
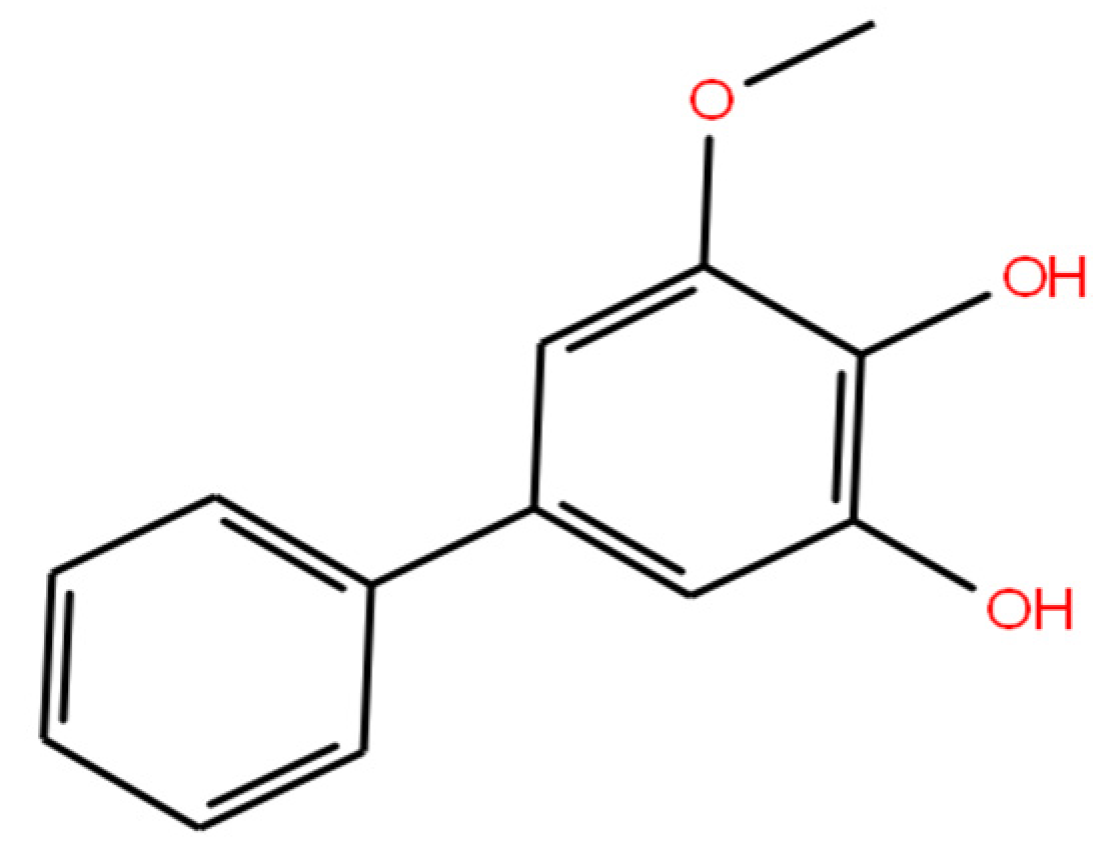
Molecular Insights into the Temperature-Dependent Binding and Conformational Dynamics of Noraucuparin with Bovine Serum Albumin: A Microsecond-Scale MD Simulation Study


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
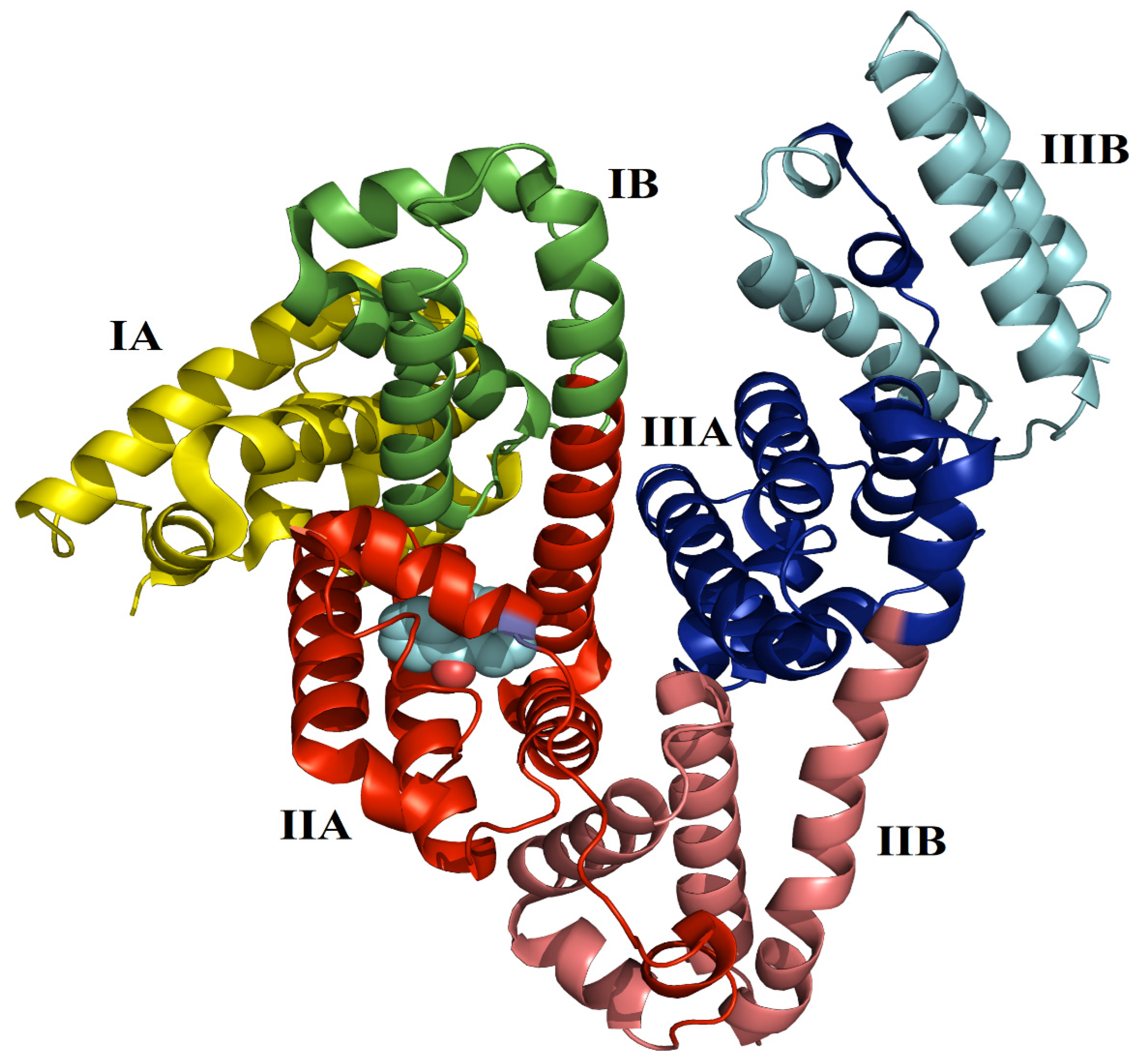
2.1. Stability of Simulated Systems
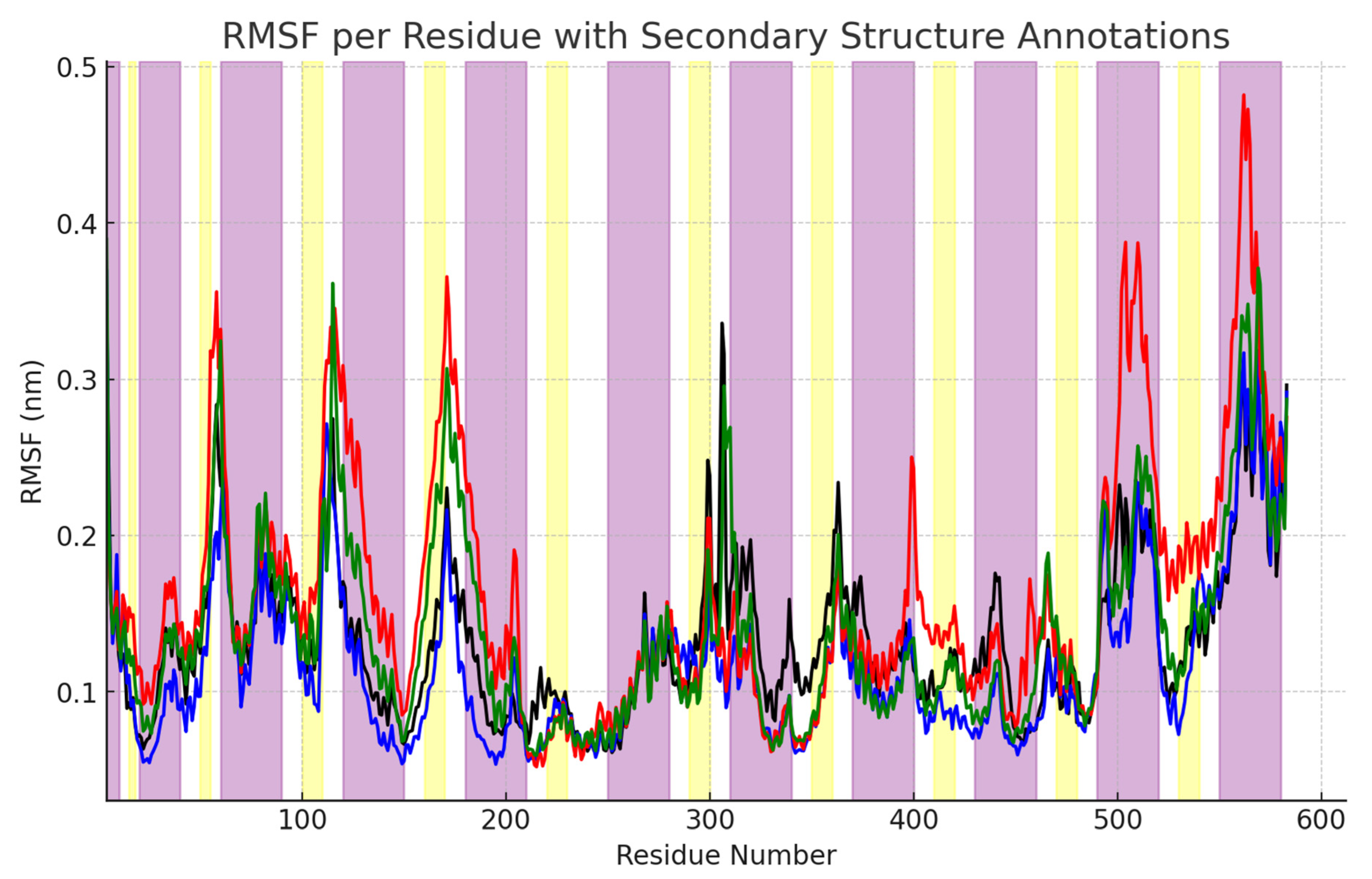
2.2. RMSF Analysis
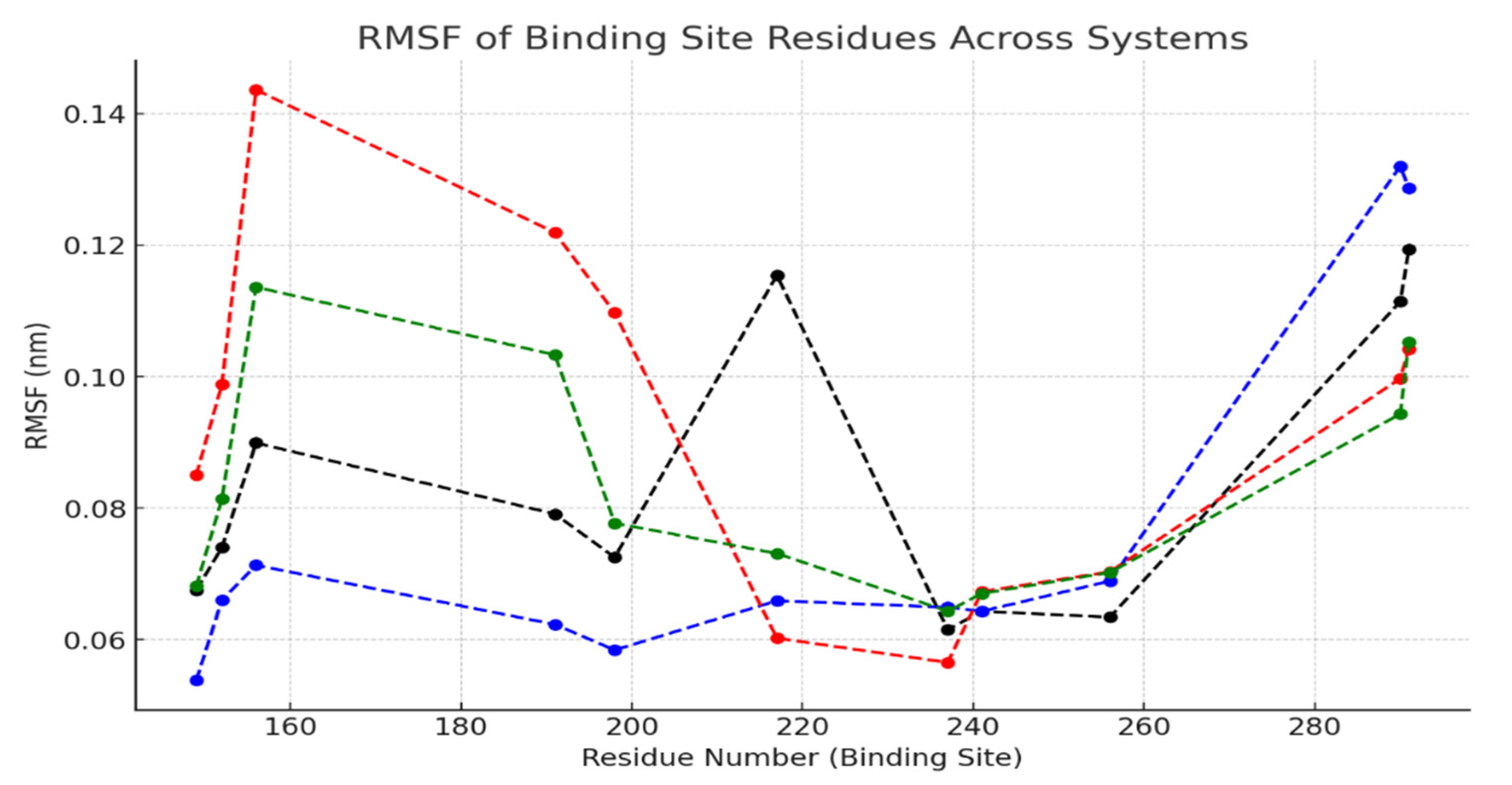
2.3. Correlation Between RMSF and Ligand Binding in the Binding Site
2.4. Phase Space Projection of Free and Bound BSA at Different Temperatures
2.5. MD Simulations of BSA–Ligand Interactions
Hydrogen Bonding and Structural Insights
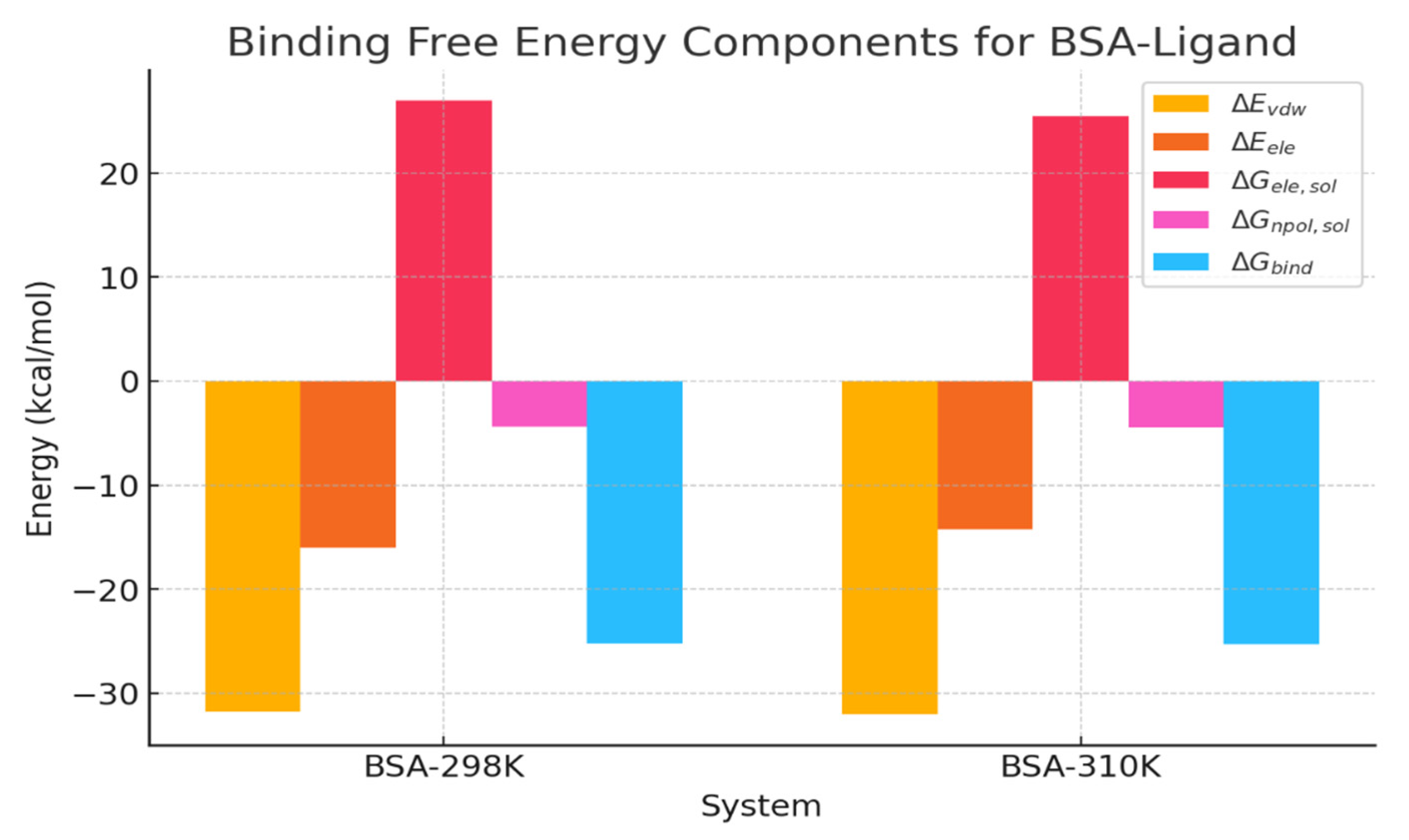
2.6. Binding-Free Energy Calculations Using MMGBSA
2.7. Per-Residue Free Energy Decomposition for Complexes Between BSA and Noraucuparin at 298 K and 310 K
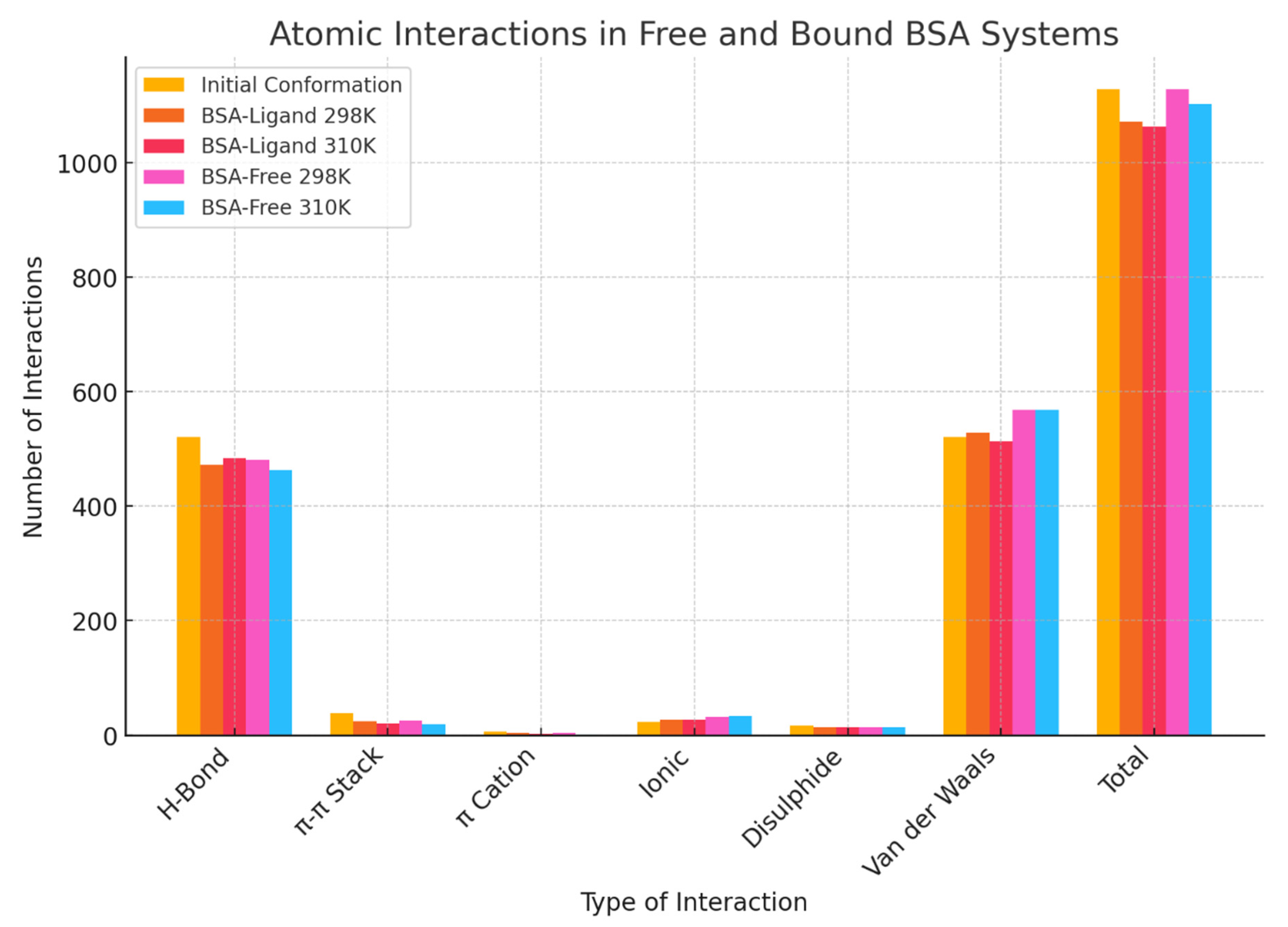
2.8. Temperature- and Ligand-Dependent Changes in Interaction Networks
3. Discussion
4. Materials and Methods
4.1. Molecular Docking
4.2. MD Simulations
4.3. Structural Interaction Network and Regional Analysis
4.4. Binding-Free Energy and Per-Residue Decomposition Calculations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liang, W.; Zhang, Z.; Zhu, Q.; Han, Z.; Huang, C.; Liang, X.; Yang, M. Molecular Interactions between Bovine Serum Albumin (BSA) and Trihalophenol: Insights from Spectroscopic, Calorimetric and Molecular Modeling Studies. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2023, 287, 122054. [Google Scholar] [CrossRef]
- Majorek, K.A.; Porebski, P.J.; Dayal, A.; Zimmerman, M.D.; Jablonska, K.; Stewart, A.J.; Chruszcz, M.; Minor, W. Structural and Immunologic Characterization of Bovine, Horse, and Rabbit Serum Albumins. Mol. Immunol. 2012, 52, 174–182. [Google Scholar] [CrossRef]
- Mavani, A.; Ray, D.; Aswal, V.K.; Bhattacharyya, J. Understanding the Molecular Interaction of BSA Protein with Antibiotic Sulfa Molecule(s) for Novel Drug Development. J. Mol. Struct. 2023, 1287, 135697. [Google Scholar] [CrossRef]
