
Tumor Niche Influences the Activity and Delivery of Anticancer Drugs: Pharmacology Meets Chemistry





Abstract
1. Introduction
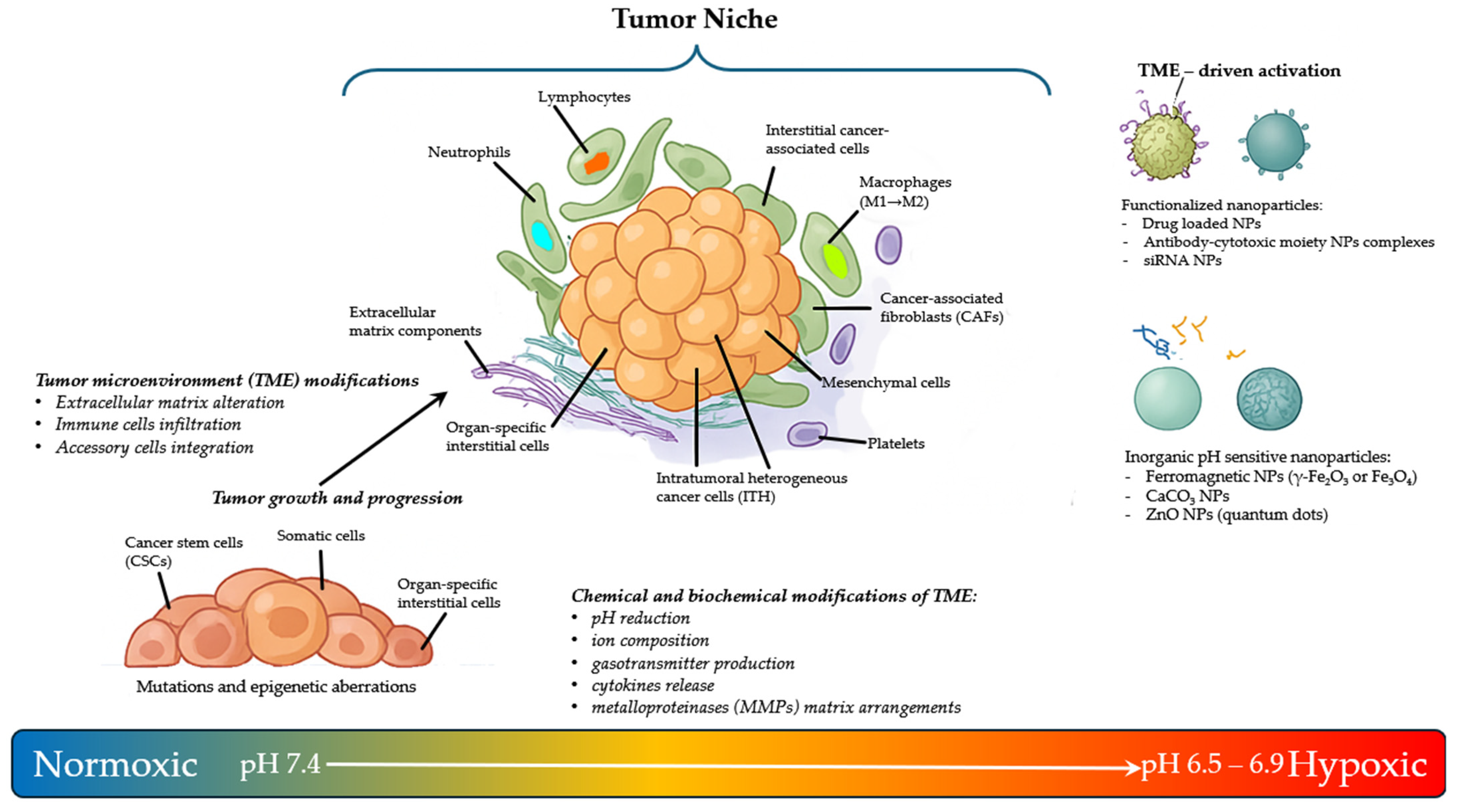
2. The Tumor Microenvironment Niche Complexity
3. The Niche Components for Integrated Pharmacological Cancer Treatments
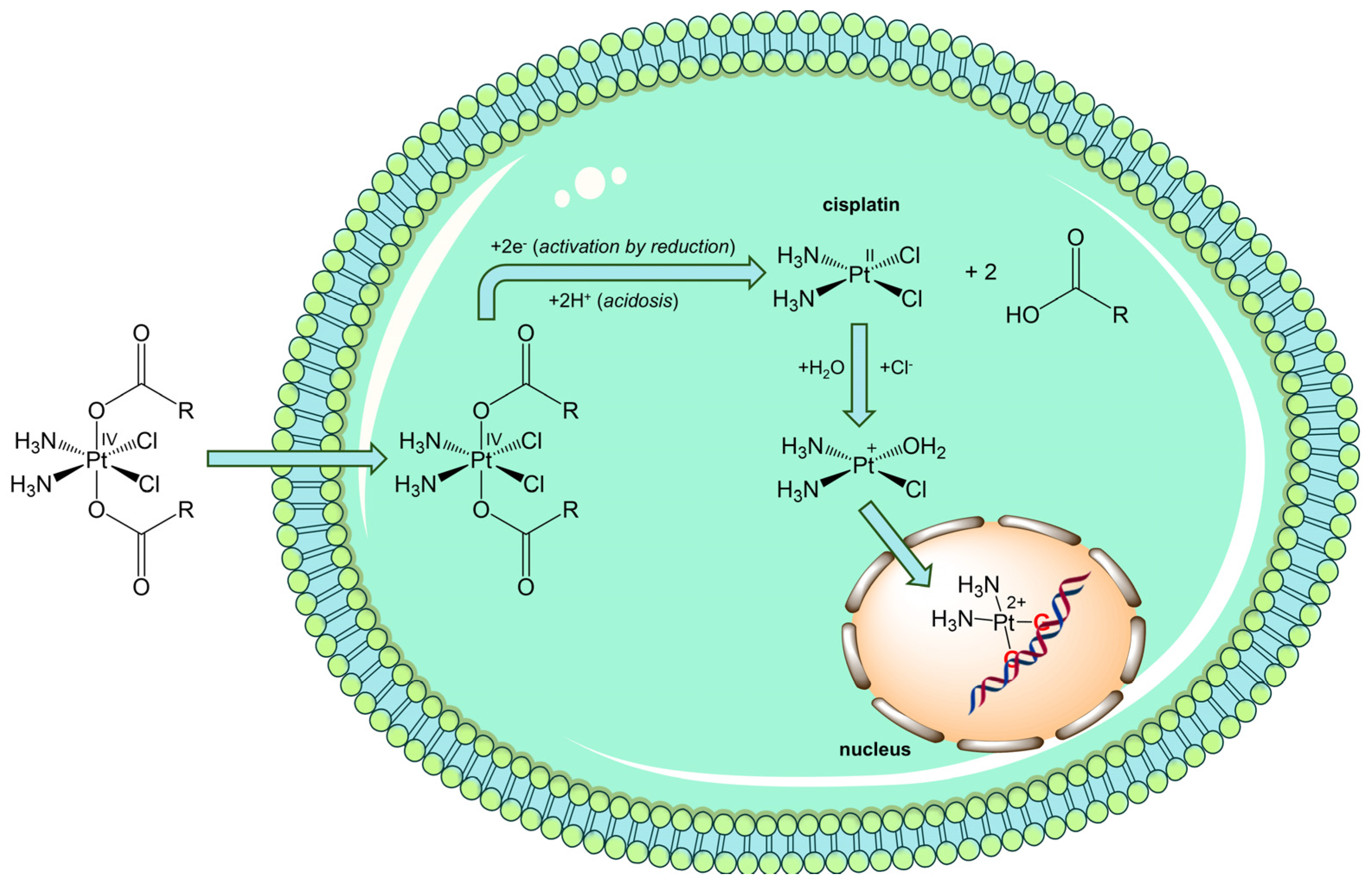
3.1. Exploiting the PH as Activator of Prodrugs
3.2. Exploiting the Hypoxia in the Niche as a Trigger for Activating Drugs
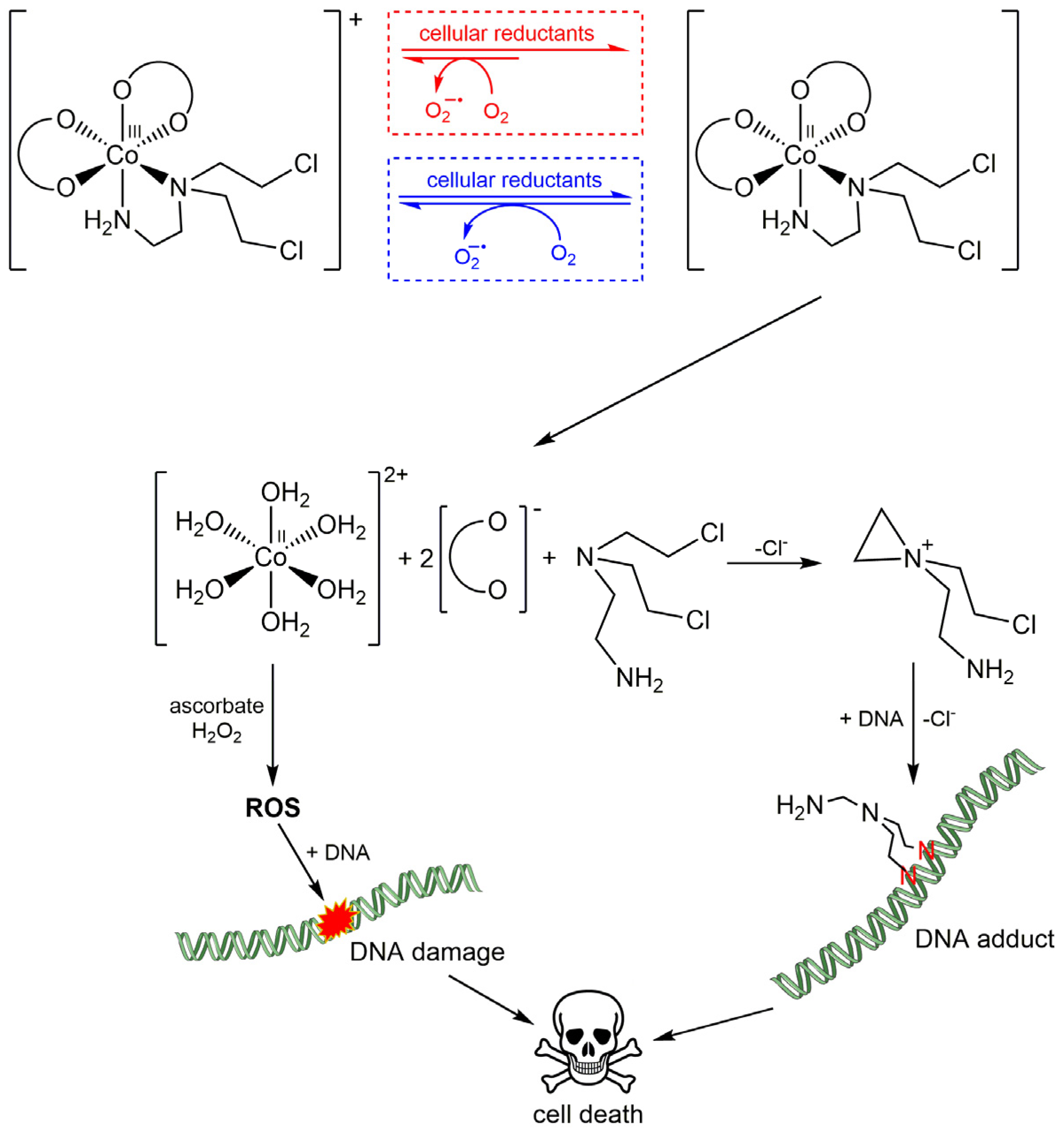
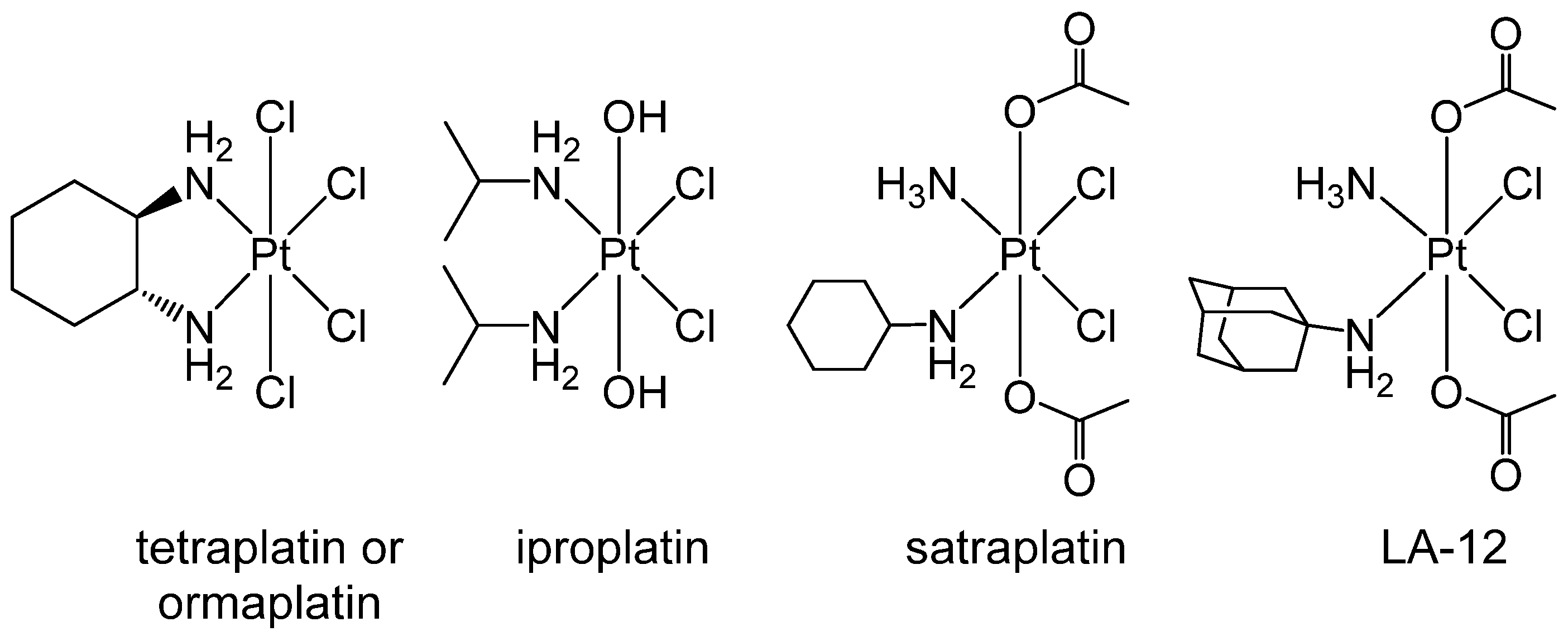
3.3. Exploiting the Potential of Metal-Based Drugs for Tumor Niche Modification
3.4. Can Gasotransmitters Disrupt Tme’s Support to Tumor Cells?
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AI | Artificial Intelligence |
| Akt | Protein Kinase B |
| ARNT | Aryl Hydrocarbon Receptor Nuclear Translocator |
| CORM | Carbon Monoxide-Releasing Molecules |
| DMSO | Dimethyl Sulfoxide |
| DOPE | 1,2-Dioleyl-sn-glycrro-3-phosphoethanolamine |
| FGF2 | Fibroblast Growth Factor 2 |
| GOD | Glucose Oxidase |
| HIF | Hypoxia-Inducible Factor |
| Hsp90 | Heat Shock Protein 90 |
| ICAM | Intercellular Adhesion Molecule |
| ITH | Intratumor Heterogeneity |
| MAPK | Mitogen-Activated Protein Kinase |
| MMP | Metalloprotease |
| mTOR | Mechanistic Target Of Rapamycin |
| NORM | Nitric Oxide-Releasing Molecules |
| NSAID | Non-Steroidal Anti-Inflammatory Drug |
| PDGF | Platelet-Derived Growth Factor |
| PE | Dioleoyl Phosphatidylethanolamine |
| PEG | Polyethylene Glycol |
| PEI | Polyethyleneimine |
| PI3K | Phosphatidylinositol 3-Kinase |
| ROS | Reactive Oxygen Species |
| TGFβ | Transforming Growth Factor Beta |
| TME | Tumor Microenvironment |
| VEGF | Vascular Endothelial Growth Factor |
References
- Wieke, J.; Jurcic, C.; Kaczorowski, A.; Böning, S.; Kirchner, M.; Schwab, C.; Schütz, V.; Hohenfellner, M.; Duensing, A.; Stenzinger, A.; et al. Extensive Genotype-Phenotype Heterogeneity in Renal Cell Carcinoma—A Proof-of-Concept Study. Front. Oncol. 2025, 15, 1551077. [Google Scholar] [CrossRef] [PubMed]
- Borchert, F.; Wullenweber, P.; Oeser, A.; Kreuzberger, N.; Karge, T.; Langer, T.; Skoetz, N.; Wieler, L.H.; Schapranow, M.-P.; Arnrich, B. High-Precision Information Retrieval for Rapid Clinical Guideline Updates. Npj Digit. Med. 2025, 8, 227. [Google Scholar] [CrossRef] [PubMed]
- Butt, R.N.; Amina, B.; Sultan, M.U.; Tanveer, Z.B.; Gondal, M.N.; Hussain, R.; Khan, S.; Akbar, R.; Nasir, Z.; Khalid, M.F.; et al. CanSeer: A Translational Methodology for Developing Personalized Cancer Models and Therapeutics. Sci. Rep. 2025, 15, 15080. [Google Scholar] [PubMed]
- Galeș, L.N.; Păun, M.-A.; Butnariu, I.; Simion, L.; Manolescu, L.S.C.; Trifănescu, O.G.; Anghel, R.M. Next-Generation Sequencing in Oncology—A Guiding Compass for Targeted Therapy and Emerging Applications. Int. J. Mol. Sci. 2025, 26, 3123. [Google Scholar] [CrossRef] [PubMed]
- Plaks, V.; Kong, N.; Werb, Z. The Cancer Stem Cell Niche: How Essential Is the Niche in Regulating Stemness of Tumor Cells? Cell Stem. Cell 2015, 16, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Lu, D.; Li, H.; Pan, S.; Zhuo, J.; Lin, Z.; He, C.; Zhang, P.; Ying, S.; Wei, X.; et al. Targeted Lung Premetastasis Niche: Mechanisms, Strategies, and Application. MedComm 2025, 6, e70248. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y. The Role of Tregs in the Tumor Microenvironment. Biomedicines. 2025, 13, 1173. [Google Scholar] [CrossRef] [PubMed]
- Giraldo, N.A.; Sanchez-Salas, R.; Peske, J.D.; Vano, Y.; Becht, E.; Petitprez, F.; Validire, P.; Ingels, A.; Cathelineau, X.; Fridman, W.H.; et al. The Clinical Role of the TME in Solid Cancer. Br. J. Cancer 2019, 120, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yang, K.; Zhang, H. Targeting Leukemia-Initiating Cells and Leukemic Niches: The Next Therapy Station for T-Cell Acute Lymphoblastic Leukemia? Cancers. 2022, 14, 5655. [Google Scholar] [CrossRef] [PubMed]
- Kolenda, T.; Przybyła, W.; Kapałczyńska, M.; Teresiak, A.; Zajączkowska, M.; Bliźniak, R.; Lamperska, K.M. Tumor Microenvironment—Unknown Niche with Powerful Therapeutic Potential. Rep. Pract. Oncol. Radiother. 2018, 23, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Hu, R.; Huang, Q.; Zhong, X.; Li, J.; Lv, M.; Yi, J.; Sun, J.; Fan, K.; Xu, Y.; et al. Decoding the Metabolic-Immune Axis: Neutrophil Glycolysis-Driven Tumor Niche Remodeling and Its Therapeutic Exploitation. Pharmacol. Res. 2025, 107811. [Google Scholar] [CrossRef] [PubMed]
- Jaguri, A.; Ahmad, A. Breaching the Fortress of Tumor Microenvironment to Control Cancer Metastasis. Cancers 2023, 15, 4562. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.; Ji, L.; Zhang, X.; Jing, S.; Li, P.; Wang, H.; Zhang, L.; Zhang, Y.; Yang, L.; Tian, J.; et al. Identification of a Novel Mesenchymal Stem Cell–Related Signature for Predicting the Prognosis and Therapeutic Responses of Bladder Cancer. Stem Cells Int. 2024, 2024, 6064671. [Google Scholar] [CrossRef] [PubMed]
- Siegler, E.L.; Kim, Y.J.; Wang, P. Nanomedicine Targeting the Tumor Microenvironment: Therapeutic Strategies to Inhibit Angiogenesis, Remodel Matrix, and Modulate Immune Responses. J. Cell. Immunother. 2016, 2, 69–78. [Google Scholar] [CrossRef]
