
Epigenetic Modulation and Bone Metastasis: Evolving Therapeutic Strategies



Abstract

1. Introduction
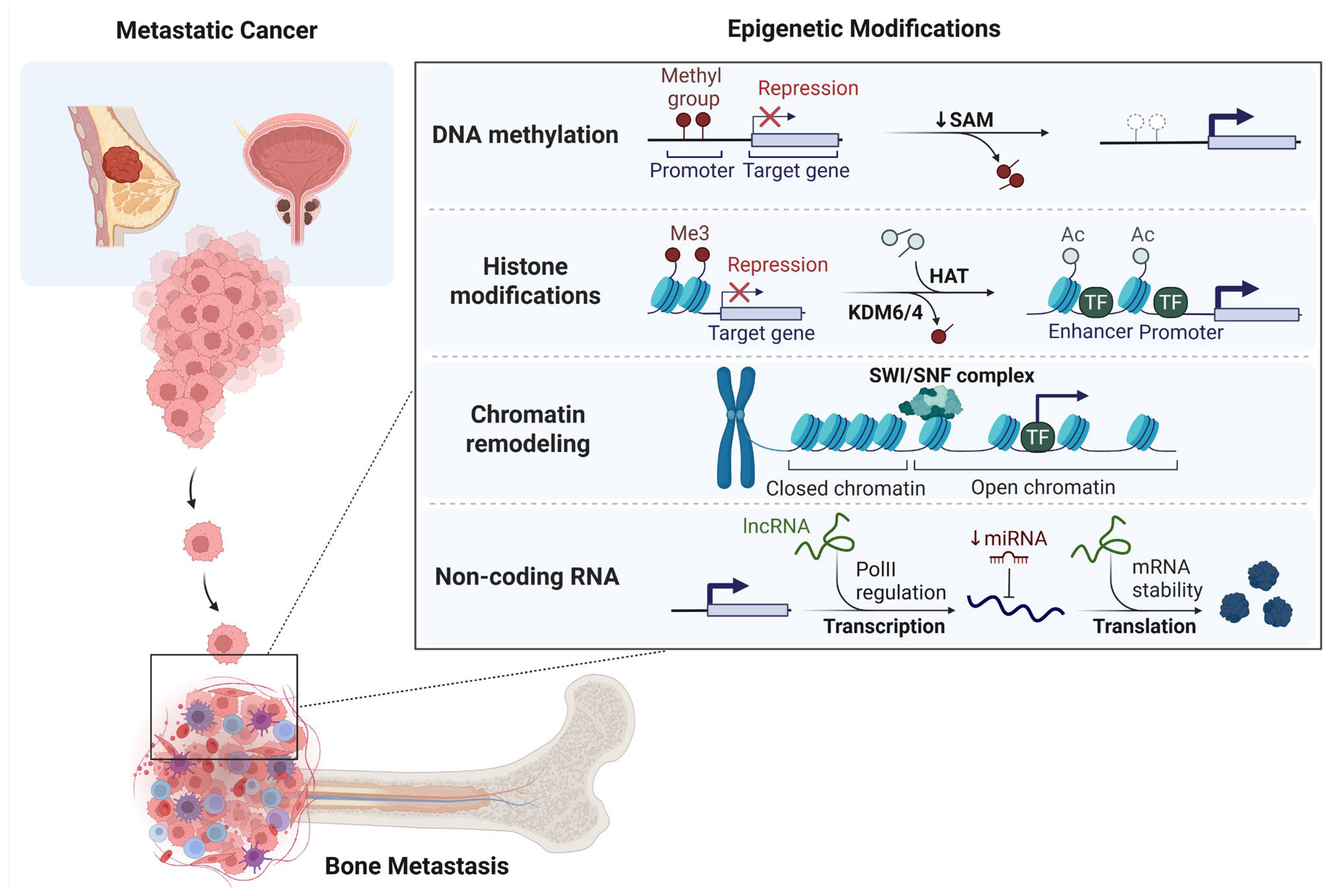
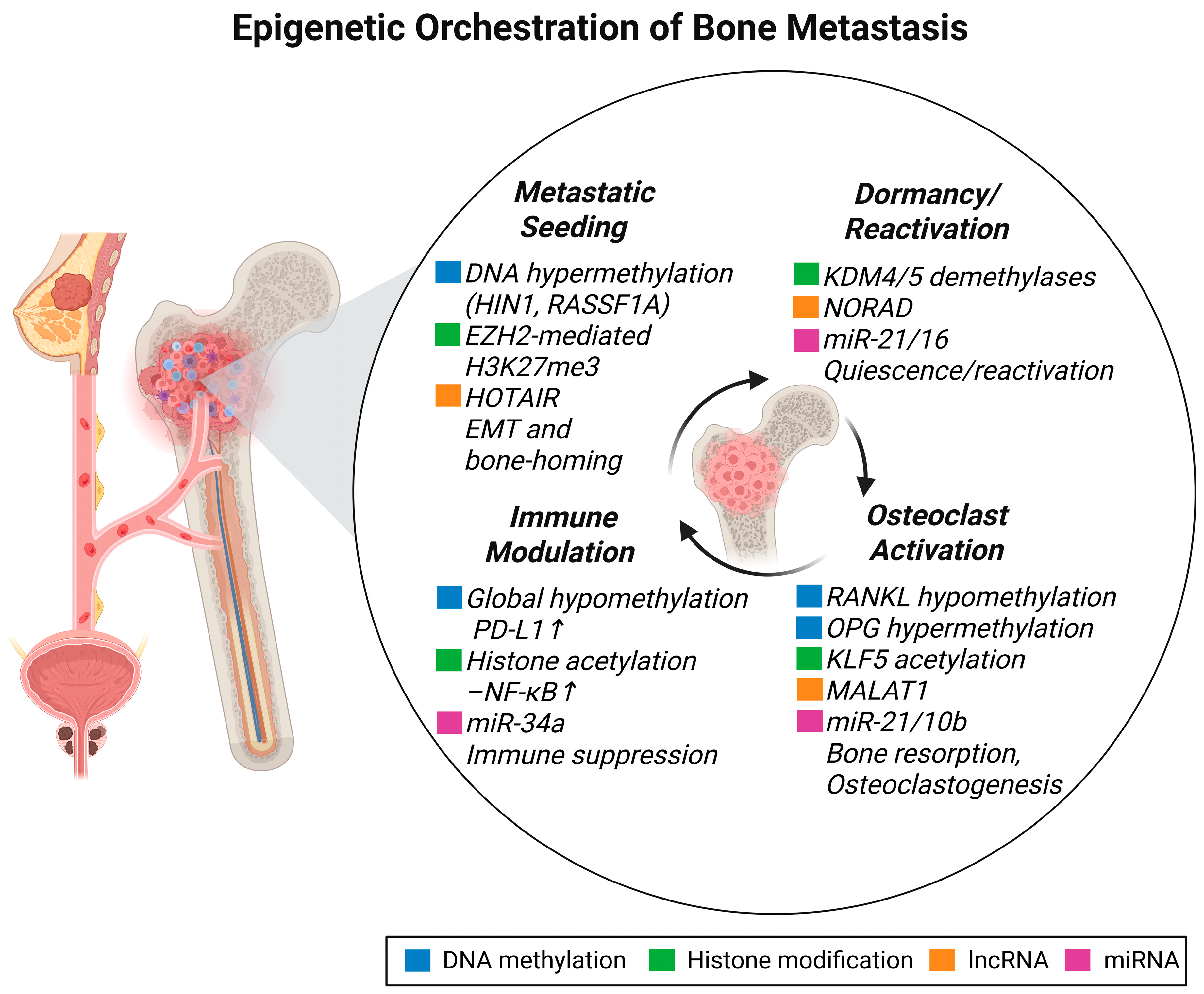
2. Epigenetic Landscape of Bone Metastasis
2.1. DNA Methylation and Bone Metastasis
2.2. Histone Modifications and Bone Metastasis
2.2.1. Histone Acetylation
2.2.2. Histone Methylation
2.3. Chromatin Remodeling and Bone Metastasis
2.4. Non-Coding RNAs and Bone Metastasis
2.4.1. Long Non-Coding RNAs (lncRNAs)
2.4.2. MicroRNAs (miRNAs)
2.4.3. Circular RNAs (circRNAs)
2.4.4. Epitranscriptomic Modifications
2.5. Epigenetic Interactions with Bone-Resident Cells
2.6. Epigenetic Modulation of Bone Homeostasis
2.7. Epigenetics of Dormancy and Lesion Heterogeneity
3. Epigenetic Therapies and Clinical Implications
3.1. DNA Methylation Inhibitors
3.2. Histone Deacetylase Inhibitors
3.3. Histone Methyltransferase and Demethylase Inhibitors
3.4. Bromodomain and Extra-Terminal Domain (BET) Inhibitors
3.5. Non-Coding RNA-Based Therapies
3.5.1. MicroRNA-Based Therapies
3.5.2. LncRNAs, circRNAs and eRNAs as Therapeutic Targets
3.5.3. CRISPR/dCas9 Epigenome Editing
3.5.4. siRNAs and snoRNAs
3.6. Epigenetic Modulation of the Bone Microenvironment by Tumor-Derived Factors
3.7. Chromatin Remodeling and Its Role in Cancer Progression and Bone Metastasis

{kind=link}
{kind=link}
{kind=link}
| Drug | Class | Cancer Type/Indication | Status | Clinical Trial Phase/Approval | Reference |
|---|---|---|---|---|---|
| 5-Azacytidine | DNMT inhibitor | MDS, AML, brain metastasis | FDA-approved | Approved | [166,175] |
| Decitabine | DNMT inhibitor | MDS, AML, bone metastasis | FDA-approved | Approved | [173] |
| Guadecitabine (SGI-110) | SGI—Second-generation DNMT inhibitor | Advanced solid tumors | In trials | Phase I | [176] |
| GSK3685032 | DNMT selective inhibitor | AML | Preclinical | Preclinical | [177] |
| Vorinostat | HDAC inhibitor | Cutaneous T-cell lymphoma | FDA-approved | Approved | [267] |
| Romidepsin | HDAC inhibitor | Peripheral T-cell lymphoma | FDA-approved | Approved | [183] |
| Panobinostat | HDAC inhibitor | Multiple myeloma | FDA-approved | Approved | [268] |
| Belinostat | HDAC inhibitor | Peripheral T-cell lymphoma | FDA-Approved | Approved | [184] |
| Entinostat | HDAC inhibitor | Breast cancer | In trials | Breakthrough therapy | [185] |
| Mocetinostat | HDAC inhibitor | Solid tumors | In trials | Phase I/II | [182] |
| Givinostat | HDAC inhibitor | Polycythemia vera, leukemia | In trials | Phase III | [187] |
| Abexinostat | HDAC inhibitor | Lymphoma, solid tumors | In trials | Phase II | [188] |
| Tubastatin A | HDAC6 inhibitor | Preclinical cancers | Preclinical | Preclinical | [84] |
| PCI-34051 | HDAC8 inhibitor | Specific cancers | Preclinical | Preclinical | [190] |
| REC-2282 (AR-42) | HDAC inhibitor | Neurofibromatosis type 2, meningioma | In trials | Phase I/II | [186] |
| Tazemetostat | EZH2 inhibitor | Follicular lymphoma, epithelioid sarcoma | FDA-approved | Approved | [198] |
| GSK126 | EZH2 inhibitor | Solid tumors | Preclinical | Preclinical | [196] |
| ORY-1001 | KDM1A inhibitor | AML, breast cancer | Preclinical | Preclinical | [201] |
| Tan IIA | KDM1A natural inhibitor | Breast cancer | Preclinical | Preclinical | [201] |
| JQ1 | BET inhibitor | Breast cancer bone metastasis | Preclinical | Preclinical | [210] |
| Molibresib | BET inhibitor | NUT midline carcinoma | In trials | Phase I | [206] |
| Birabresib (OTX015) | BET inhibitor | NHL, AML | In trials | Phase Ib | [207] |
| RO6870810 | BET inhibitor | DLBCL, solid tumors | In trials | Phase I/II | [209] |
| BI 894999 | BET inhibitor | DLBCL, solid tumors | In trials | Phase Ia/Ib | [203] |
| Pelabresib (CPI0610) | BET inhibitor | Myelofibrosis | In trials | Phase II/III | [211] |
| MRX34 | miR-34a mimic (RNA-based therapy) | Liver cancer, melanoma | Terminated | Phase I (terminated) | [220] |
| MRG-106 (Cobomarsen) | Antagomir (miR-155) | Cutaneous T-cell lymphoma | In trials | Phase I | [222,223] |
| siG12D- LODER | siRNA | Pancreatic cancer | In trials | Phase I/II | [242] |
| FHD-286 | BRG1/BRM dual ATPase inhibitor | AML, SWI/SNF-mutant cancers | In trials | Phase I | [254] |
4. Future Direction
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Arakil, N.; Akhund, S.A.; Elaasser, B.; Mohammad, K.S. Intersecting Paths: Unraveling the Complex Journey of Cancer to Bone Metastasis. Biomedicines 2024, 12, 1075. [Google Scholar] [CrossRef]
- Vičić, I.; Belev, B. The pathogenesis of bone metastasis in solid tumors: A review. Croat. Med. J. 2021, 62, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Russo, S.; Scotto di Carlo, F.; Gianfrancesco, F. The Osteoclast Traces the Route to Bone Tumors and Metastases. Front. Cell Dev. Biol. 2022, 10, 886305. [Google Scholar] [CrossRef]
- Zarrer, J.; Taipaleenmäki, H. The osteoblast in regulation of tumor cell dormancy and bone metastasis. J. Bone Oncol. 2024, 45, 100597. [Google Scholar] [CrossRef] [PubMed]
- De Leon-Oliva, D.; Barrena-Blázquez, S.; Jiménez-Álvarez, L.; Fraile-Martinez, O.; García-Montero, C.; López-González, L.; Torres-Carranza, D.; García-Puente, L.M.; Carranza, S.T.; Álvarez-Mon, M.; et al. The RANK-RANKL-OPG System: A Multifaceted Regulator of Homeostasis, Immunity, and Cancer. Medicina 2023, 59, 1752. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Xia, F.; Wei, Y.; Wei, X. Molecular mechanisms and clinical management of cancer bone metastasis. Bone Res. 2020, 8, 30. [Google Scholar] [CrossRef]
- Gu, M.; Ren, B.; Fang, Y.; Ren, J.; Liu, X.; Wang, X.; Zhou, F.; Xiao, R.; Luo, X.; You, L.; et al. Epigenetic regulation in cancer. MedComm 2024, 5, e495. [Google Scholar] [CrossRef]
- Tan, T.; Shi, P.; Abbas, M.N.; Wang, Y.; Xu, J.; Chen, Y.; Cui, H. Epigenetic modification regulates tumor progression and metastasis through EMT (Review). Int. J. Oncol. 2022, 60, 70. [Google Scholar] [CrossRef]
- Casalino, L.; Verde, P. Multifaceted Roles of DNA Methylation in Neoplastic Transformation, from Tumor Suppressors to EMT and Metastasis. Genes 2020, 11, 922. [Google Scholar] [CrossRef]
- Markouli, M.; Strepkos, D.; Basdra, E.K.; Papavassiliou, A.G.; Piperi, C. Prominent Role of Histone Modifications in the Regulation of Tumor Metastasis. Int. J. Mol. Sci. 2021, 22, 2778. [Google Scholar] [CrossRef]
- Shu, X.-S.; Lili, L.; Tao, Q. Chromatin Regulators with Tumor Suppressor Properties and their Alterations in Human Cancers. Epigenomics 2012, 4, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Humphries, B.; Wang, Z.; Yang, C. MicroRNA Regulation of Epigenetic Modifiers in Breast Cancer. Cancers 2019, 11, 897. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Shen, T.; Zeng, Y. Epigenetic Modifications in Prostate Cancer Metastasis and Microenvironment. Cancers 2023, 15, 2243. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Zhao, T.; Xu, C.; Pan, X.; Zhou, Z.; Wang, S. Epigenetic Modifiers in Cancer Metastasis. Biomolecules 2024, 14, 916. [Google Scholar] [CrossRef]
- Nickel, A.; Stadler, S.C. Role of epigenetic mechanisms in epithelial-to-mesenchymal transition of breast cancer cells. Transl. Res. 2015, 165, 126–142. [Google Scholar] [CrossRef]
- Yoodee, S.; Thongboonkerd, V. Epigenetic regulation of epithelial-mesenchymal transition during cancer development. Int. Rev. Cell Mol. Biol. 2023, 380, 1–61. [Google Scholar]
- Sun, L.; Fang, J. Epigenetic regulation of epithelial–mesenchymal transition. Cell. Mol. Life Sci. 2016, 73, 4493–4515. [Google Scholar] [CrossRef]
- Dong, B.; Qiu, Z.; Wu, Y. Tackle Epithelial-Mesenchymal Transition With Epigenetic Drugs in Cancer. Front. Pharmacol. 2020, 11, 596239. [Google Scholar] [CrossRef]
- Kravitz, C.J.; Yan, Q.; Nguyen, D.X. Epigenetic markers and therapeutic targets for metastasis. Cancer Metastasis Rev. 2023, 42, 427–443. [Google Scholar] [CrossRef]
- Mullin, B.H.; Ribet, A.B.P.; Pavlos, N.J. Bone Trans-omics: Integrating Omics to Unveil Mechanistic Molecular Networks Regulating Bone Biology and Disease. Curr. Osteoporos. Rep. 2023, 21, 493–502. [Google Scholar] [CrossRef]
- Menyhárt, O.; Győrffy, B. Multi-omics approaches in cancer research with applications in tumor subtyping, prognosis, and diagnosis. Comput. Struct. Biotechnol. J. 2021, 19, 949–960. [Google Scholar] [CrossRef]
- Tompkins, J.D. Discovering DNA Methylation, the History and Future of the Writing on DNA. J. Hist. Biol. 2022, 55, 865–887. [Google Scholar] [CrossRef]