- Qashqoosh, M.T.A.; Manea, Y.K.; Alahdal, F.A.M.; Naqvi, S. Investigation of Conformational Changes of Bovine Serum Albumin upon Binding with Benzocaine Drug: A Spectral and Computational Analysis. Bionanoscience 2019, 9, 848–858. [Google Scholar] [CrossRef]
- Bujacz, A. Structures of bovine, equine and leporine serum albumin. Acta Crystallogr. D Biol. Crystallogr. 2012, 68, 1278–1289. [Google Scholar] [CrossRef] [PubMed]
- Donini, S.; Bertoli, M.; D’Annessa, I.; Spyrakis, F.; Parisi, G.; Fossa, P.; Sturlese, M.; Cavalli, A.; Cavalli, R.; Acerbi, D.; et al. Crystal Structures of Ketoprofen Bound to Human and Bovine Serum Albumin. ACS Omega 2019, 4, 10664–10672. [Google Scholar] [CrossRef]
- Sudlow, G.; Birkett, D.J.; Wade, D.N. Spectroscopic Techniques in the Study of Protein Binding. A Fluorescence Technique for the Evaluation of the Albumin Binding and Displacement of Warfarin and Warfarin-alcohol. Clin. Exp. Pharmacol. Physiol. 1975, 2, 129–140. [Google Scholar] [CrossRef]
- Fujiwara, S.-I.; Amisaki, T. Fatty Acid Binding to Serum Albumin: Molecular Simulation Approaches. Biochim. Biophys. Acta Gen. Subj. 2013, 1830, 5427–5434. [Google Scholar] [CrossRef]
- Yuan, S.; Zhang, Y.; Liu, J.; Zhao, Y.; Tan, L.; Liu, J.; Wang, Q.; Zhang, H. Structure-Affinity Relationship of the Binding of Phenolic Acids and Their Derivatives to Bovine Serum Albumin. Food Chem. 2019, 278, 77–83. [Google Scholar] [CrossRef]
- Pawar, S.K.; Jaldappagari, S. Interaction of Repaglinide with Bovine Serum Albumin: Spectroscopic and Molecular Docking Approaches. J. Pharm. Anal. 2019, 9, 274–283. [Google Scholar] [CrossRef]
- Belinskaia, D.A.; Voronina, P.A.; Shmurak, V.I.; Jenkins, R.O.; Goncharov, N.V. Serum Albumin in Health and Disease: Esterase, Antioxidant, Transporting and Signaling Properties. Int. J. Mol. Sci. 2021, 22, 10318. [Google Scholar] [CrossRef] [PubMed]
