

Regulation of L-Lactate in Glutamate Excitotoxicity Under Cerebral Ischemia: Pathophysiology and Preventive Strategy


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Regulation of L-Lactate in Glutamate Excitotoxicity Under Cerebral Ischemia
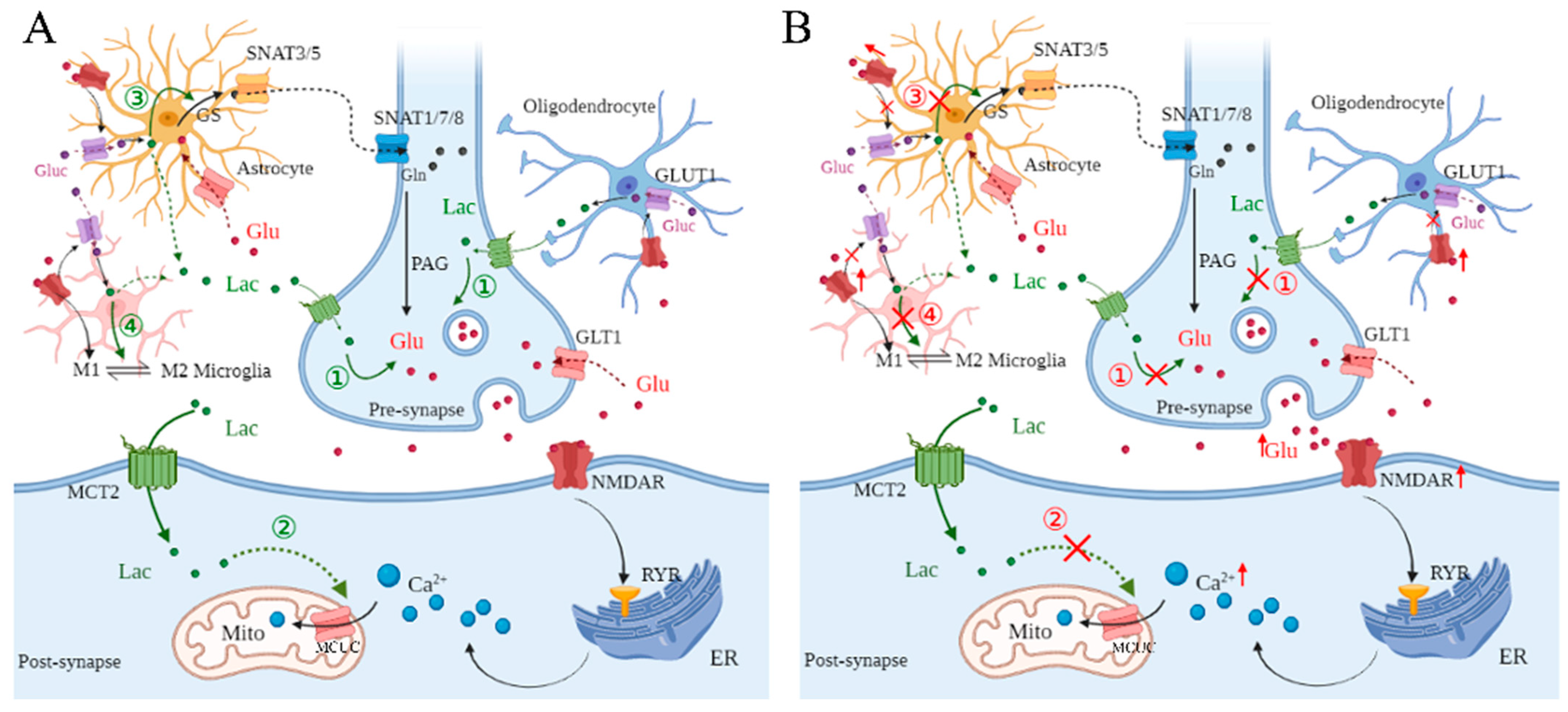
2.1. L-Lactate Regulates Glutamate Metabolism Among Neural Cells
2.2. Dysregulation of L-Lactate in Glutamate Metabolism Among Neural Cells Under Cerebral Ischemia
3. Dysregulation of L-Lactate in Postsynaptic Ca2+ Concentration Under Cerebral Ischemia
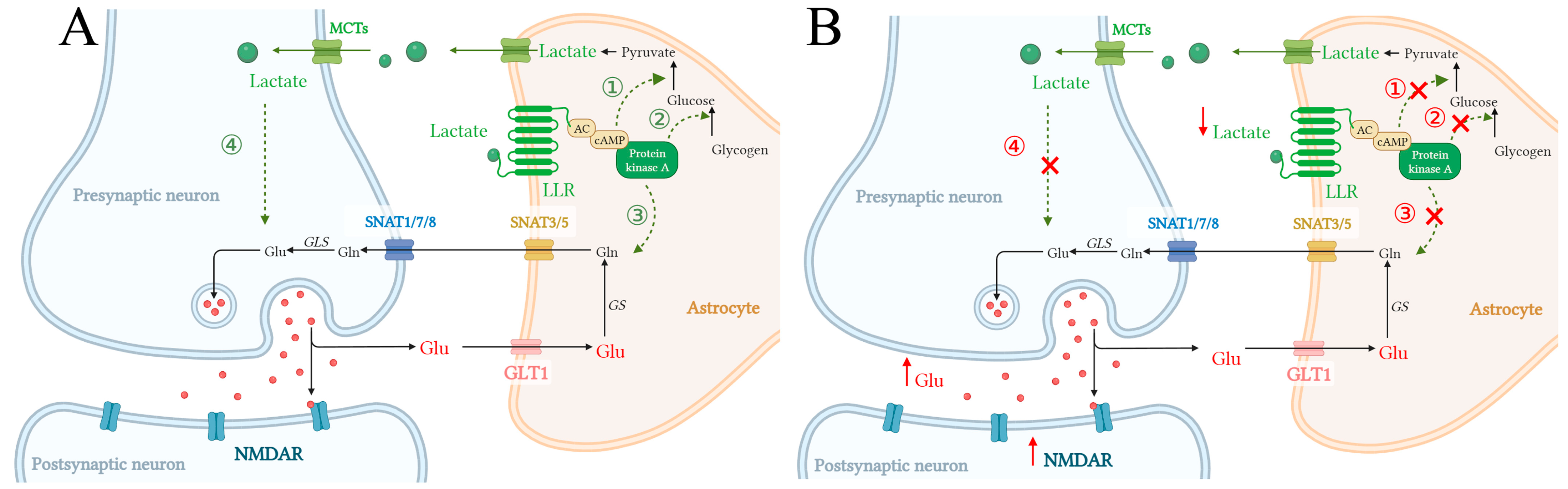
4. L-Lactate Triggers Astroglial LLR-cAMP/PKA Pathway, Promoting Glycolysis and Glutamate Uptake in Astrocytes
5. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CNS | Central nervous system |
| NMDARs | N-Methyl-D-aspartate receptors |
| Ca2+ | Calcium |
| Glu | Glutamate |
| Gln | Glutamine |
| Mito | Mitochondria |
| MCUC | Mitochondrial calcium uniporter complex |
| LLR | L-lactate-sensitive receptor |
| cAMP | Cyclic adenosine monophosphate |
| PKA | Protein kinase A |
| iGluRs | Ionotropic glutamate receptors |
| mGluRs | Metabotropic glutamate receptors |
| OLs | Oligodendrocytes |
| MCTs | Monocarboxylate transporters |
| GPCRs | G protein-coupled receptors |
| GluRs | Glutamate receptors |
| AMPARs | A-amino-3-hydroxy-5-methyl-4-isox-azolepropionic acid receptors |
| GLT1 | Glutamate transporter 1 |
| GLAST | Glutamate-aspartate transporter |
| GS | Glutamine synthetase |
| PLC | Phospholipase C |
| ATP | Adenosine triphosphate |
| GABA | Gamma-aminobutyric acid |
| OPCs | Oligodendrocyte precursor cells |
| OPC | Oligodendrocyte precursor cell |
| GLUT1 | Glucose transporter 1 |
| M0 | A “resting” state |
| M1 | “Classically activated” pro-inflammatory |
| M2 | “Alternatively activated” anti-inflammatory |
| RyRs | Ryanodine receptors |
| ER | Endoplasmic reticulum |
| Tiam1 | T lymphoma invasion and metastasis 1 |
| IP3Rs | Inositol 1,4,5-trisphosphate receptor |
| NO | Nitric oxide |
| ROS | Reactive oxygen species |
| SERCA | Sarcoendoplasmic reticulum Ca2+-ATPase |
| SPCA | Secretory protein calcium ATPase |
| PMCA | Plasma membrane Ca2+ transport ATPase |
| NCX | Na+/Ca2+ exchanger |
| MAMs | Mitochondria-associated membranes |
| VDACs | Voltage-dependent anion channels |
| DNA | Deoxyribonucleic acid |
| Olfr78 | L-lactate-sensitive olfactory receptor 78 |
| GPR4 | G-protein coupled receptor 4 |
| HMGB1 | High mobility group box 1 |
| SNAT | Sodium-coupled neutral amino acid transporter |
| GLS | Glutaminase |
| AC | Adenylate cyclase |
| ANLS | Astrocyte–neuron lactate shuttle |
References
- Sun, Y.; Feng, X.; Ding, Y.; Li, M.; Yao, J.; Wang, L.; Gao, Z. Phased treatment strategies for cerebral ischemia based on glutamate receptors. Front. Cell. Neurosci. 2019, 13, 168. [Google Scholar] [CrossRef] [PubMed]
- Giza, C.C.; Maria, N.S.; Hovda, D.A. N-Methyl-D-aspartate receptor subunit changes after traumatic injury to the developing brain. J. Neurotrauma 2006, 23, 950–961. [Google Scholar] [CrossRef] [PubMed]
- Holmes, A.; Zhou, N.; Donahue, D.L.; Balsara, R.; Castellino, F.J. A deficiency of the GluN2C subunit of the N-methyl-D-aspartate receptor is neuroprotective in a mouse model of ischemic stroke. Biochem. Biophys. Res. Commun. 2018, 495, 136–144. [Google Scholar] [CrossRef]
