
ADMET-Guided Docking and GROMACS Molecular Dynamics of Ziziphus lotus Phytochemicals Uncover Mutation-Agnostic Allosteric Stabilisers of the KRAS Switch-I/II Groove

 , , , , ,
, , , , ,
Abstract

1. Introduction
2. Results and Discussion
2.1. ADME Results
2.1.1. Physicochemical and SwissADME Bioavailability Comparison of Natural KRAS Candidates Versus AMG-510
2.1.2. In Silico ADME Profile and Interaction Risks (pkCSM Analysis)
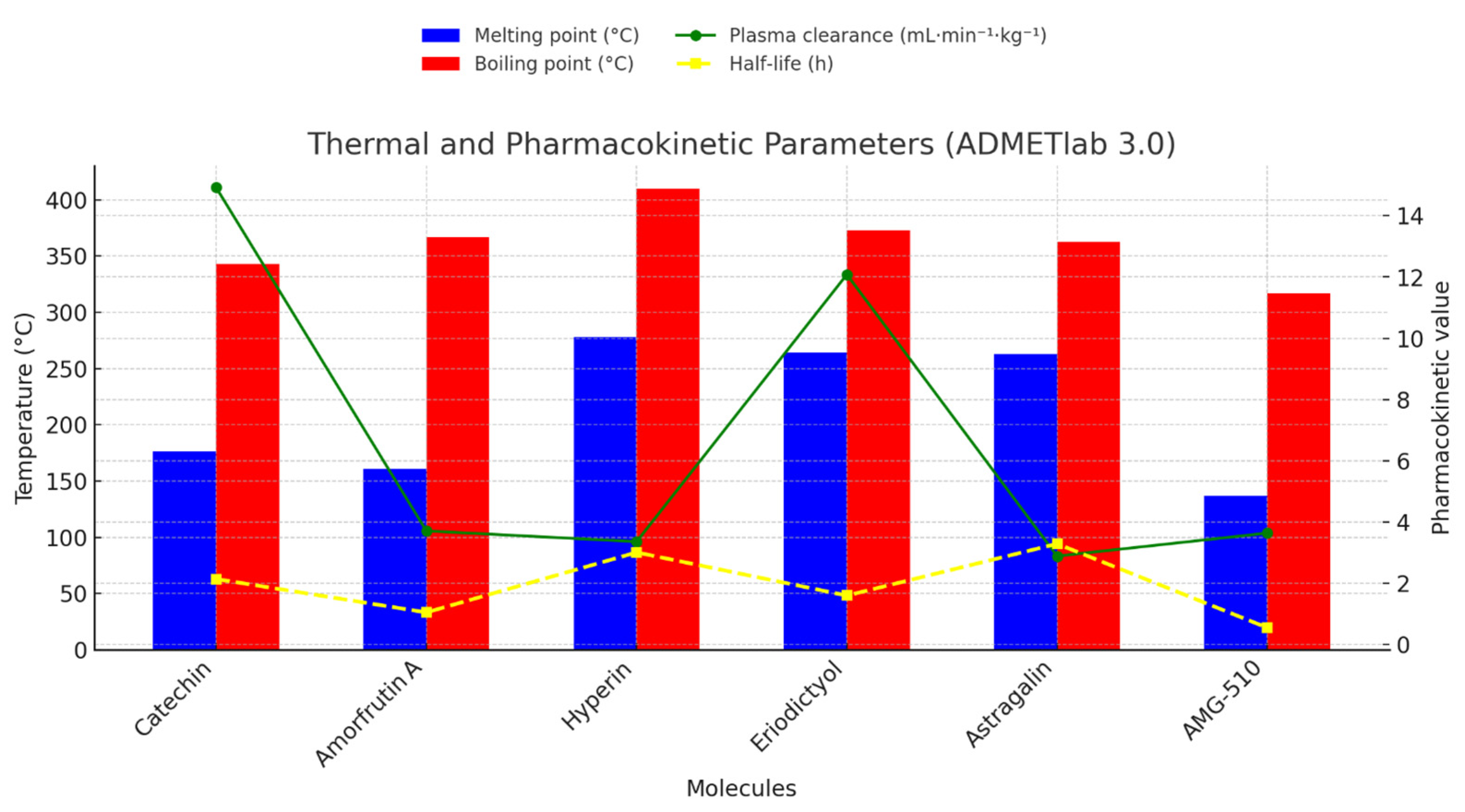
2.1.3. Integrated Thermal-Pharmacokinetic Profiling and Structure-Guided Optimisation Strategy
2.2. Predictive Acute-Toxicity Profile (Protox 3) of the Five Natural Candidates Compared with the Reference Inhibitor AMG-510
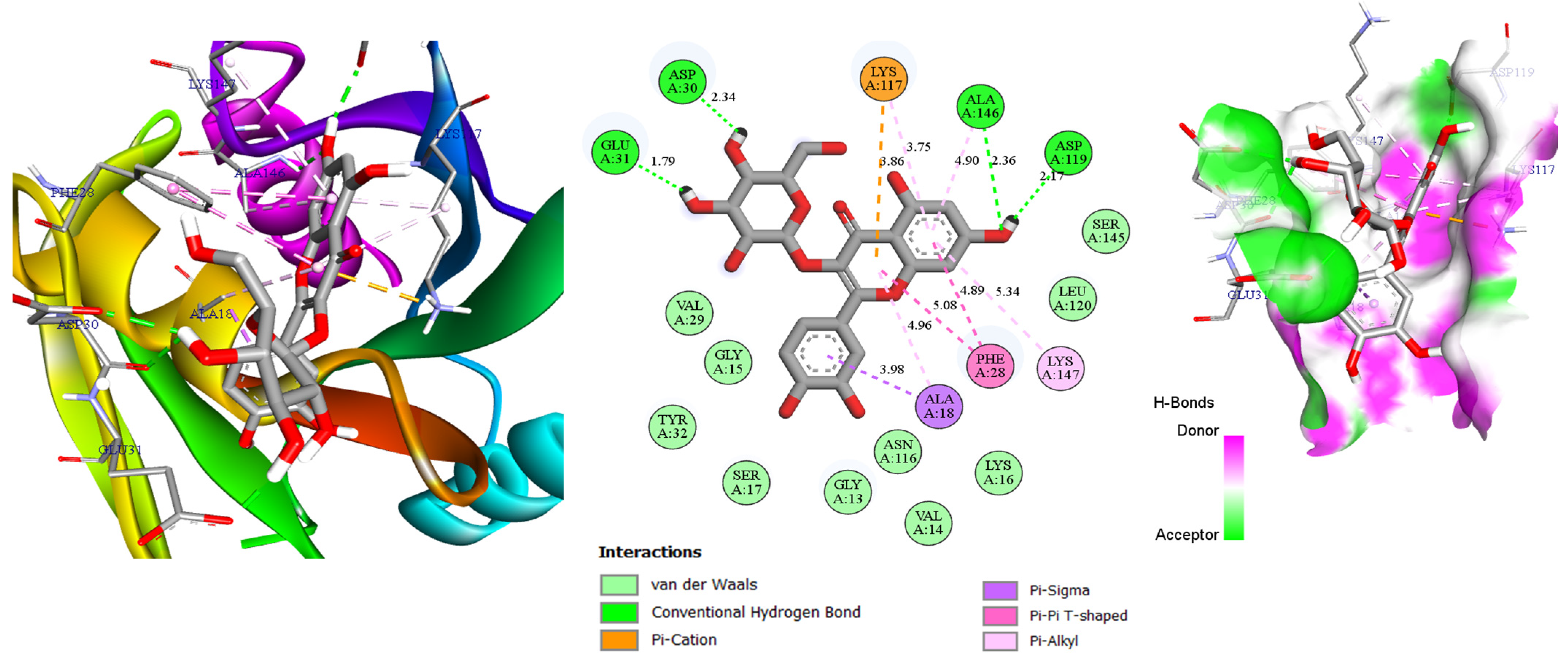
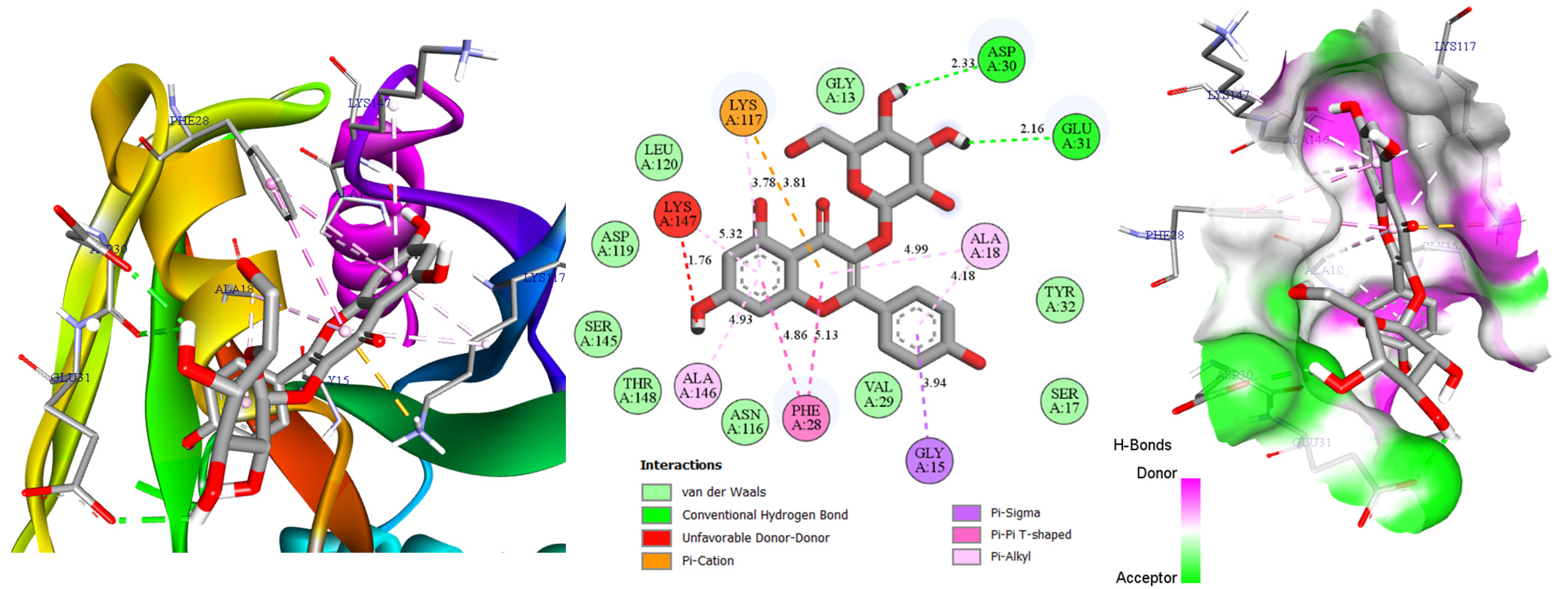
2.3. Molecular Docking
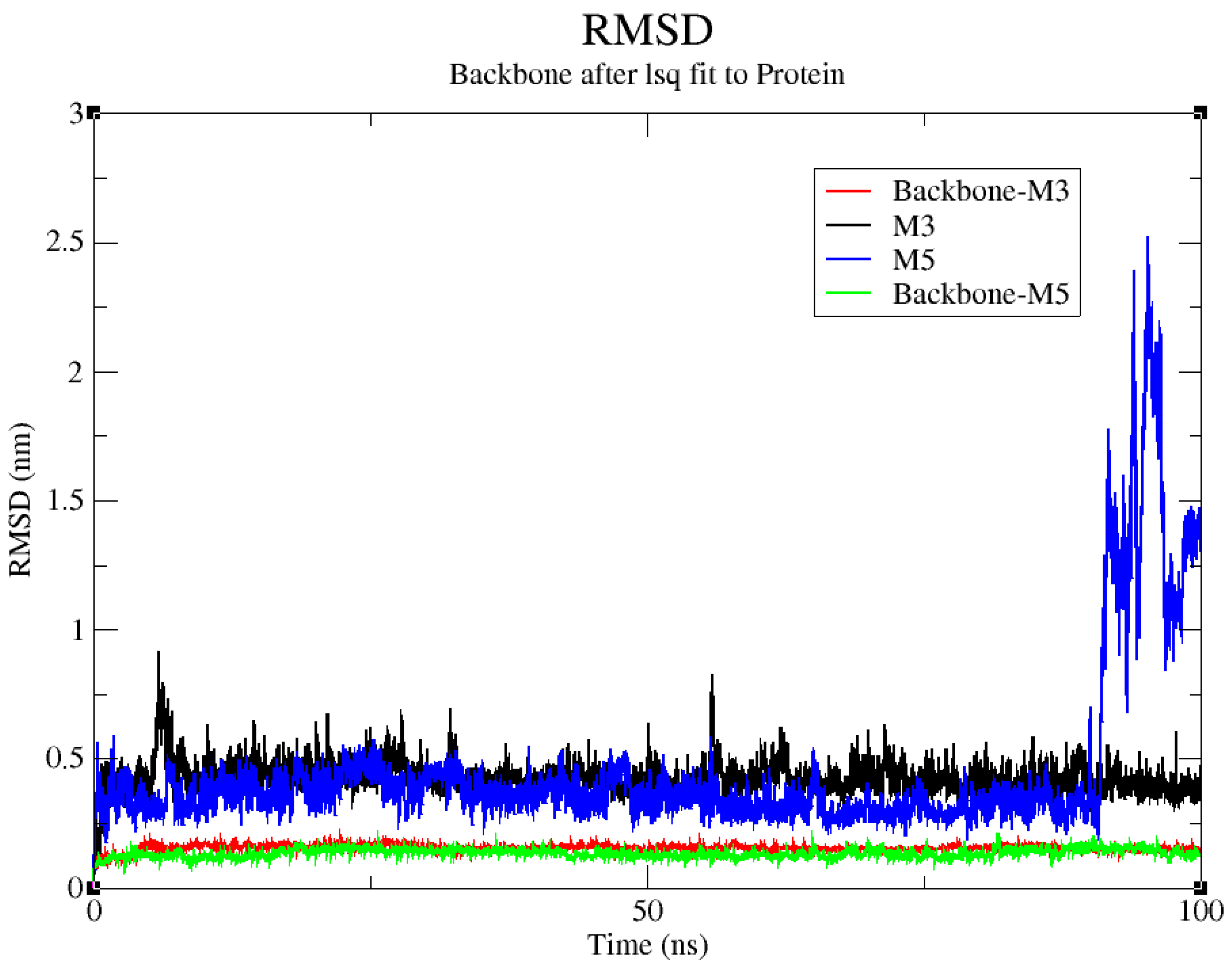
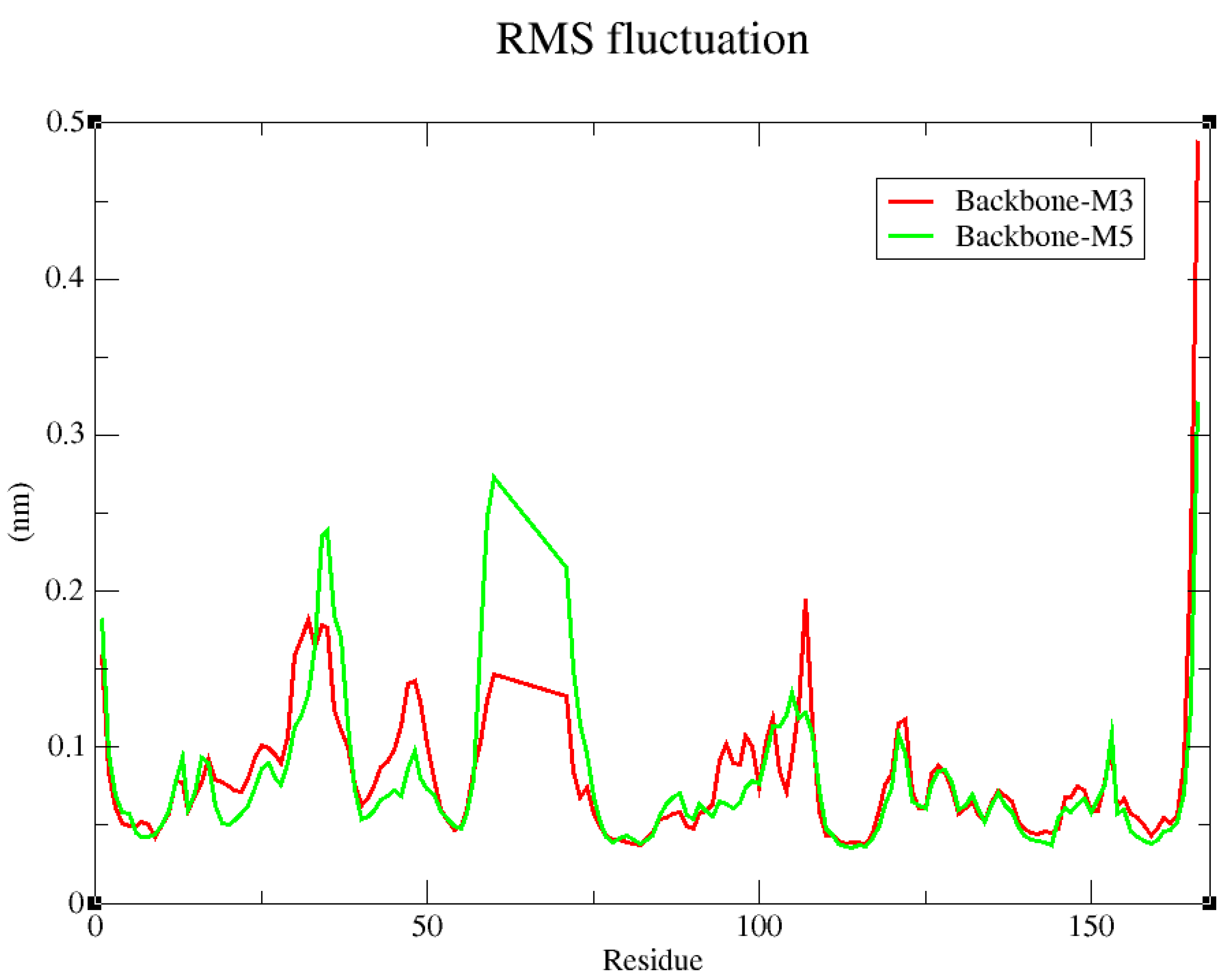
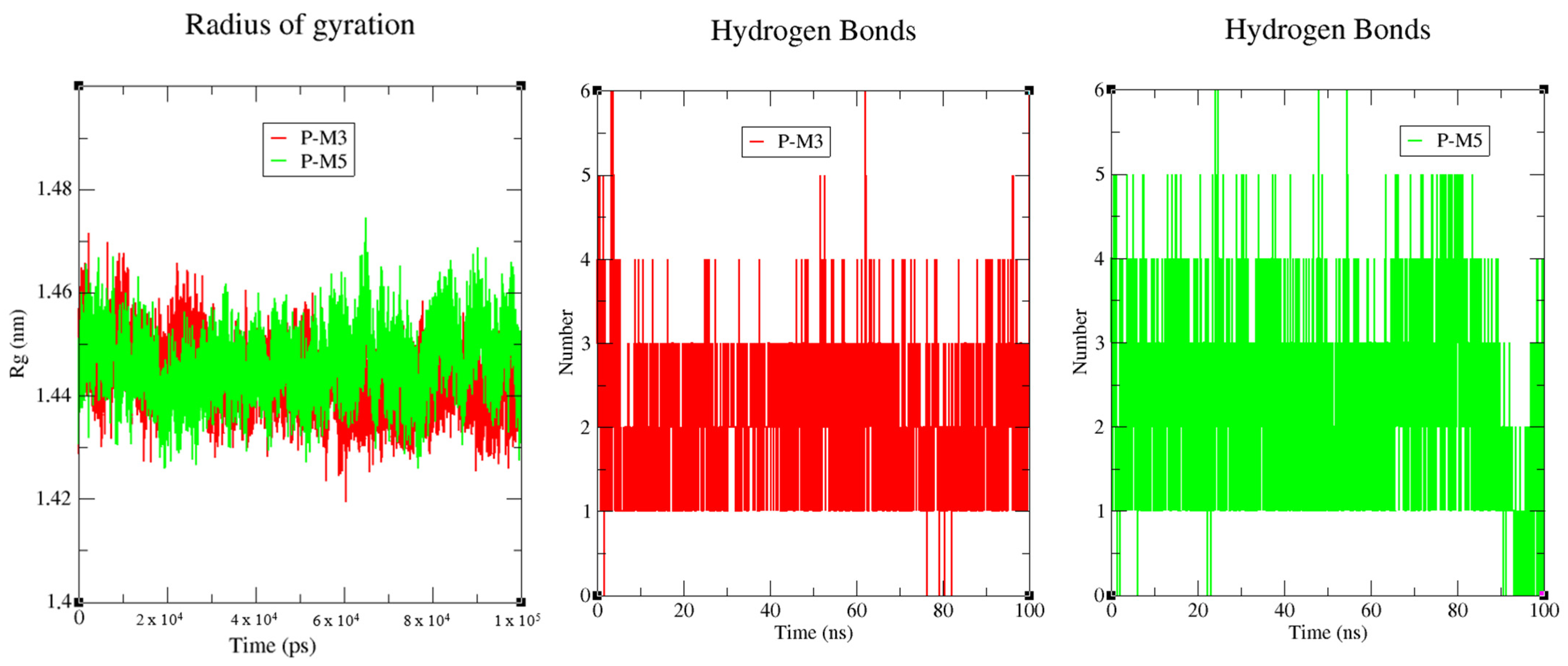
2.4. Molecular Dynamics Simulation
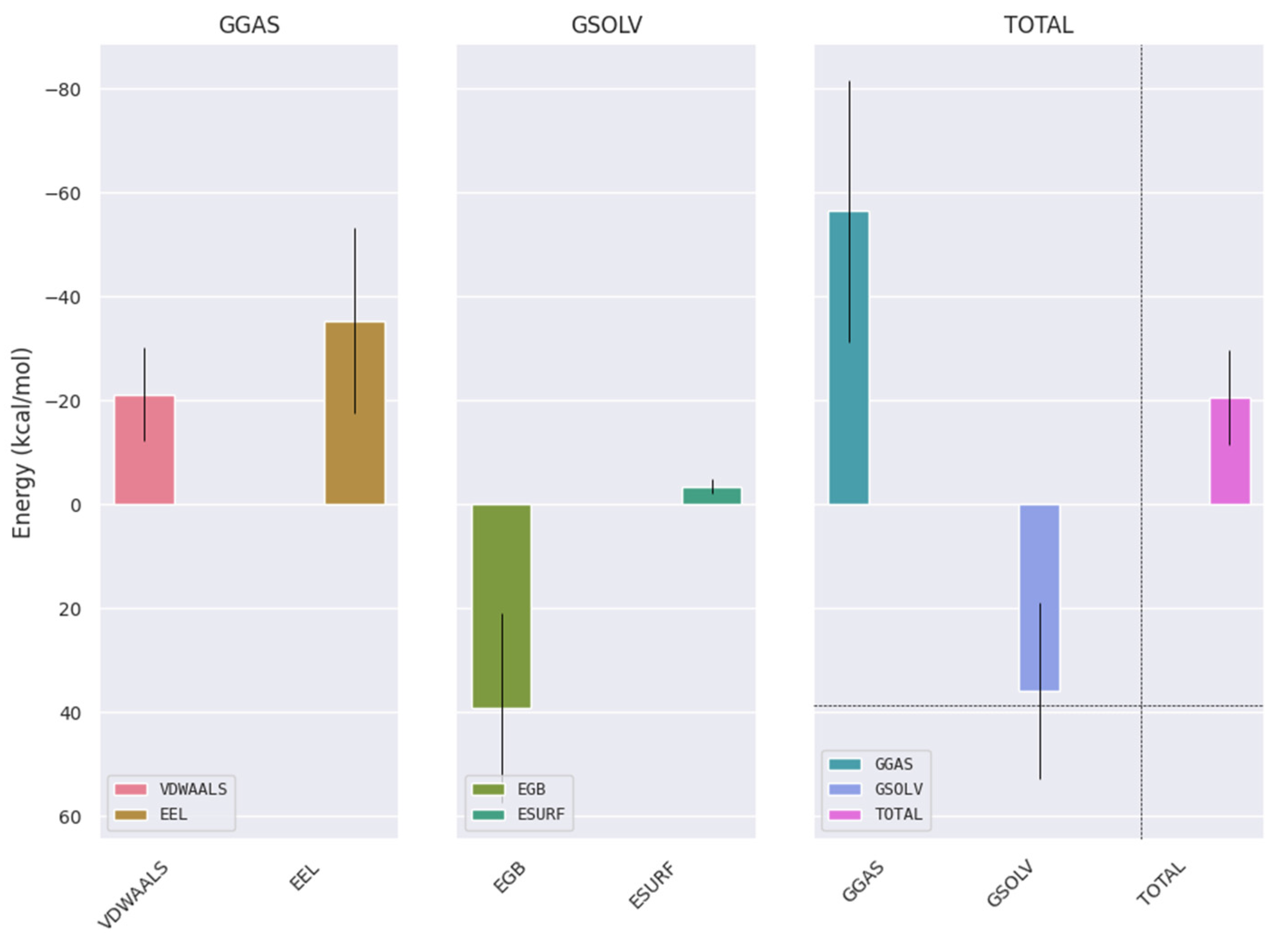
2.5. MM/GBSA Binding-Free-Energy Analysis
3. Materials and Methods
3.1. Key Bioactive Constituents Selected for In Silico Evaluation
3.2. Pharmacokinetic Analysis Using Computational Tools
3.3. Prediction of the Toxicity Analysis (Pro Tox III)
3.4. PyRx: Preparation, Configuration, and Validation of the Docking Protocol
3.5. Implementation of Molecular Dynamics Simulations Using GROMACS
3.6. MM/GBSA Calculation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| RMSD | Root-Mean-Square Deviation |
| RMSF | Root-Mean-Square Fluctuation |
| MD | Molecular Dynamics |
| ADMET | Absorption, Distribution, Metabolism, Excretion, and Toxicity |
| PDB | Protein Data Bank |
| SI/II | Switch I/Switch II |
References
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the Undruggable RAS: Mission Possible? Nat. Rev. Drug Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef] [PubMed]
- Ruscetti, M.; Morris, J.P.; Mezzadra, R.; Russell, J.; Leibold, J.; Romesser, P.B.; Simon, J.; Kulick, A.; Ho, Y.; Fennell, M. Senescence-Induced Vascular Remodeling Creates Therapeutic Vulnerabilities in Pancreas Cancer. Cell 2020, 181, 424–441. [Google Scholar] [CrossRef] [PubMed]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras (G12C) Inhibitors Allosterically Control GTP Affinity and Effector Interactions. Nature 2013, 503, 548–551. [Google Scholar] [CrossRef] [PubMed]