- Halder, K.; Sengupta, P.; Chaki, S.; Saha, R.; Dasgupta, S. Understanding Conformational Changes in Human Serum Albumin and Its Interactions with Gold Nanorods: Do Flexible Regions Play a Role in Corona Formation? Langmuir 2023, 39, 1651–1664. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X. Spectroscopic and molecular docking studies on binding interactions of camptothecin drugs with bovine serum albumin. J. Biomol. Struct. Dyn. 2025, in press. [Google Scholar]
- El Gammal, R.N.; Elmansi, H.; El-Emam, A.A.; Hammouda, M.E.A. Exploring the molecular interaction of mebendazole with bovine serum albumin using multi-spectroscopic approaches and molecular docking. Sci. Rep. 2022, 12, 11582. [Google Scholar] [CrossRef]
- Liu, J.; He, Y.; Liu, D.; He, Y.; Tang, Z.; Lou, H.; Huo, Y.; Cao, X. Characterizing the binding interaction of astilbin with bovine serum albumin: A spectroscopic study in combination with molecular docking technology. RSC Adv. 2018, 8, 13272–13282. [Google Scholar] [CrossRef]
- Khalil, M.N.A.; Brandt, W.; Beuerle, T.; Reckwell, D.; Groeneveld, J.; Hänsch, R.; Gaid, M.M.; Liu, B.; Beerhues, L. O-Methyltransferases Involved in Biphenyl and Dibenzofuran Biosynthesis. Plant J. 2015, 83, 263–276. [Google Scholar] [CrossRef]
- Sarkate, A.; Saini, S.S.; Teotia, D.; Gaid, M.; Mir, J.I.; Roy, P.; Agrawal, P.K.; Sircar, D. Comparative Metabolomics of Scab-Resistant and Susceptible Apple Cell Cultures in Response to Scab Fungus Elicitor Treatment. Sci. Rep. 2018, 8, 17844. [Google Scholar] [CrossRef]
- Zhou, L.; Yang, J.; Yang, G.; Kang, C.; Xiao, W.; Lv, C.; Wang, S.; Tang, J.; Guo, L. Biphenyl Phytoalexin in Sorbus Pohuashanensis Suspension Cell Induced by Yeast Extract. Molecules 2016, 21, 1180. [Google Scholar] [CrossRef]
- Chizzali, C.; Khalil, M.N.A.; Beuerle, T.; Schuehly, W.; Richter, K.; Flachowsky, H.; Peil, A.; Hanke, M.-V.; Liu, B.; Beerhues, L. Formation of Biphenyl and Dibenzofuran Phytoalexins in the Transition Zones of Fire Blight-Infected Stems of Malus Domestica Cv. ‘Holsteiner Cox’ and Pyrus Communis Cv. ‘Conference’. Phytochemistry 2012, 77, 179–185. [Google Scholar] [CrossRef]