- Li, S.; Peng, Y.; Panchenko, A.R. DNA methylation: Precise modulation of chromatin structure and dynamics. Curr. Opin. Struct. Biol. 2022, 75, 102430. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.; Kim, S.; An, W. Promoter hypermethylation as a novel regulator of ANO1 expression and function in prostate cancer bone metastasis. Sci. Rep. 2024, 14, 11595. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Weisenberger, D.J. DNA methylation aberrancies as a guide for surveillance and treatment of human cancers. Epigenetics 2017, 12, 416–432. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Wei, S.; Lv, X. Circulating tumor cells: From new biological insights to clinical practice. Signal Transduct. Target. Ther. 2024, 9, 226. [Google Scholar] [CrossRef]
- Besselink, N.J.M.; Keijer, J.P.; Vermeulen, C.; Boymans, S.; de Ridder, J.; van Hoeck, A.; Cuppen, E.; Kuijk, E.W. The genome-wide mutational consequences of DNA hypomethylation. Sci. Rep. 2022, 13, 6874. [Google Scholar] [CrossRef]
- Mehrotra, J.; Vali, M.; McVeigh, M.; Kominsky, S.L.; Fackler, M.J.; Lahti-Domenici, J.; Polyak, K.; Sacchi, N.; Garrett-Mayer, E.; Argani, P.; et al. Very High Frequency of Hypermethylated Genes in Breast Cancer Metastasis to the Bone, Brain, and Lung. Clin. Cancer Res. 2004, 10, 3104–3109. [Google Scholar] [CrossRef]
- Feng, W.; Orlandi, R.; Zhao, N.; Carcangiu, M.L.; Tagliabue, E.; Xu, J.; Bast, R.C.; Yu, Y. Tumor suppressor genes are frequently methylated in lymph node metastases of breast cancers. BMC Cancer 2010, 10, 378. [Google Scholar] [CrossRef]
- Grawenda, A.M.; O’Neill, E. Clinical utility of RASSF1A methylation in human malignancies. Br. J. Cancer 2015, 113, 372–381. [Google Scholar] [CrossRef]
- Tan, B.; Xu, X.; Zhang, Q.; Yuan, Z.; Dong, J. The tumor suppressive role of TIMP3 in the human osteosarcoma cells. J. Orthop. Sci. 2022, 27, 689–695. [Google Scholar] [CrossRef]
- Shinojima, T.; Yu, Q.; Huang, S.K.; Li, M.; Mizuno, R.; Liu, E.T.; Hoon, D.S.; Lessard, L. Heterogeneous epigenetic regulation of TIMP3 in prostate cancer. Epigenetics 2012, 7, 1279–1289. [Google Scholar] [CrossRef] [PubMed]
- Su, C.-W.; Chang, Y.-C.; Chien, M.-H.; Hsieh, Y.-H.; Chen, M.-K.; Lin, C.-W.; Yang, S.-F. Loss of TIMP3 by promoter methylation of Sp1 binding site promotes oral cancer metastasis. Cell Death Dis. 2019, 10, 793. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Lu, M.; Liu, B.; Huang, Y.; Wang, K. Inhibition of Ca2+-activated Cl− channel ANO1/TMEM16A expression suppresses tumor growth and invasiveness in human prostate carcinoma. Cancer Lett. 2012, 326, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Gao, J.; Guan, L.; Chen, X.; Gao, J.; Wang, K. Inhibition of ANO1/TMEM16A induces apoptosis in human prostate carcinoma cells by activating TNF-α signaling. Cell Death Dis. 2018, 9, 703. [Google Scholar] [CrossRef]
- Jia, L.; Liu, W.; Guan, L.; Lu, M.; Wang, K. Inhibition of Calcium-Activated Chloride Channel ANO1/TMEM16A Suppresses Tumor Growth and Invasion in Human Lung Cancer. PLoS ONE 2015, 10, e0136584. [Google Scholar] [CrossRef]
- Lai, Z.; Shu, Q.; Song, Y.; Tang, A.; Tian, J. Effect of DNA methylation on the osteogenic differentiation of mesenchymal stem cells: Concise review. Front. Genet. 2024, 15, 1429844. [Google Scholar] [CrossRef]
- Delgado-Calle, J.; Sañudo, C.; Bolado, A.; Fernández, A.F.; Arozamena, J.; Pascual-Carra, M.A.; Rodriguez-Rey, J.C.; Fraga, M.F.; Bonewald, L.; Riancho, J.A. DNA methylation contributes to the regulation of sclerostin expression in human osteocytes. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2012, 27, 926–937. [Google Scholar] [CrossRef]
- Sun, L.; Zhang, Y.; Chen, G.; Ji, Y.; Ma, Q.; Qiao, X.; Wu, S.; Zhou, L.; Bu, J.; Zhu, X.; et al. Targeting SOST using a small-molecule compound retards breast cancer bone metastasis. Mol. Cancer 2022, 21, 228. [Google Scholar] [CrossRef]
- Sun, M.; Cao, Y.; Yang, X.; An, F.; Wu, H.; Wang, J. DNA methylation in the OPG/RANK/RANKL pathway is associated with steroid-induced osteonecrosis of the femoral head. BMC Musculoskelet. Disord. 2021, 22, 599. [Google Scholar] [CrossRef]
- Li, B.; Wang, P.; Jiao, J.; Wei, H.; Xu, W.; Zhou, P. Roles of the RANKL–RANK Axis in Immunity—Implications for Pathogenesis and Treatment of Bone Metastasis. Front. Immunol. 2022, 13, 824117. [Google Scholar] [CrossRef]
- Brouns, A.; Hendriks, L.E.L.; Robbesom-van den Berge, I.J.; Driessen, A.; Roemen, G.; van Herpen, B.L.J.; Dekkers, Z.; Heitzer, B.; Leunissen, D.J.G.; Moonen, L.; et al. Association of RANKL and EGFR gene expression with bone metastases in patients with metastatic non-small cell lung cancer. Front. Oncol. 2023, 13, 1145001. [Google Scholar] [CrossRef] [PubMed]
- Hayat, F.; Khan, N.U.; Khan, A.U.; Ahmad, I.; Alamri, A.M.; Iftikhar, B. Risk association of RANKL and OPG gene polymorphism with breast cancer to bone metastasis in Pashtun population of Khyber Pakhtunkhwa, Pakistan. PLoS ONE 2022, 17, e0276813. [Google Scholar] [CrossRef] [PubMed]
- Sobacchi, C.; Menale, C.; Crisafulli, L.; Ficara, F. Role of RANKL Signaling in Bone Homeostasis. Physiology 2025, 40, 46–66. [Google Scholar] [CrossRef]
- Beuselinck, B.; Jean-Baptiste, J.; Couchy, G.; Job, S.; De Reynies, A.; Wolter, P.; Théodore, C.; Gravis, G.; Rousseau, B.; Albiges, L.; et al. RANK/OPG ratio of expression in primary clear-cell renal cell carcinoma is associated with bone metastasis and prognosis in patients treated with anti-VEGFR-TKIs. Br. J. Cancer 2015, 113, 1313–1322. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Calle, J.; Sañudo, C.; Fernández, A.F.; García-Renedo, R.; Fraga, M.F.; Riancho, J.A. Role of DNA methylation in the regulation of the RANKL-OPG system in human bone. Epigenetics 2012, 7, 83–91. [Google Scholar] [CrossRef]
- Yalaev, B.; Tyurin, A.; Akhiiarova, K.; Khusainova, R. Hypomethylation of the RUNX2 Gene Is a New Potential Biomarker of Primary Osteoporosis in Men and Women. Int. J. Mol. Sci. 2024, 25, 7312. [Google Scholar] [CrossRef]
- Li, X.-Q.; Lu, J.-T.; Tan, C.-C.; Wang, Q.-S.; Feng, Y.-M. RUNX2 promotes breast cancer bone metastasis by increasing integrin α5-mediated colonization. Cancer Lett. 2016, 380, 78–86. [Google Scholar] [CrossRef]
- Li, X.-Q.; Du, X.; Li, D.-M.; Kong, P.-Z.; Sun, Y.; Liu, P.-F.; Wang, Q.-S.; Feng, Y.-M. ITGBL1 Is a Runx2 Transcriptional Target and Promotes Breast Cancer Bone Metastasis by Activating the TGFβ Signaling Pathway. Cancer Res. 2015, 75, 3302–3313. [Google Scholar] [CrossRef]
- Li, X.-Q.; Zhang, R.; Lu, H.; Yue, X.-M.; Huang, Y.-F. Extracellular Vesicle–Packaged CDH11 and ITGA5 Induce the Premetastatic Niche for Bone Colonization of Breast Cancer Cells. Cancer Res. 2022, 82, 1560–1574. [Google Scholar] [CrossRef]
- Pantano, F.; Croset, M.; Driouch, K.; Bednarz-Knoll, N.; Iuliani, M.; Ribelli, G.; Bonnelye, E.; Wikman, H.; Geraci, S.; Bonin, F.; et al. Integrin alpha5 in human breast cancer is a mediator of bone metastasis and a therapeutic target for the treatment of osteolytic lesions. Oncogene 2021, 40, 1284–1299. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Pang, J.; Cheng, H.; Liu, W.-P.; Di, J.-M.; Xiao, H.-J.; Luo, Y.; Zhang, H.; Huang, W.-T.; Chen, M.-K.; et al. Manipulation of prostate cancer metastasis by locus-specific modification of the CRMP4 promoter region using chimeric TALE DNA methyltransferase and demethylase. Oncotarget 2015, 6, 10030–10044. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Aishan, N.; Zheng, Z.; Ju, S.; He, Q.; Meng, Q.; Lin, X.; Lang, J.; Zhou, J.; Chen, Y.; et al. TET-mediated 5hmC in breast cancer: Mechanism and clinical potential. Epigenetics 2025, 20, 2473250. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Li, W.; Yang, X.; Na, L.; Chen, L.; Liu, G. The Roles of Epigenetics Regulation in Bone Metabolism and Osteoporosis. Front. Cell Dev. Biol. 2021, 8, 619301. [Google Scholar] [CrossRef]
- Bellavia, D.; Costa, V.; De Luca, A.; Cordaro, A.; Fini, M.; Giavaresi, G.; Caradonna, F.; Raimondi, L. The Binomial “Inflammation-Epigenetics” in Breast Cancer Progression and Bone Metastasis: IL-1β Actions Are Influenced by TET Inhibitor in MCF-7 Cell Line. Int. J. Mol. Sci. 2022, 23, 15422. [Google Scholar] [CrossRef]
- Maroni, P.; Matteucci, E.; Bendinelli, P.; Desiderio, M.A. Functions and Epigenetic Regulation of Wwox in Bone Metastasis from Breast Carcinoma: Comparison with Primary Tumors. Int. J. Mol. Sci. 2017, 18, 75. [Google Scholar] [CrossRef]
- Zhao, Z.; Shilatifard, A. Epigenetic modifications of histones in cancer. Genome Biol. 2019, 20, 245. [Google Scholar] [CrossRef]
- Kim, J.J.; Lee, S.Y.; Miller, K.M. Preserving genome integrity and function: The DNA damage response and histone modifications. Crit. Rev. Biochem. Mol. Biol. 2019, 54, 208–241. [Google Scholar] [CrossRef]
- Mariño-Ramírez, L.; Kann, M.G.; Shoemaker, B.A.; Landsman, D. Histone structure and nucleosome stability. Expert Rev. Proteom. 2005, 2, 719–729. [Google Scholar] [CrossRef]