- Skoulidis, F.; Li, B.T.; Dy, G.K.; Price, T.J.; Falchook, G.S.; Wolf, J.; Italiano, A.; Schuler, M.; Borghaei, H.; Barlesi, F. Sotorasib for Lung Cancers with KRAS p. G12C Mutation. N. Engl. J. Med. 2021, 384, 2371–2381. [Google Scholar] [CrossRef] [PubMed]
- Palma, G.; Khurshid, F.; Lu, K.; Woodward, B.; Husain, H. Selective KRAS G12C Inhibitors in Non-Small Cell Lung Cancer: Chemistry, Concurrent Pathway Alterations, and Clinical Outcomes. NPJ Precis. Oncol. 2021, 5, 98. [Google Scholar] [CrossRef] [PubMed]
- Awad, M.M.; Liu, S.; Rybkin, I.I.; Arbour, K.C.; Dilly, J.; Zhu, V.W.; Johnson, M.L.; Heist, R.S.; Patil, T.; Riely, G.J. Acquired Resistance to KRASG12C Inhibition in Cancer. N. Engl. J. Med. 2021, 384, 2382–2393. [Google Scholar] [CrossRef] [PubMed]
- Fedele, C.; Li, S.; Teng, K.W.; Foster, C.J.R.; Peng, D.; Ran, H.; Mita, P.; Geer, M.J.; Hattori, T.; Koide, A. SHP2 Inhibition Diminishes KRASG12C Cycling and Promotes Tumor Microenvironment Remodeling. J. Exp. Med. 2020, 218, e20201414. [Google Scholar] [CrossRef] [PubMed]
- Angappulige, D.H.; Mahajan, N.P.; Mahajan, K. Epigenetic Underpinnings of Tumor-Immune Dynamics in Prostate Cancer Immune Suppression. Trends Cancer 2024, 10, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Kessler, D.; Gmachl, M.; Mantoulidis, A.; Martin, L.J.; Zoephel, A.; Mayer, M.; Gollner, A.; Covini, D.; Fischer, S.; Gerstberger, T. Drugging an Undruggable Pocket on KRAS. Proc. Natl. Acad. Sci. USA 2019, 116, 15823–15829. [Google Scholar] [CrossRef] [PubMed]
- Issahaku, A.R.; Aljoundi, A.; Soliman, M.E.S. Establishing the Mutational Effect on the Binding Susceptibility of AMG510 to KRAS Switch II Binding Pocket: Computational Insights. Inform. Med. Unlocked 2022, 30, 100952. [Google Scholar] [CrossRef]
- Vasta, J.D.; Peacock, D.M.; Zheng, Q.; Walker, J.A.; Zhang, Z.; Zimprich, C.A.; Thomas, M.R.; Beck, M.T.; Binkowski, B.F.; Corona, C.R. KRAS Is Vulnerable to Reversible Switch-II Pocket Engagement in Cells. Nat. Chem. Biol. 2022, 18, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Herdeis, L.; Rudolph, D.; Zhao, Y.; Böttcher, J.; Vides, A.; Ayala-Santos, C.I.; Pourfarjam, Y.; Cuevas-Navarro, A.; Xue, J.Y. Pan-KRAS Inhibitor Disables Oncogenic Signalling and Tumour Growth. Nature 2023, 619, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Xue, J.Y.; Lito, P. Suppressing Nucleotide Exchange to Inhibit KRAS-Mutant Tumors. Cancer Discov. 2021, 11, 17–19. [Google Scholar] [CrossRef] [PubMed]
