide dismutase leads to cell death via the nitric oxide-peroxynitrite pathway. J. Neurosci. 1996, 16, 253–261. [Google Scholar]
- Pooga, M.; Soomets, U.; Hallbrink, M.; Valkna, A.; Saar, K.; Rezaei, K.; Kahl, U.; Hao, J.X.; Xu, X.J.; Wiesenfeld-Hallin, Z.; Hokfelt, T.; Bartfai, T.; Langel, U. Cell penetrating PNA constructs regulate galanin receptor levels and modify pain transmission in vivo. Nat. Biotechnol. 1998, 16, 857–861. [Google Scholar] [PubMed]
- Good, L.; Awasthi, S.K.; Dryselius, R.; Larsson, O.; Nielsen, P.E. Bactericidal antisense effects of peptide-PNA conjugates. Nat. Biotechnol. 2001, 19, 360–364. [Google Scholar]
- Meade, B.R.; Dowdy, S.F. Exogenous siRNA delivery using peptide transduction domains/cell penetrating peptides. Adv. Drug Delivery Rev. 2007, 59, 134–140. [Google Scholar]
- Eguchi, A.; Dowdy, S.F. siRNA delivery using peptide transduction domains. Trends Pharmacol. Sci. 2009, 30, 341–345. [Google Scholar]
- Lewin, M.; Carlesso, N.; Tung, C.H.; Tang, X.W.; Cory, D.; Scadden, D.T.; Weissleder, R. Tat peptide-derivatized magnetic nanoparticles allow in vivo tracking and recovery of progenitor cells. Nat. Biotechnol. 2000, 18, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Torchilin, V.P.; Rammohan, R.; Weissig, V.; Levchenko, T.S. TAT peptide on the surface of liposomes affords their efficient intracellular delivery even at low temperature and in the presence of metabolic inhibitors. Proc. Natl. Acad. Sci. USA 2001, 98, 8786–8791. [Google Scholar]
- Rajarao, G.K.; Nekhotiaeva, N.; Good, L. Peptide-mediated delivery of green fluorescent protein into yeasts and bacteria. FEMS Microbiol. Lett. 2002, 215, 267–272. [Google Scholar]
- Corradin, S.; Ransijn, A.; Corradin, G.; Bouvier, J.; Delgado, M.B.; Fernandez-Carneado, J.; Mottram, J.C.; Vergeres, G.; Mauel, J. Novel peptide inhibitors of Leishmania gp63 based on the cleavage site of MARCKS (myristoylated alanine-rich C kinase substrate)-related protein. Biochem. J. 2002, 367, 761–769. [Google Scholar]
- Lee, H.; Jefferies, R.; Watt, P.; Hopkins, R.; Sotzik, F.; Reid, S.; Armson, A.; Boxell, A.; Ryan, U. In vitro analysis of the TAT protein transduction domain as a drug delivery vehicle in protozoan parasites. Exp. Parasitol. 2008, 118, 303–307. [Google Scholar] [CrossRef] [PubMed]
- Schwarze, S.R.; Hruska, K.A.; Dowdy, S.F. Protein transduction: Unrestricted delivery into all cells? Trends Cell Biol. 2000, 10, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Schwarze, S.R.; Ho, A.; Vocero-Akbani, A.; Dowdy, S.F. In vivo protein transduction: Delivery of a biologically active protein into the mouse. Science 1999, 285, 1569–1572. [Google Scholar] [PubMed]
- Harada, H.; Hiraoka, M.; Kizaka-Kondoh, S. Antitumor effect of TAT-oxygen-dependent degradation-caspase-3 fusion protein specifically stabilized and activated in hypoxic tumor cells. Cancer Res. 2002, 62, 2013–2018. [Google Scholar]
- Bian, J.; Popovic, Z.B.; Benejam, C.; Kiedrowski, M.; Rodriguez, L.L.; Penn, M.S. Effect of cell-based intercellular delivery of transcription factor GATA4 on ischemic cardiomyopathy. Circ. Res. 2007, 100, 1626–1633. [Google Scholar]