- Gao, Y.; Yang, J.; Yang, X.; Zhang, L.; Wang, J.; Li, Q.; Lin, D.-M.; Zhang, M.; Xia, S.-N.; Xu, L.-L.; et al. Novel Dibenzofuran and Biphenyl Phytoalexins from Sorbus Pohuashanensis Suspension Cell and Their Antimicrobial Activities. Fitoterapia 2021, 152, 104914. [Google Scholar] [CrossRef]
- Saini, S.S.; Teotia, D.; Gaid, M.; Thakur, A.; Beerhues, L.; Sircar, D. Benzaldehyde Dehydrogenase-Driven Phytoalexin Biosynthesis in Elicitor-Treated Pyrus Pyrifolia Cell Cultures. J. Plant Physiol. 2017, 215, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Weiß, S.; Liu, B.; Reckwell, D.; Beerhues, L.; Winkelmann, T. Impaired Defense Reactions in Apple Replant Disease-Affected Roots of Malus Domestica ‘M26’. Tree Physiol. 2017, 37, 1672–1685. [Google Scholar] [CrossRef]
- Sarkate, A.; Saini, S.S.; Gaid, M.; Teotia, D.; Mir, J.I.; Agrawal, P.K.; Beerhues, L.; Sircar, D. Molecular Cloning and Functional Analysis of a Biphenyl Phytoalexin-Specific O-Methyltransferase from Apple Cell Suspension Cultures. Planta 2019, 249, 677–691. [Google Scholar] [CrossRef]
- Teotia, D.; Gaid, M.; Saini, S.S.; Verma, A.; Yennamalli, R.M.; Khare, S.P.; Ambatipudi, K.; Mir, J.I.; Beuerle, T.; Hänsch, R.; et al. Cinnamate-CoA Ligase Is Involved in Biosynthesis of Benzoate-derived Biphenyl Phytoalexin in Malus × Domestica ‘Golden Delicious’ Cell Cultures. Plant J. 2019, 100, 1176–1192. [Google Scholar] [CrossRef] [PubMed]
- Teotia, D.; Saini, S.S.; Gaid, M.; Beuerle, T.; Beerhues, L.; Sircar, D. Development and Validation of a New HPLC Method for the Determination of Biphenyl and Dibenzofuran Phytoalexins in Rosaceae. J. Chromatogr. Sci. 2016, 54, 918–922. [Google Scholar] [CrossRef]
- Dai, L.; Yu, F.; Zhu, J.; Xu, J.; Chen, J.; Zhou, T.; Yu, B.; Han, X.-L. Structural Insights into the Binding Interaction between Noraucuparin and Bovine Serum Albumin: Implications for Antibiotic Design and Function. J. Mol. Struct. 2025, 1319, 139392. [Google Scholar] [CrossRef]