- Peng, Y.; Li, S.; Landsman, D.; Panchenko, A.R. Histone tails as signaling antennas of chromatin. Curr. Opin. Struct. Biol. 2021, 67, 153–160. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Barnes, C.E.; English, D.M.; Cowley, S.M. Acetylation & Co: An expanding repertoire of histone acylations regulates chromatin and transcription. Essays Biochem. 2019, 63, 97–107. [Google Scholar] [CrossRef]
- Zhao, M.; Tao, Y.; Peng, G.H. The Role of Histone Acetyltransferases and Histone Deacetylases in Photoreceptor Differentiation and Degeneration. Int. J. Med. Sci. 2020, 17, 1307–1314. [Google Scholar] [CrossRef] [PubMed]
- Hai, R.; He, L.; Shu, G.; Yin, G. Characterization of Histone Deacetylase Mechanisms in Cancer Development. Front. Oncol. 2021, 11, 700947. [Google Scholar] [CrossRef] [PubMed]
- Dang, F.; Wei, W. Targeting the acetylation signaling pathway in cancer therapy. Semin. Cancer Biol. 2022, 85, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, T.; Pagnotti, G.M.; Guise, T.A.; Mohammad, K.S. The Role of TGF-β in Bone Metastases. Biomolecules 2021, 11, 1643. [Google Scholar] [CrossRef]
- Buijs, J.T.; Stayrook, K.R.; Guise, T.A. TGF-β in the Bone Microenvironment: Role in Breast Cancer Metastases. Cancer Microenviron. 2011, 4, 261–281. [Google Scholar] [CrossRef]
- Zhang, B.; Li, Y.; Wu, Q.; Xie, L.; Barwick, B.; Fu, C.; Li, X.; Wu, D.; Xia, S.; Chen, J.; et al. Acetylation of KLF5 maintains EMT and tumorigenicity to cause chemoresistant bone metastasis in prostate cancer. Nat. Commun. 2021, 12, 1714. [Google Scholar] [CrossRef]
- Zhang, B.; Zhang, Z.; Xia, S.; Xing, C.; Ci, X.; Li, X.; Zhao, R.; Tian, S.; Ma, G.; Zhu, Z.; et al. KLF5 activates microRNA 200 transcription to maintain epithelial characteristics and prevent induced epithelial-mesenchymal transition in epithelial cells. Mol. Cell. Biol. 2013, 33, 4919–4935. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, B.; Xiang, L.; Xia, S.; Kucuk, O.; Deng, X.; Boise, L.H.; Dong, J.T. TGF-β causes Docetaxel resistance in Prostate Cancer via the induction of Bcl-2 by acetylated KLF5 and Protein Stabilization. Theranostics 2020, 10, 7656–7670. [Google Scholar] [CrossRef]
- Huang, Q.; Liu, M.; Zhang, D.; Lin, B.-B.; Fu, X.; Zhang, Z.; Zhang, B.; Dong, J.-T. Nitazoxanide inhibits acetylated KLF5-induced bone metastasis by modulating KLF5 function in prostate cancer. BMC Med. 2023, 21, 68. [Google Scholar] [CrossRef]
- Hyun, K.; Jeon, J.; Park, K.; Kim, J. Writing, erasing and reading histone lysine methylations. Exp. Mol. Med. 2017, 49, e324. [Google Scholar] [CrossRef] [PubMed]
- Yi, X.; Jiang, X.; Li, X.; Jiang, D.-S. Histone methyltransferases: Novel targets for tumor and developmental defects. Am. J. Transl. Res. 2015, 7, 2159–2175. [Google Scholar] [PubMed]
- Hu, Y.; Zhao, Z.; Xie, Q.; Li, H.; Zhang, C.; He, X.; Ma, Y.; Zhang, C.; Li, Q.; Shi, C. JARID1D-dependent Androgen Receptor and JunD Signaling Activation of Osteoclast Differentiation Inhibits Prostate Cancer Bone Metastasis Through Demethylating H3K4. Theranostics 2025, 15, 1320–1337. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Dhar, S.S.; Chen, T.Y.; Kan, P.Y.; Wei, Y.; Kim, J.H.; Chan, C.H.; Lin, H.K.; Hung, M.C.; Lee, M.G. JARID1D Is a Suppressor and Prognostic Marker of Prostate Cancer Invasion and Metastasis. Cancer Res. 2016, 76, 831–843. [Google Scholar] [CrossRef]
- Lemster, A.-L.; Sievers, E.; Pasternack, H.; Lazar-Karsten, P.; Klümper, N.; Sailer, V.; Offermann, A.; Brägelmann, J.; Perner, S.; Kirfel, J. Histone Demethylase KDM5C Drives Prostate Cancer Progression by Promoting EMT. Cancers 2022, 14, 1894. [Google Scholar] [CrossRef]
- Li, G.; Kanagasabai, T.; Lu, W.; Zou, M.R.; Zhang, S.M.; Celada, S.I.; Izban, M.G.; Liu, Q.; Lu, T.; Ballard, B.R.; et al. KDM5B Is Essential for the Hyperactivation of PI3K/AKT Signaling in Prostate Tumorigenesis. Cancer Res. 2020, 80, 4633–4643. [Google Scholar] [CrossRef]
- Yi, S.-J.; Jang, Y.-J.; Kim, H.-J.; Lee, K.; Lee, H.; Kim, Y.; Kim, J.; Hwang, S.Y.; Song, J.S.; Okada, H.; et al. The KDM4B–CCAR1–MED1 axis is a critical regulator of osteoclast differentiation and bone homeostasis. Bone Res. 2021, 9, 27. [Google Scholar] [CrossRef]
- Deng, P.; Yuan, Q.; Cheng, Y.; Li, J.; Liu, Z.; Liu, Y.; Li, Y.; Su, T.; Wang, J.; Salvo, M.E.; et al. Loss of KDM4B exacerbates bone-fat imbalance and mesenchymal stromal cell exhaustion in skeletal aging. Cell Stem Cell 2021, 28, 1057–1073.e1057. [Google Scholar] [CrossRef]
- Wu, M.J.; Chen, C.J.; Lin, T.Y.; Liu, Y.Y.; Tseng, L.L.; Cheng, M.L.; Chuu, C.P.; Tsai, H.K.; Kuo, W.L.; Kung, H.J.; et al. Targeting KDM4B that coactivates c-Myc-regulated metabolism to suppress tumor growth in castration-resistant prostate cancer. Theranostics 2021, 11, 7779–7796. [Google Scholar] [CrossRef]
- Wang, Z.; Cai, H.; Zhao, E.; Cui, H. The Diverse Roles of Histone Demethylase KDM4B in Normal and Cancer Development and Progression. Front. Cell Dev. Biol. 2022, 9, 790129. [Google Scholar] [CrossRef]
- Li, S.; Wu, L.; Wang, Q.; Li, Y.; Wang, X. KDM4B promotes epithelial-mesenchymal transition through up-regulation of ZEB1 in pancreatic cancer. Acta Biochim. Biophys. Sin. 2015, 47, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Lagunas-Rangel, F.A. KDM6B (JMJD3) and its dual role in cancer. Biochimie 2021, 184, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Xun, J.; Gao, R.; Wang, B.; Li, Y.; Ma, Y.; Guan, J.; Zhang, Q. Histone demethylase KDM6B inhibits breast cancer metastasis by regulating Wnt/β-catenin signaling. FEBS Open Bio 2021, 11, 2273–2281. [Google Scholar] [CrossRef] [PubMed]
- Qin, M.; Han, F.; Wu, J.; Gao, F.-x.; Li, Y.; Yan, D.-x.; He, X.-m.; Long, Y.; Tang, X.-p.; Ren, D.-l.; et al. KDM6B promotes ESCC cell proliferation and metastasis by facilitating C/EBPβ transcription. BMC Cancer 2021, 21, 559. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, F.; Gao, B.; Ma, M.; Chen, M.; Wu, Y.; Zhang, W.; Sun, Y.; Liu, S.; Shen, H. KDM6B-mediated histone demethylation of LDHA promotes lung metastasis of osteosarcoma. Theranostics 2021, 11, 3868–3881. [Google Scholar] [CrossRef]
- Salz, T.; Deng, C.; Pampo, C.; Siemann, D.; Qiu, Y.; Brown, K.; Huang, S. Histone Methyltransferase hSETD1A Is a Novel Regulator of Metastasis in Breast Cancer. Mol. Cancer Res. 2015, 13, 461–469. [Google Scholar] [CrossRef]
- Jin, M.L.; Kim, Y.W.; Jin, H.L.; Kang, H.; Lee, E.K.; Stallcup, M.R.; Jeong, K.W. Aberrant expression of SETD1A promotes survival and migration of estrogen receptor α-positive breast cancer cells. Int. J. Cancer 2018, 143, 2871–2883. [Google Scholar] [CrossRef]
- Batham, J.; Lim, P.S.; Rao, S. SETDB-1: A Potential Epigenetic Regulator in Breast Cancer Metastasis. Cancers 2019, 11, 1143. [Google Scholar] [CrossRef]
- Cock-Rada, A.M.; Medjkane, S.; Janski, N.; Yousfi, N.; Perichon, M.; Chaussepied, M.; Chluba, J.; Langsley, G.; Weitzman, J.B. SMYD3 promotes cancer invasion by epigenetic upregulation of the metalloproteinase MMP-9. Cancer Res. 2012, 72, 810–820. [Google Scholar] [CrossRef]
- Nair, S.S.; Kumar, R. Chromatin remodeling in cancer: A gateway to regulate gene transcription. Mol. Oncol. 2012, 6, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Moreno, T.; Monterde, B.; González-Silva, L.; Betancor-Fernández, I.; Revilla, C.; Agraz-Doblas, A.; Freire, J.; Isidro, P.; Quevedo, L.; Blanco, R.; et al. ARID2 deficiency promotes tumor progression and is associated with higher sensitivity to chemotherapy in lung cancer. Oncogene 2021, 40, 2923–2935. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, X.; Qiao, X.; Xie, Y.; Guo, D.; Li, B.; Cao, J.; Tao, Z.; Hu, X. Chromatin Remodelling Molecule ARID1A Determines Metastatic Heterogeneity in Triple-Negative Breast Cancer by Competitively Binding to YAP. Cancers 2023, 15, 2447. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Li, N.; Dong, B.; Guo, W.; Wei, H.; Chen, Q.; Yuan, H.; Han, Y.; Chang, H.; Kan, S.; et al. Chromatin remodeling ATPase BRG1 and PTEN are synthetic lethal in prostate cancer. J. Clin. Investig. 2019, 129, 759–773. [Google Scholar] [CrossRef]
- Puppo, M.; Taipaleenmäki, H.; Hesse, E.; Clézardin, P. Non-coding RNAs in bone remodelling and bone metastasis: Mechanisms of action and translational relevance. Br. J. Pharmacol. 2021, 178, 1936–1954. [Google Scholar] [CrossRef]
- Sanjeev, G.; Pranavkrishna, S.; Akshaya, R.L.; Rohini, M.; Selvamurugan, N. Regulation of bone metastasis and metastasis suppressors by non-coding RNAs in breast cancer. Biochimie 2021, 187, 14–24. [Google Scholar] [CrossRef]
- Yan, H.; Bu, P. Non-coding RNA in cancer. Essays Biochem. 2021, 65, 625–639. [Google Scholar] [CrossRef]