- Chen, Y.; Yu, Y.; Fan, Y.; Lu, W.; Wei, Y.; Wu, J.; Ruan, B.; Wan, Z.; Zhao, Y.; Xie, K.; et al. Biomimetic Nanoplatforms for Combined DDR2 Inhibition and Photothermal Therapy in Dense Breast Cancer Treatment. Biomaterials 2025, 324, 123497. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Lin, X.; Ying, Y.; Fan, G.; Shi, J.; Zheng, X.; Hu, B.; Che, H.; Chen, H.; Yang, W.; et al. A Dual-Targeting Strategy to Inhibit Colorectal Cancer Liver Metastasis via Tumor Cell Ferroptosis and Cancer-Associated Fibroblast Reprogramming. Bioact. Mater. 2025, 52, 73–91. [Google Scholar] [CrossRef] [PubMed]
- Marks, D.L.; Olson, R.L.; Fernandez-Zapico, M.E. Epigenetic Control of the Tumor Microenvironment. Epigenomics 2016, 8, 1671–1687. [Google Scholar] [CrossRef] [PubMed]
- Dratkiewicz, E.; Simiczyjew, A.; Mazurkiewicz, J.; Ziętek, M.; Matkowski, R.; Nowak, D. Hypoxia and Extracellular Acidification as Drivers of Melanoma Progression and Drug Resistance. Cells 2021, 10, 862. [Google Scholar] [CrossRef] [PubMed]
- Vander Linden, C.; Corbet, C. Therapeutic Targeting of Cancer Stem Cells: Integrating and Exploiting the Acidic Niche. Front. Oncol. 2019, 9, 159. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-T.; Jin, W.-L.; Li, X. Intercellular Communication in the Tumour Microecosystem: Mediators and Therapeutic Approaches for Hepatocellular Carcinoma. Biochim. Biophys. Acta Mol. Basis. Dis. 2022, 1868, 166528. [Google Scholar] [CrossRef] [PubMed]
- Kather, J.N. Artificial Intelligence in Oncology: Chances and Pitfalls. J. Cancer Res. Clin. Oncol 2023, 149, 7995–7996. [Google Scholar] [CrossRef] [PubMed]
- Reitsam, N.G.; Enke, J.S.; Vu Trung, K.; Märkl, B.; Kather, J.N. Artificial Intelligence in Colorectal Cancer: From Patient Screening over Tailoring Treatment Decisions to Identification of Novel Biomarkers. Digestion 2024, 105, 331–344. [Google Scholar] [CrossRef] [PubMed]
- Rigamonti, A.; Viatore, M.; Polidori, R.; Rahal, D.; Erreni, M.; Fumagalli, M.R.; Zanini, D.; Doni, A.; Putignano, A.R.; Bossi, P.; et al. Integrating AI-Powered Digital Pathology and Imaging Mass Cytometry Identifies Key Classifiers of Tumor Cells, Stroma, and Immune Cells in Non–Small Cell Lung Cancer. Cancer Res. 2024, 84, 1165–1177. [Google Scholar] [CrossRef] [PubMed]
- Casanova-Acebes, M.; Dalla, E.; Leader, A.M.; LeBerichel, J.; Nikolic, J.; Morales, B.M.; Brown, M.; Chang, C.; Troncoso, L.; Chen, S.T.; et al. Tissue-Resident Macrophages Provide a pro-Tumorigenic Niche to Early NSCLC Cells. Nature 2021, 595, 578–584. [Google Scholar] [CrossRef] [PubMed]
- Rab, S.O.; Altalbawy, F.M.A.; Bishoyi, A.K.; Ballal, S.; Kareem, M.; Singh, A.; Kubaev, A.; Soleimani Samarkhazan, H.; Bagheri, S. Targeting Platelet-Tumor Cell Interactions: A Novel Approach to Cancer Therapy. Med. Oncol. 2025, 42, 232. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.M.; Simon, M.C. The Tumor Microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef] [PubMed]
- Baldominos, P.; Barbera-Mourelle, A.; Barreiro, O.; Huang, Y.; Wight, A.; Cho, J.-W.; Zhao, X.; Estivill, G.; Adam, I.; Sanchez, X.; et al. Quiescent Cancer Cells Resist T Cell Attack by Forming an Immunosuppressive Niche. Cell 2022, 185, 1694–1708.e19. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Ferrer, S.; Bonnet, D.; Steensma, D.P.; Hasserjian, R.P.; Ghobrial, I.M.; Gribben, J.G.; Andreeff, M.; Krause, D.S. Bone Marrow Niches in Haematological Malignancies. Nat. Rev. Cancer 2020, 20, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Eble, J.A.; Niland, S. The Extracellular Matrix in Tumor Progression and Metastasis. Clin. Exp. Metastasis 2019, 36, 171–198. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Weaver, V.M.; Werb, Z. The Extracellular Matrix: A Dynamic Niche in Cancer Progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Patras, L.; Shaashua, L.; Matei, I.; Lyden, D. Immune Determinants of the Pre-Metastatic Niche. Cancer Cell 2023, 41, 546–572. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Qin, C.; Dewanjee, S.; Bhattacharya, H.; Chakraborty, P.; Jha, N.K.; Gangopadhyay, M.; Jha, S.K.; Liu, Q. Tumor-Derived Small Extracellular Vesicles in Cancer Invasion and Metastasis: Molecular Mechanisms, and Clinical Significance. Mol. Cancer 2024, 23, 18. [Google Scholar] [CrossRef] [PubMed]
- Dagogo-Jack, I.; Shaw, A.T. Tumour Heterogeneity and Resistance to Cancer Therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Grzes, M.; Oron, M.; Staszczak, Z.; Jaiswar, A.; Nowak-Niezgoda, M.; Walerych, D. A Driver Never Works Alone—Interplay Networks of Mutant P53, MYC, RAS, and Other Universal Oncogenic Drivers in Human Cancer. Cancers 2020, 12, 1532. [Google Scholar] [CrossRef] [PubMed]
- Panda, S.S.; Sahoo, R.K.; Patra, S.K.; Biswal, S.; Biswal, B.K. Molecular Insights to Therapeutic in Cancer: Role of Exosomes in Tumor Microenvironment, Metastatic Progression and Drug Resistance. Drug Discov. Today 2024, 29, 104061. [Google Scholar] [CrossRef] [PubMed]