- Engin, A.; Engin, A.B. N-Methyl-D-Aspartate receptor signaling-protein kinases crosstalk in cerebral ischemia. Adv. Exp. Med. Biol. 2021, 1275, 259–283. [Google Scholar]
- Stefani, M.A.; Modkovski, R.; Hansel, G.; Zimmer, E.R.; Kopczynski, A.; Muller, A.P.; Strogulski, N.R.; Rodolphi, M.S.; Carteri, R.K.; Schmidt, A.P.; et al. Elevated glutamate and lactate predict brain death after severe head trauma. Ann. Clin. Transl. Neurol. 2017, 4, 392–402. [Google Scholar] [CrossRef]
- Quintard, H.; Patet, C.; Zerlauth, J.-B.; Suys, T.; Bouzat, P.; Pellerin, L.; Meuli, R.; Magistretti, P.J.; Oddo, M. Improvement of neuroenergetics by hypertonic lactate therapy in patients with traumatic brain injury is dependent on baseline cerebral lactate/pyruvate ratio. J. Neurotrauma 2016, 33, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Montana, V.; Flint, D.; Waagepetersen, H.S.; Schousboe, A.; Parpura, V. Two metabolic fuels, glucose and lactate, differentially modulate exocytotic glutamate release from cultured astrocytes. Neurochem. Res. 2021, 46, 2551–2579. [Google Scholar] [CrossRef]
- Proia, P.; Di Liegro, C.M.; Schiera, G.; Fricano, A.; Di Liegro, I. Lactate as a metabolite and a regulator in the central nervous system. Int. J. Mol. Sci. 2016, 17, 1450. [Google Scholar] [CrossRef]
- Yamagata, K. Lactate supply from astrocytes to neurons and its role in ischemic stroke-induced neurodegeneration. Neuroscience 2022, 481, 219–231. [Google Scholar] [CrossRef]
- Magistretti, P.J.; Allaman, I. Lactate in the brain: From metabolic end-product to signalling molecule. Nat. Rev. Neurosci. 2018, 19, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Li, H.; Chen, J.; Qian, Q. Lactic acid: No longer an inert and end-product of glycolysis. Physiology 2017, 32, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Pan, R.-Y.; He, L.; Zhang, J.; Liu, X.; Liao, Y.; Gao, J.; Liao, Y.; Yan, Y.; Li, Q.; Zhou, X.; et al. Positive feedback regulation of microglial glucose metabolism by histone H4 lysine 12 lactylation in Alzheimer’s disease. Cell Metab. 2022, 34, 634–648. [Google Scholar] [CrossRef]
- Ribeiro, F.M.; Vieira, L.B.; Pires, R.G.; Olmo, R.P.; Ferguson, S.S. Metabotropic glutamate receptors and neurodegenerative diseases. Pharmacol. Res. 2017, 115, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Plaitakis, A.; Sidiropoulou, K.; Kotzamani, D.; Litso, I.; Zaganas, I.; Spanaki, C. Evolution of glutamate metabolism via GLUD2 enhances lactate-dependent synaptic plasticity and complex cognition. Int. J. Mol. Sci. 2024, 25, 5297. [Google Scholar] [CrossRef]
- Andersen, J.V.; Markussen, K.H.; Jakobsen, E.; Schousboe, A.; Waagepetersen, H.S.; Rosenberg, P.A.; Aldana, B.I. Glutamate metabolism and recycling at the excitatory synapse in health and neurodegeneration. Neuropharmacology 2021, 196, 108719. [Google Scholar] [CrossRef]