- Klinge, C.M. Non-Coding RNAs in Breast Cancer: Intracellular and Intercellular Communication. Non-Coding RNA 2018, 4, 40. [Google Scholar] [CrossRef]
- Maroni, P.; Gomarasca, M.; Lombardi, G. Long non-coding RNAs in bone metastasis: Progresses and perspectives as potential diagnostic and prognostic biomarkers. Front. Endocrinol. 2023, 14, 1156494. [Google Scholar] [CrossRef]
- Gilyazova, I.; Gimalova, G.; Nizamova, A.; Galimova, E.; Ishbulatova, E.; Pavlov, V.; Khusnutdinova, E. Non-Coding RNAs as Key Regulators in Lung Cancer. Int. J. Mol. Sci. 2024, 25, 560. [Google Scholar] [CrossRef]
- Akshaya, R.L.; Rohini, M.; Selvamurugan, N. Regulation of Breast Cancer Progression by Noncoding RNAs. Curr. Cancer Drug Targets 2020, 20, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, W.; Zhu, W.; Dong, J.; Cheng, Y.; Yin, Z.; Shen, F. Mechanisms and Functions of Long Non-Coding RNAs at Multiple Regulatory Levels. Int. J. Mol. Sci. 2019, 20, 5573. [Google Scholar] [CrossRef] [PubMed]
- Asma, V.; Zahra, S.; Ahmad, M.; Soheila, M.; Sima, F.; Hamid, R.M.; Afshin, N.; Amir, S.; Hamed, M. Long Non-Coding RNAs As Epigenetic Regulators in Cancer. Curr. Pharm. Des. 2019, 25, 3563–3577. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Sun, W.; Liu, Y.; Dong, X. The role of lncRNA MALAT1 in bone metastasis in patients with non-small cell lung cancer. Oncol. Rep. 2016, 36, 1679–1685. [Google Scholar] [CrossRef]
- Tang, Y.; Xiao, G.; Chen, Y.; Deng, Y. LncRNA MALAT1 promotes migration and invasion of non-small-cell lung cancer by targeting miR-206 and activating Akt/mTOR signaling. Anti-Cancer Drugs 2018, 29, 725–735. [Google Scholar] [CrossRef]
- Liu, C.; Li, H.; Jia, J.; Ruan, X.; Liu, Y.; Zhang, X. High Metastasis-Associated Lung Adenocarcinoma Transcript 1 (MALAT1) Expression Promotes Proliferation, Migration, and Invasion of Non-Small Cell Lung Cancer via ERK/Mitogen-Activated Protein Kinase (MAPK) Signaling Pathway. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2019, 25, 5143–5149. [Google Scholar] [CrossRef]
- Zhao, Y.; Ning, J.; Teng, H.; Deng, Y.; Sheldon, M.; Shi, L.; Martinez, C.; Zhang, J.; Tian, A.; Sun, Y.; et al. Long noncoding RNA Malat1 protects against osteoporosis and bone metastasis. Nat. Commun. 2024, 15, 2384. [Google Scholar] [CrossRef]
- Hu, C.-y.; Chen, J.; Qin, X.-h.; You, P.; Ma, J.; Zhang, J.; Zhang, H.; Xu, J.-d. Long non-coding RNA NORAD promotes the prostate cancer cell extracellular vesicle release via microRNA-541-3p-regulated PKM2 to induce bone metastasis of prostate cancer. J. Exp. Clin. Cancer Res. 2021, 40, 98. [Google Scholar] [CrossRef]
- Liu, W.; Zhou, X.; Li, Y.; Jiang, H.; Chen, A. Long Non-Coding RNA NORAD Inhibits Breast Cancer Cell Proliferation and Metastasis by Regulating miR-155-5p/SOCS1 Axis. J. Breast Cancer 2021, 24, 330–343. [Google Scholar] [CrossRef]
- Venneri, M.; Passantino, A. MiRNA: What clinicians need to know. Eur. J. Intern. Med. 2023, 113, 6–9. [Google Scholar] [CrossRef]
- Budakoti, M.; Panwar, A.S.; Molpa, D.; Singh, R.K.; Büsselberg, D.; Mishra, A.P.; Coutinho, H.D.M.; Nigam, M. Micro-RNA: The darkhorse of cancer. Cell. Signal. 2021, 83, 109995. [Google Scholar] [CrossRef]
- Puppo, M.; Valluru, M.K.; Clézardin, P. MicroRNAs and Their Roles in Breast Cancer Bone Metastasis. Curr. Osteoporos. Rep. 2021, 19, 256–263. [Google Scholar] [CrossRef]
- Kinget, L.; Roussel, E.; Lambrechts, D.; Boeckx, B.; Vanginderhuysen, L.; Albersen, M.; Rodríguez-Antona, C.; Graña-Castro, O.; Inglada-Pérez, L.; Verbiest, A.; et al. MicroRNAs Possibly Involved in the Development of Bone Metastasis in Clear-Cell Renal Cell Carcinoma. Cancers 2021, 13, 1554. [Google Scholar] [CrossRef] [PubMed]
- Kitayama, K.; Kawamoto, T.; Kawakami, Y.; Hara, H.; Takemori, T.; Fujiwara, S.; Yahiro, S.; Miyamoto, T.; Mifune, Y.; Hoshino, Y.; et al. Regulatory roles of miRNAs 16, 133a, and 223 on osteoclastic bone destruction caused by breast cancer metastasis. Int. J. Oncol. 2021, 59, 97. [Google Scholar] [CrossRef] [PubMed]
- Puppo, M.; Jaafar, M.; Diaz, J.-J.; Marcel, V.; Clézardin, P. MiRNAs and snoRNAs in Bone Metastasis: Functional Roles and Clinical Potential. Cancers 2023, 15, 242. [Google Scholar] [CrossRef] [PubMed]
- Gomarasca, M.; Maroni, P.; Banfi, G.; Lombardi, G. microRNAs in the Antitumor Immune Response and in Bone Metastasis of Breast Cancer: From Biological Mechanisms to Therapeutics. Int. J. Mol. Sci. 2020, 21, 2805. [Google Scholar] [CrossRef]
- Zhou, R.; Wu, Y.; Wang, W.; Su, W.; Liu, Y.; Wang, Y.; Fan, C.; Li, X.; Li, G.; Li, Y.; et al. Circular RNAs (circRNAs) in cancer. Cancer Lett. 2018, 425, 134–142. [Google Scholar] [CrossRef]
- Zhao, W.; Dong, M.; Pan, J.; Wang, Y.; Zhou, J.; Ma, J.; Liu, S. Circular RNAs: A novel target among non-coding RNAs with potential roles in malignant tumors (Review). Mol. Med. Rep. 2019, 20, 3463–3474. [Google Scholar] [CrossRef]
- Xu, S.; Zhou, L.; Ponnusamy, M.; Zhang, L.; Dong, Y.; Zhang, Y.; Wang, Q.; Liu, J.; Wang, K. A comprehensive review of circRNA: From purification and identification to disease marker potential. PeerJ 2018, 6, e5503. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, S.; Liao, X.; Li, M.; Chen, S.; Li, X.; Wu, X.; Yang, M.; Tang, M.; Hu, Y.; et al. Circular RNA circIKBKB promotes breast cancer bone metastasis through sustaining NF-κB/bone remodeling factors signaling. Mol. Cancer 2021, 20, 98. [Google Scholar] [CrossRef]
- Jin, R.; Sterling, J.A.; Edwards, J.R.; DeGraff, D.J.; Lee, C.; Park, S.I.; Matusik, R.J. Activation of NF-kappa B Signaling Promotes Growth of Prostate Cancer Cells in Bone. PLoS ONE 2013, 8, e60983. [Google Scholar] [CrossRef]
- Zhang, J.; Mao, W.; Chen, Z.; Gu, H.; Lian, C. Clinical Significance of Has_circ_0060937 in Bone Metastasis of NSCLC. Int. J. Gen. Med. 2020, 13, 1115–1121. [Google Scholar] [CrossRef]
- Xu, Y.; Li, X.; Zhang, S.; Tang, M.; Yu, R.; Liao, X.; Li, Z.; Li, M.; Chen, S.; Qian, W.; et al. CircMMP2(6,7) Cooperates with β-Catenin and PRMT5 to Disrupt Bone Homeostasis and Promote Breast Cancer Bone Metastasis. Cancer Res. 2024, 84, 328–343. [Google Scholar] [CrossRef]
- Zhou, Y.; Cao, P.; Zhu, Q. The regulatory role of m6A in cancer metastasis. Front. Cell Dev. Biol. 2025, 13, 1539678. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Huang, X.; Zhang, J.; Lin, D.; Zheng, J. Roles and implications of mRNA N(6)-methyladenosine in cancer. Cancer Commun. 2023, 43, 729–748. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.-Y.; Shi, L.; Li, Z.; Yang, H.-Y.; Wei, J.-F.; Ding, Q. Main N6-Methyladenosine Readers: YTH Family Proteins in Cancers. Front. Oncol. 2021, 11, 635329. [Google Scholar] [CrossRef] [PubMed]
- Lang, C.; Yin, C.; Lin, K.; Li, Y.; Yang, Q.; Wu, Z.; Du, H.; Ren, D.; Dai, Y.; Peng, X. m6A modification of lncRNA PCAT6 promotes bone metastasis in prostate cancer through IGF2BP2-mediated IGF1R mRNA stabilization. Clin. Transl. Med. 2021, 11, e426. [Google Scholar] [CrossRef]
- Wang, S.; Xu, L.; Wang, D.; Zhao, S.; Li, K.; Ma, F.; Yao, Q.; Zhang, Y.; Wu, Z.; Shao, Y.; et al. YTHDF1 promotes the osteolytic bone metastasis of breast cancer via inducing EZH2 and CDH11 translation. Cancer Lett. 2024, 597, 217047. [Google Scholar] [CrossRef]
- Liu, J.; Chen, X.; Yu, X. Unraveling the Role of N6-Methylation Modification: From Bone Biology to Osteoporosis. Int. J. Med. Sci. 2025, 22, 2545–2559. [Google Scholar] [CrossRef]
- Kitazawa, R.; Haraguchi, R.; Murata, Y.; Takaoka, Y.; Kitazawa, S. CpG Methylation of Receptor Activator NF-κB (RANK) Gene Promoter Region Delineates Senescence-Related Decrease of RANK Gene Expression. Acta Histochem. Cytochem. 2024, 57, 137–147. [Google Scholar] [CrossRef]
- Ölken, E.A.; Aszodi, A.; Taipaleenmäki, H.; Saito, H.; Schönitzer, V.; Chaloupka, M.; Apfelbeck, M.; Böcker, W.; Saller, M.M. SFRP2 Overexpression Induces an Osteoblast-like Phenotype in Prostate Cancer Cells. Cells 2022, 11, 4081. [Google Scholar] [CrossRef]
- Stegen, S.; Moermans, K.; Stockmans, I.; Thienpont, B.; Carmeliet, G. The serine synthesis pathway drives osteoclast differentiation through epigenetic regulation of NFATc1 expression. Nat. Metab. 2024, 6, 141–152. [Google Scholar] [CrossRef]
- Li, K.; Han, J.; Wang, Z. Histone modifications centric-regulation in osteogenic differentiation. Cell Death Discov. 2021, 7, 91. [Google Scholar] [CrossRef] [PubMed]
- Montecino, M.; Carrasco, M.E.; Nardocci, G. Epigenetic Control of Osteogenic Lineage Commitment. Front. Cell Dev. Biol. 2021, 8, 611197. [Google Scholar] [CrossRef] [PubMed]
