

Yinchenhao Decoction Mitigates Cholestatic Liver Injury in Mice via Gut Microbiota Regulation and Activation of FXR-FGF15 Pathway

Abstract

1. Introduction
2. Results
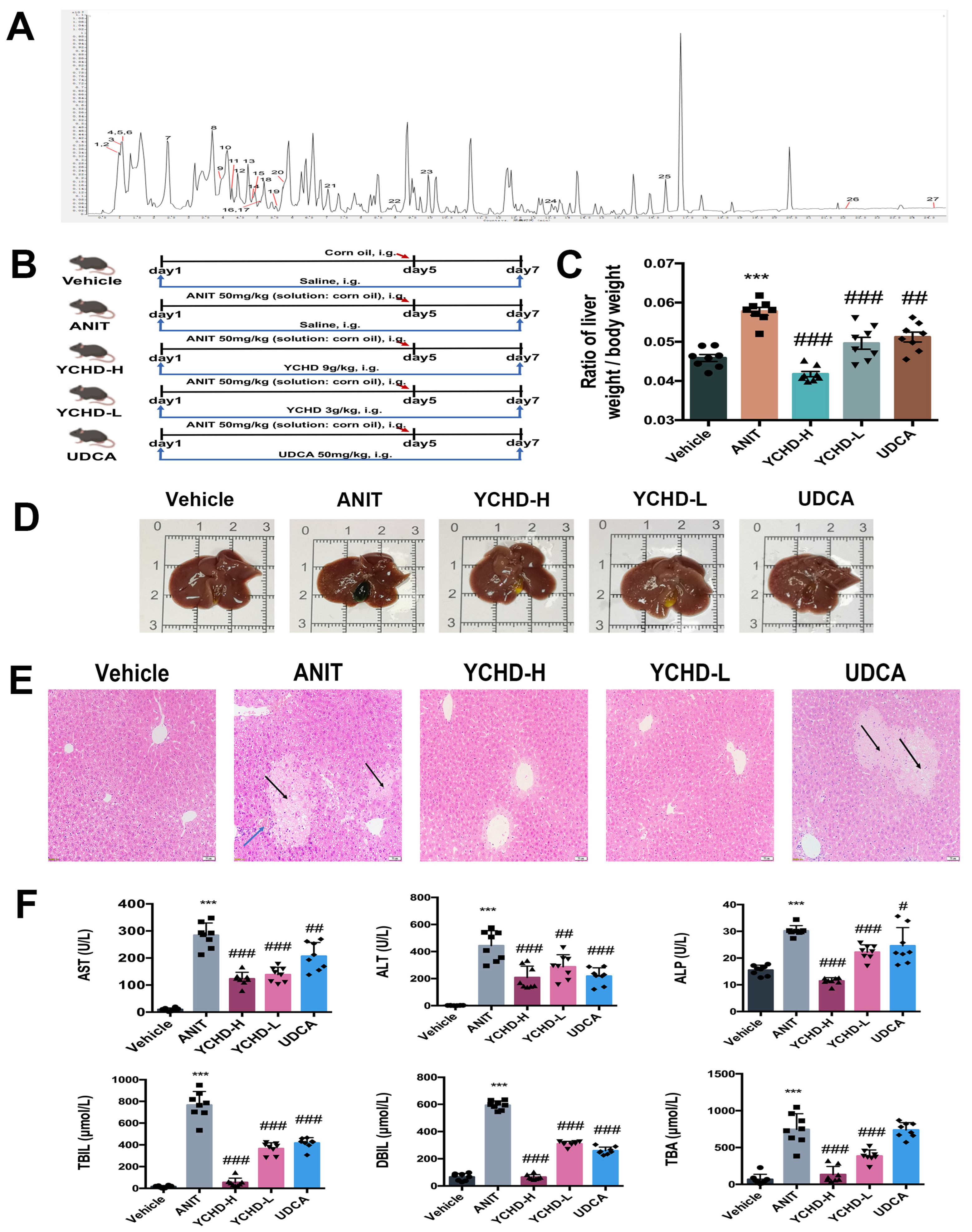
2.1. Chemical Characteristics of YCHD
2.2. YCHD Ameliorates Cholestatic Liver Injury in ANIT-Induced Mice
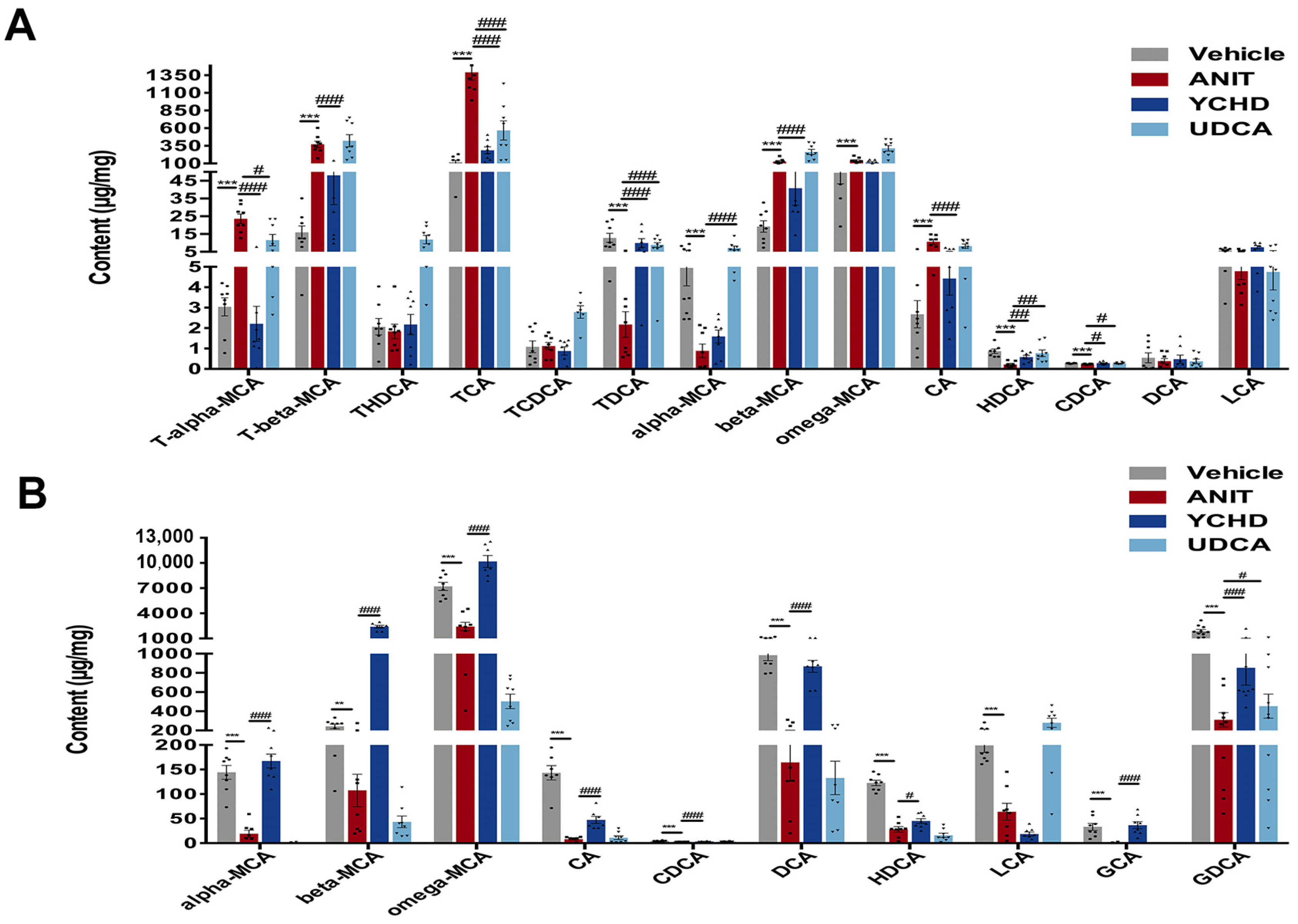
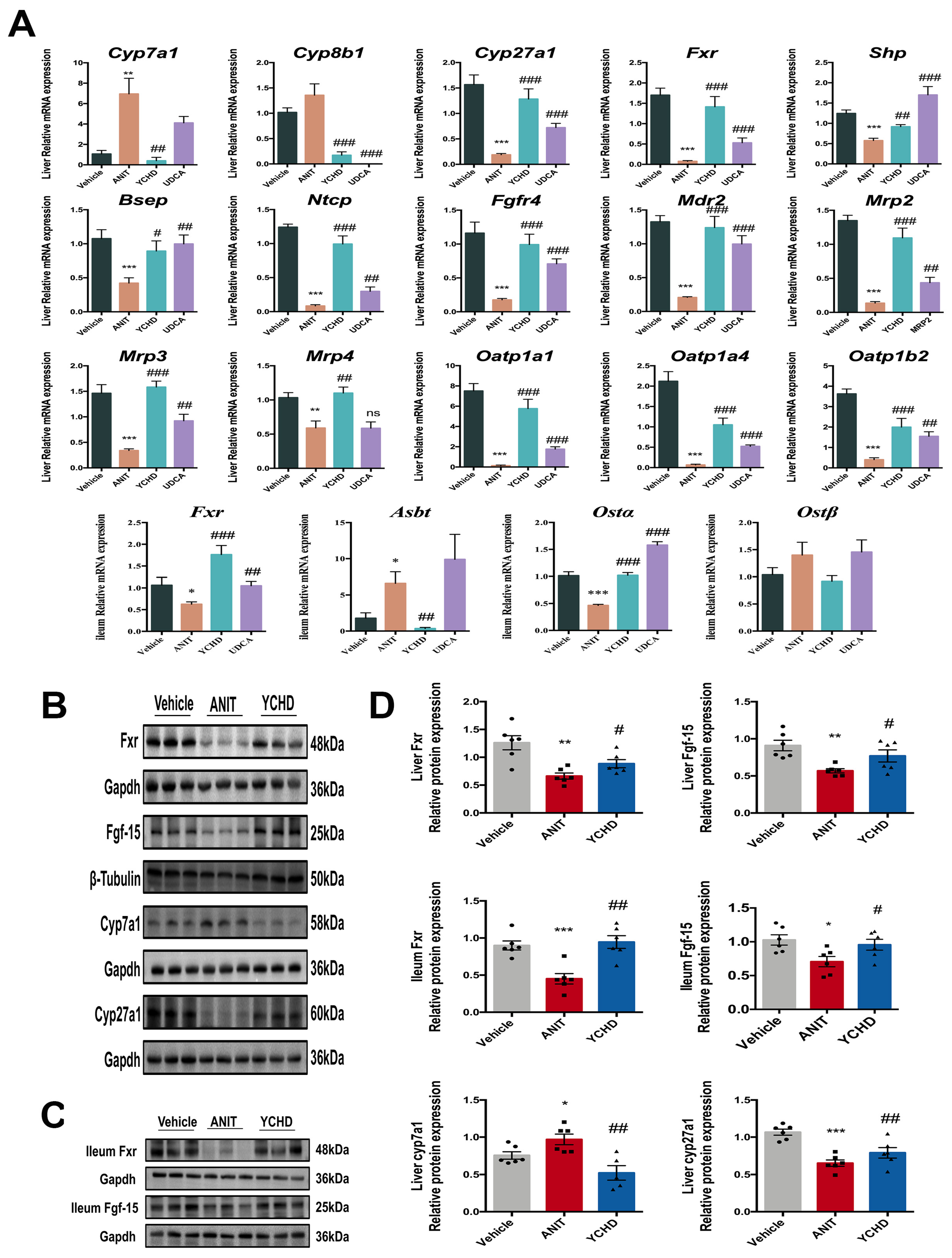
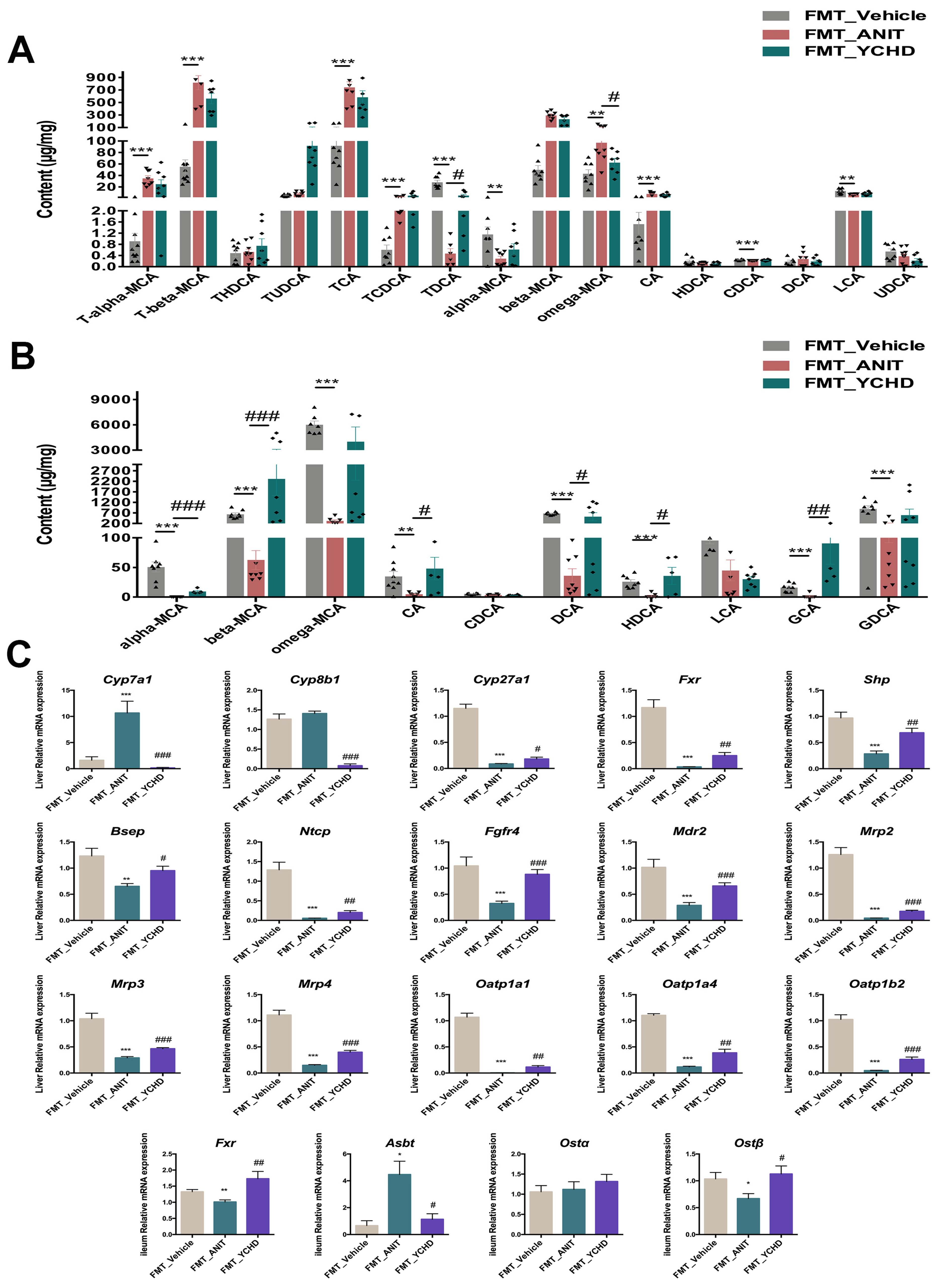
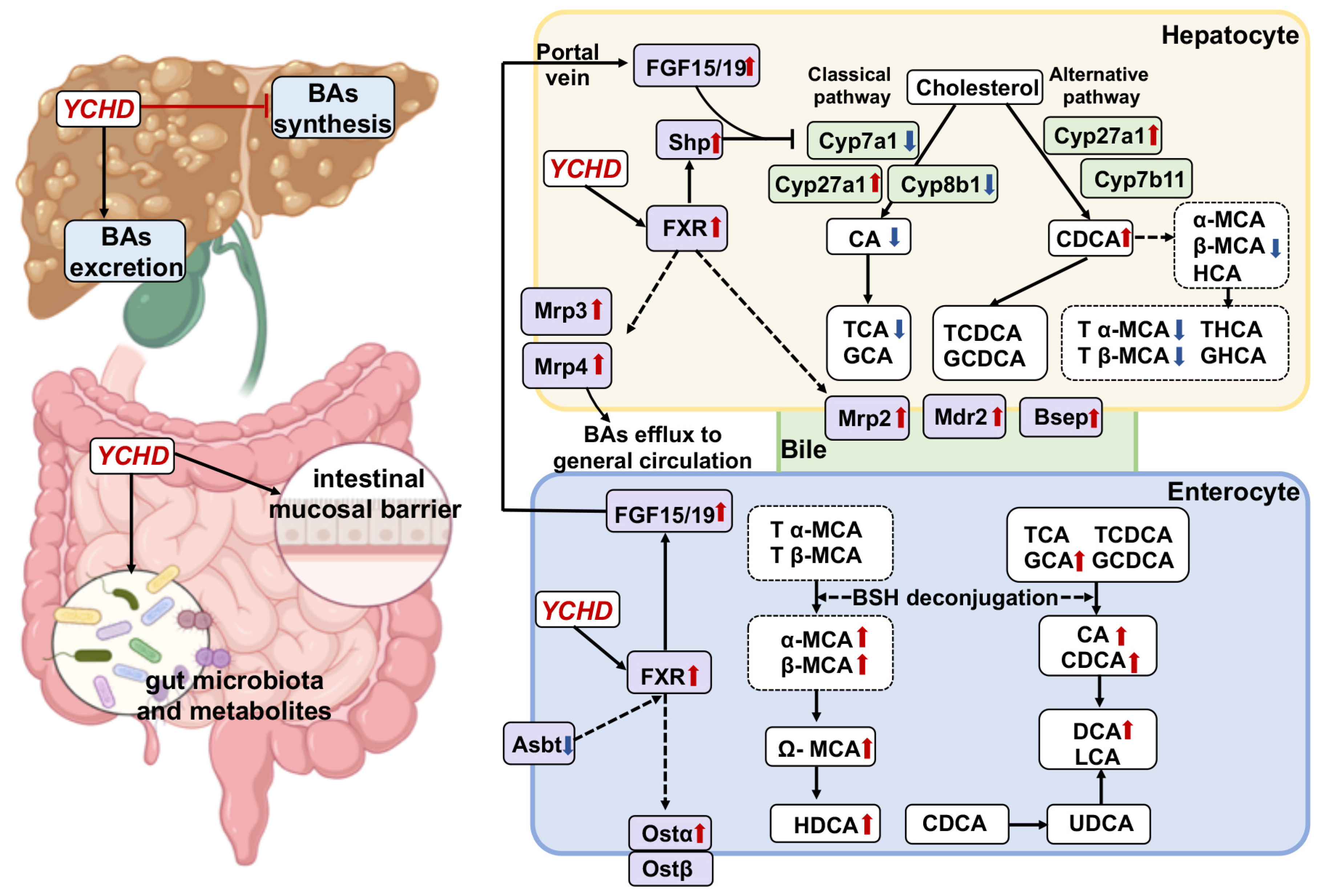
2.3. YCHD Enhanced BA Balance in Cholestatic Mice via FXR Pathway Modulation
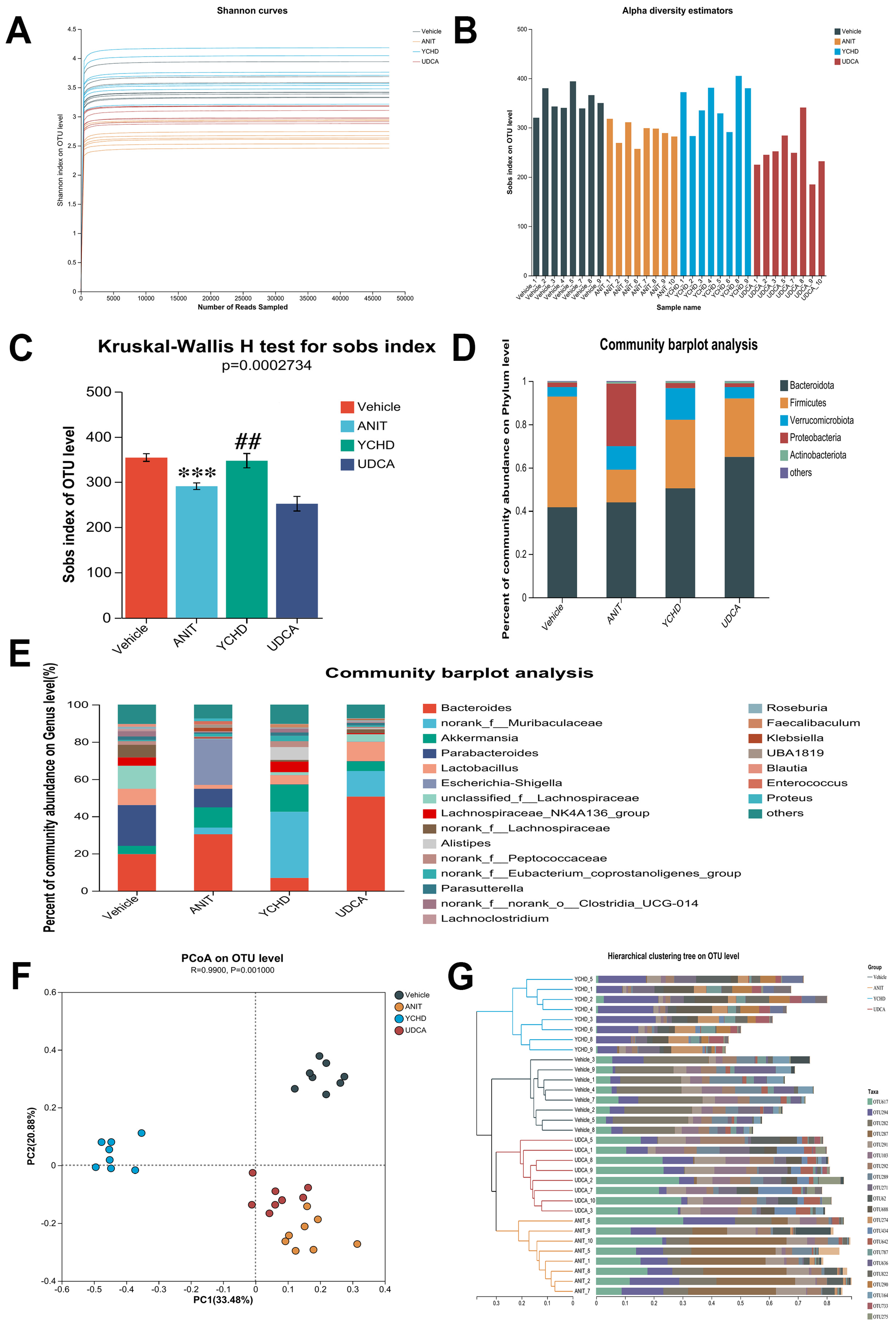
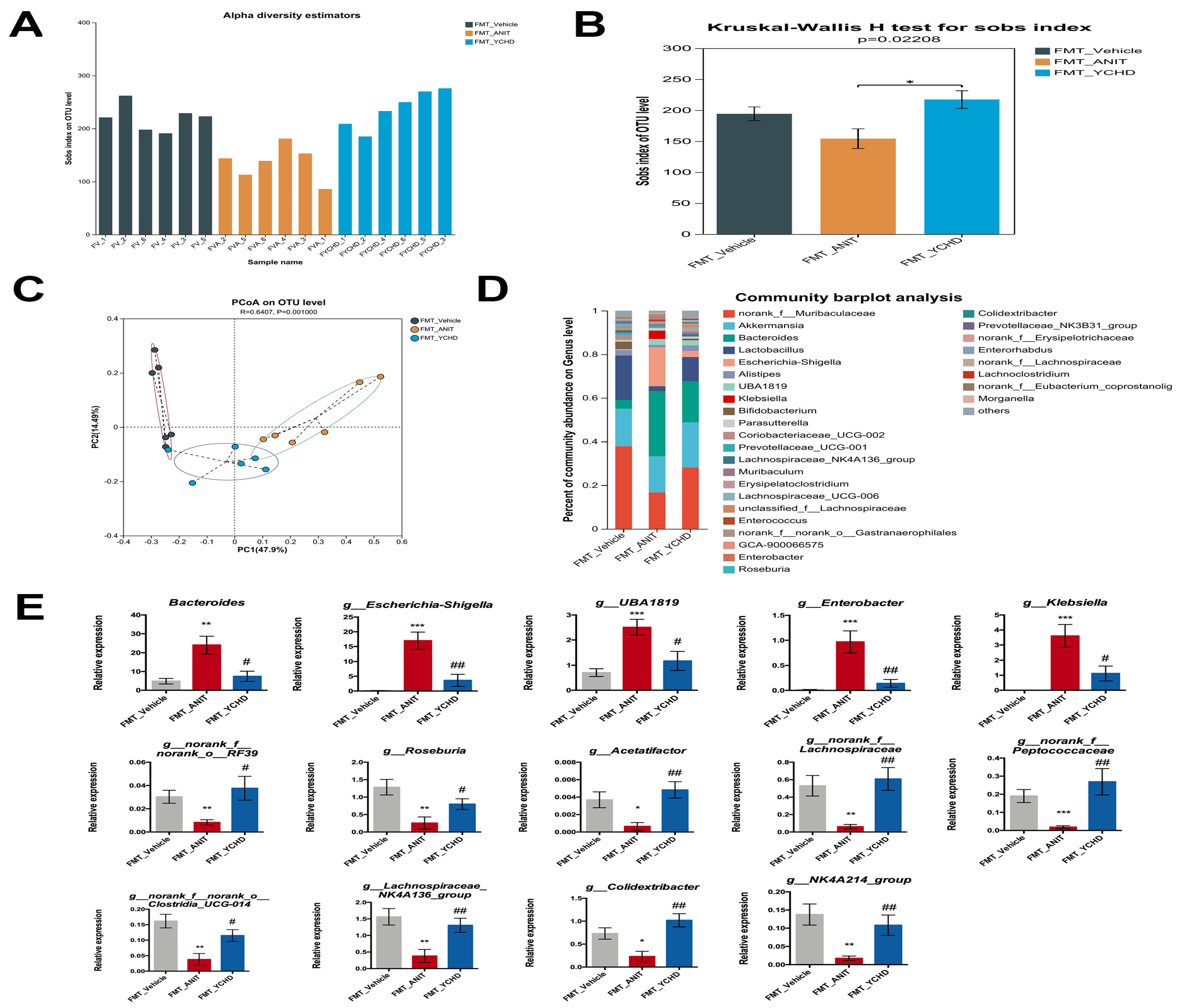
2.4. YCHD Alleviated Gut Microbiota Dysbiosis in Mice with Cholestasis
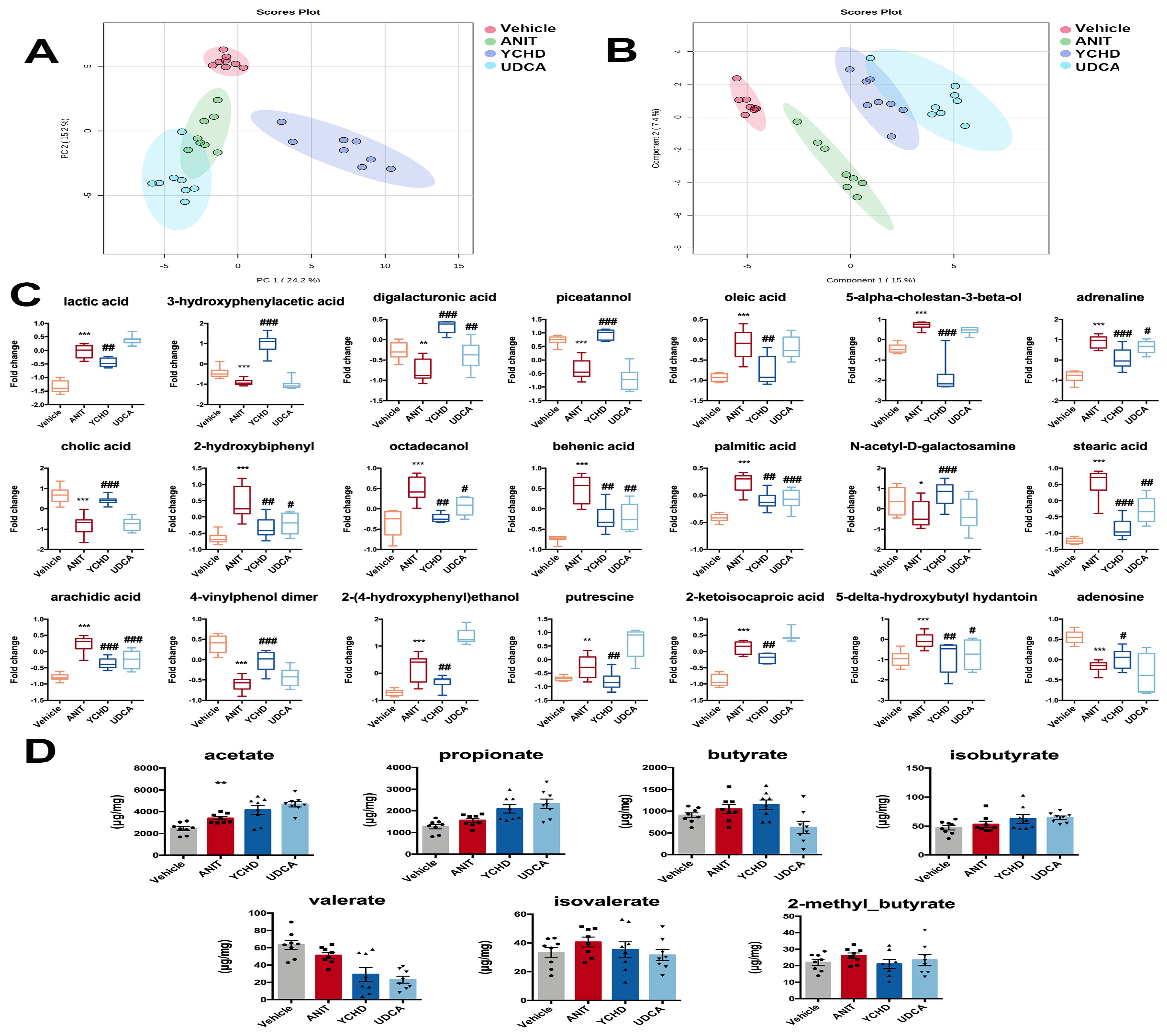
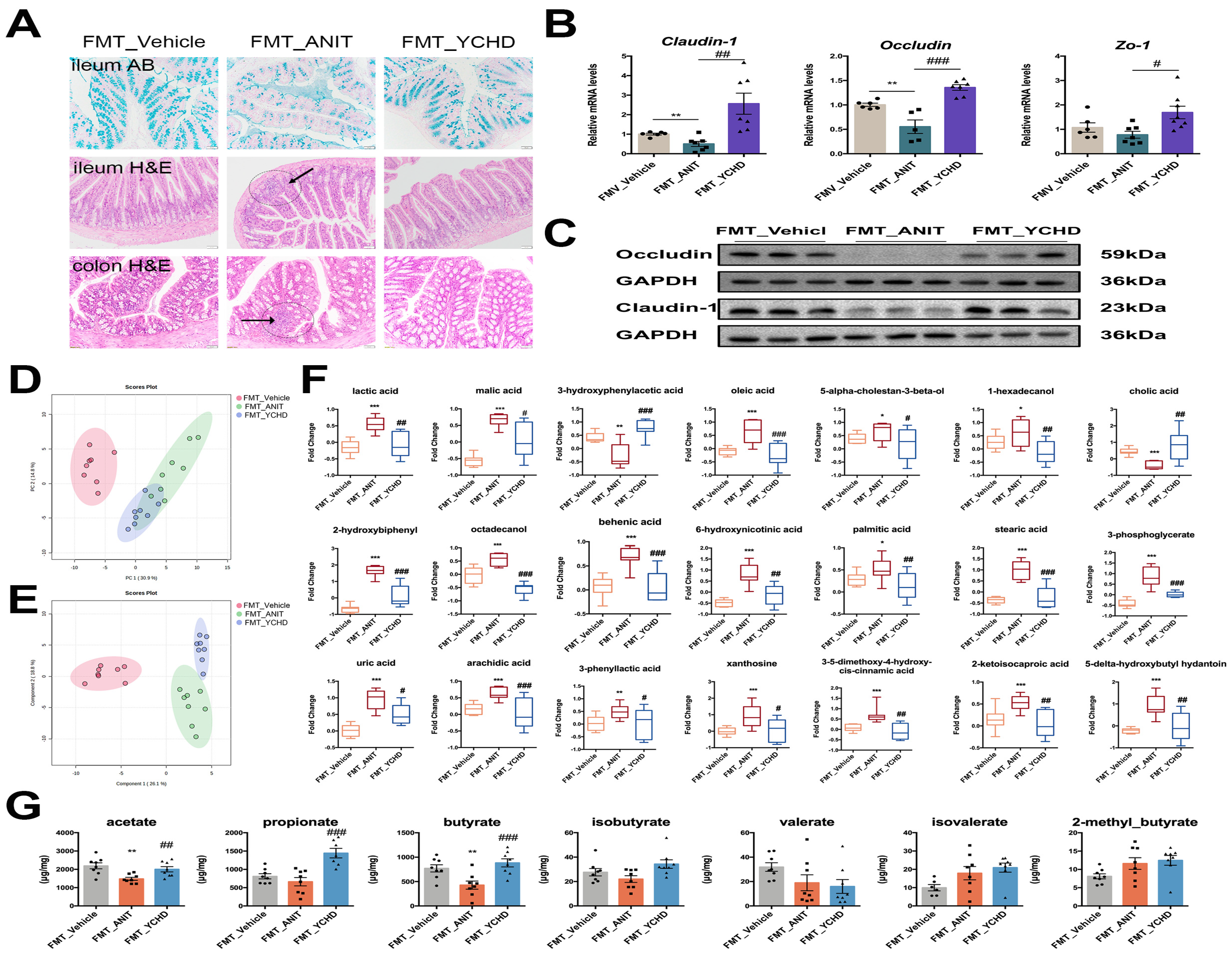
2.5. YCHD Improved Intestinal Metabolite Disorders in Mice with Cholestasis
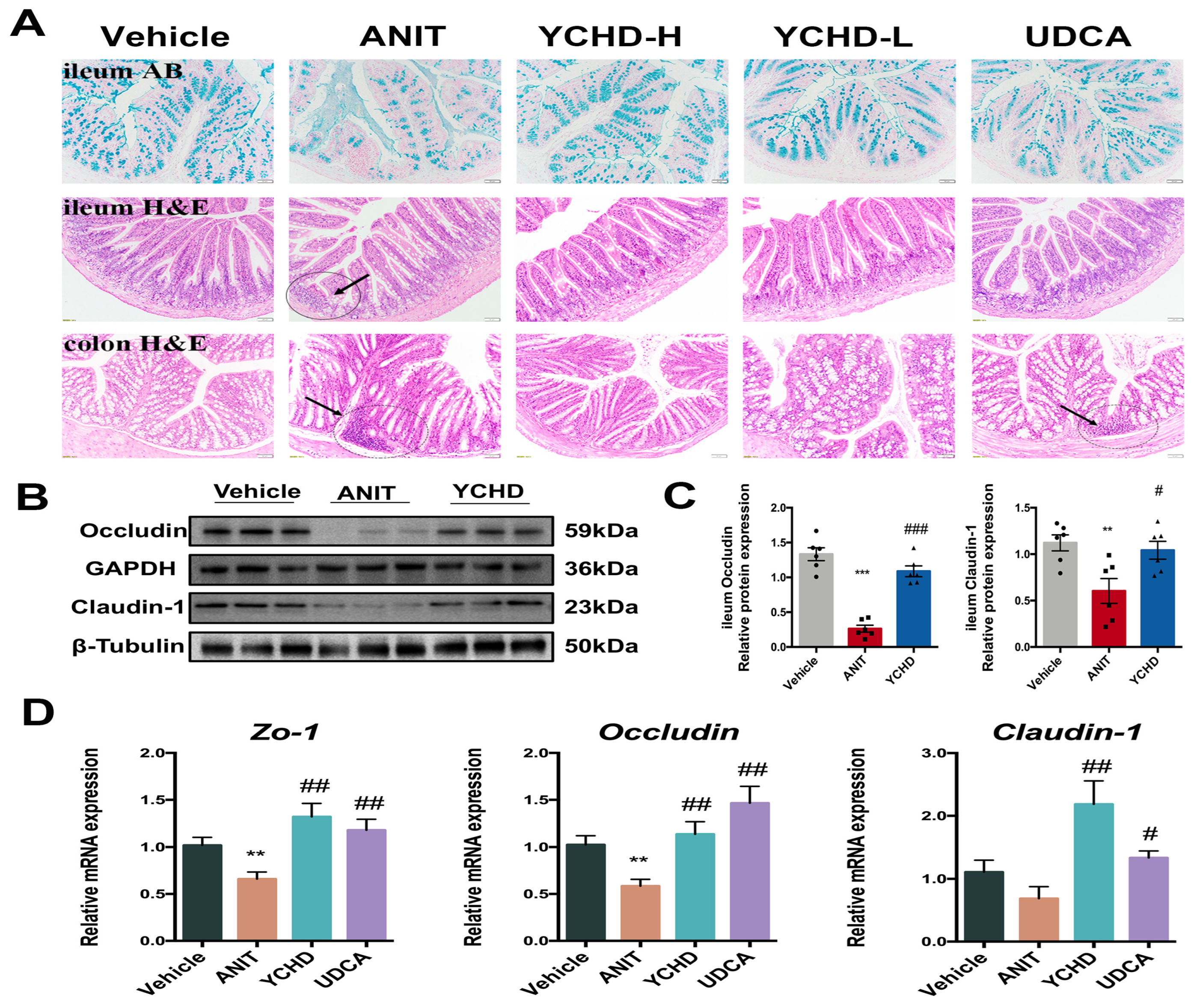
2.6. YCHD Restored Intestinal Mucosal Barrier Disruption in Mice with Cholestasis
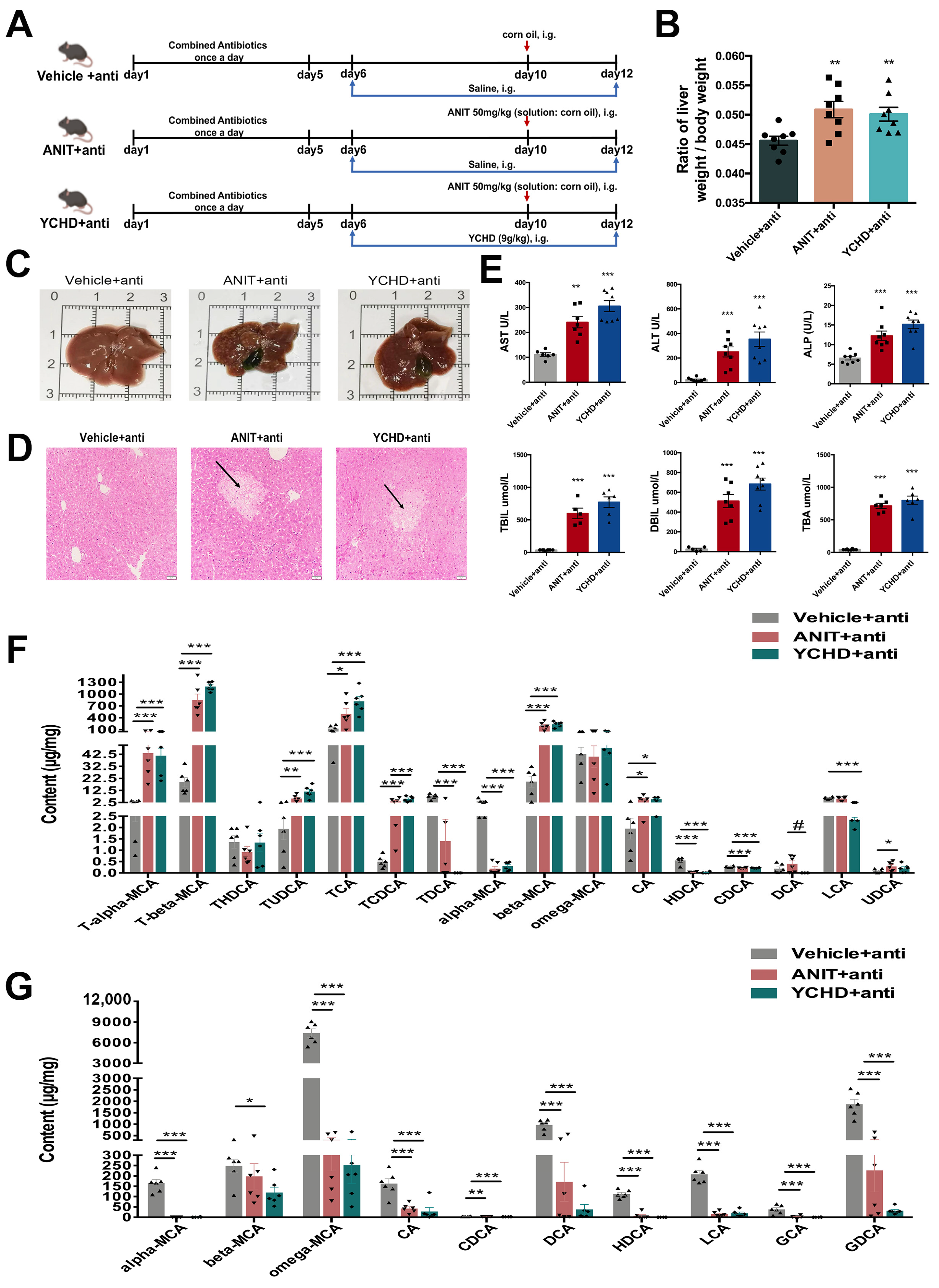
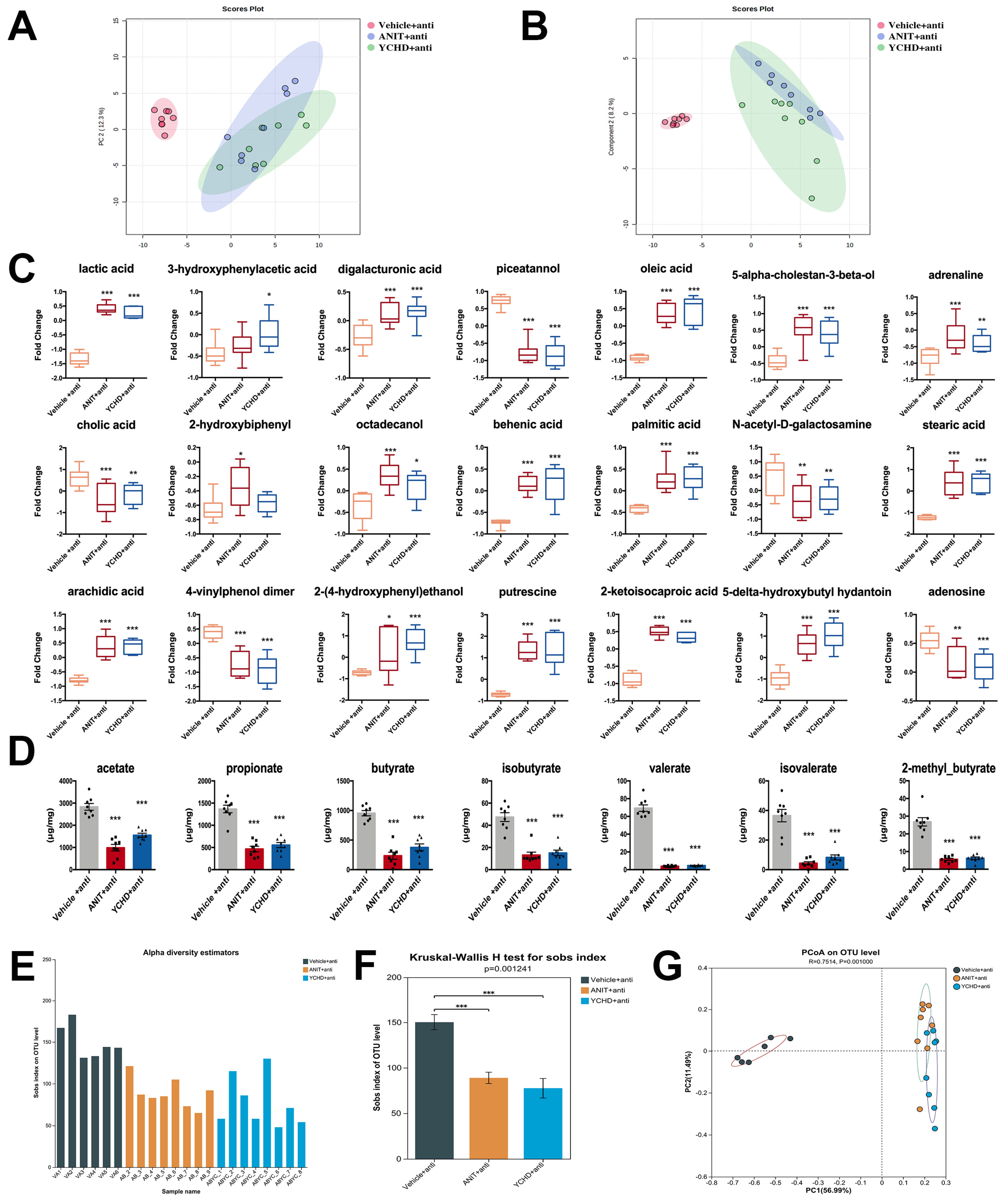
2.7. Antibiotic Therapy Reduced Efficacy of YCHD in Cholestatic Mice
2.8. FMT from YCHD Alleviated Cholestatic Liver Damage
2.9. FMT Improved BA Disorder in Mice with Cholestasis
2.10. FMT from YCHD Significantly Restored Intestinal Homeostasis in Cholestatic Mice
3. Discussion
4. Materials and Methods
4.1. Preparation of YCHD
4.2. Identification of Constituents in YCHD by Ultrahigh-Performance Liquid Chromatography–Mass Spectrometry (UHPLC-MS)
4.3. Mice
4.4. Analysis of Serum Biochemical Markers
4.5. Histopathology
4.6. Western Blot Analysis
4.7. Quantitative Real-Time Polymerase Chain Reaction (RT-PCR)
4.8. BAs Assessment
4.9. Quantification of Short-Chain Fatty Acid (SCFA) Profiles
4.10. GC-MS-Based Metabolomics
4.11. Fecal Microbiota Transplantation (FMT)
4.11.1. Antibiotic Pretreatment of Recipient Mice
4.11.2. Donor Fecal Material Processing
4.11.3. FMT Procedure and Cholestasis Induction
4.12. Antibiotic Cocktail Experiment
4.13. 16S rRNA Gene Sequencing
4.13.1. DNA Extraction and PCR Amplification
4.13.2. Illumina Sequencing
4.13.3. Data Processing and Statistical Analysis
4.14. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Jeyaraj, R.; Maher, E.R.; Kelly, D. Paediatric research sets new standards for therapy in paediatric and adult cholestasis. Lancet Child Adolesc. Health 2024, 8, 75–84. [Google Scholar] [CrossRef]
- Nevens, F.; Andreone, P.; Mazzella, G.; Strasser, S.I.; Bowlus, C.; Invernizzi, P.; Drenth, J.P.; Pockros, P.J.; Regula, J.; Beuers, U.; et al. A Placebo-Controlled Trial of Obeticholic Acid in Primary Biliary Cholangitis. N. Engl. J. Med. 2016, 375, 631–643. [Google Scholar] [CrossRef]
- Goldstein, J.; Levy, C. Novel and emerging therapies for cholestatic liver diseases. Liver. Int. 2018, 38, 1520–1535. [Google Scholar] [CrossRef] [PubMed]