- Maurer, T.; Garrenton, L.S.; Oh, A.; Pitts, K.; Anderson, D.J.; Skelton, N.J.; Fauber, B.P.; Pan, B.; Malek, S.; Stokoe, D. Small-Molecule Ligands Bind to a Distinct Pocket in Ras and Inhibit SOS-Mediated Nucleotide Exchange Activity. Proc. Natl. Acad. Sci. USA 2012, 109, 5299–5304. [Google Scholar] [CrossRef] [PubMed]
- Hillig, R.C.; Sautier, B.; Schroeder, J.; Moosmayer, D.; Hilpmann, A.; Stegmann, C.M.; Werbeck, N.D.; Briem, H.; Boemer, U.; Weiske, J. Discovery of Potent SOS1 Inhibitors That Block RAS Activation via Disruption of the RAS–SOS1 Interaction. Proc. Natl. Acad. Sci. USA 2019, 116, 2551–2560. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef] [PubMed]
- Scalbert, A.; Johnson, I.T.; Saltmarsh, M. Polyphenols: Antioxidants and Beyond. Am. J. Clin. Nutr. 2005, 81, 215S–217S. [Google Scholar] [CrossRef] [PubMed]
- Manach, C.; Scalbert, A.; Morand, C.; Rémésy, C.; Jiménez, L. Polyphenols: Food Sources and Bioavailability. Am. J. Clin. Nutr. 2004, 79, 727–747. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.B.; Holloway, G.A. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [PubMed]
- Touam, S.; Nacer, H.; Ziani, N.; Lebrazi, S.; Kertiou, N. Prediction of Surface Tension of Propane Derivatives Using QSPR Approach and Artificial Neural Networks Prediction of Surface Tension of Propane Derivatives Using QSPR Approach and Artificial Neural Networks. Moroccan J. Chem. 2025, 13, 498–940. [Google Scholar] [CrossRef]
- Hollingsworth, S.A.; Dror, R.O. Molecular Dynamics Simulation for All. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef] [PubMed]
- Homeyer, N.; Gohlke, H. Free Energy Calculations by the Molecular Mechanics Poisson− Boltzmann Surface Area Method. Mol. Inform. 2012, 31, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cui, H.; Zhang, R.; Zhang, H.; Huang, W. Nanoparticulation of Prodrug into Medicines for Cancer Therapy. Adv. Sci. 2021, 8, 2101454. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Sun, Q.; Wu, F.; Dai, Y.; Chen, X. Polyphenol-containing Nanoparticles: Synthesis, Properties, and Therapeutic Delivery. Adv. Mater. 2021, 33, 2007356. [Google Scholar] [CrossRef] [PubMed]