- Islamoğlu, F. Molecular Docking, Bioactivity, ADME, Toxicity Risks, and Quantum Mechanical Parameters of Some 1,2-Dihydroquinoline Derivatives Were Calculated Theoretically for Investigation of Its Use as a Pharmaceutical Active Ingredient in the Treatment of Multiple Sclerosis (MS). Prospect. Pharm. Sci. 2024, 22, 168–187. [Google Scholar] [CrossRef]
- Bojarski, K.K.; Donatan, S.; Koziara, K.; Samsonov, S.A. Molecular Dynamics Insights into Protein–Glycosaminoglycan Systems from Microsecond-Scale Simulations. Biopolymers 2019, 110, e23252. [Google Scholar] [CrossRef]
- Bhati, A.P.; Hoti, A.; Potterton, A.; Bieniek, M.K.; Coveney, P.V. Long Time Scale Ensemble Methods in Molecular Dynamics: Ligand–Protein Interactions and Allostery in SARS-CoV-2 Targets. J. Chem. Theory Comput. 2023, 19, 3359–3378. [Google Scholar] [CrossRef]
- Nury, H.; Poitevin, F.; Van Renterghem, C.; Changeux, J.P.; Delarue, M.; Corringer, P.J. One-Microsecond Molecular Dynamics Simulation of Channel Gating in a Nicotinic Receptor Homologue. Proc. Natl. Acad. Sci. USA 2010, 107, 6275–6280. [Google Scholar] [CrossRef]
- Cruz-León, S.; Arreguín-Espinosa, R.; Ibarra, J.A.; Becerra-Flores, G. Conformational Analysis and Druggability Predictions in Long MD Simulations of Viral Proteins. Food Funct. 2020, 11, 8060–8070. [Google Scholar]
- Cino, E.A.; Wong-Ekkabut, J.; Karttunen, M.; Choy, W.Y. Microsecond Molecular Dynamics Simulations of Intrinsically Disordered Proteins Involved in the Oxidative Stress Response. PLoS ONE 2011, 6, e27371. [Google Scholar] [CrossRef]
- Freddolino, P.L.; Harrison, C.B.; Liu, Y.; Schulten, K. Ten-Microsecond Molecular Dynamics Simulation of a Fast-Folding WW Domain. Biophys. J. 2008, 94, L75–L77. [Google Scholar] [CrossRef] [PubMed]
- Dyson, H.J.; Wright, P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005, 6, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Changeux, J.-P.; Edelstein, S.J. Allosteric mechanisms of signal transduction. Science 2005, 308, 1424–1428. [Google Scholar] [CrossRef]