- Xiang, J.; Zhang, Z.; Xie, H.; Zhang, C.; Bai, Y.; Cao, H.; Che, Q.; Guo, J.; Su, Z. Effect of different bile acids on the intestine through enterohepatic circulation based on FXR. Gut Microbes 2021, 13, 1949095. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Yang, L.; Wang, Z.; Huang, W. Bile acid nuclear receptor FXR and digestive system diseases. Acta Pharm. Sin. B 2015, 5, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Trauner, M.; Gulamhusein, A.; Hameed, B.; Caldwell, S.; Shiffman, M.L.; Landis, C.; Eksteen, B.; Agarwal, K.; Muir, A.; Rushbrook, S.; et al. The Nonsteroidal Farnesoid X Receptor Agonist Cilofexor (GS-9674) Improves Markers of Cholestasis and Liver Injury in Patients with Primary Sclerosing Cholangitis. Hepatology 2019, 70, 788–801. [Google Scholar] [CrossRef]
- Erstad, D.J.; Farrar, C.T.; Ghoshal, S.; Masia, R.; Ferreira, D.S.; Chen, Y.I.; Choi, J.K.; Wei, L.; Waghorn, P.A.; Rotile, N.J.; et al. Molecular magnetic resonance imaging accurately measures the antifibrotic effect of EDP-305, a novel farnesoid X receptor agonist. Hepatol. Commun. 2018, 2, 821–835. [Google Scholar] [CrossRef]
- Zhou, M.; Learned, R.M.; Rossi, S.J.; DePaoli, A.M.; Tian, H.; Ling, L. Engineered fibroblast growth factor 19 reduces liver injury and resolves sclerosing cholangitis in Mdr2-deficient mice. Hepatology 2016, 63, 914–929. [Google Scholar] [CrossRef]
- Wei, X.; Wu, H.; Wang, Z.; Zhu, J.; Wang, W.; Wang, J.; Wang, Y.; Wang, C. Rumen-protected lysine supplementation improved amino acid balance, nitrogen utilization and altered hindgut microbiota of dairy cows. Anim. Nutr. 2023, 15, 320–331. [Google Scholar] [CrossRef]
- Dong, L.; Dong, F.; Guo, P.; Li, T.; Fang, Y.; Dong, Y.; Xu, X.; Cai, T.; Liang, S.; Song, X.; et al. Gut microbiota as a new target for hyperuricemia: A perspective from natural plant products. Phytomedicine 2025, 138, 156402. [Google Scholar] [CrossRef]
- Fan, J.; Wang, L.; Yang, T.; Liu, J.; Ge, W.; Shen, J.; Wang, L. Comparative analysis of gut microbiota in incident and prevalent peritoneal dialysis patients with peritoneal fibrosis, correlations with peritoneal equilibration test data in the peritoneal fibrosis cohort. Ther. Apher. Dial. 2024; online ahead of print. [Google Scholar] [CrossRef]
- Liu, Q.; Li, B.; Li, Y.; Wei, Y.; Huang, B.; Liang, J.; You, Z.; Li, Y.; Qian, Q.; Wang, R.; et al. Altered faecal microbiome and metabolome in IgG4-related sclerosing cholangitis and primary sclerosing cholangitis. Gut 2022, 71, 899–909. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Tang, L.; Li, S.; Liu, S.; He, J.; Li, P.; Wang, S.; Yang, M.; Zhang, L.; Lei, Y.; et al. Gut microbiota alters host bile acid metabolism to contribute to intrahepatic cholestasis of pregnancy. Nat. Commun. 2023, 14, 1305. [Google Scholar] [CrossRef] [PubMed]