- Cao, G.D.; Pei, W.; Ge, H.L.; Liang, Q.H.; Luo, Y.M.; Sharp, F.R.; Lu, A.G.; Ran, R.Q.; Graham, S.H.; Chen, J. In vivo delivery of a Bcl-xL fusion protein containing the TAT protein transduction domain protects against ischemic brain injury and neuronal apoptosis. J. Neurosci. 2002, 22, 5423–5431. [Google Scholar] [PubMed]
- Jearawiriyapaisarn, N.; Moulton, H.M.; Buckley, B.; Roberts, J.; Sazani, P.; Fucharoen, S.; Iversen, P.L.; Kole, R. Sustained dystrophin expression induced by peptide-conjugated morpholino oligomers in the muscles of mdx mice. Mol. Ther. 2008, 16, 1624–1629. [Google Scholar]
- Price, V.H. Therapy of alopecia areata: On the cusp and in the future. J. Invest. Dermat. Symposium Proc. 2003, 8, 207–211. [Google Scholar]
- Stephenson, M.L.; Zamecnik, P.C. Inhibition of Rous-sarcoma viral-RNA translation by a specific oligodeoxyribonucleotide. Proc. Natl. Acad. Sci. USA 1978, 75, 285–288. [Google Scholar]
- Zamecnik, P.C.; Stephenson, M.L. Inhibition of Rous-sarcoma virus-replication and cell transformation by a specific oligodeoxynucleotide. Proc. Natl. Acad. Sci. USA 1978, 75, 280–284. [Google Scholar]
- Summerton, J.; Weller, D. Antisense properties of morpholino oligomers. Nucleosides Nucleotides 1997, 16, 889–898. [Google Scholar]
- Summerton, J.E. Morpholino, siRNA, and S-DNA compared: Impact of structure and mechanism of action on off-target effects and sequence specificity. Curr. Top. Med. Chem. 2007, 7, 651–660. [Google Scholar] [CrossRef] [PubMed]
- Moulton, H.M.; Nelson, M.H.; Hatlevig, S.A.; Reddy, M.T.; Iversen, P.L. Cellular uptake of antisense morpholino oligomers conjugated to arginine-rich peptides. Bioconjugate Chem. 2004, 15, 290–299. [Google Scholar]
- Amantana, A.; Moulton, H.M.; Cate, M.L.; Reddy, M.T.; Whitehead, T.; Hassinger, J.N.; Youngblood, D.S.; Iversen, P.L. Pharmacokinetics, biodistribution, stability and toxicity of a cell-penetrating peptide-morpholino oligomer conjugate. Bioconjug. Chem. 2007, 18, 1325–1331. [Google Scholar] [PubMed]
- Moulton, J.D.; Jiang, S. Gene Knockdowns in Adult Animals: PPMOs and Vivo-Morpholinos. Molecules 2009, 14, 1304–1323. [Google Scholar]
- Neuman, B.W.; Stein, D.A.; Kroeker, A.D.; Paulino, A.D.; Moulton, H.M.; Iversen, P.L.; Buchmeier, M.J. Antisense morpholino-oligomers directed against the 5' end of the genome inhibit coronavirus proliferation and growth. J. Virol. 2004, 78, 5891–5899. [Google Scholar]
- Stein, D.A. Inhibition of RNA Virus Infections with Peptide-Conjugated Morpholino Oligomers. Curr. Pharm. Des. 2008, 14, 2619–2634. [Google Scholar]
- Abes, S.; Moulton, H.M.; Clair, P.; Prevot, P.; Youngblood, D.S.; Wu, R.P.; Iversen, P.L.; Lebleu, B. Vectorization of morpholino oligomers by the (R-Ahx-R)(4) peptide allows efficient splicing correction in the absence of endosomolytic agents. J. Control. Release 2006, 116, 304–313. [Google Scholar]
- Abes, R.; Moulton, H.M.; Clair, P.; Yang, S.T.; Abes, S.; Melikov, K.; Prevot, P.; Youngblood, D.S.; Iversen, P.L.; Chernomordik, L.V.; Lebleu, B. Delivery of steric block morpholino oligomers by (R-X-R)(4) peptides: Structure-activity studies. Nucleic Acids Res. 2008, 36, 6343–6354. [Google Scholar]
- Burrer, R.; Neuman, B.W.; Ting, J.P.C.; Stein, D.A.; Moulton, H.M.; Iversen, P.L.; Kuhn, P.; Buchmeier, M.J. Antiviral effects of antisense morpholino oligomers in murine coronavirus infection models. J. Virol. 2007, 81, 5637–5648. [Google Scholar]