- Rodriguez Chavez, V.; Moran, J.; Molina Salinas, G.; Zepeda Ruiz, W.A.; Rodriguez, M.C.; Picazo, O.; Cerbon, M. Participation of Glutamatergic Ionotropic Receptors in Excitotoxicity: The Neuroprotective Role of Prolactin. Neuroscience 2021, 461, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Reiner, A.; Levitz, J. Glutamatergic signaling in the central nervous system: Ionotropic and metabotropic receptors in concert. Neuron 2018, 98, 1080–1098. [Google Scholar] [CrossRef]
- Shen, Z.; Xiang, M.; Chen, C.; Ding, F.; Wang, Y.; Shang, C.; Xin, L.; Zhang, Y.; Cui, X. Glutamate excitotoxicity: Potential therapeutic target for ischemic stroke. Biomed. Pharmacother. 2022, 151, 113125. [Google Scholar] [CrossRef]
- Lai, T.W.; Zhang, S.; Wang, Y.T. Excitotoxicity and stroke: Identifying novel targets for neuroprotection. Prog. Neurobiol. 2014, 115, 157–188. [Google Scholar] [CrossRef]
- Andersen, J.V.; Schousboe, A.; Verkhratsky, A. Astrocyte energy and neurotransmitter metabolism in Alzheimer’s disease: Integration of the glutamate/GABA-glutamine cycle. Prog. Neurobiol. 2022, 217, 102331. [Google Scholar] [CrossRef]
- Albrecht, J.; Zielińska, M.; Norenberg, M.D. Glutamine as a mediator of ammonia neurotoxicity: A critical appraisal. Biochem. Pharmacol. 2010, 80, 1303–1308. [Google Scholar] [CrossRef]
- Perrillat Mercerot, A.; Bourmeyster, N.; Guillevin, C.; Miranville, A.; Guillevin, R. Analysis of a mathematical model for the glutamate/glutamine cycle in the brain. Bull. Math. Biol. 2019, 81, 4251–4270. [Google Scholar] [CrossRef]
- Sonnewald, U.; Schousboe, A. Introduction to the glutamate-glutamine cycle. Adv. Neurobiol. 2016, 13, 1–7. [Google Scholar] [PubMed]
- Limón, I.D.; Angulo Cruz, I.; Sánchez Abdon, L.; Patricio Martínez, A. Disturbance of the glutamate-glutamine cycle, secondary to hepatic damage, compromises memory function. Front. Neurosci. 2021, 15, 578922. [Google Scholar] [CrossRef] [PubMed]
- Kofuji, P.; Araque, A. G-protein-coupled receptors in astrocyte-neuron communication. Neuroscience 2021, 456, 71–84. [Google Scholar] [CrossRef]
- Habermacher, C.; Angulo, M.C.; Benamer, N. Glutamate versus GABA in neuron-oligodendroglia communication. Glia 2019, 67, 2092–2106. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.-J.; Kula, B.; Nagy, B.; Barzan, R.; Gall, A.; Ehrlich, I.; Kukley, M. In Vivo Regulation of Oligodendrocyte Precursor Cell Proliferation and Differentiation by the AMPA-Receptor Subunit GluA2. Cell Rep. 2018, 25, 852–861.e7. [Google Scholar] [CrossRef]
- Saab, A.S.; Tzvetavona, I.D.; Trevisiol, A.; Baltan, S.; Dibaj, P.; Kusch, K.; Möbius, W.; Goetze, B.; Jahn, H.M.; Huang, W.; et al. Oligodendroglial NMDA receptors regulate glucose import and axonal energy metabolism. Neuron 2016, 91, 119–132. [Google Scholar] [CrossRef]
- Jha, M.K.; Morrison, B.M. Lactate transporters mediate glia-neuron metabolic crosstalk in homeostasis and disease. Front. Cell. Neurosci. 2020, 14, 589582. [Google Scholar] [CrossRef]
- Li, Y.; Du, X.F.; Du, J.L. Resting microglia respond to and regulate neuronal activity in vivo. Commun. Integr. Biol. 2013, 6, e24493. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, D.; Zhang, B.; Zhu, J.; Zhou, Z.; Cui, L. Regulation of microglia by glutamate and its signal pathway in neurodegenerative diseases. Drug Discov. 2020, 25, 1074–1085. [Google Scholar] [CrossRef] [PubMed]