- Juanola, O.; Hassan, M.; Kumar, P.; Yilmaz, B.; Keller, I.; Simillion, C.; Engelmann, C.; Tacke, F.; Dufour, J.F.; De Gottardi, A.; et al. Intestinal microbiota drives cholestasis-induced specific hepatic gene expression patterns. Gut Microbes 2021, 13, 1911534. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, T.; Liang, X.; Zhu, L.; Fang, Y.; Dong, L.; Zheng, Y.; Xu, X.; Li, M.; Cai, T.; et al. A decrease in Flavonifractor plautii and its product, phytosphingosine, predisposes individuals with phlegm-dampness constitution to metabolic disorders. Cell. Discov. 2025, 11, 25. [Google Scholar] [CrossRef]
- Yan, Y.; Li, B.; Gao, Q.; Wu, M.; Ma, H.; Bai, J.; Ma, C.; Xie, X.; Gong, Y.; Xu, L.; et al. Intestine-Decipher Engineered Capsules Protect Against Sepsis-induced Intestinal Injury via Broad-spectrum Anti-inflammation and Parthanatos Inhibition. Adv. Sci. 2025, 12, e2412799. [Google Scholar] [CrossRef]
- Wang, C.; Liu, Z.; Zhou, T.; Wu, J.; Feng, F.; Wang, S.; Chi, H.; Sha, Y.; Zha, S.; Shu, S.; et al. Gut microbiota-derived butyric acid regulates calcific aortic valve disease pathogenesis by modulating GAPDH lactylation and butyrylation. iMeta 2025, e70048. [Google Scholar] [CrossRef]
- Li, J.Y.; Cao, H.Y.; Sun, L.; Sun, R.F.; Wu, C.; Bian, Y.Q.; Dong, S.; Liu, P.; Sun, M.Y. Therapeutic mechanism of Yin-Chen-Hao decoction in hepatic diseases. World J. Gastroenterol. 2017, 23, 1125–1138. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, H.; Song, D.; Ye, P.; Xu, N.; Chen, Y.; Zhang, W.; Hu, Q.; Ma, X.; Wen, J.; et al. Comparative Evidence for Intrahepatic Cholestasis of Pregnancy Treatment with Traditional Chinese Medicine Therapy: A Network Meta-Analysis. Front. Pharmacol. 2021, 12, 774884. [Google Scholar] [CrossRef]
- Chen, Z.; Ma, X.; Zhao, Y.; Wang, J.; Zhang, Y.; Li, J.; Wang, R.; Zhu, Y.; Wang, L.; Xiao, X. Yinchenhao decoction in the treatment of cholestasis: A systematic review and meta-analysis. J. Ethnopharmacol. 2015, 168, 208–216. [Google Scholar] [CrossRef]
- Song, G.; Zou, B.; Zhao, J.; Weng, F.; Li, Y.; Xu, X.; Zhang, S.; Yan, D.; Jin, J.; Sun, X.; et al. Yinchen decoction protects against cholic acid diet-induced cholestatic liver injury in mice through liver and ileal FXR signaling. J. Ethnopharmacol. 2023, 313, 116560. [Google Scholar] [CrossRef]
- Li, H.; Xi, Y.; Xin, X.; Feng, Q.; Hu, Y. Geniposide plus chlorogenic acid reverses non-alcoholic steatohepatitis via regulation of gut microbiota and bile acid signaling in a mouse model in vivo. Front. Pharmacol. 2023, 14, 1148737. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Sun, Z.; Hu, P.; Tian, Q.; Xu, Z.; Li, Z.; Tian, X.; Chen, M.; Huang, C. Determining the protective effects of Yin-Chen-Hao Tang against acute liver injury induced by carbon tetrachloride using 16S rRNA gene sequencing and LC/MS-based metabolomics. J. Pharm. Biomed. Anal. 2019, 174, 567–577. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; An, H.; Gui, M.; Qiu, Y.; Xu, W.; Chen, L.; Li, Q.; Yao, S.; Lin, S.; Khrustaleva, T.; et al. Qingjie Fuzheng Granule prevents colitis-associated colorectal cancer by inhibiting abnormal activation of NOD2/NF-κB signaling pathway mediated by gut microbiota disorder. Chin. Herb. Med. 2025; in press. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, X.; Cao, D.; Yang, J.; Mao, H.; Sun, L.; Wang, C. Integrated multi-omics reveals the relationship between growth performance, rumen microbes and metabolic status of Hu sheep with different residual feed intakes. Anim. Nutr. 2024, 18, 284–295. [Google Scholar] [CrossRef]
- Zhang, L.; Su, H.; Li, Y.; Fan, Y.; Wang, Q.; Jiang, J.; Hu, Y.; Chen, G.; Tan, B.; Qiu, F. Different effects of ursodeoxycholic acid on intrahepatic cholestasis in acute and recovery stages induced by alpha-naphthylisothiocyanate in mice. Toxicol. Appl. Pharmacol. 2018, 342, 69–78. [Google Scholar] [CrossRef]
- Jia, W.; Xie, G.; Jia, W. Bile acid-microbiota crosstalk in gastrointestinal inflammation and carcinogenesis. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 111–128. [Google Scholar] [CrossRef]
- Gadaleta, R.M.; Cariello, M.; Sabba, C.; Moschetta, A. Tissue-specific actions of FXR in metabolism and cancer. Biochim. Biophys. Acta 2015, 1851, 30–39. [Google Scholar] [CrossRef]
- Song, P.; Rockwell, C.E.; Cui, J.Y.; Klaassen, C.D. Individual bile acids have differential effects on bile acid signaling in mice. Toxicol. Appl. Pharmacol. 2015, 283, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Vaquero, J.; Monte, M.J.; Dominguez, M.; Muntane, J.; Marin, J.J. Differential activation of the human farnesoid X receptor depends on the pattern of expressed isoforms and the bile acid pool composition. Biochem. Pharmacol. 2013, 86, 926–939. [Google Scholar] [CrossRef]
- Wahlstrom, A.; Sayin, S.I.; Marschall, H.U.; Backhed, F. Intestinal Crosstalk between Bile Acids and Microbiota and Its Impact on Host Metabolism. Cell. Metab. 2016, 24, 41–50. [Google Scholar] [CrossRef]
- Hui, Y.; Wang, X.; Yu, Z.; Fan, X.; Cui, B.; Zhao, T.; Mao, L.; Feng, H.; Lin, L.; Yu, Q.; et al. Scoparone as a therapeutic drug in liver diseases: Pharmacology, pharmacokinetics and molecular mechanisms of action. Pharmacol. Res. 2020, 160, 105170. [Google Scholar] [CrossRef]
- Liu, J.; Li, Y.; Sun, C.; Liu, S.; Yan, Y.; Pan, H.; Fan, M.; Xue, L.; Nie, C.; Zhang, H.; et al. Geniposide reduces cholesterol accumulation and increases its excretion by regulating the FXR-mediated liver-gut crosstalk of bile acids. Pharmacol. Res. 2020, 152, 104631. [Google Scholar] [CrossRef] [PubMed]
- Wen, M.; Liu, Y.; Chen, R.; He, P.; Wu, F.; Li, R.; Lin, Y. Geniposide suppresses liver injury in a mouse model of DDC-induced sclerosing cholangitis. Phytother. Res. 2021, 35, 3799–3811. [Google Scholar] [CrossRef]