- Stark, V.A.; Facey, C.O.B.; Viswanathan, V.; Boman, B.M. The Role of miRNAs, miRNA Clusters, and isomiRs in Development of Cancer Stem Cell Populations in Colorectal Cancer. Int. J. Mol. Sci. 2021, 22, 1424. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Zou, J.; Li, S.; Topper, M.J.; Tao, Y.; Zhang, H.; Jiao, X.; Xie, W.; Kong, X.; Vaz, M.; et al. Epigenetic Therapy Inhibits Metastases by Disrupting Premetastatic Niches. Nature 2020, 579, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Korkaya, H.; Liu, S.; Wicha, M.S. Breast Cancer Stem Cells, Cytokine Networks, and the Tumor Microenvironment. J. Clin. Investig. 2011, 121, 3804–3809. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jia, J.; Wang, F.; Fang, Y.; Yang, Y.; Zhou, Q.; Yuan, W.; Gu, X.; Hu, J.; Yang, S. Pre-Metastatic Niche: Formation, Characteristics and Therapeutic Implication. Signal Transduct. Target. Ther. 2024, 9, 236. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L. The Role of Mesenchymal Stem Cells in Modulating the Breast Cancer Microenvironment. Cell Transplant. 2023, 32, 09636897231220073. [Google Scholar] [CrossRef] [PubMed]
- Mittal, S.; Brown, N.J.; Holen, I. The Breast Tumor Microenvironment: Role in Cancer Development, Progression and Response to Therapy. Expert Rev. Mol. Diagn. 2018, 18, 227–243. [Google Scholar] [CrossRef] [PubMed]
- Paget, S. The distribution of secondary growths in cancer of the breast. The Lancet 1889, 133, 571–573. [Google Scholar] [CrossRef]
- Gomez, K.; Miura, S.; Huuki, L.A.; Spell, B.S.; Townsend, J.P.; Kumar, S. Somatic Evolutionary Timings of Driver Mutations. BMC Cancer 2018, 18, 85. [Google Scholar] [CrossRef] [PubMed]
- Salvadores, M.; Mas-Ponte, D.; Supek, F. Passenger Mutations Accurately Classify Human Tumors. PLoS Comput. Biol. 2019, 15, e1006953. [Google Scholar] [CrossRef] [PubMed]
- Muto, S.; Enta, A.; Maruya, Y.; Inomata, S.; Yamaguchi, H.; Mine, H.; Takagi, H.; Ozaki, Y.; Watanabe, M.; Inoue, T.; et al. Wnt/β-Catenin Signaling and Resistance to Immune Checkpoint Inhibitors: From Non-Small-Cell Lung Cancer to Other Cancers. Biomedicines 2023, 11, 190. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Xi, M.; Li, J. Cancer Metastasis: Molecular Mechanisms and Therapeutic Interventions. Mol. Biomed. 2025, 6, 20. [Google Scholar] [CrossRef] [PubMed]
- Wortzel, I.; Dror, S.; Kenific, C.M.; Lyden, D. Exosome-Mediated Metastasis: Communication from a Distance. Dev. Cell 2019, 49, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Sellner, F.; Compérat, E.; Klimpfinger, M. Genetic and Epigenetic Characteristics in Isolated Pancreatic Metastases of Clear-Cell Renal Cell Carcinoma. Int. J. Mol. Sci. 2023, 24, 16292. [Google Scholar] [CrossRef] [PubMed]
- Casey, J.R.; Grinstein, S.; Orlowski, J. Sensors and Regulators of Intracellular pH. Nat. Rev. Mol. Cell Biol. 2010, 11, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P.; Multhoff, G. Revisiting the Warburg Effect: Historical Dogma versus Current Understanding. J. Physiol. 2021, 599, 1745–1757. [Google Scholar] [CrossRef] [PubMed]
- Chiche, J.; Brahimi-Horn, M.C.; Pouysségur, J. Tumour Hypoxia Induces a Metabolic Shift Causing Acidosis: A Common Feature in Cancer. J. Cell. Mol. Med. 2010, 14, 771–794. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Ozawa, S.; Miyamoto, C.; Maehata, Y.; Suzuki, A.; Maeda, T.; Baba, Y. Acidic Extracellular Microenvironment and Cancer. Cancer Cell Int. 2013, 13, 89. [Google Scholar] [CrossRef] [PubMed]
- Damaghi, M.; Wojtkowiak, J.W.; Gillies, R.J. pH Sensing and Regulation in Cancer. Front. Physiol. 2013, 4, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Corbet, C.; Feron, O. Tumour Acidosis: From the Passenger to the Driver’s Seat. Nat. Rev. Cancer 2017, 17, 577–593. [Google Scholar] [CrossRef] [PubMed]
- Boedtkjer, E.; Pedersen, S.F. The Acidic Tumor Microenvironment as a Driver of Cancer. Annu. Rev. Physiol. 2020, 82, 103–126. [Google Scholar] [CrossRef] [PubMed]
- Swietach, P.; Vaughan-Jones, R.D.; Harris, A.L.; Hulikova, A. The Chemistry, Physiology and Pathology of pH in Cancer. Phil. Trans. R. Soc. B 2014, 369, 20130099. [Google Scholar] [CrossRef] [PubMed]
- Mann, M.; Kumar, S.; Sharma, A.; Chauhan, S.S.; Bhatla, N.; Kumar, S.; Bakhshi, S.; Gupta, R.; Kumar, L. PARP-1 Inhibitor Modulate β-Catenin Signaling to Enhance Cisplatin Sensitivity in Cancer Cervix. Oncotarget 2019, 10, 4262–4275. [Google Scholar] [CrossRef] [PubMed]
- Wojtkowiak, J.W.; Verduzco, D.; Schramm, K.J.; Gillies, R.J. Drug Resistance and Cellular Adaptation to Tumor Acidic pH Microenvironment. Mol. Pharm. 2011, 8, 2032–2038. [Google Scholar] [CrossRef] [PubMed]
- Pathak, L.; Pal, B.; Talukdar, J.; Saikia, P.J.; Sandhya, S.; Tasabehji, W.; Li, H.; Phukan, J.; Bhuyan, A.; Patra, S.; et al. Hypoxia-Driven Mobilization of Altruistic Cancer Stem Cells in Platinum-Treated Head and Neck Cancer. Front. Immunol. 2025, 15, 1336882. [Google Scholar] [CrossRef] [PubMed]
- Edvall, C.; Kale, N.; Tani, S.; Ambhore, S.; Hossain, R.; Ozoude, C.; Van Horsen, K.; Mohammad, J.; Tuvin, D.M.; Kalathingal, S.; et al. Hypoxia-Responsive Polymersomes for Stemness Reduction in Patient-Derived Solid Tumor Spheroids. ACS Appl. Bio Mater. 2025, 8, 2916–2926. [Google Scholar] [CrossRef] [PubMed]
- Prankerd, R.J. Critical Compilation of pKa Values for Pharmaceutical Substances. Profiles Drug Subst. Excip. Relat. Methodol. 2007, 33, 1–33. [Google Scholar] [PubMed]