- Swenson, D.L.; Warfield, K.L.; Warren, T.K.; Lovejoy, C.; Hassinger, J.N.; Ruthel, G.; Blouch, R.E.; Moulton, H.M.; Weller, D.D.; Iversen, P.L.; Bavari, S. Chemical modifications of antisense morpholino oligomers enhance their efficacy against Ebola virus infection. Antimicrob. Agents Chemother. 2009, 53, 2089–2099. [Google Scholar]
- Youngblood, D.S.; Hatlevig, S.A.; Hassinger, J.N.; Iversen, P.L.; Moulton, H.M. Stability of cell-penetrating peptide-morpholino oligomer conjugates in human serum and in cells. Bioconjugate Chem. 2007, 18, 50–60. [Google Scholar]
- Deas, T.S.; Binduga-Gajewska, I.; Tilgner, M.; Ren, P.; Stein, D.A.; Moulton, H.M.; Iversen, P.L.; Kauffman, E.B.; Kramer, L.D.; Shi, P.Y. Inhibition of flavivirus infections by antisense oligorners specifically suppressing viral translation and RNA replication. J. Virol. 2005, 79, 4599–4609. [Google Scholar]
- Deas, T.S.; Bennett, C.J.; Jones, S.A.; Tilgner, M.; Ren, P.; Behr, M.J.; Stein, D.A.; Iversen, P.L.; Kramer, L.D.; Bernard, K.A.; Shi, P.Y. In vitro resistance selection and in vivo efficacy of morpholino oligomers against West Nile virus. Antimicrob. Agents Chemother. 2007, 51, 2470–2482. [Google Scholar] [PubMed]
- Kinney, R.; Huang, C.; Rose, B.; Kroeker, A.; Iversen, P.; Stein, D. Inhibition of Dengue virus serotypes 1-4 in cell culture with morpholino oligomers. Antiviral Res. 2005, 65, A87–A87. [Google Scholar]
- Holden, K.L.; Stein, D.A.; Pierson, T.C.; Ahmed, A.A.; Clyde, K.; Iversen, P.L.; Harris, E. Inhibition of dengue virus translation and RNA synthesis by a morpholino oligomer targeted to the top of the terminal 3 ' stem-loop structure. Virology 2006, 344, 439–452. [Google Scholar]
- Stein, D.A.; Huang, C.Y.H.; Silengo, S.; Amantana, A.; Crumley, S.; Blouch, R.E.; Iversen, P.L.; Kinney, R.M. Treatment of AG129 mice with antisense morpholino oligomers increases survival time following challenge with dengue 2 virus. J. Antimicrob. Chemother. 2008, 62, 555–565. [Google Scholar]
- Neuman, B.W.; Stein, D.A.; Kroeker, A.D.; Churchill, M.J.; Kim, A.M.; Kuhn, P.; Dawson, P.; Moulton, H.M.; Bestwick, R.K.; Iversen, P.L.; Buchmeier, M.J. Inhibition, escape, and attenuated growth of severe acute respiratory syndrome coronavirus treated with antisense morpholino oligomers. J. Virol. 2005, 79, 9665–9676. [Google Scholar] [PubMed]
- van den Born, E.; Stein, D.A.; Iversen, P.L.; Snijder, E.J. Antiviral activity of morpholino oligomers designed to block various aspects of Equine arteritis virus amplification in cell culture. J. Gen. Virol. 2005, 86, 3081–3090. [Google Scholar]
- Zhang, Y.J.; Stein, D.A.; Fan, S.M.; Wang, K.Y.; Kroeker, A.D.; Meng, X.J.; Iversen, P.L.; Matson, D.O. Suppression of porcine reproductive and respiratory syndrome virus replication by morpholino antisense oligomers. Vet. Microbiol. 2006, 117, 117–129. [Google Scholar]
- Vagnozzi, A.; Stein, D.A.; Iversen, P.L.; Rieder, E. Inhibition of foot-and-mouth disease virus infections in cell cultures with antisense morpholino oligorners. J. Virol. 2007, 81, 11669–11680. [Google Scholar]