- Qin, T.; Hasnat, M.; Wang, Z.; Hassan, H.M.; Zhou, Y.; Yuan, Z.; Zhang, W. Geniposide alleviated bile acid-associated NLRP3 inflammasome activation by regulating SIRT1/FXR signaling in bile duct ligation-induced liver fibrosis. Phytomedicine 2023, 118, 154971. [Google Scholar] [CrossRef] [PubMed]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B. Bile salt biotransformations by human intestinal bacteria. J. Lipid. Res. 2006, 47, 241–259. [Google Scholar] [CrossRef]
- Xiang, H.; Liu, Z.; Xiang, H.; Xiang, D.; Xiao, S.; Xiao, J.; Shen, W.; Hu, P.; Ren, H.; Peng, M. Dynamics of the gut-liver axis in rats with varying fibrosis severity. Int. J. Biol. Sci. 2022, 18, 3390–3404. [Google Scholar] [CrossRef]
- Quraishi, M.N.; Cheesbrough, J.; Rimmer, P.; Mullish, B.H.; Sharma, N.; Efstathiou, E.; Acharjee, A.; Gkoutus, G.; Patel, A.; Marchesi, J.R.; et al. Open Label Vancomycin in Primary Sclerosing Cholangitis-Inflammatory Bowel Disease: Improved Colonic Disease Activity and Associations with Changes in Host-Microbiome-Metabolomic Signatures. J. Crohns. Colitis 2025, 19, jjae189. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, Y.; Li, W.; Zhang, B.; Yin, J.; Liuqi, S.; Wang, J.; Peng, B.; Wang, S. Fucoidan Ameliorated Dextran Sulfate Sodium-Induced Ulcerative Colitis by Modulating Gut Microbiota and Bile Acid Metabolism. J. Agric. Food Chem. 2022, 70, 14864–14876. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhang, K.X.; Bu, Q.Y.; Song, S.X.; Chen, Y.; Zou, H.; You, X.Y.; Zhao, G.P. Ginsenosides From Panax ginseng Improves Hepatic Lipid Metabolism Disorders in HFD-Fed Rats by Regulating Gut Microbiota and Cholesterol Metabolism Signaling Pathways. Phytother. Res. 2025, 39, 714–732. [Google Scholar] [CrossRef]
- Sun, H.; Su, X.; Liu, Y.; Li, G.; Du, Q. Roseburia intestinalis relieves intrahepatic cholestasis of pregnancy through bile acid/FXR-FGF15 in rats. iScience 2023, 26, 108392. [Google Scholar] [CrossRef]
- Shi, H.; Li, X.; Hou, C.; Chen, L.; Zhang, Y.; Li, J. Effects of Pomegranate Peel Polyphenols Combined with Inulin on Gut Microbiota and Serum Metabolites of High-Fat-Induced Obesity Rats. J. Agric. Food Chem. 2023, 71, 5733–5744. [Google Scholar] [CrossRef]
- Albillos, A.; de Gottardi, A.; Rescigno, M. The gut-liver axis in liver disease: Pathophysiological basis for therapy. J. Hepatol. 2020, 72, 558–577. [Google Scholar] [CrossRef]
- Moreno-Gonzalez, M.; Hampton, K.; Ruiz, P.; Beasy, G.; Nagies, F.S.; Parker, A.; Lazenby, J.; Bone, C.; Alava-Arteaga, A.; Patel, M.; et al. Regulation of intestinal senescence during cholestatic liver disease modulates barrier function and liver disease progression. JHEP Rep. 2024, 6, 101159. [Google Scholar] [CrossRef]
- Zuhl, M.; Schneider, S.; Lanphere, K.; Conn, C.; Dokladny, K.; Moseley, P. Exercise regulation of intestinal tight junction proteins. Br. J. Sports Med. 2014, 48, 980–986. [Google Scholar] [CrossRef]