- Webb, B.A.; Chimenti, M.; Jacobson, M.P.; Barber, D.L. Dysregulated pH: A Perfect Storm for Cancer Progression. Nat. Rev. Cancer 2011, 11, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.; Spugnini, E.P.; Assaraf, Y.G.; Azzarito, T.; Rauch, C.; Fais, S. Microenvironment Acidity as a Major Determinant of Tumor Chemoresistance: Proton Pump Inhibitors (PPIs) as a Novel Therapeutic Approach. Drug Resist. Updates 2015, 23, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Anemone, A.; Consolino, L.; Arena, F.; Capozza, M.; Longo, D.L. Imaging Tumor Acidosis: A Survey of the Available Techniques for Mapping in Vivo Tumor pH. Cancer Metastasis Rev. 2019, 38, 25–49. [Google Scholar] [CrossRef] [PubMed]
- Palanisamy, B.; Mandal, A.K.A. Unlocking the Potential: Receptor-Mediated Targeted Drug Delivery in Cancer Therapy. Pathol. Res. Pract. 2025, 270, 155955. [Google Scholar] [CrossRef] [PubMed]
- García Rubia, G.; Peigneux, A.; Jabalera, Y.; Puerma, J.; Oltolina, F.; Elert, K.; Colangelo, D.; Gómez Morales, J.; Prat, M.; Jimenez-Lopez, C. pH-Dependent Adsorption Release of Doxorubicin on MamC-Biomimetic Magnetite Nanoparticles. Langmuir 2018, 34, 13713–13724. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Ruiz, I.; Delgado-López, J.M.; Durán-Olivencia, M.A.; Iafisco, M.; Tampieri, A.; Colangelo, D.; Prat, M.; Gómez-Morales, J. pH-Responsive Delivery of Doxorubicin from Citrate–Apatite Nanocrystals with Tailored Carbonate Content. Langmuir 2013, 29, 8213–8221. [Google Scholar] [CrossRef] [PubMed]
- Cano Plá, S.M.; D’Urso, A.; Fernández-Sánchez, J.F.; Colangelo, D.; Choquesillo-Lazarte, D.; Ferracini, R.; Bosetti, M.; Prat, M.; Gómez-Morales, J. Biomimetic Citrate-Coated Luminescent Apatite Nanoplatforms for Diclofenac Delivery in Inflammatory Environments. Nanomaterials 2022, 12, 562. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Qin, C.; Wang, X.; Shen, C.; Li, S.; Liu, T.; Li, W.; Chen, Z.; Wang, Y.; Liu, L.; et al. Hybrid Prodrug Nanoassembly for Hypoxia-Triggered Immunogenic Chemotherapy and Immune Modulation. J. Control. Release 2025, 379, 221–235. [Google Scholar] [CrossRef] [PubMed]
- Huai, Y.; Hossen, M.N.; Wilhelm, S.; Bhattacharya, R.; Mukherjee, P. Nanoparticle Interactions with the Tumor Microenvironment. Bioconjug. Chem. 2019, 30, 2247–2263. [Google Scholar] [CrossRef] [PubMed]
- Huo, M.; Wang, L.; Chen, Y.; Shi, J. Tumor-Selective Catalytic Nanomedicine by Nanocatalyst Delivery. Nat. Commun 2017, 8, 357. [Google Scholar] [CrossRef] [PubMed]
- D’Urso, A.; Oltolina, F.; Borsotti, C.; Prat, M.; Colangelo, D.; Follenzi, A. Macrophage Reprogramming via the Modulation of Unfolded Protein Response with siRNA-Loaded Magnetic Nanoparticles in a TAM-like Experimental Model. Pharmaceutics 2023, 15, 1711. [Google Scholar] [CrossRef] [PubMed]
- Maleki Dizaj, S.; Barzegar-Jalali, M.; Zarrintan, M.H.; Adibkia, K.; Lotfipour, F. Calcium Carbonate Nanoparticles as Cancer Drug Delivery System. Expert Opin. Drug Deliv. 2015, 12, 1649–1660. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wang, R.; Chen, L. Drug Delivery System Targeting Cancer-Associated Fibroblast for Improving Immunotherapy. Int. J. Nanomed. 2025, 20, 483–503. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Luo, Y.; Zhang, W.; Du, D.; Lin, Y. pH-Sensitive ZnO Quantum Dots–Doxorubicin Nanoparticles for Lung Cancer Targeted Drug Delivery. ACS Appl. Mater. Interfaces 2016, 8, 22442–22450. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhang, F.; Su, Y.; Zhou, J.; Wang, W. Nanoparticles designed to regulate tumor microenvironment for cancer therapy. Life Sci. 2018, 201, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Torchilin, V.P. Fundamentals of Stimuli-Responsive Drug and Gene Delivery Systems. In Stimuli-Responsive Drug Delivery Systems; Singh, A., Amiji, M.M., Eds.; The Royal Society of Chemistry: London, UK, 2018; pp. 1–32. [Google Scholar]
- Prabaharan, M.; Grailer, J.J.; Pilla, S.; Steeber, D.A.; Gong, S. Amphiphilic Multi-Arm-Block Copolymer Conjugated with Doxorubicin via pH-Sensitive Hydrazone Bond for Tumor-Targeted Drug Delivery. Biomaterials 2009, 30, 5757–5766. [Google Scholar] [CrossRef] [PubMed]
- Ganta, S.; Devalapally, H.; Shahiwala, A.; Amiji, M. A Review of Stimuli-Responsive Nanocarriers for Drug and Gene Delivery. J. Control. Release 2008, 126, 187–204. [Google Scholar] [CrossRef] [PubMed]
- Sawant, R.R.; Sriraman, S.K.; Navarro, G.; Biswas, S.; Dalvi, R.A.; Torchilin, V.P. Polyethyleneimine-Lipid Conjugate-Based pH-Sensitive Micellar Carrier for Gene Delivery. Biomaterials 2012, 33, 3942–3951. [Google Scholar] [CrossRef] [PubMed]
- Fan, R.; Cai, L.; Liu, H.; Chen, H.; Chen, C.; Guo, G.; Xu, J. Enhancing Metformin-Induced Tumor Metabolism Destruction by Glucose Oxidase for Triple-Combination Therapy. J. Pharm Anal. 2024, 14, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Ren, Z.; Zhang, Z.; Yang, K.; Jin, Y.; Deng, H.; Liu, Y.; Wang, J.; Ji, P.; Liu, P. Triple Synergistic Enhancement of Breast Cancer Treatment via Chemotherapy, Chemodynamic Therapy, and Tumor Starvation Therapy Driven by Lipid-COF Nanoparticles. Sci. Rep. 2025, 15, 9791. [Google Scholar] [CrossRef] [PubMed]
- Paredes, F.; Williams, H.C.; San Martin, A. Metabolic Adaptation in Hypoxia and Cancer. Cancer Lett. 2021, 502, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Xiao, C.; Zhou, R.; Yao, X.; Chen, Q.; Xu, T.; Cao, F.; Wang, Y.; Li, X.; Yan, O.; et al. Inhibiting Autophagy Selectively Prunes Dysfunctional Tumor Vessels and Optimizes the Tumor Immune Microenvironment. Theranostics 2025, 15, 258–276. [Google Scholar] [CrossRef] [PubMed]