- Illes, P.; Rubini, P.; Ulrich, H.; Zhao, Y.; Tang, Y. Regulation of microglial functions by purinergic mechanisms in the healthy and diseased CNS. Cells 2020, 9, 1108. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.Y.; Liu, L.; Yang, Q.W. Functions and mechanisms of microglia/ macrophages in neuroinflammation and neurogenesis after stroke. Prog. Neurobiol. 2016, 142, 23–44. [Google Scholar] [CrossRef]
- Hu, X.; Leak, R.K.; Shi, Y.; Suenaga, J.; Gao, Y.; Zheng, P.; Chen, J. Microglial and macrophage polarization-new prospects for brain repair. Nat. Rev. Neurol. 2015, 11, 56–64. [Google Scholar] [CrossRef]
- Liu, X.; Wen, S.; Yan, F.; Liu, K.; Liu, L.; Wang, L.; Zhao, S.; Ji, X. Salidroside provides neuroprotection by modulating microglial polarization after cerebral ischemia. J. Neuroinflamm. 2018, 15, 39. [Google Scholar] [CrossRef] [PubMed]
- Beard, E.; Lengacher, S.; Dias, S.; Magistretti, P.J.; Finsterwald, C. Astrocytes as key regulators of brain energy metabolism: New therapeutic perspectives. Front. Physiol. 2022, 12, 825816. [Google Scholar] [CrossRef]
- Dienel, G.A. Brain glucose metabolism: Integration of energetics with function. Physiol. Rev. 2019, 99, 949–1045. [Google Scholar] [CrossRef]
- Ramírez, O.A.; Córdova, A.; Cerda, M.; Lobos, P.; Härtel, S.; Couve, A.; Hidalgo, C. Ryanodine receptor-mediated Ca2+ release and atlastin-2 GTPase activity contribute to IP3-induced dendritic Ca2+ signals in primary hippocampal neurons. Cell Calcium 2021, 96, 102399. [Google Scholar] [CrossRef]
- Kamer, K.J.; Mootha, V.K. The molecular era of the mitochondrial calcium uniporter. Nat Rev Mol. Cell Biol. 2015, 16, 545–553. [Google Scholar]
- Wang, N.; Wang, W.; Wang, X.; Mang, G.; Chen, J.; Yan, X.; Tong, Z.; Yang, Q.; Wang, M.; Chen, L.; et al. Histone lactylation boosts reparative gene activation post-myocardial infarction. Circ. Res. 2022, 131, 893–908. [Google Scholar] [CrossRef]
- Zhou, Y.; Yang, L.; Liu, X.; Wang, H. Lactylation may be a novel posttranslational modification in inflammation in neonatal hypoxic-ischemic encephalopathy. Front. Pharmacol. 2022, 13, 926802. [Google Scholar] [CrossRef] [PubMed]
- Ciszewski, W.M.; Sobierajska, K.; Stasiak, A.; Wagner, W. Lactate drives cellular DNA repair capacity: Role of lactate and related short-chain fatty acids in cervical cancer chemoresistance and viral infection. Front. Cell Dev. Biol. 2022, 10, 1012254. [Google Scholar] [CrossRef]
- Llorente-Folch, I.; Rueda, C.B.; Pérez-Liébana, I.; Satrústegui, J.; Pardo, B. L-Lactate-Mediated Neuroprotection against Glutamate-Induced Excitotoxicity Requires ARALAR/AGC1. J. Neurosci. 2016, 36, 4443–4456. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Ji, T.; Li, G.; Zhang, H.; Zheng, Y.; Li, M.; Ma, J.; Li, Y.; Chi, G. Lactate attenuates astrocytic inflammation by inhibiting ubiquitination and degradation of NDRG2 under oxygen-glucose deprivation conditions. J. Neuroinflamm. 2022, 19, 314. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, S.; Yang, L.; Zhang, Y.; Cheng, Y.; Jia, P.; Lv, Y.; Wang, K.; Fan, P.; Zhang, P.; et al. Lactate modulates microglial inflammatory responses through HIF-α-mediated CCL7 signaling after cerebral ischemia in mice. Int. Immunopharmacol. 2024, 146, 113801. [Google Scholar] [CrossRef] [PubMed]