- Sorribas, M.; Jakob, M.O.; Yilmaz, B.; Li, H.; Stutz, D.; Noser, Y.; de Gottardi, A.; Moghadamrad, S.; Hassan, M.; Albillos, A.; et al. FXR modulates the gut-vascular barrier by regulating the entry sites for bacterial translocation in experimental cirrhosis. J. Hepatol. 2019, 71, 1126–1140. [Google Scholar] [CrossRef] [PubMed]
- Rooks, M.G.; Garrett, W.S. Gut microbiota, metabolites and host immunity. Nat. Rev. Immunol. 2016, 16, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Cong, Y. Gut microbiota-derived metabolites in the regulation of host immune responses and immune-related inflammatory diseases. Cell. Mol. Immunol. 2021, 18, 866–877. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Che, D.; Adams, S.; Guo, N.; Han, R.; Zhang, C.; Qin, G.; Farouk, M.H.; Jiang, H. N-Acetyl-d-galactosamine prevents soya bean agglutinin-induced intestinal barrier dysfunction in intestinal porcine epithelial cells. J. Anim. Physiol. Anim. Nutr. 2019, 103, 1198–1206. [Google Scholar] [CrossRef]
- Xu, H.; Feng, R.; Ye, M.L.; Hu, J.C.; Lu, J.Y.; Wang, J.Y.; Zuo, H.T.; Zhao, Y.; Song, J.Y.; Jiang, J.D.; et al. Multiple Enzymes Expressed by the Gut Microbiota Can Transform Typhaneoside and Are Associated with Improving Hyperlipidemia. Adv. Sci. 2025, 12, e2411770. [Google Scholar] [CrossRef]
- Li, W.; Zhu, J.; Zhou, T.; Jin, Z. Exploring the mechanisms of Yinchenhao decoction against ANIT-induced cholestatic liver injury by lipidomics, metabolomics and network pharmacology. J. Pharm. Biomed. Anal. 2025, 258, 116736. [Google Scholar] [CrossRef]
- Zhang, C.; Zhen, Y.; Weng, Y.; Lin, J.; Xu, X.; Ma, J.; Zhong, Y.; Wang, M. Research progress on the microbial metabolism and transport of polyamines and their roles in animal gut homeostasis. J. Anim. Sci. Biotechnol. 2025, 16, 57. [Google Scholar] [CrossRef]
- Yang, S.; Kuang, G.; Jiang, R.; Wu, S.; Zeng, T.; Wang, Y.; Xu, F.; Xiong, L.; Gong, X.; Wan, J. Geniposide protected hepatocytes from acetaminophen hepatotoxicity by down-regulating CYP 2E1 expression and inhibiting TLR 4/NF-kappaB signaling pathway. Int. Immunopharmacol. 2019, 74, 105625. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Yang, J.; Shi, D.; Deng, R.; Yan, B. Scoparone potentiates transactivation of the bile salt export pump gene and this effect is enhanced by cytochrome P450 metabolism but abolished by a PKC inhibitor. Br. J. Pharmacol. 2011, 164, 1547–1557. [Google Scholar] [CrossRef]
- Masubuchi, N.; Nishiya, T.; Imaoka, M.; Mizumaki, K.; Okazaki, O. Promising toxicological biomarkers for the diagnosis of liver injury types: Bile acid metabolic profiles and oxidative stress marker as screening tools in drug development. Chem. Biol. Interact. 2016, 255, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Dinan, K.; Dinan, T. Antibiotics and mental health: The good, the bad and the ugly. J. Intern. Med. 2022, 292, 858–869. [Google Scholar] [CrossRef]