- Chodera, J.D.; Mobley, D.L.; Shirts, M.R.; Dixon, R.W.; Branson, K.; Pande, V.S. Alchemical free energy methods for drug discovery: Progress and challenges. Curr. Opin. Struct. Biol. 2011, 21, 150–160. [Google Scholar] [CrossRef]
- Wang, W.; Donini, O.; Reyes, C.M.; Kollman, P.A. Biomolecular simulations: Recent developments in force fields, simulations of enzyme catalysis, protein–ligand, protein–protein, and protein–nucleic acid noncovalent interactions. Annu. Rev. Biophys. Biomol. Struct. 2001, 30, 211–243. [Google Scholar] [CrossRef]
- Bahena Culhuac, E.; Bello, M. Unveiling the Mechanisms of EGCG–P53 Interactions through Molecular Dynamics Simulations. ACS Omega 2024, 9, 20066–20085. [Google Scholar] [CrossRef]
- Del Conte, A.; Camagni, G.F.; Clementel, D.; Minervini, G.; Monzon, A.M.; Ferrari, C.; Piovesan, D.; Tosatto, S.C.E. RING 4.0: Faster Residue Interaction Networks with Novel Interaction Types across over 35,000 Different Chemical Structures. Nucleic Acids Res. 2024, 52, W306–W312. [Google Scholar] [CrossRef]
- Morrisett, J.D.; Pownall, H.J.; Gotto, A.M., Jr. Bovine Serum Albumin. Study of the Fatty Acid and Steroid Binding Sites Using Spin-Labeled Lipids. J. Biol. Chem. 1975, 250, 2487–2494. [Google Scholar] [CrossRef]
- Preininger, A.M.; Meiler, J.; Hamm, H.E. Conformational Flexibility and Structural Dynamics in GPCR-Mediated G Protein Activation: A Perspective. J. Mol. Biol. 2013, 425, 2288–2298. [Google Scholar] [CrossRef]
- Glasgow, A.; Hobbs, H.T.; Perry, Z.R.; Wells, M.L.; Marqusee, S.; Kortemme, T. Ligand-Specific Changes in Conformational Flexibility Mediate Long-Range Allostery in the Lac Repressor. Nat. Commun. 2023, 14, 1179. [Google Scholar] [CrossRef] [PubMed]