- Bröker, J.; Waterson, A.G.; Smethurst, C.; Kessler, D.; Böttcher, J.; Mayer, M.; Gmaschitz, G.; Phan, J.; Little, A.; Abbott, J.R. Fragment Optimization of Reversible Binding to the Switch II Pocket on KRAS Leads to a Potent, in Vivo Active KRASG12C Inhibitor. J. Med. Chem. 2022, 65, 14614–14629. [Google Scholar] [CrossRef] [PubMed]
- Bagdanoff, J.T.; Chen, Z.; Acker, M.; Chen, Y.N.; Chan, H.; Dore, M.; Firestone, B.; Fodor, M.; Fortanet, J.; Hentemann, M.; et al. Optimization of Fused Bicyclic Allosteric SHP2 Inhibitors. J. Med. Chem. 2019, 62, 1781–1792. [Google Scholar] [CrossRef] [PubMed]
- Vakili, M.G.; Gorgulla, C.; Snider, J.; Nigam, A.; Bezrukov, D.; Varoli, D.; Aliper, A.; Polykovsky, D.; Das, K.M.P.; Iii, H.C. Quantum-Computing-Enhanced Algorithm Unveils Potential KRAS Inhibitors. Nat. Biotechnol. 2025. [Google Scholar] [CrossRef]
- Lucchetti, D.; Luongo, F.; Colella, F.; Gurreri, E.; Artemi, G.; Desiderio, C.; Serra, S.; Giuliante, F.; De Maria, R.; Sgambato, A. Exploiting Bioactive Natural Products of Marine Origin: Evaluation of the Meroterpenoid Metachromin V as a Novel Potential Therapeutic Drug for Colorectal Cancer. Biomed. Pharmacother. 2023, 162, 114679. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Bai, Y.; Cui, J.; Zong, Z.; Gao, Y.; Zheng, Z. Computer-Aided Drug Design Boosts RAS Inhibitor Discovery. Molecules 2022, 27, 5710. [Google Scholar] [CrossRef] [PubMed]
- Yin, G.; Huang, J.; Petela, J.; Jiang, H.; Zhang, Y.; Gong, S.; Wu, J.; Liu, B.; Shi, J.; Gao, Y. Targeting Small GTPases: Emerging Grasps on Previously Untamable Targets, Pioneered by KRAS. Signal Transduct. Target. Ther. 2023, 8, 212. [Google Scholar] [CrossRef] [PubMed]
- Bencheikh, N.; Radi, F.Z.; Fakchich, J.; Elbouzidi, A.; Ouahhoud, S.; Ouasti, M.; Bouhrim, M.; Ouasti, I.; Hano, C.; Elachouri, M. Ethnobotanical, Phytochemical, Toxicological, and Pharmacological Properties of Ziziphus lotus (L.) Lam.: A Comprehensive Review. Pharmaceuticals 2023, 16, 575. [Google Scholar] [CrossRef] [PubMed]