- Zoni, E.; van der Pluijm, G. The role of microRNAs in bone metastasis. J. Bone Oncol. 2016, 5, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Oka, S.; Li, X.; Zhang, F.; Tewari, N.; Ma, R.; Zhong, L.; Makishima, M.; Liu, Y.; Bhawal, U.K. MicroRNA-21 facilitates osteoblast activity. Biochem. Biophys. Rep. 2021, 25, 100894. [Google Scholar] [CrossRef]
- Wang, S.; Liu, Z.; Wang, J.; Ji, X.; Yao, Z.; Wang, X. miR-21 promotes osteoclastogenesis through activation of PI3K/Akt signaling by targeting Pten in RAW264.7 cells. Mol. Med. Rep. 2020, 21, 1125–1132. [Google Scholar] [CrossRef]
- Bonci, D.; Coppola, V.; Patrizii, M.; Addario, A.; Cannistraci, A.; Francescangeli, F.; Pecci, R.; Muto, G.; Collura, D.; Bedini, R.; et al. A microRNA code for prostate cancer metastasis. Oncogene 2016, 35, 1180–1192. [Google Scholar] [CrossRef]
- Kim, O.; Tran, P.T.; Gal, M.; Lee, S.J.; Na, S.H.; Hwangbo, C.; Lee, J.-H. RAS-stimulated release of exosomal miR-494-3p promotes the osteolytic bone metastasis of breast cancer cells. Int. J. Mol. Med. 2023, 52, 84. [Google Scholar] [CrossRef]
- Ma, Q.; Liang, M.; Wu, Y.; Dou, C.; Xu, J.; Dong, S.; Luo, F. Small extracellular vesicles deliver osteolytic effectors and mediate cancer-induced osteolysis in bone metastatic niche. J. Extracell. Vesicles 2021, 10, e12068. [Google Scholar] [CrossRef]
- Alečković, M.; Kang, Y. Bone marrow stroma-derived miRNAs as regulators, biomarkers and therapeutic targets of bone metastasis. BoneKEy Rep. 2015, 4, 671. [Google Scholar] [CrossRef]
- Adamik, J.; Roodman, G.D.; Galson, D.L. Epigenetic-Based Mechanisms of Osteoblast Suppression in Multiple Myeloma Bone Disease. JBMR Plus 2019, 3, e10183. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, L.; De Luca, A.; Morelli, E.; Giavaresi, G.; Tagliaferri, P.; Tassone, P.; Amodio, N. MicroRNAs: Novel Crossroads between Myeloma Cells and the Bone Marrow Microenvironment. BioMed Res. Int. 2016, 2016, 6504593. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Maruo, A.; Fujii, K.; Nomi, M.; Nakamura, T.; Eto, S.; Minami, Y. Intercellular adhesion molecule 1 discriminates functionally different populations of human osteoblasts: Characteristic involvement of cell cycle regulators. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2000, 15, 1912–1923. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Fan, Y.; Zhao, Z.; Le, C.; Liu, W. Expression of ODF and ICAM-1 of Bone Marrow Mesenchymal Stem Cells is Enhanced with Osteogenic Differentiation. Key Eng. Mater. 2007, 361–363, 1173–1176. [Google Scholar] [CrossRef]
- Del Real, A.; Pérez-Campo, F.M.; Fernández, A.F.; Sañudo, C.; Ibarbia, C.G.; Pérez-Núñez, M.I.; Criekinge, W.V.; Braspenning, M.; Alonso, M.A.; Fraga, M.F.; et al. Differential analysis of genome-wide methylation and gene expression in mesenchymal stem cells of patients with fractures and osteoarthritis. Epigenetics 2017, 12, 113–122. [Google Scholar] [CrossRef]
- Fernandez-Rebollo, E.; Eipel, M.; Seefried, L.; Hoffmann, P.; Strathmann, K.; Jakob, F.; Wagner, W. Primary Osteoporosis Is Not Reflected by Disease-Specific DNA Methylation or Accelerated Epigenetic Age in Blood. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2018, 33, 356–361. [Google Scholar] [CrossRef]
- Huang, Y.; Wang, H.; Yue, X.; Li, X. Bone serves as a transfer station for secondary dissemination of breast cancer. Bone Res. 2023, 11, 21. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, K.; Kim, I.; Seong, S.; Kook, H.; Kim, K.K.; Koh, J.-T.; Kim, N. Bifunctional Role of CrkL during Bone Remodeling. Int. J. Mol. Sci. 2021, 22, 7007. [Google Scholar] [CrossRef]
- Rahim, F.; Hajizamani, S.; Mortaz, E.; Ahmadzadeh, A.; Shahjahani, M.; Shahrabi, S.; Saki, N. Molecular regulation of bone marrow metastasis in prostate and breast cancer. Bone Marrow Res. 2014, 2014, 405920. [Google Scholar] [CrossRef]
- Sharma, S.; Xing, F.; Liu, Y.; Wu, K.; Said, N.; Pochampally, R.; Shiozawa, Y.; Lin, H.K.; Balaji, K.C.; Watabe, K. Secreted Protein Acidic and Rich in Cysteine (SPARC) Mediates Metastatic Dormancy of Prostate Cancer in Bone. J. Biol. Chem. 2016, 291, 19351–19363. [Google Scholar] [CrossRef] [PubMed]
- Sosa, M.S.; Parikh, F.; Maia, A.G.; Estrada, Y.; Bosch, A.; Bragado, P.; Ekpin, E.; George, A.; Zheng, Y.; Lam, H.M.; et al. NR2F1 controls tumour cell dormancy via SOX9- and RARβ-driven quiescence programmes. Nat. Commun. 2015, 6, 6170. [Google Scholar] [CrossRef] [PubMed]
- Byrne, N.M.; Summers, M.A.; McDonald, M.M. Tumor Cell Dormancy and Reactivation in Bone: Skeletal Biology and Therapeutic Opportunities. JBMR Plus 2019, 3, e10125. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.K.; Patel, V.G.; Oh, W.K.; Aguirre-Ghiso, J.A. Prostate Cancer Dormancy and Reactivation in Bone Marrow. J. Clin. Med. 2021, 10, 2648. [Google Scholar] [CrossRef]
- McGrath, J.; Panzica, L.; Ransom, R.; Withers, H.G.; Gelman, I.H. Identification of Genes Regulating Breast Cancer Dormancy in 3D Bone Endosteal Niche Cultures. Mol. Cancer Res. 2019, 17, 860–869. [Google Scholar] [CrossRef]
- Dawalibi, A.; Bakir, M.; Mohammad, K.S. The genetic architecture of bone metastases: Unveiling the role of epigenetic and genetic modifications in drug resistance. Cancer Drug Resist. 2025, 8, 19. [Google Scholar] [CrossRef]
- Hall, C.L.; Bafico, A.; Dai, J.; Aaronson, S.A.; Keller, E.T. Prostate Cancer Cells Promote Osteoblastic Bone Metastases through Wnts. Cancer Res. 2005, 65, 7554–7560. [Google Scholar] [CrossRef]
- Larson, S.R.; Zhang, X.; Dumpit, R.; Coleman, I.; Lakely, B.; Roudier, M.; Higano, C.S.; True, L.D.; Lange, P.H.; Montgomery, B.; et al. Characterization of osteoblastic and osteolytic proteins in prostate cancer bone metastases. Prostate 2013, 73, 932–940. [Google Scholar] [CrossRef]
- Sharma, G.; Sultana, A.; Abdullah, K.M.; Pothuraju, R.; Nasser, M.W.; Batra, S.K.; Siddiqui, J.A. Epigenetic regulation of bone remodeling and bone metastasis. Semin. Cell Dev. Biol. 2024, 154, 275–285. [Google Scholar] [CrossRef]
- Jones, P.A.; Ohtani, H.; Chakravarthy, A.; De Carvalho, D.D. Epigenetic therapy in immune-oncology. Nat. Rev. Cancer 2019, 19, 151–161. [Google Scholar] [CrossRef]
- Lee, A.V.; Nestler, K.A.; Chiappinelli, K.B. Therapeutic targeting of DNA methylation alterations in cancer. Pharmacol. Ther. 2024, 258, 108640. [Google Scholar] [CrossRef]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef]
- Baby, S.; Gurukkala Valapil, D.; Shankaraiah, N. Unravelling KDM4 histone demethylase inhibitors for cancer therapy. Drug Discov. Today 2021, 26, 1841–1856. [Google Scholar] [CrossRef] [PubMed]
- Aspeslagh, S.; Morel, D.; Soria, J.C.; Postel-Vinay, S. Epigenetic modifiers as new immunomodulatory therapies in solid tumours. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2018, 29, 812–824. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Li, H.Q.; Liu, F. DNA Methyltransferase Inhibitors and their Therapeutic Potential. Curr. Top. Med. Chem. 2018, 18, 2448–2457. [Google Scholar] [CrossRef] [PubMed]
- Fenaux, P.; Mufti, G.J.; Hellstrom-Lindberg, E.; Santini, V.; Finelli, C.; Giagounidis, A.; Schoch, R.; Gattermann, N.; Sanz, G.; List, A.; et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: A randomised, open-label, phase III study. Lancet Oncol. 2009, 10, 223–232. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Döhner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef]
- Yu, G.; Wu, Y.; Wang, W.; Xu, J.; Lv, X.; Cao, X.; Wan, T. Low-dose decitabine enhances the effect of PD-1 blockade in colorectal cancer with microsatellite stability by re-modulating the tumor microenvironment. Cell. Mol. Immunol. 2019, 16, 401–409. [Google Scholar] [CrossRef]
- Wong, K.K.; Hassan, R.; Yaacob, N.S. Hypomethylating Agents and Immunotherapy: Therapeutic Synergism in Acute Myeloid Leukemia and Myelodysplastic Syndromes. Front. Oncol. 2021, 11, 624742. [Google Scholar] [CrossRef]
- Alexandraki, A.; Strati, K. Decitabine Treatment Induces a Viral Mimicry Response in Cervical Cancer Cells and Further Sensitizes Cells to Chemotherapy. Int. J. Mol. Sci. 2022, 23, 14042. [Google Scholar] [CrossRef]
- Momparler, R.L.; Momparler, L.F.; Samson, J. Comparison of the antileukemic activity of 5-AZA-2′-deoxycytidine, 1-beta-D-arabinofuranosylcytosine and 5-azacytidine against L1210 leukemia. Leuk. Res. 1984, 8, 1043–1049. [Google Scholar] [CrossRef] [PubMed]
- Ateeq, B.; Unterberger, A.; Szyf, M.; Rabbani, S.A. Pharmacological inhibition of DNA methylation induces proinvasive and prometastatic genes in vitro and in vivo. Neoplasia 2008, 10, 266–278. [Google Scholar] [CrossRef] [PubMed]
- Guan, H.; Mi, B.; Li, Y.; Wu, W.; Tan, P.; Fang, Z.; Li, J.; Zhang, Y.; Li, F. Decitabine represses osteoclastogenesis through inhibition of RANK and NF-κB. Cell. Signal. 2015, 27, 969–977. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.K. DNMT1: A key drug target in triple-negative breast cancer. Semin. Cancer Biol. 2021, 72, 198–213. [Google Scholar] [CrossRef]