- Kumari, M.; Kumari, P.; Kashyap, H.K. Structural adaptations in the bovine serum albumin protein in archetypal deep eutectic solvent reline and its aqueous mixtures. Phys. Chem. Chem. Phys. 2022, 24, 5627–5637. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; He, H.; Wang, J.; Wang, J.; Chang, C.E.A. Uncovering water effects in protein–ligand recognition: Importance in the second hydration shell and binding kinetics. Phys. Chem. Chem. Phys. 2023, 25, 2098–2109. [Google Scholar] [CrossRef] [PubMed]
- Maffucci, I.; Contini, A. Explicit ligand hydration shells improve the correlation between MM-PB/GBSA binding energies and experimental activities. J. Chem. Theory Comput. 2013, 9, 2706–2717. [Google Scholar] [CrossRef]
- Qu, S.S.; Liu, Y.; Wang, T.Z.; Gao, W.Y. Thermodynamics of binding of cadmium to bovine serum albumin. Chemosphere 2002, 46, 1211–1214. [Google Scholar] [CrossRef]
- de Paula, H.M.C.; Coelho, Y.L.; Agudelo, A.J.P.; de Paula Rezende, J.; Ferreira, G.M.D.; Ferreira, G.M.D.; Pires, A.C.D.S.; da Silva, L.H.M. Kinetics and thermodynamics of bovine serum albumin interactions with Congo red dye. Colloids Surf. B Biointerfaces 2017, 159, 737–742. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology Modelling of Protein Structures and Complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Kumar, M.; Rathore, R.S. RamPlot: A Webserver to Draw 2D, 3D, and Assorted Ramachandran (φ, ψ) Maps. J. Appl. Crystallogr. 2025, 58, 630–636. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef]
- Grosdidier, A.; Zoete, V.; Michielin, O. SwissDock, a Protein-Small Molecule Docking Web Service Based on EADock DSS. Nucleic Acids Res. 2011, 39, W270–W277. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. The PyMOL Molecular Graphics System. 2002. Available online: http://www.pymol.org/ (accessed on 20 May 2024).
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Uberuaga, B.P.; Anghel, M.; Voter, A.F. Synchronization of Trajectories in Canonical Molecular-Dynamics Simulations: Observation, Explanation, and Exploitation. J. Chem. Phys. 2004, 120, 6363–6374. [Google Scholar] [CrossRef]