- Zhai, X.; Li, J.; Li, L.; Sun, Y.; Zhang, X.; Xue, Y.; Lv, J.; Gao, Y.; Li, S.; Yan, W.; et al. L-lactate preconditioning promotes plasticity-related proteins expression and reduces neurological deficits by potentiating GPR81 signaling in rat traumatic brain injury model. Brain Res. 2020, 1746, 146945. [Google Scholar] [CrossRef]
- Hu, X.; De Silva, T.M.; Chen, J.; Faraci, F.M. Cerebral vascular disease and neurovascular injury in ischemic stroke. Circ. Res. 2017, 120, 449–471. [Google Scholar] [CrossRef]
- Amantea, D.; Bagetta, G. Excitatory and inhibitory amino acid neurotransmitters in stroke: From neurotoxicity to ischemic tolerance. Curr. Opin. Pharmacol. 2017, 35, 111–119. [Google Scholar] [CrossRef]
- Yuan, Q.; Yuan, Y.; Zheng, Y.; Sheng, R.; Liu, L.; Xie, F.; Tan, J. Anti-cerebral ischemia reperfusion injury of polysaccharides: A review of the mechanisms. Biomed. Pharmacother. 2021, 137, 111303. [Google Scholar] [CrossRef]
- Kumagai, A.; Sasaki, T.; Matsuoka, K.; Abe, M.; Tabata, T.; Itoh, Y.; Fuchino, H.; Wugangerile, S.; Suga, M.; Yamaguchi, T.; et al. Monitoring of glutamate-induced excitotoxicity by mitochondrial oxygen consumption. Synapse 2019, 73, e22067. [Google Scholar] [CrossRef]
- Zaric, M.; Drakulic, D.; Stojanovic, I.G.; Mitrovic, N.; Grkovic, I.; Martinovic, J. Regional-specific effects of cerebral ischemia/reperfusion and dehydroepiandrosterone on synaptic NMDAR/PSD-95 complex in male Wistar rats. Brain Res. 2018, 1688, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Ohta, K.; Iwai, M.; Sato, K.; Omori, N.; Nagano, I.; Shoji, M.; Abe, K. Dissociative increase of oligodendrocyte progenitor cells between young and aged rats after transient cerebral ischemia. Neurosci. Lett. 2003, 335, 159–162. [Google Scholar] [CrossRef]
- Xiao, L.; Hu, C.; Yang, W.; Guo, D.; Li, C.; Shen, W.; Liu, X.; Aijun, H.; Dan, W.; He, C. NMDA receptor couples Rac1-GEF Tiam1 to direct oligodendrocyte precursor cell migration. Glia 2013, 61, 2078–2099. [Google Scholar] [CrossRef]
- Nagy, E.E.; Frigy, A.; Szász, J.A.; Horváth, E. Neuroinflammation and microglia/macrophage phenotype modulate the molecular background of post-stroke depression: A literature review. Exp. Ther. Med. 2020, 20, 2510–2523. [Google Scholar] [CrossRef] [PubMed]
- Byrnes, K.R.; Stoica, B.; Loane, D.J.; Riccio, A.; Davis, M.I.; Faden, A.I. Metabotropic glutamate receptor 5 activation inhibits microglial associated inflammation and neurotoxicity. Glia 2009, 57, 550–560. [Google Scholar] [CrossRef] [PubMed]
- Byrnes, K.R.; Loane, D.J.; Faden, A.I. Metabotropic glutamate receptors as targets for multipotential treatment of neurological disorders. Neurotherapeutics 2009, 6, 94–107. [Google Scholar] [CrossRef]
- Durand, D.; Carniglia, L.; Turati, J.; Ramírez, D.; Saba, J.; Caruso, C.; Lasaga, M. Amyloid-beta neurotoxicity and clearance are both regulated by glial group II metabotropic glutamate receptors. Neuropharmacology 2017, 123, 274–286. [Google Scholar] [CrossRef]
- Noda, M. Dysfunction of glutamate receptors in microglia may cause neurodegeneration. Curr. Alzheimer Res. 2016, 13, 381–386. [Google Scholar] [CrossRef]