- McKeown, S.R. Defining Normoxia, Physoxia and Hypoxia in Tumours—Implications for Treatment Response. Br. J. Radiol. 2014, 87, 20130676. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Defining the Role of Hypoxia-Inducible Factor 1 in Cancer Biology and Therapeutics. Oncogene 2010, 29, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Gilkes, D.M.; Semenza, G.L.; Wirtz, D. Hypoxia and the Extracellular Matrix: Drivers of Tumour Metastasis. Nat. Rev. Cancer 2014, 14, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-Inducible Factors in Physiology and Medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-Inducible Factors: Mediators of Cancer Progression and Targets for Cancer Therapy. Trends Pharmacol. Sci. 2012, 33, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Unwith, S.; Zhao, H.; Hennah, L.; Ma, D. The Potential Role of HIF on Tumour Progression and Dissemination. Intl. J. Cancer 2015, 136, 2491–2503. [Google Scholar] [CrossRef] [PubMed]
- Schito, L.; Semenza, G.L. Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. Trends Cancer 2016, 2, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Petrova, V.; Annicchiarico-Petruzzelli, M.; Melino, G.; Amelio, I. The Hypoxic Tumour Microenvironment. Oncogenesis 2018, 7, 10. [Google Scholar] [CrossRef] [PubMed]
- Jing, X.; Yang, F.; Shao, C.; Wei, K.; Xie, M.; Shen, H.; Shu, Y. Role of Hypoxia in Cancer Therapy by Regulating the Tumor Microenvironment. Mol. Cancer 2019, 18, 157. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M.; Wilson, W.R. Exploiting Tumour Hypoxia in Cancer Treatment. Nat. Rev. Cancer 2004, 4, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Rohwer, N.; Cramer, T. Hypoxia-Mediated Drug Resistance: Novel Insights on the Functional Interaction of HIFs and Cell Death Pathways. Drug Resist. Updates 2011, 14, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Albadari, N.; Deng, S.; Li, W. The Transcriptional Factors HIF-1 and HIF-2 and Their Novel Inhibitors in Cancer Therapy. Expert Opin. Drug Discov. 2019, 14, 667–682. [Google Scholar] [CrossRef] [PubMed]
- Bertout, J.A.; Patel, S.A.; Simon, M.C. The Impact of O2 Availability on Human Cancer. Nat. Rev. Cancer 2008, 8, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Thews, O.; Gassner, B.; Kelleher, D.K.; Schwerdt, G.; Gekle, M. Impact Of Hypoxic And Acidic Extracellular Conditions On Cytotoxicity Of Chemotherapeutic Drugs. In Oxygen Transport to Tissue XXVIII; Maguire, D.J., Bruley, D.F., Harrison, D.K., Eds.; Advances in Experimental Medicine and Biology; Springer: Boston, MA, USA, 2007; Volume 599, pp. 155–161. [Google Scholar]
- Tang, Z.; Luo, W.; Xu, M.; Liu, Y.; Yu, Q.; Wang, L. A TME-Responsive Oxygen-Self-Supplying Hybridized Polymersome for Synergistic Triple-Modal Therapy and Precision Theranostics in Hypoxic Tumors. J. Mater. Chem. B. 2025, 13, 8136–8148. [Google Scholar] [CrossRef] [PubMed]
- Mori, Y.; Okimoto, Y.; Sakai, H.; Kanda, Y.; Ohata, H.; Shiokawa, D.; Suzuki, M.; Yoshida, H.; Ueda, H.; Sekizuka, T.; et al. Targeting PDGF Signaling of Cancer-Associated Fibroblasts Blocks Feedback Activation of HIF-1α and Tumor Progression of Clear Cell Ovarian Cancer. Cell Rep. Med. 2024, 5, 101532. [Google Scholar] [CrossRef] [PubMed]
- Hajizadeh, F.; Okoye, I.; Esmaily, M.; Ghasemi Chaleshtari, M.; Masjedi, A.; Azizi, G.; Irandoust, M.; Ghalamfarsa, G.; Jadidi-Niaragh, F. Hypoxia Inducible Factors in the Tumor Microenvironment as Therapeutic Targets of Cancer Stem Cells. Life Sci. 2019, 237, 116952. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Gonzalez, J.A.; Russell, J.; Rouzaut, A.; Gil-Bazo, I.; Montuenga, L. Targeting Hypoxia and Angiogenesis through HIF-1alpha Inhibition. Cancer Biol. Ther. 2005, 4, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.-S.; Zheng, J.Z.; Reed, E.; Jiang, B.-H. SU5416 Inhibited VEGF and HIF-1α Expression through the PI3K/AKT/p70S6K1 Signaling Pathway. Biochem. Biophys. Res. Commun. 2004, 324, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Egawa, N.; Tanaka, T.; Matsufuji, S.; Yamada, K.; Ito, K.; Kitagawa, H.; Okuyama, K.; Kitajima, Y.; Noshiro, H. Antitumor Effects of Low-dose Tipifarnib on the mTOR Signaling Pathway and Reactive Oxygen Species Production in HIF-1α-expressing Gastric Cancer Cells. FEBS Open Bio 2021, 11, 1465–1475. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Chen, W.; Wei, L.; Su, Y.; Liang, J.; Lian, H.; Wang, H.; Long, F.; Yang, F.; Gao, S.; et al. Lonafarnib Inhibits Farnesyltransferase via Suppressing ERK Signaling Pathway to Prevent Osteoclastogenesis in Titanium Particle-Induced Osteolysis. Front. Pharmacol. 2022, 13, 848152. [Google Scholar] [CrossRef] [PubMed]