- Butler, C.; Sprowls, S.; Szalai, G.; Arsiwala, T.; Saralkar, P.; Straight, B.; Hatcher, S.; Tyree, E.; Yost, M.; Kohler, W.J.; et al. Hypomethylating Agent Azacitidine Is Effective in Treating Brain Metastasis Triple-Negative Breast Cancer Through Regulation of DNA Methylation of Keratin 18 Gene. Transl. Oncol. 2020, 13, 100775. [Google Scholar] [CrossRef]
- Daher-Reyes, G.S.; Merchan, B.M.; Yee, K.W.L. Guadecitabine (SGI-110): An investigational drug for the treatment of myelodysplastic syndrome and acute myeloid leukemia. Expert Opin. Investig. Drugs 2019, 28, 835–849. [Google Scholar] [CrossRef]
- Pappalardi, M.B.; Keenan, K.; Cockerill, M.; Kellner, W.A.; Stowell, A.; Sherk, C.; Wong, K.; Pathuri, S.; Briand, J.; Steidel, M.; et al. Discovery of a first-in-class reversible DNMT1-selective inhibitor with improved tolerability and efficacy in acute myeloid leukemia. Nat. Cancer 2021, 2, 1002–1017. [Google Scholar] [CrossRef]
- Wiseman, A.K.; Tiedemann, R.L.; Fan, H.; Shen, H.; Madaj, Z.; McCabe, M.T.; Pappalardi, M.B.; Jones, P.A. Chromosome-specific retention of cancer-associated DNA hypermethylation following pharmacological inhibition of DNMT1. Commun. Biol. 2022, 5, 528. [Google Scholar] [CrossRef]
- Crunkhorn, S. A safe and effective DNA hypomethylating agent. Nat. Rev. Drug Discov. 2021, 20, 816. [Google Scholar] [CrossRef]
- Vendetti, F.P.; Topper, M.; Huang, P.; Dobromilskaya, I.; Easwaran, H.; Wrangle, J.; Baylin, S.B.; Poirier, J.T.; Rudin, C.M. Evaluation of azacitidine and entinostat as sensitization agents to cytotoxic chemotherapy in preclinical models of non-small cell lung cancer. Oncotarget 2015, 6, 56–70. [Google Scholar] [CrossRef]
- Bondarev, A.D.; Attwood, M.M.; Jonsson, J.; Chubarev, V.N.; Tarasov, V.V.; Schiöth, H.B. Recent developments of HDAC inhibitors: Emerging indications and novel molecules. Br. J. Clin. Pharmacol. 2021, 87, 4577–4597. [Google Scholar] [CrossRef]
- McClure, J.J.; Li, X.; Chou, C.J. Advances and Challenges of HDAC Inhibitors in Cancer Therapeutics. Adv. Cancer Res. 2018, 138, 183–211. [Google Scholar] [CrossRef]
- Stuver, R.; Epstein-Peterson, Z.D.; Johnson, W.T.; Khan, N.; Lewis, N.; Moskowitz, A.J.; Sauter, C.S.; Horwitz, S. Current Treatment of Peripheral T-cell Lymphoma. Oncology 2022, 36, 293–305. [Google Scholar] [CrossRef]
- Lu, G.; Jin, S.; Lin, S.; Gong, Y.; Zhang, L.; Yang, J.; Mou, W.; Du, J. Update on histone deacetylase inhibitors in peripheral T-cell lymphoma (PTCL). Clin. Epigenetics 2023, 15, 124. [Google Scholar] [CrossRef]
- Roussos Torres, E.T.; Ho, W.J.; Danilova, L.; Tandurella, J.A.; Leatherman, J.; Rafie, C.; Wang, C.; Brufsky, A.; LoRusso, P.; Chung, V.; et al. Entinostat, nivolumab and ipilimumab for women with advanced HER2-negative breast cancer: A phase Ib trial. Nat. Cancer 2024, 5, 866–879. [Google Scholar] [CrossRef] [PubMed]
- Liva, S.; Chen, M.; Mortazavi, A.; Walker, A.; Wang, J.; Dittmar, K.; Hofmeister, C.; Coss, C.C.; Phelps, M.A. Population Pharmacokinetic Analysis from First-in-Human Data for HDAC Inhibitor, REC-2282 (AR-42), in Patients with Solid Tumors and Hematologic Malignancies: A Case Study for Evaluating Flat vs. Body Size Normalized Dosing. Eur. J. Drug Metab. Pharmacokinet. 2021, 46, 807–816. [Google Scholar] [CrossRef]
- Mercuri, E.; Vilchez, J.J.; Boespflug-Tanguy, O.; Zaidman, C.M.; Mah, J.K.; Goemans, N.; Müller-Felber, W.; Niks, E.H.; Schara-Schmidt, U.; Bertini, E.; et al. Safety and efficacy of givinostat in boys with Duchenne muscular dystrophy (EPIDYS): A multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Neurol. 2024, 23, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Ribrag, V.; Kim, W.S.; Bouabdallah, R.; Lim, S.T.; Coiffier, B.; Illes, A.; Lemieux, B.; Dyer, M.J.S.; Offner, F.; Felloussi, Z.; et al. Safety and efficacy of abexinostat, a pan-histone deacetylase inhibitor, in non-Hodgkin lymphoma and chronic lymphocytic leukemia: Results of a phase II study. Haematologica 2017, 102, 903–909. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, C.; Göder, A.; Beyer, M.; Kiweler, N.; Mahendrarajah, N.; Rauch, A.; Nikolova, T.; Stojanovic, N.; Wieczorek, M.; Reich, T.R.; et al. Class I histone deacetylases regulate p53/NF-κB crosstalk in cancer cells. Cell. Signal. 2017, 29, 218–225. [Google Scholar] [CrossRef]
- Kim, J.Y.; Han, S.Y.; Yoo, J.; Kim, G.W.; Jeon, Y.H.; Lee, S.W.; Park, J.; Kwon, S.H. HDAC8-Selective Inhibition by PCI-34051 Enhances the Anticancer Effects of ACY-241 in Ovarian Cancer Cells. Int. J. Mol. Sci. 2022, 23, 8645. [Google Scholar] [CrossRef]
- Clements, M.E.; Holtslander, L.; Johnson, J.R.; Johnson, R.W. Select HDAC Inhibitors Enhance Osteolysis and Bone Metastasis Outgrowth but Can Be Mitigated With Bisphosphonate Therapy. JBMR Plus 2023, 7, e10694. [Google Scholar] [CrossRef]
- Edwards, C.M.; Johnson, R.W. Targeting Histone Modifications in Bone and Lung Metastatic Cancers. Curr. Osteoporos. Rep. 2021, 19, 230–246. [Google Scholar] [CrossRef]
- Jarrold, J.; Davies, C.C. PRMTs and Arginine Methylation: Cancer’s Best-Kept Secret? Trends Mol. Med. 2019, 25, 993–1009. [Google Scholar] [CrossRef]
- Taylor-Papadimitriou, J.; Burchell, J.M. Histone Methylases and Demethylases Regulating Antagonistic Methyl Marks: Changes Occurring in Cancer. Cells 2022, 11, 1113. [Google Scholar] [CrossRef]
- Straining, R.; Eighmy, W. Tazemetostat: EZH2 Inhibitor. J. Adv. Pract. Oncol. 2022, 13, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Singh, A.; Singh, M.P.; Nengroo, M.A.; Saini, K.K.; Satrusal, S.R.; Khan, M.A.; Chaturvedi, P.; Sinha, A.; Meena, S.; et al. EZH2-H3K27me3 mediated KRT14 upregulation promotes TNBC peritoneal metastasis. Nat. Commun. 2022, 13, 7344. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Qu, J.; Qi, Y.; Duan, Y.; Huang, Y.W.; Zhou, Z.; Li, P.; Yao, J.; Huang, B.; Zhang, S.; et al. EZH2 engages TGFβ signaling to promote breast cancer bone metastasis via integrin β1-FAK activation. Nat. Commun. 2022, 13, 2543. [Google Scholar] [CrossRef]
- Italiano, A.; Soria, J.C.; Toulmonde, M.; Michot, J.M.; Lucchesi, C.; Varga, A.; Coindre, J.M.; Blakemore, S.J.; Clawson, A.; Suttle, B.; et al. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: A first-in-human, open-label, phase 1 study. Lancet Oncol. 2018, 19, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Varghese, B.; Del Gaudio, N.; Cobellis, G.; Altucci, L.; Nebbioso, A. KDM4 Involvement in Breast Cancer and Possible Therapeutic Approaches. Front. Oncol. 2021, 11, 750315. [Google Scholar] [CrossRef]
- Taube, J.H.; Sphyris, N.; Johnson, K.S.; Reisenauer, K.N.; Nesbit, T.A.; Joseph, R.; Vijay, G.V.; Sarkar, T.R.; Bhangre, N.A.; Song, J.J.; et al. The H3K27me3-demethylase KDM6A is suppressed in breast cancer stem-like cells, and enables the resolution of bivalency during the mesenchymal-epithelial transition. Oncotarget 2017, 8, 65548–65565. [Google Scholar] [CrossRef]
- Luo, N.; Zhang, K.; Li, X.; Hu, Y.; Guo, L. Tanshinone IIA destabilizes SLC7A11 by regulating PIAS4-mediated SUMOylation of SLC7A11 through KDM1A, and promotes ferroptosis in breast cancer. J. Adv. Res. 2025, 69, 313–327. [Google Scholar] [CrossRef]
- Alqahtani, A.; Choucair, K.; Ashraf, M.; Hammouda, D.M.; Alloghbi, A.; Khan, T.; Senzer, N.; Nemunaitis, J. Bromodomain and extra-terminal motif inhibitors: A review of preclinical and clinical advances in cancer therapy. Future Sci. OA 2019, 5, Fso372. [Google Scholar] [CrossRef] [PubMed]
- Lauer, U.M.; Awada, A.; Postel-Vinay, S.; Shapiro, G.I.; Thieblemont, C.; Piha-Paul, S.A.; Paik, P.K.; Shepard, D.R.; Docampo, L.I.; Galot, R.; et al. Final results from the phase Ia/Ib study of the novel bromodomain and extra-terminal domain inhibitor, BI 894999, in patients with advanced solid tumors or diffuse large B-cell lymphoma. ESMO Open 2025, 10, 104499. [Google Scholar] [CrossRef] [PubMed]
- Sahai, V.; Redig, A.J.; Collier, K.A.; Eckerdt, F.D.; Munshi, H.G. Targeting BET bromodomain proteins in solid tumors. Oncotarget 2016, 7, 53997–54009. [Google Scholar] [CrossRef] [PubMed]
- Asangani, I.A.; Dommeti, V.L.; Wang, X.; Malik, R.; Cieslik, M.; Yang, R.; Escara-Wilke, J.; Wilder-Romans, K.; Dhanireddy, S.; Engelke, C.; et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature 2014, 510, 278–282. [Google Scholar] [CrossRef]
- Parikh, S.A.; French, C.A.; Costello, B.A.; Marks, R.S.; Dronca, R.S.; Nerby, C.L.; Roden, A.C.; Peddareddigari, V.G.; Hilton, J.; Shapiro, G.I.; et al. NUT midline carcinoma: An aggressive intrathoracic neoplasm. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2013, 8, 1335–1338. [Google Scholar] [CrossRef]