- Fannon, J.; Tarmier, W.; Fulton, D. Neuronal activity and AMPA-type glutamate receptor activation regulates the morphological development of oligodendrocyte precursor cells. Glia 2015, 63, 1021–1035. [Google Scholar] [CrossRef]
- Radak, D.; Katsiki, N.; Resanovic, I.; Jovanovic, A.; Sudar-Milovanovic, E.; Zafirovic, S.; Mousad, S.A.; Isenovic, E.R. Apoptosis and acute brain ischemia in ischemic stroke. Curr. Vasc. Pharmacol. 2017, 15, 115–122. [Google Scholar] [CrossRef]
- Vallese, F.; Barazzuol, L.; Maso, L.; Brini, M.; Cali, T. ER-mitochondria calcium transfer, organelle contacts and neurodegenerative diseases. Adv. Exp. Med. Biol. 2020, 1131, 719–746. [Google Scholar] [PubMed]
- Gaspers, L.D.; Bartlett, P.J.; Politi, A.; Burnett, P.; Metzger, W.; Johnston, J.; Joseph, S.K.; Hofer, T.; Thomas, A.P. Hormone-induced calcium oscillations depend on cross-coupling with inositol 1,4,5-trisphosphate oscillations. Cell Rep. 2014, 9, 1209–1218. [Google Scholar] [CrossRef]
- Zalk, R.; Clarke, O.B.; des Georges, A.; Grassucci, R.A.; Reiken, S.; Mancia, F.; Hendrickson, W.A.; Frank, J.; Marks, A.R. Structure of a mammalian ryanodine receptor. Nature 2015, 517, 44–49. [Google Scholar] [CrossRef]
- Vandecaetsbeek, I.; Vangheluwe, P.; Raeymaekers, L.; Wuytack, F.; Vanoevelen, J. The Ca2+ pumps of the endoplasmic reticulum and Golgi apparatus. Cold Spring Harb. Perspect. Biol. 2011, 3, a004184. [Google Scholar] [CrossRef]
- Bruce, J.I.E. Metabolic regulation of the PMCA: Role in cell death and survival. Cell Calcium 2018, 69, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Trebak, M.; Perocchi, F.; Khananshvili, D.; Sekler, I. Crosslink between calcium and sodium signalling. Exp. Physiol. 2018, 103, 157–169. [Google Scholar] [CrossRef]
- Zhang, D.; Tang, Z.; Huang, H.; Zhou, G.; Cui, C.; Weng, Y.; Liu, W.; Kim, S.; Lee, S.; Perez-Neut, M.; et al. Metabolic regulation of gene expression by histone lactylation. Nature 2019, 574, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; Missiroli, S.; Patergnani, S.; Duszynski, J.; Wieckowski, M.R.; Pinton, P. Mitochondria-associated membranes: Composition, molecular mechanisms, and physiopathological implications. Antioxid. Redox Signal. 2015, 22, 995–1019. [Google Scholar] [CrossRef]
- Williams, G.S.; Boyman, L.; Lederer, W.J. Mitochondrial calcium and the regulation of metabolism in the heart. J. Mol. Cell Cardiol. 2015, 78, 35–45. [Google Scholar] [CrossRef]
- Varughese, J.T.; Buchanan, S.K.; Pitt, A.S. The role of voltage-dependent anion channel in mitochondrial dysfunction and human disease. Cells 2012, 10, 1737. [Google Scholar] [CrossRef]
- Oxenoid, K.; Dong, Y.; Cao, C.; Cui, T.; Sancak, Y.; Markhard, A.L.; Grabarek, Z.; Kong, L.; Liu, Z.; Ouyang, B.; et al. Architecture of the mitochondrial calcium uniporter. Nature 2016, 533, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Skowrońska, K.; Kozłowska, H.; Albrecht, J. Neuron-derived factors negatively modulate ryanodine receptor-mediated calcium release in cultured mouse astrocytes. Cell Calcium 2020, 92, 102304. [Google Scholar] [CrossRef]
- Zhao, Q.; Wang, S.; Li, Y.; Wang, P.; Li, S.; Guo, Y.; Yao, R. The role of the mitochondrial calcium uniporter in cerebral ischemia/reperfusion injury in rats involves regulation of mitochondrial energy metabolism. Mol. Med. Rep. 2013, 7, 1073–1080. [Google Scholar] [CrossRef] [PubMed]