- Hammouti, Y.; Elbouzidi, A.; Taibi, M.; Bellaouchi, R.; Loukili, E.H.; Bouhrim, M.; Noman, O.M.; Mothana, R.A.; Ibrahim, M.N.; Asehraou, A.; et al. Screening of Phytochemical, Antimicrobial, and Antioxidant Properties of Juncus Acutus from Northeastern Morocco. Life 2023, 13, 2135. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Jahan, S.; Redhu, N.S.; Siddiqui, A.J.; Iqbal, D.; Khan, J.; Banawas, S.; Alaidarous, M.; Alshehri, B.; Mir, S.A.; Adnan, M.; et al. Nobiletin as a Neuroprotectant against NMDA Receptors: An In Silico Approach. Pharmaceutics 2022, 14, 1123. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, R.; Kawata, T.; Ueda, S.; Shinbo, T.; Higashimori, M.; Natsume-Kitatani, Y.; Mizuguchi, K. Prediction of the Contribution Ratio of a Target Metabolic Enzyme to Clearance from Chemical Structure Information. Mol. Pharm. 2023, 20, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Delage, C.; Darnaud, L.; Etain, B.; Vignes, M.; Ly, T.K.; Frapsauce, A.; Veyrier, M.; Delavest, M.; Marlinge, E.; Hennion, V.; et al. Cytochromes P450 and P-Glycoprotein Phenotypic Assessment to Optimize Psychotropic Pharmacotherapy: A Retrospective Analysis of Four Years of Practice in Psychiatry. J. Pers. Med. 2022, 12, 1869. [Google Scholar] [CrossRef] [PubMed]
- Kobus, M.; Friedrich, T.; Zorn, E.; Burmeister, N.; Maison, W. Medicinal Chemistry of Drugs with N-Oxide Functionalities. J. Med. Chem. 2024, 67, 5168–5184. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Martínez, J.D.; Valdés, A.; Gallego, R.; Suárez-Montenegro, Z.J.; Alarcón, M.; Ibañez, E.; Alvarez-Rivera, G.; Cifuentes, A. Blood–Brain Barrier Permeability Study of Potential Neuroprotective Compounds Recovered from Plants and Agri-Food by-Products. Front. Nutr. 2022, 9, 924596. [Google Scholar] [CrossRef] [PubMed]
- Korzekwa, K.; Nagar, S. Process and System Clearances in Pharmacokinetic Models: Our Basic Clearance Concepts Are Correct. Drug Metab. Dispos. 2023, 51, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Ghasemitabar, H.; Movagharnejad, K. Estimation of the Normal Boiling Point of Organic Compounds via a New Group Contribution Method. Fluid Phase Equilib. 2016, 411, 13–23. [Google Scholar] [CrossRef]
- Nannoolal, Y.; Rarey, J.; Ramjugernath, D.; Cordes, W. Estimation of Pure Component Properties: Part 1. Estimation of the Normal Boiling Point of Non-Electrolyte Organic Compounds via Group Contributions and Group Interactions. Fluid Phase Equilib. 2004, 226, 45–63. [Google Scholar] [CrossRef]