- Minet, E.; Mottet, D.; Michel, G.; Roland, I.; Raes, M.; Remacle, J.; Michiels, C. Hypoxia-induced Activation of HIF-1: Role of HIF-1α-Hsp90 Interaction. FEBS Lett. 1999, 460, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Serwetnyk, M.A.; Blagg, B.S.J. The Disruption of Protein−protein Interactions with Co-Chaperones and Client Substrates as a Strategy towards Hsp90 Inhibition. Acta. Pharm. Sin. B 2021, 11, 1446–1468. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhu, M.; Dong, R.; Cao, D.; Li, Y.; Chen, Z.; Cai, J.; Zuo, X. TH-302-Loaded Nanodrug Reshapes the Hypoxic Tumour Microenvironment and Enhances PD-1 Blockade Efficacy in Gastric Cancer. J. Nanobiotechnol. 2023, 21, 440. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Liang, J.; Zhang, D.; Xu, X.; Cheng, Q.; Xiao, Z.; Shi, C.; Luo, L. Monitoring Treatment Efficacy of Antiangiogenic Therapy Combined With Hypoxia-Activated Prodrugs Online Using Functional MRI. Front. Oncol. 2021, 11, 672047. [Google Scholar] [CrossRef] [PubMed]
- Perche, F.; Biswas, S.; Wang, T.; Zhu, L.; Torchilin, V.P. Hypoxia-Targeted siRNA Delivery. Angew. Chem. Int. Ed. 2014, 53, 3362–3366. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Ji, J.; Liu, Z. Multifunctional MnO2 Nanoparticles for Tumor Microenvironment Modulation and Cancer Therapy. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2021, 13, e1720. [Google Scholar] [CrossRef] [PubMed]
- Gordijo, C.R.; Abbasi, A.Z.; Amini, M.A.; Lip, H.Y.; Maeda, A.; Cai, P.; O’Brien, P.J.; DaCosta, R.S.; Rauth, A.M.; Wu, X.Y. Design of Hybrid MnO2-Polymer-Lipid Nanoparticles with Tunable Oxygen Generation Rates and Tumor Accumulation for Cancer Treatment. Adv. Funct. Mater. 2015, 25, 1858–1872. [Google Scholar] [CrossRef]
- Voskuil, F.J.; Steinkamp, P.J.; Zhao, T.; Van Der Vegt, B.; Koller, M.; Doff, J.J.; Jayalakshmi, Y.; Hartung, J.P.; Gao, J.; Sumer, B.D.; et al. Exploiting Metabolic Acidosis in Solid Cancers Using a Tumor-Agnostic pH-Activatable Nanoprobe for Fluorescence-Guided Surgery. Nat. Commun. 2020, 11, 3257. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Dirisala, A.; Ge, Z.; Wang, Y.; Yin, W.; Ke, W.; Toh, K.; Xie, J.; Matsumoto, Y.; Anraku, Y.; et al. Therapeutic Vesicular Nanoreactors with Tumor-Specific Activation and Self-Destruction for Synergistic Tumor Ablation. Angew. Chem. Int. Ed. 2017, 56, 14025–14030. [Google Scholar] [CrossRef] [PubMed]
- Jungwirth, U.; Kowol, C.R.; Keppler, B.K.; Hartinger, C.G.; Berger, W.; Heffeter, P. Anticancer Activity of Metal Complexes: Involvement of Redox Processes. Antioxid. Redox. Signal. 2011, 15, 1085–1127. [Google Scholar] [CrossRef] [PubMed]
- Graf, N.; Lippard, S.J. Redox Activation of Metal-Based Prodrugs as a Strategy for Drug Delivery. Adv. Drug Deliv Rev. 2012, 64, 993–1004. [Google Scholar] [CrossRef] [PubMed]
- Zeng, G.; Mao, J.; Xing, H.; Xu, Z.; Cao, Z.; Kang, Y.; Liu, G.; Xue, P. Gold Nanodots-Anchored Cobalt Ferrite Nanoflowers as Versatile Tumor Microenvironment Modulators for Reinforced Redox Dyshomeostasis. Adv. Sci. 2024, 2406683. [Google Scholar] [CrossRef] [PubMed]
- Karmakar, S.; Maji, M.; Mukherjee, A. Modulation of the Reactivity of Nitrogen Mustards by Metal Complexation: Approaches to Modify Their Therapeutic Properties. Dalton. Trans. 2019, 48, 1144–1160. [Google Scholar] [CrossRef] [PubMed]
- Wexselblatt, E.; Gibson, D. What Do We Know about the Reduction of Pt(IV) pro-Drugs? J. Inorg. Biochem. 2012, 117, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Gibson, D. Platinum(iv) Anticancer Prodrugs—Hypotheses and Facts. Dalton. Trans. 2016, 45, 12983–12991. [Google Scholar] [CrossRef] [PubMed]
- Ravera, M.; Gabano, E.; McGlinchey, M.J.; Osella, D. Pt(IV) Antitumor Prodrugs: Dogmas, Paradigms, and Realities. Dalton. Trans. 2022, 51, 2121–2134. [Google Scholar] [CrossRef] [PubMed]
- Pia Rigobello, M.; Messori, L.; Marcon, G.; Agostina Cinellu, M.; Bragadin, M.; Folda, A.; Scutari, G.; Bindoli, A. Gold Complexes Inhibit Mitochondrial Thioredoxin Reductase: Consequences on Mitochondrial Functions. J. Inorg. Biochem. 2004, 98, 1634–1641. [Google Scholar] [CrossRef] [PubMed]
- Prast-Nielsen, S.; Cebula, M.; Pader, I.; Arnér, E.S.J. Noble Metal Targeting of Thioredoxin Reductase—Covalent Complexes with Thioredoxin and Thioredoxin-Related Protein of 14 kDa Triggered by Cisplatin. Free Radic. Biol. Med. 2010, 49, 1765–1778. [Google Scholar] [CrossRef] [PubMed]
- Navas, F.; Chocarro-Calvo, A.; Iglesias-Hernández, P.; Fernández-García, P.; Morales, V.; García-Martínez, J.M.; Sanz, R.; De La Vieja, A.; García-Jiménez, C.; García-Muñoz, R.A. Promising Anticancer Prodrugs Based on Pt(IV) Complexes with Bis-Organosilane Ligands in Axial Positions. J. Med. Chem. 2024, 67, 6410–6424. [Google Scholar] [CrossRef] [PubMed]
- Liaw, B.C.; Tsao, C.-K.; Seng, S.; Jun, T.; Gong, Y.; Galsky, M.D.; Oh, W.K. Biomarker Development Trial of Satraplatin in Patients with Metastatic Castration-Resistant Prostate Cancer. Oncologist 2023, 28, 366-e224. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkova, D.; Ivanova, S. Application of Approved Cisplatin Derivatives in Combination Therapy against Different Cancer Diseases. Molecules 2022, 27, 2466. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Guo, Z. Targeting and Delivery of Platinum-Based Anticancer Drugs. Chem. Soc. Rev. 2013, 42, 202–224. [Google Scholar] [CrossRef] [PubMed]