- Brooks, G.A. Cell-cell and intracellular lactate shuttles. J. Physiol. 2019, 587, 5591–5600. [Google Scholar] [CrossRef] [PubMed]
- Horvat, A.; Zorec, R.; Vardjan, N. Lactate as an astroglial signal augmenting aerobic glycolysis and lipid metabolism. Front. Physiol. 2021, 12, 735532. [Google Scholar] [CrossRef] [PubMed]
- Orecchioni, M.; Matsunami, H.; Ley, K. Olfactory receptors in macrophages and inflammation. Front. Immunol. 2022, 13, 1029244. [Google Scholar] [CrossRef]
- Mosienko, V.; Teschemacher, A.G.; Kasparov, S. Is L-lactate a novel signaling molecule in the brain? J. Cereb. Blood Flow Metab. 2015, 35, 1069–1075. [Google Scholar] [CrossRef]
- Abi-Saab, W.M.; Maggs, D.G.; Jones, T.; Jacob, R.; Srihari, V.; Thompson, J.; Kerr, D.; Leone, P.; Krystal, J.H.; Spencer, D.D.; et al. Striking differences in glucose and lactate levels between brain extracellular fluid and plasma in conscious human subjects: Effects of hyperglycemia and hypoglycemia. J. Cereb. Blood Flow Metab. 2022, 22, 271–279. [Google Scholar] [CrossRef]
- Bonvento, G.; Bolaños, J.P. Astrocyte-neuron metabolic cooperation shapes brain activity. Cell Metab. 2021, 33, 1546–1564. [Google Scholar] [CrossRef]
- Yang, K.; Fan, M.; Wang, X.; Xu, J.; Wang, Y.; Tu, F.; Gill, P.S.; Ha, T.; Liu, L.; Williams, D.L.; et al. Lactate promotes macrophage HMGB1 lactylation, acetylation, and exosomal release in polymicrobial sepsis. Cell Death Differ. 2022, 29, 133–146. [Google Scholar] [CrossRef]
- Yao, X.; Li, C. Lactate dehydrogenase A mediated histone lactylation induced the pyroptosis through targeting HMGB1. Metab. Brain Dis. 2023, 38, 1543–1553. [Google Scholar] [CrossRef]
- Zhang, Q.; Jia, M.; Wang, Y.; Wang, Q.; Wu, J. Cell death mechanisms in cerebral ischemia-reperfusion injury. Neurochem. Res. 2022, 47, 3525–3542. [Google Scholar] [CrossRef] [PubMed]
- Menyhárt, Á.; Frank, R.; Farkas, A.E.; Süle, Z.; Varga, V.É.; Nyúl-Tóth, Á.; Meiller, A.; Ivánkovits-Kiss, O.; Lemale, C.L.; Szabó, Í.; et al. Malignant astrocyte swelling and impaired glutamate clearance drive the expansion of injurious spreading depolarization foci. J. Cereb. Blood Flow Metab. 2022, 42, 584–599. [Google Scholar] [CrossRef]
- Dejakaisaya, H.; Kwan, P.; Jones, N.C. Astrocyte and glutamate involvement in the pathogenesis of epilepsy in Alzheimer’s disease. Epilepsia 2021, 62, 1485–1493. [Google Scholar] [CrossRef]
- McKenna, M.C. The glutamate-glutamine cycle is not stoichiometric: Fates of glutamate in brain. J. Neurosci. Res. 2007, 85, 3347–3358. [Google Scholar] [CrossRef] [PubMed]
- Gheni, G.; Ogura, M.; Iwasaki, M.; Yokoi, N.; Minami, K.; Nakayama, Y.; Harada, K.; Hastoy, B.; Wu, X.; Takahashi, H.; et al. Glutamate acts as a key signal linking glucose metabolism to incretin/cAMP action to amplify insulin secretion. Cell Rep. 2014, 9, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Sami, A.; Noristani, H.N.; Slattery, K.; Qiu, J.; Groves, T.; Wang, S.; Veerasammy, K.; Chen, Y.X.; Morales, J.; et al. Glial metabolic rewiring promotes axon regeneration and functional recovery in the central nervous system. Cell Metab. 2020, 32, 767–785. [Google Scholar] [CrossRef]