- Sindhikara, D.J.; Kim, S.; Voter, A.F.; Roitberg, A.E. Bad Seeds Sprout Perilous Dynamics: Stochastic Thermostat Induced Trajectory Synchronization in Biomolecules. J. Chem. Theory Comput. 2009, 5, 1624–1631. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: Particle Mesh Ewald: An N·log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber Biomolecular Simulation Programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef]
- Jendele, L.; Krivak, R.; Skoda, P.; Novotny, M.; Hoksza, D. PrankWeb: A Web Server for Ligand Binding Site Prediction and Visualization. Nucleic Acids Res. 2019, 47, W345–W349. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.Py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Bello, M.; Bandala, C. Evaluating the Ability of End-Point Methods to Predict the Binding Affinity Tendency of Protein Kinase Inhibitors. RSC Adv. 2023, 13, 25118–25128. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bahena-Culhuac, E.; Bello, M. Molecular Insights into the Temperature-Dependent Binding and Conformational Dynamics of Noraucuparin with Bovine Serum Albumin: A Microsecond-Scale MD Simulation Study. Pharmaceuticals 2025, 18, 1048. https://doi.org/10.3390/ph18071048
Bahena-Culhuac E, Bello M. Molecular Insights into the Temperature-Dependent Binding and Conformational Dynamics of Noraucuparin with Bovine Serum Albumin: A Microsecond-Scale MD Simulation Study. Pharmaceuticals. 2025; 18(7):1048. https://doi.org/10.3390/ph18071048
Chicago/Turabian StyleBahena-Culhuac, Erick, and Martiniano Bello. 2025. "Molecular Insights into the Temperature-Dependent Binding and Conformational Dynamics of Noraucuparin with Bovine Serum Albumin: A Microsecond-Scale MD Simulation Study" Pharmaceuticals 18, no. 7: 1048. https://doi.org/10.3390/ph18071048
APA StyleBahena-Culhuac, E., & Bello, M. (2025). Molecular Insights into the Temperature-Dependent Binding and Conformational Dynamics of Noraucuparin with Bovine Serum Albumin: A Microsecond-Scale MD Simulation Study. Pharmaceuticals, 18(7), 1048. https://doi.org/10.3390/ph18071048