- Mao, F.; Kong, Q.; Ni, W.; Xu, X.; Ling, D.; Lu, Z.; Li, J. Melting Point Distribution Analysis of Globally Approved and Discontinued Drugs: A Research for Improving the Chance of Success of Drug Design and Discovery. ChemistryOpen 2016, 5, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Gadaleta, D.; Vuković, K.; Toma, C.; Lavado, G.J.; Karmaus, A.L.; Mansouri, K.; Kleinstreuer, N.C.; Benfenati, E.; Roncaglioni, A. SAR and QSAR Modeling of a Large Collection of LD50 Rat Acute Oral Toxicity Data. J. Cheminform. 2019, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Tong, G.C.; Cornwell, W.K.; Means, G.E. Reactions of Acrylamide with Glutathione and Serum Albumin. Toxicol. Lett. 2004, 147, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Danyaya, A.I.; Kumar, A.; Hamza, U.A.; Yahaya, N.L. Virtual Screening, Molecular Docking, and ADME/T Analysis of Natural Product Library Against Cell Invasion Protein SipB from Salmonella enterica serotype typhi: In Silico Analysis. Acta Sci. Pharm. Sci. 2020, 4, 22–26. [Google Scholar] [CrossRef]
- Tripathi, S.K.; Muttineni, R.; Singh, S.K. Extra Precision Docking, Free Energy Calculation and Molecular Dynamics Simulation Studies of CDK2 Inhibitors. J. Theor. Biol. 2013, 334, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Et-tazy, L.; Fedeli, R.; Khibech, O.; Lamiri, A.; Challioui, A. Effects of Monoterpene-Based Biostimulants on Chickpea (Cicer arietinum L.) Plants: Functional and Molecular Insights. Biology 2025, 14, 657. [Google Scholar] [CrossRef] [PubMed]
- Merzouki, M.; Khibech, O.; Fraj, E.; Bouammali, H. Computational engineering of malonate and tetrazole derivatives targeting SARS-CoV-2 main protease: Pharmacokinetics, docking, and molecular dynamics insights to support the sustainable development goals (SDGs), with a bibliometric analysis. Indonesian J. Sci. Technol. 2025, 10, 399–418. [Google Scholar]
- Khan, A.; Khan, S.U.; Khan, A.; Shal, B.; Rehman, S.U.; Rehman, S.U.; Htar, T.T.; Khan, S.; Anwar, S.; Alafnan, A.; et al. Anti-Inflammatory and Anti-Rheumatic Potential of Selective Plant Compounds by Targeting TLR-4/AP-1 Signaling: A Comprehensive Molecular Docking and Simulation Approaches. Molecules 2022, 27, 4319. [Google Scholar] [CrossRef] [PubMed]
- Imran, M.; Alshrari, A.S.; Hafiz, M.N.; Jawad, M.M.; Khan, A.; Alanazi, F.J.; Asdaq, S.M.B. Exploring Therapeutic Paradigm Focusing on Genes, Proteins, and Pathways to Combat Leprosy and Tuberculosis: A Network Medicine and Drug Repurposing Approach. J. Infect. Public Health 2025, 18, 102763. [Google Scholar] [CrossRef] [PubMed]