- Stone, J.K.; Rijnbrand, R.; Stein, D.A.; Ma, Y.H.; Yang, Y.; Iversen, P.L.; Andino, R. A morpholino oligomer targeting highly conserved internal ribosome entry site sequence is able to inhibit multiple species of picornavirus. Antimicrob. Agents Chemother. 2008, 52, 1970–1981. [Google Scholar]
- Yuan, J.; Stein, D.A.; Lim, T.; Qiu, D.X.; Coughlin, S.; Liu, Z.; Wang, Y.J.; Blouch, R.; Moulton, H.M.; Iversen, P.L.; Yang, D.C. Inhibition of coxsackievirus B3 in cell cultures and in mice by peptide-conjugated morpholino oligomers targeting the internal ribosome entry site. J. Virol. 2006, 80, 11510–11519. [Google Scholar]
- Paessler, S.; Rijnbrand, R.; Stein, D.A.; Ni, H.L.; Yun, N.E.; Dziuba, N.; Borisevich, V.; Seregin, A.; Ma, Y.H.; Blouch, R.; Iversen, P.L.; Zacks, M.A. Inhibition of alphavirus infection in cell culture and in mice with antisense morpholino oligomers. Virology 2008, 376, 357–370. [Google Scholar]
- Enterlein, S.; Warfield, K.L.; Swenson, D.L.; Stein, D.A.; Smith, J.L.; Gamble, C.S.; Kroeker, A.D.; Iversen, P.L.; Bavari, S.; Muhlberger, E. VP35 knockdown inhibits Ebola virus amplification and protects against lethal infection in mice. Antimicrob. Agents Chemother. 2006, 50, 984–993. [Google Scholar]
- Lai, S.H.; Stein, D.A.; Guerrero-Plata, A.; Liao, S.L.; Ivanciuc, T.; Hong, C.; Iversen, P.L.; Casola, A.; Garofalo, R.P. Inhibition of respiratory syncytial virus infections with morpholino oligomers in cell cultures and in mice. Mol. Ther. 2008, 16, 1120–1128. [Google Scholar]
- Sleeman, K.; Stein, D.A.; Tamin, A.; Reddish, M.; Iversen, P.L.; Rota, P.A. Inhibition of measles virus infections in cell cultures by peptide-conjugated morpholino oligomers. Virus Res. 2009, 140, 49–56. [Google Scholar]
- Ge, Q.; Pastey, M.; Kobasa, D.; Puthavathana, P.; Lupfer, C.; Bestwick, R.K.; Iversen, P.L.; Chen, J.; Stein, D.A. Inhibition of multiple subtypes of influenza A virus in cell cultures with morpholino oligomers. Antimicrob. Agents Chemother. 2006, 50, 3724–3733. [Google Scholar]
- Gabriel, G.; Nordmann, A.; Stein, D.A.; Iversen, P.L.; Klenk, H.D. Morpholino oligomers targeting the PB1 and NP genes enhance the survival of mice infected with highly pathogenic influenza A H7N7 virus. J. Gen. Virol. 2008, 89, 939–948. [Google Scholar]
- Lupfer, C.; Stein, D.A.; Mourich, D.V.; Tepper, S.E.; Iversen, P.L.; Pastey, M. Inhibition of influenza A H3N8 virus infections in mice by morpholino oligomers. Arch. Virol. 2008, 153, 929–937. [Google Scholar]
- Zhang, Y.J.; Wang, K.Y.; Stein, D.A.; Patel, D.; Watkins, R.; Moulton, H.M.; Iversen, P.L.; Matson, D.O. Inhibition of replication and transcription activator and latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus by morpholino oligomers. Antiviral Res. 2007, 73, 12–23. [Google Scholar]
- Zhang, Y.J.; Bonaparte, R.S.; Patel, D.; Stein, D.A.; Iversen, P.L. Blockade of viral interleukin-6 expression of Kaposi's sarcoma-associated herpesvirus. Mol. Cancer Ther. 2008, 7, 712–720. [Google Scholar]
- Moerdyk-Schauwecker, M.; Stein, D.A.; Eide, K.; Blouch, R.E.; Bildfell, R.; Iversen, P.; Jin, L. Inhibition of HSV-1 ocular infection with morpholino oligomers targeting ICP0 and ICP27. Antiviral Res. 2009, 84, 131–141. [Google Scholar]