- Butler, J.S.; Sadler, P.J. Targeted Delivery of Platinum-Based Anticancer Complexes. Curr. Opin. Chem. Biol. 2013, 17, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Gabano, E.; Ravera, M.; Osella, D. Pros and Cons of Bifunctional Platinum(Iv) Antitumor Prodrugs: Two Are (Not Always) Better than One. Dalton. Trans. 2014, 43, 9813. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, T.C.; Suntharalingam, K.; Lippard, S.J. The Next Generation of Platinum Drugs: Targeted Pt(II) Agents, Nanoparticle Delivery, and Pt(IV) Prodrugs. Chem. Rev. 2016, 116, 3436–3486. [Google Scholar] [CrossRef] [PubMed]
- Kenny, R.G.; Chuah, S.W.; Crawford, A.; Marmion, C.J. Platinum(IV) Prodrugs—A Step Closer to Ehrlich’s Vision? Eur. J. Inorg. Chem. 2017, 2017, 1596–1612. [Google Scholar] [CrossRef]
- Kenny, R.G.; Marmion, C.J. Toward Multi-Targeted Platinum and Ruthenium Drugs—A New Paradigm in Cancer Drug Treatment Regimens? Chem. Rev. 2019, 119, 1058–1137. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Deng, Z.; Zhu, G. Emerging Platinum(IV) Prodrugs to Combat Cisplatin Resistance: From Isolated Cancer Cells to Tumor Microenvironment. Dalton. Trans. 2019, 48, 2536–2544. [Google Scholar] [CrossRef] [PubMed]
- Ravera, M.; Gabano, E.; McGlinchey, M.J.; Osella, D. A View on Multi-Action Pt(IV) Antitumor Prodrugs. Inorg. Chim. Acta. 2019, 492, 32–47. [Google Scholar] [CrossRef]
- Jia, C.; Deacon, G.B.; Zhang, Y.; Gao, C. Platinum(IV) Antitumor Complexes and Their Nano-Drug Delivery. Coord. Chem. Rev. 2021, 429, 213640. [Google Scholar] [CrossRef]
- Jiang, X.; Yang, Q.; Qi, R.; Yan, L. Nanoparticle-Mediated Delivery of Satraplatin to Overcome Cisplatin Drug Resistance. J. Funct. Biomater. 2023, 14, 387. [Google Scholar] [CrossRef] [PubMed]
- Kudo, S.; Nagasaki, Y. A Novel Nitric Oxide-Based Anticancer Therapeutics by Macrophage-Targeted Poly(l-Arginine)-Based Nanoparticles. J. Control. Release 2015, 217, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Do, Q.N.; Ratnakar, J.S.; Kovács, Z.; Sherry, A.D. Redox- and Hypoxia-Responsive MRI Contrast Agents. Chem. Med. Chem. 2014, 9, 1116–1129. [Google Scholar] [CrossRef] [PubMed]
- Pinto, S.M.; Tomé, V.; Calvete, M.J.F.; Castro, M.M.C.A.; Tóth, É.; Geraldes, C.F.G.C. Metal-Based Redox-Responsive MRI Contrast Agents. Coord. Chem. Rev. 2019, 390, 1–31. [Google Scholar] [CrossRef]
- Mi, P. Stimuli-Responsive Nanocarriers for Drug Delivery, Tumor Imaging, Therapy and Theranostics. Theranostics 2020, 10, 4557–4588. [Google Scholar] [CrossRef] [PubMed]
- Das, S.S.; Bharadwaj, P.; Bilal, M.; Barani, M.; Rahdar, A.; Taboada, P.; Bungau, S.; Kyzas, G.Z. Stimuli-Responsive Polymeric Nanocarriers for Drug Delivery, Imaging, and Theragnosis. Polymers 2020, 12, 1397. [Google Scholar] [CrossRef] [PubMed]
- Lou, Z.; Li, P.; Han, K. Redox-Responsive Fluorescent Probes with Different Design Strategies. Acc. Chem. Res. 2015, 48, 1358–1368. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Thompson, D.H. Stimuli-responsive Liposomes for Drug Delivery. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2017, 9, e1450. [Google Scholar] [CrossRef] [PubMed]
- Kwon, N.; Kim, D.; Swamy, K.M.K.; Yoon, J. Metal-Coordinated Fluorescent and Luminescent Probes for Reactive Oxygen Species (ROS) and Reactive Nitrogen Species (RNS). Coord. Chem. Rev. 2021, 427, 213581. [Google Scholar] [CrossRef]
- Wang, R. Two’s Company, Three’s a Crowd: Can H2 S Be the Third Endogenous Gaseous Transmitter? FASEB J. 2002, 16, 1792–1798. [Google Scholar] [CrossRef] [PubMed]
- Wang, R. Gasotransmitters: Growing Pains and Joys. Trends Biochem. Sci. 2014, 39, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Hao, M.-Y.; Li, H.-J.; Han, H.-S.; Chu, T.; Wang, Y.-W.; Si, W.-R.; Jiang, Q.-Y.; Wu, D.-D. Recent Advances in the Role of Gasotransmitters in Necroptosis. Apoptosis 2025, 30, 616–635. [Google Scholar] [CrossRef] [PubMed]
- Papapetropoulos, A.; Foresti, R.; Ferdinandy, P. Pharmacology of the ‘Gasotransmitters’ NO, CO and H2S: Translational Opportunities. Br. J. Pharm. 2015, 172, 1395–1396. [Google Scholar] [CrossRef] [PubMed]
- Salihi, A.; Al-Naqshabandi, M.; Khudhur, Z.; Housein, Z.; Hama, H.; Abdullah, R.; Hussen, B.; Alkasalias, T. Gasotransmitters in the Tumor Microenvironment: Impacts on Cancer Chemotherapy (Review). Mol. Med. Rep. 2022, 26, 233. [Google Scholar] [CrossRef] [PubMed]
- Lechner, M.; Lirk, P.; Rieder, J. Inducible Nitric Oxide Synthase (iNOS) in Tumor Biology: The Two Sides of the Same Coin. Semin. Cancer Biol. 2005, 15, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Burke, A.J.; Sullivan, F.J.; Giles, F.J.; Glynn, S.A. The Yin and Yang of Nitric Oxide in Cancer Progression. Carcinogenesis 2013, 34, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Forstermann, U.; Sessa, W.C. Nitric Oxide Synthases: Regulation and Function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Bhowmik, R.; Roy, M. Recent Advances on the Development of NO-Releasing Molecules (NORMs) for Biomedical Applications. Eur. J. Med. Chem. 2024, 268, 116217. [Google Scholar] [CrossRef] [PubMed]
- Alimoradi, H.; Greish, K.; Gamble, A.B.; Giles, G.I. Controlled Delivery of Nitric Oxide for Cancer Therapy. Pharm. Nanotechnol. 2019, 7, 279–303. [Google Scholar] [CrossRef] [PubMed]
- Ampßler, T.; Monsch, G.; Popp, J.; Riggenmann, T.; Salvador, P.; Schröder, D.; Klüfers, P. Not Guilty on Every Count: The “Non-Innocent” Nitrosyl Ligand in the Framework of IUPAC’s Oxidation-State Formalism. Angew. Chem. Int. Ed. 2020, 59, 12381–12386. [Google Scholar] [CrossRef] [PubMed]