- Amala, M.; Rajamanikandan, S.; Prabhu, D.; Surekha, K.; Jeyakanthan, J. Identification of Anti-Filarial Leads against Aspartate Semialdehyde Dehydrogenase of Wolbachia Endosymbiont of Brugia Malayi: Combined Molecular Docking and Molecular Dynamics Approaches. J. Biomol. Struct. Dyn. 2019, 37, 394–410. [Google Scholar] [CrossRef] [PubMed]
- Camargo, P.G.; da Silva, R.B.; Zuma, A.A.; Garden, S.J.; Albuquerque, M.G.; Rodrigues, C.R.; da Silva Lima, C.H. In Silico Evaluation of N-Aryl-1,10-Phenanthroline-2-Amines as Potential Inhibitors of T. Cruzi GP63 Zinc-Metalloprotease by Docking and Molecular Dynamics Simulations. Sci. Rep. 2025, 15, 6036. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. PkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Shi, S.; Yi, J.; Wang, N.; He, Y.; Wu, Z.; Peng, J.; Deng, Y.; Wang, W.; Wu, C. ADMETlab 3.0: An Updated Comprehensive Online ADMET Prediction Platform Enhanced with Broader Coverage, Improved Performance, API Functionality and Decision Support. Nucleic Acids Res. 2024, 52, gkae236. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.; Kemmler, E.; Dunkel, M.; Preissner, R. ProTox 3.0: A Webserver for the Prediction of Toxicity of Chemicals. Nucleic Acids Res. 2024, 52, gkae303. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. Gromacs: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecules | Catechin | Amorfrutin A | Hyperin | Eriodictyol | Astragalin | AMG-510 |
|---|---|---|---|---|---|---|
| Molecular WEIGHT (g/mol) | 290.27 | 340.41 | 464.38 | 288.25 | 448.38 | 560.59 |
| H-bond acceptors | 6 | 4 | 12 | 6 | 11 | 8 |
| H-bond donors | 5 | 2 | 8 | 4 | 7 | 1 |
| Rotatable bonds | 1 | 7 | 4 | 1 | 4 | 6 |
| TPSA Å2 | 110.38 | 66.76 | 210.51 | 107.22 | 190.28 | 104.45 |
| Consensus Log (Po/w) | 0.85 | 4.37 | −0.34 | 1.45 | −0.09 | 4.05 |
| Lipinski: violations | 0 | 0 | 2 | 0 | 2 | 1 |
| Veber: violations | 0 | 0 | 1 | 0 | 1 | 0 |
| Bioavailability Score | 0.55 | 0.85 | 0.17 | 0.55 | 0.17 | 0.55 |
| Molecules | Catechin | Amorfrutin A | Hyperin | Eriodictyol | Astragalin | AMG-510 |
|---|---|---|---|---|---|---|
| Intestinal absorption (human) % | 72.06 | 93.13 | 35.179 | 79.404 | 42.437 | 88.641 |
| BBB permeability | −1.159 | −0.263 | −1.897 | −1.246 | −1.683 | −1.484 |