- Reagan-Shaw, S.; Nihal, M.; Ahmad, N. Dose translation from animal to human studies revisited. FASEB J. 2008, 22, 659–661. [Google Scholar] [CrossRef] [PubMed]
- Shi, K.; Wen, J.; Zeng, J.; Guo, Y.; Hu, J.; Li, C.; Zhao, Y.; Ma, X. Preclinical evidence of Yinchenhao decoction on cholestasis: A systematic review and meta-analysis of animal studies. Phytother. Res. 2021, 35, 138–154. [Google Scholar] [CrossRef]
- Li, W.; Chen, H.; Qian, Y.; Wang, S.; Luo, Z.; Shan, J.; Kong, X.; Gao, Y. Integrated Lipidomics and Metabolomics Study of Four Chemically Induced Mouse Models of Acute Intrahepatic Cholestasis. Front. Pharmacol. 2022, 13, 907271. [Google Scholar] [CrossRef]
- Li, W.W.; Yang, Y.; Dai, Q.G.; Lin, L.L.; Xie, T.; He, L.L.; Tao, J.L.; Shan, J.J.; Wang, S.C. Non-invasive urinary metabolomic profiles discriminate biliary atresia from infantile hepatitis syndrome. Metabolomics 2018, 14, 90. [Google Scholar] [CrossRef]
- Barcena, C.; Valdes-Mas, R.; Mayoral, P.; Garabaya, C.; Durand, S.; Rodriguez, F.; Fernandez-Garcia, M.T.; Salazar, N.; Nogacka, A.M.; Garatachea, N.; et al. Healthspan and lifespan extension by fecal microbiota transplantation into progeroid mice. Nat. Med. 2019, 25, 1234–1242. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Chemical Formula | Retention Time (min) | m/z | Chemical Name | PubChem Compound Identifier |
|---|---|---|---|---|---|
| 1 | C7H12O6 | 0.858 | 215.0517 | Quinic acid | 6508 |
| 2 | C16H14O4 | 0.858 | 309.0494 | Ammidin | 10212 |
| 3 | C16H24O11 | 0.958 | 431.0919 | Caryoptosidic acid | 131751540 |
| 4 | C17H22O10 | 0.992 | 425.0876 | 1-O-Sinapoylglucose | 6168296 |
| 5 | C16H26O9 | 0.992 | 401.1162 | Villosolside | 156599111 |
| 6 | C17H14O8 | 0.992 | 347.079 | Eupatolitin | 5317291 |
| 7 | C17H24O11 | 2.533 | 443.0963 | Scandoside methyl ester | 442433 |
| 8 | C17H24O10 | 3.638 | 427.1032 | Geniposide | 107848 |
| 9 | C27H30O16 | 3.739 | 611.1631 | Rheinoside A | 13888123 |
| 10 | C8H8O2 | 4.241 | 137.0605 | 4-Hydroxyacetophenone | 7469 |
| 11 | C5H7NO3 | 4.342 | 147.0788 | Pyroglutamic acid | 7405 |
| 12 | C15H10O5 | 4.442 | 271.0631 | Aloe-emodin | 10207 |
| 13 | C21H20O12 | 4.542 | 465.105 | Quercetin-3-glycoside | 5280804 |
| 14 | C22H22O11 | 4.576 | 463.1259 | Cinnamoyl-glucogallin | 53355598 |
| 15 | C10H8O4 | 4.61 | 193.0513 | Scopoletin | 5280460 |
| 16 | C22H22O12 | 5.146 | 479.1204 | Isorhamnetin-3-glucoside | 5318645 |
| 17 | C16H12O7 | 5.146 | 317.0676 | Capillarisin | 5281342 |
| 18 | C28H32O15 | 5.28 | 609.1833 | Physciondiglucoside | 73981703 |
| 19 | C15H10O6 | 5.614 | 287.0573 | Demethoxycapillarisin | 5316511 |
| 20 | C11H10O4 | 5.815 | 207.0665 | Scoparone | 8417 |
| 21 | C20H24O4 | 7.591 | 329.1769 | Crocetin | 5281232 |
| 22 | C17H14O7 | 8.998 | 331.0842 | Eupalitin | 5748611 |