- Nelson, M.H.; Stein, D.A.; Kroeker, A.D.; Hatlevig, S.A.; Iversen, P.L.; Moulton, H.M. Arginine-rich peptide conjugation to morpholino oligomers: Effects on antisense activity and specificity. Bioconjug. Chem. 2005, 16, 959–966. [Google Scholar]
- Warfield, K.L.; Swenson, D.L.; Olinger, G.G.; Nichols, D.K.; Pratt, W.D.; Blouch, R.; Stein, D.A.; Aman, M.J.; Iversen, P.L.; Bavari, S. Gene-specific countermeasures against Ebola virus based on antisense phosphorodiamidate morpholino oligomers. PLoS Pathog. 2006, 2, 5–13. [Google Scholar]
- Smith, A.W.; Iversen, P.L.; O'Hanley, P.D.; Skilling, D.E.; Christensen, J.R.; Weaver, S.S.; Longley, K.; Stone, M.A.; Poet, S.E.; Matson, D.O. Virus-specific antiviral treatment for controlling severe and fatal outbreaks of feline calicivirus infection. Am. J. Vet. Res. 2008, 69, 23–32. [Google Scholar]
- Buchardt, O.; Egholm, M.; Berg, R.H.; Nielsen, P.E. Peptide Nucleic-Acids and Their Potential Applications in Biotechnology. Trends Biotechnol. 1993, 11, 384–386. [Google Scholar]
- Kaushik, N.; Basu, A.; Palumbo, P.; Myers, R.L.; Pandey, V.N. Anti-TAR polyamide nucleotide analog conjugated with a membrane-permeating peptide inhibits human immunodeficiency virus type 1 production. J. Virol. 2002, 76, 3881–3891. [Google Scholar]
- Chaubey, B.; Tripathi, S.; Ganguly, S.; Harris, D.; Casale, R.A.; Pandey, V.N. A PNA-transportan conjugate targeted to the TAR region of the HIV-1 genome exhibits both antiviral and virucidal properties. Virology 2005, 331, 418–428. [Google Scholar]
- Tripathi, S.; Chaubey, B.; Ganguly, S.; Harris, D.; Casale, R.A.; Pandey, V.N. Anti-HIV-1 activity of anti-TAR polyamide nucleic acid coniugated with various membrane transducing peptides. Nucleic Acids Res. 2005, 33, 4345–4356. [Google Scholar]
- Tripathi, S.; Chaubey, B.; Barton, B.E.; Pandey, V.N. Anti HIV-1 virucidal activity of polyamide nucleic acid-membrane transducing peptide conjugates targeted to primer binding site of HIV-1 genome. Virology 2007, 363, 91–103. [Google Scholar]
- Turner, J.J.; Ivanova, G.D.; Verbeure, B.; Williams, D.; Arzumanov, A.A.; Abes, S.; Lebleu, B.; Gait, M.J. Cell-penetrating peptide conjugates of peptide nucleic acids (PNA) as inhibitors of HIV-1 Tat-dependent trans-activation in cells. Nucleic Acids Res. 2005, 33, 6837–6849. [Google Scholar]
- Yoo, J.S.; Kim, C.M.; Kim, J.H.; Kim, J.Y.; Oh, J.W. Inhibition of Japanese encephalitis virus replication by peptide nucleic acids targeting cis-acting elements on the plus- and minus-strands of viral RNA. Antiviral Res. 2009, 82, 122–133. [Google Scholar]
- Uchil, P.D.; Satchidanandam, V. Architecture of the flaviviral replication complex - Protease, nuclease, and detergents reveal encasement within double-layered membrane compartments. J. Biol. Chem. 2003, 278, 24388–24398. [Google Scholar]
- Lebleu, B.; Moulton, H.M.; Abes, R.; Ivanova, G.D.; Abes, S.; Stein, D.A.; Iversen, P.L.; Arzumanov, A.A.; Gait, M.J. Cell penetrating peptide conjugates of steric block oligonucleotides. Adv. Drug Delivery Rev. 2008, 60, 517–529. [Google Scholar]
- Chaubey, B.; Tripathi, S.; Pandey, V.N. Single acute-dose and repeat-doses toxicity of anti-HIV-1 PNA(TAR)-penetratin conjugate after intraperitoneal administration to mice. Oligonucleotides 2008, 18, 9–20. [Google Scholar]