| CNS permeability | −3.388 | −2.171 | −5.127 | −3.259 | −4.841 | −3.303 |
| CYP2D6 inhibitor | No | No | No | No | No | No |
| CYP3A4 inhibitor | No | No | No | No | No | Yes |
| Renal OCT2 substrate | No | No | No | No | No | Yes |
| Molecules | Docking Score (Kcal/mol) | Hydrogen Bonds | Distance (Å) |
|---|---|---|---|
| Catechin | −8.5 | Asp119, Ser145, Ala146, Lys147 | 2.49; 2.63; 2.48; 2.51 |
| Amorfrutin A | −7.4 | Asp30 | 2.41 |
| Hyperin | −8.6 | Asp30, Glu31, Asp119, Ala146 | 2.34; 1.79; 2.17; 2.36 |
| Eriodictyol | −8.4 | Asn116, Asp119, Ala146 | 2.51; 1.91; 2.72 |
| Astragalin | −8.6 | Asp30, Glu31 | 2.33; 2.16 |
| AMG-510 | −8.1 | Gly13 | 3.05 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahimi, A.; Khibech, O.; Benabbou, A.; Merzouki, M.; Bouhrim, M.; Al-Zharani, M.; Nasr, F.A.; Ahmed Qurtam, A.; Abadi, S.; Challioui, A.; et al. ADMET-Guided Docking and GROMACS Molecular Dynamics of Ziziphus lotus Phytochemicals Uncover Mutation-Agnostic Allosteric Stabilisers of the KRAS Switch-I/II Groove. Pharmaceuticals 2025, 18, 1110. https://doi.org/10.3390/ph18081110
Rahimi A, Khibech O, Benabbou A, Merzouki M, Bouhrim M, Al-Zharani M, Nasr FA, Ahmed Qurtam A, Abadi S, Challioui A, et al. ADMET-Guided Docking and GROMACS Molecular Dynamics of Ziziphus lotus Phytochemicals Uncover Mutation-Agnostic Allosteric Stabilisers of the KRAS Switch-I/II Groove. Pharmaceuticals. 2025; 18(8):1110. https://doi.org/10.3390/ph18081110
Chicago/Turabian StyleRahimi, Abdessadek, Oussama Khibech, Abdessamad Benabbou, Mohammed Merzouki, Mohamed Bouhrim, Mohammed Al-Zharani, Fahd A. Nasr, Ashraf Ahmed Qurtam, Said Abadi, Allal Challioui, and et al. 2025. "ADMET-Guided Docking and GROMACS Molecular Dynamics of Ziziphus lotus Phytochemicals Uncover Mutation-Agnostic Allosteric Stabilisers of the KRAS Switch-I/II Groove" Pharmaceuticals 18, no. 8: 1110. https://doi.org/10.3390/ph18081110
APA StyleRahimi, A., Khibech, O., Benabbou, A., Merzouki, M., Bouhrim, M., Al-Zharani, M., Nasr, F. A., Ahmed Qurtam, A., Abadi, S., Challioui, A., Mimouni, M., & Elbekay, M. (2025). ADMET-Guided Docking and GROMACS Molecular Dynamics of Ziziphus lotus Phytochemicals Uncover Mutation-Agnostic Allosteric Stabilisers of the KRAS Switch-I/II Groove. Pharmaceuticals, 18(8), 1110. https://doi.org/10.3390/ph18081110