| 23 | C17H14O6 | 10.103 | 315.0879 | Cirsimaritin | 188323 |
| 24 | C16H12O5 | 13.52 | 285.0773 | Genkwanin | 5281617 |
| 25 | C20H36O2 | 16.669 | 326.3056 | Mandenol | 5282184 |
| 26 | C30H50 | 22.13 | 411.3948 | Supraene | 638072 |
| 27 | C29H48O | 24.575 | 413.3792 | Stigmasterol | 5280794 |
| No. | Target Genes | Primer Forward (5′->3′) | Primer Reverse (5′->3′) |
|---|---|---|---|
| 1 | Cyp7a1 | GGGATTGCTGTGGTAGTGAGC | GGTATGGAATCAACCCGTTGTC |
| 2 | Cyp8b1 | CCTCTGGACAAGGGTTTTGTG | GCACCGTGAAGACATCCCC |
| 3 | Cyp27a1 | CCAGGCACAGGAGAGTACG | GGGCAAGTGCAGCACATAG |
| 4 | Fxr | GGCAGAATCTGGATTTGGAATCG | GCCCAGGTTGGAATAGTAAGACG |
| 5 | Shp | CTCATGGCCTCTACCCTCAA | GGTCACCTCAGCAAAAGCAT |
| 6 | Bsep | CAATGTTCAGTTCCTCCGTTCA | TCTCTTTGGTGTTGTCCCCATA |
| 7 | Ntcp | ACTGGCTTCCTGATGGGCTAC | GAGTTGGACGTTTTGGAATCCT |
| 8 | Fgfr4 | CTGACTCGCAGACGACATGAG | AGGCCATGATCTTGAGATGAGA |
| 9 | Mdr2 | CGGCGACTTTGAACTAGGCA | CAGAGTATCGGAACAGTGTCAAC |
| 10 | Mrp2 | TCTTCGTCTCCTATGGTTTCCA | CGTGTGTTGAGTCGCTTGATT |
| 11 | Mrp3 | CTGGGTCCCCTGCATCTAC | GCCGTCTTGAGCCTGGATAAC |
| 12 | Mrp4 | GGCACTCCGGTTAAGTAACTC | TGTCACTTGGTCGAATTTGTTCA |
| 13 | Oatp1a1 | GTGCATACCTAGCCAAATCACT | CCAGGCCCATAACCACACATC |
| 14 | Oatp1a4 | ATAGCTTCAGGCGCATTTAC | TTCTCCATCATTCTGCATCG |
| 15 | Oatp1b2 | GGGAACATGCTTCGTGGGATA | GGAGTTATGCGGACACTTCTC |
| 16 | Asbt | GTCTGTCCCCCAAATGCAACT | CACCCCATAGAAAACATCACCA |
| 17 | Osta | AGGCAGGACTCATATCAAACTTG | TGAGGGCTATGTCCACTGGG |
| 18 | Ostβ | AGATGCGGCTCCTTGGAATTA | TGGCTGCTTCTTTCGATTTCTG |
| 19 | Zo-1 | GCCGCTAAGAGCACAGCAA | GCCCTCCTTTTAACACATCAGA |
| 20 | Occludin | TTGAAAGTCCACCTCCTTACAGA | CCGGATAAAAAGAGTACGCTGG |
| 21 | Claudin-1 | GGGGACAACATCGTGACCG | AGGAGTCGAAGACTTTGCACT |
| 22 | β-actin | CGTTGACATCCGTAAAGACC | AACAGTCCGCCTAGAAGCAC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, W.; Huang, D.; Luo, Z.; Zhou, T.; Jin, Z. Yinchenhao Decoction Mitigates Cholestatic Liver Injury in Mice via Gut Microbiota Regulation and Activation of FXR-FGF15 Pathway. Pharmaceuticals 2025, 18, 932. https://doi.org/10.3390/ph18070932
Li W, Huang D, Luo Z, Zhou T, Jin Z. Yinchenhao Decoction Mitigates Cholestatic Liver Injury in Mice via Gut Microbiota Regulation and Activation of FXR-FGF15 Pathway. Pharmaceuticals. 2025; 18(7):932. https://doi.org/10.3390/ph18070932
Chicago/Turabian StyleLi, Weiwei, Doudou Huang, Zichen Luo, Ting Zhou, and Ziwen Jin. 2025. "Yinchenhao Decoction Mitigates Cholestatic Liver Injury in Mice via Gut Microbiota Regulation and Activation of FXR-FGF15 Pathway" Pharmaceuticals 18, no. 7: 932. https://doi.org/10.3390/ph18070932
APA StyleLi, W., Huang, D., Luo, Z., Zhou, T., & Jin, Z. (2025). Yinchenhao Decoction Mitigates Cholestatic Liver Injury in Mice via Gut Microbiota Regulation and Activation of FXR-FGF15 Pathway. Pharmaceuticals, 18(7), 932. https://doi.org/10.3390/ph18070932