- Upadhyay, A.; Ponzio, N.M.; Pandey, V.N. Immunological Response to Peptide Nucleic Acid and its Peptide Conjugate Targeted to Transactivation Response (TAR) Region of HIV-1 RNA Genome. Oligonucleotides 2008, 18, 329–335. [Google Scholar]
- Ganguly, S.; Chaubey, B.; Tripathi, S.; Upadhyay, A.; Neti, P.; Howell, R.W.; Pandey, V.N. Pharmacokinetic analysis of polyamide nucleic-acid-cell penetrating peptide conjugates targeted against HIV-1 transactivation response element. Oligonucleotides 2008, 18, 277–286. [Google Scholar]
- Meng, S.; Wei, B.J.; Xu, R.H.; Zhang, K.; Wang, L.N.; Zhang, R.; Li, J.M. TAT Peptides Mediated Small Interfering RNA Delivery to Huh-7 Cells and Efficiently Inhibited Hepatitis C Virus RNA Replication. Intervirology 2009, 52, 135–140. [Google Scholar]
- Vocero-Akbani, A.M.; Vander Heyden, N.; Lissy, N.A.; Ratner, L.; Dowdy, S.F. Killing HIV-infected cells by transduction with an HIV protease-activated caspase-3 protein. Nat. Med. 1999, 5, 29–33. [Google Scholar]
- Darbinian, N.; Popov, Y.; Khaliji, K.; Amini, S. Creation of a bi-directional protein transduction system for suppression of HIV-1 expression by p27SJ. Antiviral Res. 2008, 79, 136–141. [Google Scholar]
- Roisin, A.; Robin, J.P.; Dereuddre-Bosquet, N.; Vitte, A.L.; Dormont, D.; Clayette, P.; Jalinot, P. Inhibition of HIV-1 replication by cell-penetrating peptides binding Rev. J. Biol. Chem. 2004, 279, 9208–9214. [Google Scholar]
- Mino, T.; Mori, T.; Aoyama, Y.; Sera, T. Cell-permeable artificial zinc-finger proteins as potent antiviral drugs for human papillomaviruses. Arch. Virol. 2008, 153, 1291–1298. [Google Scholar]
- Bultmann, H.; Busse, J.S.; Brandt, C.R. Modified FGF4 signal peptide inhibits entry of herpes simplex virus type 1. J. Virol. 2001, 75, 2634–2645. [Google Scholar]
- Bultmann, H.; Brandt, C.R. Peptides containing membrane-transiting motifs inhibit virus entry. J. Biol. Chem. 2002, 277, 36018–36023. [Google Scholar]
- Bultmann, H.; Teuton, J.; Brandt, C.R. Addition of a C-terminal cysteine improves the anti-herpes simplex virus activity of a peptide containing the human immunodeficiency virus type 1 TAT protein transduction domain. Antimicrob. Agents Chemother. 2007, 51, 1596–1607. [Google Scholar]
- Klotman, M.E.; Chang, T.L. Defensins in innate antiviral immunity. Nat. Rev. Immunol. 2006, 6, 447–456. [Google Scholar]
- Krajewski, K.; Marchand, C.; Long, Y.Q.; Pommier, Y.; Roller, P.P. Synthesis and HIV-1 integrase inhibitory activity of dimeric and tetrameric analogs of indolicidin. Bioorg. Med. Chem. Lett. 2004, 14, 5595–5598. [Google Scholar]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Delcroix, M.; Riley, L.W. Cell-Penetrating Peptides for Antiviral Drug Development. Pharmaceuticals 2010, 3, 448-470. https://doi.org/10.3390/ph3030448
Delcroix M, Riley LW. Cell-Penetrating Peptides for Antiviral Drug Development. Pharmaceuticals. 2010; 3(3):448-470. https://doi.org/10.3390/ph3030448
Chicago/Turabian StyleDelcroix, Melaine, and Lee W. Riley. 2010. "Cell-Penetrating Peptides for Antiviral Drug Development" Pharmaceuticals 3, no. 3: 448-470. https://doi.org/10.3390/ph3030448
APA StyleDelcroix, M., & Riley, L. W. (2010). Cell-Penetrating Peptides for Antiviral Drug Development. Pharmaceuticals, 3(3), 448-470. https://doi.org/10.3390/ph3030448