- Amorim, S.; Stathis, A.; Gleeson, M.; Iyengar, S.; Magarotto, V.; Leleu, X.; Morschhauser, F.; Karlin, L.; Broussais, F.; Rezai, K.; et al. Bromodomain inhibitor OTX015 in patients with lymphoma or multiple myeloma: A dose-escalation, open-label, pharmacokinetic, phase 1 study. Lancet Haematol. 2016, 3, e196–e204. [Google Scholar] [CrossRef]
- Berthon, C.; Raffoux, E.; Thomas, X.; Vey, N.; Gomez-Roca, C.; Yee, K.; Taussig, D.C.; Rezai, K.; Roumier, C.; Herait, P.; et al. Bromodomain inhibitor OTX015 in patients with acute leukaemia: A dose-escalation, phase 1 study. Lancet Haematol. 2016, 3, e186–e195. [Google Scholar] [CrossRef]
- Moreno, V.; Sepulveda, J.M.; Vieito, M.; Hernández-Guerrero, T.; Doger, B.; Saavedra, O.; Ferrero, O.; Sarmiento, R.; Arias, M.; De Alvaro, J.; et al. Phase I study of CC-90010, a reversible, oral BET inhibitor in patients with advanced solid tumors and relapsed/refractory non-Hodgkin’s lymphoma. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2020, 31, 780–788. [Google Scholar] [CrossRef]
- Moreno, V.; Vieito, M.; Sepulveda, J.M.; Galvao, V.; Hernández-Guerrero, T.; Doger, B.; Saavedra, O.; Carlo-Stella, C.; Michot, J.M.; Italiano, A.; et al. BET inhibitor trotabresib in heavily pretreated patients with solid tumors and diffuse large B-cell lymphomas. Nat. Commun. 2023, 14, 1359. [Google Scholar] [CrossRef]
- Ferreira Gomes, G.; Harrison, C. Pelabresib (CPI-0610): An Exciting Novel Drug for the Treatment of Myelofibrosis. Curr. Hematol. Malig. Rep. 2023, 18, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Zou, J.; Li, S.; Topper, M.J.; Tao, Y.; Zhang, H.; Jiao, X.; Xie, W.; Kong, X.; Vaz, M.; et al. Epigenetic therapy inhibits metastases by disrupting premetastatic niches. Nature 2020, 579, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Alexandrova, E.; Smal, M.; Di Rosa, D.; Brancaccio, R.N.; Parisi, R.; Russo, F.; Tarallo, R.; Nassa, G.; Giurato, G.; Weisz, A.; et al. BRPF1 inhibition reduces migration and invasion of metastatic ovarian cancer cells, representing a potential therapeutic target. Sci. Rep. 2025, 15, 7602. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Liu, P.; Chen, W.; Liu, X.; Tong, F.; Sun, J.; Zhou, Y.; Lei, T.; Yang, W.; Ma, D.; et al. Hypoxia-cleavable and specific targeted nanomedicine delivers epigenetic drugs for enhanced treatment of breast cancer and bone metastasis. J. Nanobiotechnol. 2023, 21, 221. [Google Scholar] [CrossRef]
- Grillone, K.; Caridà, G.; Luciano, F.; Cordua, A.; Di Martino, M.T.; Tagliaferri, P.; Tassone, P. A systematic review of non-coding RNA therapeutics in early clinical trials: A new perspective against cancer. J. Transl. Med. 2024, 22, 731. [Google Scholar] [CrossRef]
- Nappi, F. Non-Coding RNA-Targeted Therapy: A State-of-the-Art Review. Int. J. Mol. Sci. 2024, 25, 3630. [Google Scholar] [CrossRef]
- Beylerli, O.; Shi, H.; Begliarzade, S.; Shumadalova, A.; Ilyasova, T.; Sufianov, A. MiRNAs as new potential biomarkers and therapeutic targets in brain metastasis. Non-Coding RNA Res. 2024, 9, 678–686. [Google Scholar] [CrossRef]
- Ma, W.; Xiao, G.G.; Mao, J.; Lu, Y.; Song, B.; Wang, L.; Fan, S.; Fan, P.; Hou, Z.; Li, J.; et al. Dysregulation of the miR-34a-SIRT1 axis inhibits breast cancer stemness. Oncotarget 2015, 6, 10432–10444. [Google Scholar] [CrossRef]
- Wen, Y.; Huang, H.; Huang, B.; Liao, X. HSA-miR-34a-5p regulates the SIRT1/TP53 axis in prostate cancer. Am. J. Transl. Res. 2022, 14, 4493–4504. [Google Scholar]
- Hong, D.S.; Kang, Y.K.; Borad, M.; Sachdev, J.; Ejadi, S.; Lim, H.Y.; Brenner, A.J.; Park, K.; Lee, J.L.; Kim, T.Y.; et al. Phase 1 study of MRX34, a liposomal miR-34a mimic, in patients with advanced solid tumours. Br. J. Cancer 2020, 122, 1630–1637. [Google Scholar] [CrossRef]
- Soghli, N.; Ferns, G.A.; Sadeghsoltani, F.; Qujeq, D.; Yousefi, T.; Vaghari-Tabari, M. MicroRNAs and osteosarcoma: Potential targets for inhibiting metastasis and increasing chemosensitivity. Biochem. Pharmacol. 2022, 201, 115094. [Google Scholar] [CrossRef]
- Seto, A.G.; Beatty, X.; Lynch, J.M.; Hermreck, M.; Tetzlaff, M.; Duvic, M.; Jackson, A.L. Cobomarsen, an oligonucleotide inhibitor of miR-155, co-ordinately regulates multiple survival pathways to reduce cellular proliferation and survival in cutaneous T-cell lymphoma. Br. J. Haematol. 2018, 183, 428–444. [Google Scholar] [CrossRef] [PubMed]
- Witten, L.; Slack, F.J. miR-155 as a novel clinical target for hematological malignancies. Carcinogenesis 2020, 41, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Anfossi, S.; Giordano, A.; Gao, H.; Cohen, E.N.; Tin, S.; Wu, Q.; Garza, R.J.; Debeb, B.G.; Alvarez, R.H.; Valero, V.; et al. High serum miR-19a levels are associated with inflammatory breast cancer and are predictive of favorable clinical outcome in patients with metastatic HER2+ inflammatory breast cancer. PLoS ONE 2014, 9, e83113. [Google Scholar] [CrossRef] [PubMed]
- Vimalraj, S.; Miranda, P.J.; Ramyakrishna, B.; Selvamurugan, N. Regulation of breast cancer and bone metastasis by microRNAs. Dis. Markers 2013, 35, 369–387. [Google Scholar] [CrossRef]
- Shaath, H.; Vishnubalaji, R.; Elango, R.; Kardousha, A.; Islam, Z.; Qureshi, R.; Alam, T.; Kolatkar, P.R.; Alajez, N.M. Long non-coding RNA and RNA-binding protein interactions in cancer: Experimental and machine learning approaches. Semin. Cancer Biol. 2022, 86, 325–345. [Google Scholar] [CrossRef]
- Tang, X.; Ren, H.; Guo, M.; Qian, J.; Yang, Y.; Gu, C. Review on circular RNAs and new insights into their roles in cancer. Comput. Struct. Biotechnol. J. 2021, 19, 910–928. [Google Scholar] [CrossRef]
- He, W.; Li, D.; Zhang, X. LncRNA HOTAIR promotes the proliferation and invasion/metastasis of breast cancer cells by targeting the miR-130a-3p/Suv39H1 axis. Biochem. Biophys. Rep. 2022, 30, 101279. [Google Scholar] [CrossRef]
- Rajagopal, T.; Talluri, S.; Akshaya, R.L.; Dunna, N.R. HOTAIR LncRNA: A novel oncogenic propellant in human cancer. Clin. Chim. Acta Int. J. Clin. Chem. 2020, 503, 1–18. [Google Scholar] [CrossRef]
- Zhang, J.J.; Zhou, X.H.; Zhou, Y.; Wang, Y.G.; Qian, B.Z.; He, A.N.; Shen, Z.; Hu, H.Y.; Yao, Y. Bufalin suppresses the migration and invasion of prostate cancer cells through HOTAIR, the sponge of miR-520b. Acta Pharmacol. Sin. 2019, 40, 1228–1236. [Google Scholar] [CrossRef]
- Fasciano, S.; Luo, S.; Wang, S. Long non-coding RNA (lncRNA) MALAT1 in regulating osteogenic and adipogenic differentiation using a double-stranded gapmer locked nucleic acid nanobiosensor. Analyst 2023, 148, 6261–6273. [Google Scholar] [CrossRef]
- Yang, K.; Zhang, W.; Zhong, L.; Xiao, Y.; Sahoo, S.; Fassan, M.; Zeng, K.; Magee, P.; Garofalo, M.; Shi, L. Long non-coding RNA HIF1A-As2 and MYC form a double-positive feedback loop to promote cell proliferation and metastasis in KRAS-driven non-small cell lung cancer. Cell Death Differ. 2023, 30, 1533–1549. [Google Scholar] [CrossRef] [PubMed]
- Abdelgwad, M.; Zakaria, R.; Marzouk, S.; Sabry, D.; Ahmed, R.; Badary, H.A.; Samir, M. The Emerging Role of Circular RNA Homeodomain Interacting Protein Kinase 3 and Circular RNA 0046367 through Wnt/Beta-Catenin Pathway on the Pathogenesis of Nonalcoholic Steatohepatitis in Egyptian Patients. Rep. Biochem. Mol. Biol. 2023, 11, 614–625. [Google Scholar] [CrossRef]
- Bai, F.; Zuo, C.; Ouyang, Y.; Xiao, K.; He, Z.; Yang, Z. Circular RNA 0001666 inhibits colorectal cancer cell proliferation, invasion and stemness by inactivating the Wnt/β-catenin signaling pathway and targeting microRNA-1229. Oncol. Lett. 2022, 23, 153. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Yu, F.; Li, P.; Wang, K. Emerging Function and Clinical Significance of Exosomal circRNAs in Cancer. Mol. Ther. Nucleic Acids 2020, 21, 367–383. [Google Scholar] [CrossRef] [PubMed]
- Gong, Z.; Hu, W.; Zhou, C.; Guo, J.; Yang, L.; Wang, B. Recent advances and perspectives on the development of circular RNA cancer vaccines. NPJ Vaccines 2025, 10, 41. [Google Scholar] [CrossRef]
- Li, P.; Lin, Z.; Liu, Q.; Chen, S.; Gao, X.; Guo, W.; Gong, F.; Wei, J.; Lin, H. Enhancer RNA SLIT2 Inhibits Bone Metastasis of Breast Cancer Through Regulating P38 MAPK/c-Fos Signaling Pathway. Front. Oncol. 2021, 11, 743840. [Google Scholar] [CrossRef]
- Gu, P.; Zhao, J.; Zhang, W.; Ruan, X.; Hu, L.; Zeng, Y.; Hou, X.; Zheng, X.; Gao, M.; Chi, J. An Inducible CRISPR-dCas9-Based Transcriptional Repression System for Cancer Therapy. Small Methods 2024, 8, e2301310. [Google Scholar] [CrossRef]
- Qian, J.; Liu, S.X. CRISPR/dCas9-Tet1-Mediated DNA Methylation Editing. Bio-Protocol 2024, 14, e4976. [Google Scholar] [CrossRef]