- Díaz García, C.M.; Mongeon, R.; Lahmann, C.; Koveal, D.; Zucker, H.; Yellen, G. Neuronal stimulation triggers neuronal glycolysis and not lactate uptake. Cell Metab. 2017, 26, 361–374. [Google Scholar] [CrossRef]
- Kirdajova, D.B.; Kriska, J.; Tureckova, J.; Anderova, M. Ischemia-triggered glutamate excitotoxicity from the perspective of glial cells. Front. Cell. Neurosci. 2020, 14, 51. [Google Scholar] [CrossRef]
- Moraga-Amaro, R.; Jerez-Baraona, J.; Simon, F.; Stehberg, J. Role of astrocytes in memory and psychiatric disorders. J. Physiol. Paris 2014, 108, 240–251. [Google Scholar] [CrossRef]
- Hirrlinger, J.; Nave, K.A. Adapting brain metabolism to myelination and long-range signal transduction. Glia 2014, 62, 1749–1761. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.N.; Hao, L.; Guo, Y.S.; Wang, H.Y.; Li, L.L.; Liu, L.Z.; Li, W.B. Are glutamate transporters neuroprotective or neurodegenerative during cerebral ischemia? J. Mol. Med. 2019, 97, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Gascón, S.; Sobrado, M.; Roda, J.M.; Rodríguez-Peña, A.; Díaz-Guerra, M. Excitotoxicity and focal cerebral ischemia induce truncation of the NR2A and NR2B subunits of the NMDA receptor and cleavage of the scaffolding protein PSD-95. Mol. Psychiatry 2008, 13, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Bading, H. Nuclear calcium signalling in the regulation of brain function. Nat. Rev. Neurosci. 2013, 14, 593–608. [Google Scholar] [CrossRef]
- Zhou, J.J.; Shao, J.Y.; Chen, S.R.; Li, D.P.; Pan, H.L. α2δ-1-dependent NMDA receptor activity in the hypothalamus is an effector of genetic-environment interactions that drive persistent hypertension. J. Neurosci. 2021, 41, 6551–6563. [Google Scholar] [CrossRef]
- Calvo, M.; Villalobos, C.; Núñez, L. Calcium imaging in neuron cell death. Methods Mol. Biol. 2015, 1254, 73–85. [Google Scholar]
- Jourdain, P.; Rothenfusser, K.; Ben-Adiba, C.; Allaman, I.; Marquet, P.; Magistretti, P.J. Dual action of L-Lactate on the activity of NR2B-containing NMDA receptors: From potentiation to neuroprotection. Sci. Rep. 2018, 8, 13472. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, M.; Wang, Y.; Gong, Z.; Jiang, W.; Ge, G.; Guo, H. Regulation of L-Lactate in Glutamate Excitotoxicity Under Cerebral Ischemia: Pathophysiology and Preventive Strategy. Pharmaceuticals 2025, 18, 935. https://doi.org/10.3390/ph18070935
Zhang M, Wang Y, Gong Z, Jiang W, Ge G, Guo H. Regulation of L-Lactate in Glutamate Excitotoxicity Under Cerebral Ischemia: Pathophysiology and Preventive Strategy. Pharmaceuticals. 2025; 18(7):935. https://doi.org/10.3390/ph18070935
Chicago/Turabian StyleZhang, Mao, Yanyan Wang, Zili Gong, Wen Jiang, Guodong Ge, and Hong Guo. 2025. "Regulation of L-Lactate in Glutamate Excitotoxicity Under Cerebral Ischemia: Pathophysiology and Preventive Strategy" Pharmaceuticals 18, no. 7: 935. https://doi.org/10.3390/ph18070935
APA StyleZhang, M., Wang, Y., Gong, Z., Jiang, W., Ge, G., & Guo, H. (2025). Regulation of L-Lactate in Glutamate Excitotoxicity Under Cerebral Ischemia: Pathophysiology and Preventive Strategy. Pharmaceuticals, 18(7), 935. https://doi.org/10.3390/ph18070935