- An, C.; Wang, I.; Li, X.; Xia, R.; Deng, F. Long non-coding RNA in prostate cancer. Am. J. Clin. Exp. Urol. 2022, 10, 170–179. [Google Scholar]
- Ito, M.; Miyata, Y.; Okada, M. Current clinical trials with non-coding RNA-based therapeutics in malignant diseases: A systematic review. Transl. Oncol. 2023, 31, 101634. [Google Scholar] [CrossRef] [PubMed]
- Golan, T.; Khvalevsky, E.Z.; Hubert, A.; Gabai, R.M.; Hen, N.; Segal, A.; Domb, A.; Harari, G.; David, E.B.; Raskin, S.; et al. RNAi therapy targeting KRAS in combination with chemotherapy for locally advanced pancreatic cancer patients. Oncotarget 2015, 6, 24560–24570. [Google Scholar] [CrossRef] [PubMed]
- Escuin, D.; Bell, O.; García-Valdecasas, B.; Clos, M.; Larrañaga, I.; López-Vilaró, L.; Mora, J.; Andrés, M.; Arqueros, C.; Barnadas, A. Small Non-Coding RNAs and Their Role in Locoregional Metastasis and Outcomes in Early-Stage Breast Cancer Patients. Int. J. Mol. Sci. 2024, 25, 3982. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Feng, C.; Wang, S.; Shi, L.; Gu, Q.; Zhang, H.; Lan, X.; Zhao, Y.; Qiang, W.; Ji, M.; et al. The noncoding RNAs SNORD50A and SNORD50B-mediated TRIM21-GMPS interaction promotes the growth of p53 wild-type breast cancers by degrading p53. Cell Death Differ. 2021, 28, 2450–2464. [Google Scholar] [CrossRef]
- Xi, X.; Hu, Z.; Wu, Q.; Hu, K.; Cao, Z.; Zhou, J.; Liao, J.; Zhang, Z.; Hu, Y.; Zhong, X.; et al. High expression of small nucleolar RNA host gene 3 predicts poor prognosis and promotes bone metastasis in prostate cancer by activating transforming growth factor-beta signaling. Bioengineered 2022, 13, 1895–1907. [Google Scholar] [CrossRef]
- Yang, J.; Xu, J.; Wang, W.; Zhang, B.; Yu, X.; Shi, S. Epigenetic regulation in the tumor microenvironment: Molecular mechanisms and therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 210. [Google Scholar] [CrossRef]
- Zhang, Y.; Liang, J.; Liu, P.; Wang, Q.; Liu, L.; Zhao, H. The RANK/RANKL/OPG system and tumor bone metastasis: Potential mechanisms and therapeutic strategies. Front. Endocrinol. 2022, 13, 1063815. [Google Scholar] [CrossRef]
- Chen, X.; Song, E. Turning foes to friends: Targeting cancer-associated fibroblasts. Nat. Rev. Drug Discov. 2019, 18, 99–115. [Google Scholar] [CrossRef]
- Kehrberg, R.J.; Bhyravbhatla, N.; Batra, S.K.; Kumar, S. Epigenetic regulation of cancer-associated fibroblast heterogeneity. Biochim. Biophys. Acta Rev. Cancer 2023, 1878, 188901. [Google Scholar] [CrossRef]
- Lamoureux, F.; Baud’huin, M.; Rodriguez Calleja, L.; Jacques, C.; Berreur, M.; Rédini, F.; Lecanda, F.; Bradner, J.E.; Heymann, D.; Ory, B. Selective inhibition of BET bromodomain epigenetic signalling interferes with the bone-associated tumour vicious cycle. Nat. Commun. 2014, 5, 3511. [Google Scholar] [CrossRef]
- Mazzucchelli, S.; Signati, L.; Messa, L.; Franceschini, A.; Bonizzi, A.; Castagnoli, L.; Gasparini, P.; Consolandi, C.; Mangano, E.; Pelucchi, P.; et al. Breast cancer patient-derived organoids for the investigation of patient-specific tumour evolution. Cancer Cell Int. 2024, 24, 220. [Google Scholar] [CrossRef]
- Suraweera, A.; O’Byrne, K.J.; Richard, D.J. Epigenetic drugs in cancer therapy. Cancer Metastasis Rev. 2025, 44, 37. [Google Scholar] [CrossRef]
- Centore, R.C.; Sandoval, G.J.; Soares, L.M.M.; Kadoch, C.; Chan, H.M. Mammalian SWI/SNF Chromatin Remodeling Complexes: Emerging Mechanisms and Therapeutic Strategies. Trends Genet. 2020, 36, 936–950. [Google Scholar] [CrossRef]
- Fiskus, W.; Piel, J.; Collins, M.; Hentemann, M.; Cuglievan, B.; Mill, C.P.; Birdwell, C.E.; Das, K.; Davis, J.A.; Hou, H.; et al. BRG1/BRM inhibitor targets AML stem cells and exerts superior preclinical efficacy combined with BET or menin inhibitor. Blood 2024, 143, 2059–2072. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, K.; Wang, L.; Lu, C.; Liu, Z.; Yang, D.; Zhong, H.; Zou, J.; Fahira, A.; Wang, J.; Huang, Z. Assessment of SWI/SNF chromatin remodeling complex related genes as potential biomarkers and therapeutic targets in pan-cancer. Mol. Cancer 2024, 23, 176. [Google Scholar] [CrossRef]
- Vaswani, R.G.; Huang, D.S.; Anthony, N.; Xu, L.; Centore, R.; Schiller, S.; Li, Z.; Fan, H.; Setser, J.; Zawadzke, L.E.; et al. Discovery of FHD-286, a First-in-Class, Orally Bioavailable, Allosteric Dual Inhibitor of the Brahma Homologue (BRM) and Brahma-Related Gene 1 (BRG1) ATPase Activity for the Treatment of SWItch/Sucrose Non-Fermentable (SWI/SNF) Dependent Cancers. J. Med. Chem. 2025, 68, 1772–1792. [Google Scholar] [CrossRef] [PubMed]
- Duplaquet, L.; So, K.; Ying, A.W.; Pal Choudhuri, S.; Li, X.; Xu, G.D.; Li, Y.; Qiu, X.; Li, R.; Singh, S.; et al. Mammalian SWI/SNF complex activity regulates POU2F3 and constitutes a targetable dependency in small cell lung cancer. Cancer Cell 2024, 42, 1352–1369.e1313. [Google Scholar] [CrossRef] [PubMed]
- Baud’huin, M.; Lamoureux, F.; Jacques, C.; Rodriguez Calleja, L.; Quillard, T.; Charrier, C.; Amiaud, J.; Berreur, M.; Brounais-LeRoyer, B.; Owen, R.; et al. Inhibition of BET proteins and epigenetic signaling as a potential treatment for osteoporosis. Bone 2017, 94, 10–21. [Google Scholar] [CrossRef]
- Khandekar, D.; Tiriveedhi, V. Role of BET Inhibitors in Triple Negative Breast Cancers. Cancers 2020, 12, 784. [Google Scholar] [CrossRef]
- Park-Min, K.H.; Lim, E.; Lee, M.J.; Park, S.H.; Giannopoulou, E.; Yarilina, A.; van der Meulen, M.; Zhao, B.; Smithers, N.; Witherington, J.; et al. Inhibition of osteoclastogenesis and inflammatory bone resorption by targeting BET proteins and epigenetic regulation. Nat. Commun. 2014, 5, 5418. [Google Scholar] [CrossRef]
- Swami, A.; Reagan, M.R.; Basto, P.; Mishima, Y.; Kamaly, N.; Glavey, S.; Zhang, S.; Moschetta, M.; Seevaratnam, D.; Zhang, Y.; et al. Engineered nanomedicine for myeloma and bone microenvironment targeting. Proc. Natl. Acad. Sci. USA 2014, 111, 10287–10292. [Google Scholar] [CrossRef]
- Huang, M.; Qiu, Q.; Xiao, Y.; Zeng, S.; Zhan, M.; Shi, M.; Zou, Y.; Ye, Y.; Liang, L.; Yang, X.; et al. BET Bromodomain Suppression Inhibits VEGF-induced Angiogenesis and Vascular Permeability by Blocking VEGFR2-mediated Activation of PAK1 and eNOS. Sci. Rep. 2016, 6, 23770. [Google Scholar] [CrossRef]
- Zhang, L.; Cai, T.; Lin, X.; Huang, X.; Bui, M.H.; Plotnik, J.P.; Bellin, R.J.; Faivre, E.J.; Kuruvilla, V.M.; Lam, L.T.; et al. Selective Inhibition of the Second Bromodomain of BET Family Proteins Results in Robust Antitumor Activity in Preclinical Models of Acute Myeloid Leukemia. Mol. Cancer Ther. 2021, 20, 1809–1819. [Google Scholar] [CrossRef] [PubMed]
- Xing, L.; Ebetino, F.H.; Boeckman, R.K., Jr.; Srinivasan, V.; Tao, J.; Sawyer, T.K.; Li, J.; Yao, Z.; Boyce, B.F. Targeting anti-cancer agents to bone using bisphosphonates. Bone 2020, 138, 115492. [Google Scholar] [CrossRef] [PubMed]
- Fatima, H.; Rangwala, H.S.; Riaz, F.; Ali, L.; Abbas, S.R.; Haque, S.U. Castration resistant prostate cancer: Recent advances in novel therapeutic treatments. IJS Glob. Health 2024, 7, e0400. [Google Scholar] [CrossRef]
- Aimiuwu, J.; Wang, H.; Chen, P.; Xie, Z.; Wang, J.; Liu, S.; Klisovic, R.; Mims, A.; Blum, W.; Marcucci, G.; et al. RNA-dependent inhibition of ribonucleotide reductase is a major pathway for 5-azacytidine activity in acute myeloid leukemia. Blood 2012, 119, 5229–5238. [Google Scholar] [CrossRef]
- Lv, Z.; Ji, T.; Liu, J.; Sun, X.; Liang, H. Synthetic approaches and clinical applications of representative HDAC inhibitors for cancer therapy: A review. Eur. J. Med. Chem. 2025, 283, 117185. [Google Scholar] [CrossRef]
- Moore, D.C.; Arnall, J.R.; Harvey, R.D. Incidence and management of adverse events associated with panobinostat in the treatment of relapsed/refractory multiple myeloma. J. Oncol. Pharm. Pract. Off. Publ. Int. Soc. Oncol. Pharm. Pract. 2019, 25, 613–622. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhra, M.; Holail, J.H.; Mohammad, K.S. Epigenetic Modulation and Bone Metastasis: Evolving Therapeutic Strategies. Pharmaceuticals 2025, 18, 1140. https://doi.org/10.3390/ph18081140
Zhra M, Holail JH, Mohammad KS. Epigenetic Modulation and Bone Metastasis: Evolving Therapeutic Strategies. Pharmaceuticals. 2025; 18(8):1140. https://doi.org/10.3390/ph18081140
Chicago/Turabian StyleZhra, Mahmoud, Jasmine Hanafy Holail, and Khalid S. Mohammad. 2025. "Epigenetic Modulation and Bone Metastasis: Evolving Therapeutic Strategies" Pharmaceuticals 18, no. 8: 1140. https://doi.org/10.3390/ph18081140
APA StyleZhra, M., Holail, J. H., & Mohammad, K. S. (2025). Epigenetic Modulation and Bone Metastasis: Evolving Therapeutic Strategies. Pharmaceuticals, 18(8), 1140. https://doi.org/10.3390/ph18081140