- Manoharan, P.T.; Gray, H.B. Electronic Structure of Nitroprusside Ion. J. Am. Chem. Soc. 1965, 87, 3340–3348. [Google Scholar] [CrossRef]
- Crespo, P.M.; Odio, O.F.; Reguera, E. Photochemistry of Metal Nitroprussides: State-of-the-Art and Perspectives. Photochem 2022, 2, 390–404. [Google Scholar] [CrossRef]
- Hirst, D.; Robson, T. Targeting Nitric Oxide for Cancer Therapy. J. Pharm. Pharmacol. 2007, 59, 3–13. [Google Scholar] [CrossRef] [PubMed]
- De La Rosa, Á.J.; Rodríguez-Hernández, Á.; González, R.; Romero-Brufau, S.; Navarro-Villarán, E.; Barrera-Pulido, L.; Pereira, S.; Marín, L.M.; López-Bernal, F.; Álamo, J.M.; et al. Antitumoral Gene-Based Strategy Involving Nitric Oxide Synthase Type III Overexpression in Hepatocellular Carcinoma. Gene Ther. 2016, 23, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Iyer, A.K.V.; Azad, N.; Wang, L.; Rojanasakul, Y. Role of S-Nitrosylation in Apoptosis Resistance and Carcinogenesis. Nitric. Oxide. 2008, 19, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Iyer, A.K.; Khaled, G.; Fang, J.; Maeda, H. Exploiting the Enhanced Permeability and Retention Effect for Tumor Targeting. Drug Discov. Today 2006, 11, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.J.; Barbougli, P.A.; Espreafico, E.M.; Tfouni, E. Trans-[Ru(NO)(NH3)4(Py)](BF4)3·H2O Encapsulated in PLGA Microparticles for Delivery of Nitric Oxide to B16-F10 Cells: Cytotoxicity and Phototoxicity. J. Inorg. Biochem. 2008, 102, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Alimoradi, H.; Greish, K.; Barzegar-Fallah, A.; ALshaibani, L.; Pittalà, V. Nitric Oxide-Releasing Nanoparticles Improve Doxorubicin Anticancer Activity. Int. J. Nanomed. 2018, 13, 7771–7787. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Zhang, J.; Chen, W.; Miao, C. Prospects of Molecular Hydrogen in Cancer Prevention and Treatment. J. Cancer Res. Clin. Oncol. 2024, 150, 170. [Google Scholar] [CrossRef] [PubMed]
- Tien Vo, T.T.; Vo, Q.C.; Tuan, V.P.; Wee, Y.; Cheng, H.-C.; Lee, I.-T. The Potentials of Carbon Monoxide-Releasing Molecules in Cancer Treatment: An Outlook from ROS Biology and Medicine. Redox Biol. 2021, 46, 102124. [Google Scholar] [CrossRef] [PubMed]
- Adach, W.; Olas, B. Carbon Monoxide and Its Donors—Their Implications for Medicine. Future Med. Chem. 2019, 11, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Bansal, S.; Liu, D.; Mao, Q.; Bauer, N.; Wang, B. Carbon Monoxide as a Potential Therapeutic Agent: A Molecular Analysis of Its Safety Profiles. J. Med. Chem. 2024, 67, 9789–9815. [Google Scholar] [CrossRef] [PubMed]
- Juszczak, M.; Kluska, M.; Wysokiński, D.; Woźniak, K. DNA Damage and Antioxidant Properties of CORM-2 in Normal and Cancer Cells. Sci. Rep. 2020, 10, 12200. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.; Liu, C.; Wang, S.; Liu, J.; Wang, L.; Lv, L.; Zou, Y. The Impact of Exogenous CO Releasing Molecule CORM-2 on Inflammation and Signaling of Orthotopic Lung Cancer. Oncol. Lett. 2018, 16, 3223–3230. [Google Scholar] [CrossRef] [PubMed]
- Kourti, M.; Jiang, W.G.; Cai, J. Aspects of Carbon Monoxide in Form of CO-Releasing Molecules Used in Cancer Treatment: More Light on the Way. Oxid. Med. Cell. Longev. 2017, 2017, 9326454. [Google Scholar] [CrossRef] [PubMed]
- Bauer, N.; Yuan, Z.; Yang, X.; Wang, B. Plight of CORMs: The Unreliability of Four Commercially Available CO-Releasing Molecules, CORM-2, CORM-3, CORM-A1, and CORM-401, in Studying CO Biology. Biochem. Pharm. 2023, 214, 115642. [Google Scholar] [CrossRef] [PubMed]
- Zanellato, I.; Bonarrigo, I.; Ravera, M.; Gabano, E.; Gust, R.; Osella, D. The Hexacarbonyldicobalt Derivative of Aspirin Acts as a CO-Releasing NSAID on Malignant Mesothelioma Cells. Metallomics 2013, 5, 1604. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.C.; Kim, D.W.; Shim, Y.H.; Bang, J.S.; Oh, H.S.; Kim, S.W.; Seo, M.H. In Vivo Evaluation of Polymeric Micellar Paclitaxel Formulation: Toxicity and Efficacy. J. Control. Release 2001, 72, 191–202. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strategy | Mechanism of Action | TME Trigger | Selectivity | Development Stage | Key Challenges |
|---|---|---|---|---|---|
| pH-sensitive nanocarriers | Drug release under acidic extracellular pH | Acidic pHe (6.5–6.9) | High (TME-specific) | Preclinical/early clinical | Scale-up, EPR heterogeneity, immune response |
| Hypoxia-activated prodrugs | Enzymatic or chemical activation in low O2 | Hypoxia (<2% O2) | Moderate–High | Preclinical/clinical | Heterogeneous oxygen levels, off-target effects |
| Pt(IV) complexes | Reductive activation in hypoxic/reductive areas | Hypoxia, Reductive TME | Moderate | Clinical trials (e.g., satraplatin) | Neurotoxicity, limited reduction selectivity |
| Gasotransmitters (NO, CO) | Redox modulation and signaling disruption | ROS, hypoxia, enzymes | Variable | Preclinical | Dosing window, systemic toxicity, targeted delivery systems |
| NCT Number | Study Title | Conditions | Sponsor | Study Type |
|---|---|---|---|---|
| NCT06790797 | Investigating the Biomarkers in Tumor and Peripheral Blood to Evaluate the Efficacy of Cancer Immunotherapy in Chest Cancer Patients | Lung cancer; Esophageal cancer; In vitro stimulation of PBMCs with tumor antigen nanoparticles | Zhao Jun | Observational |
| NCT06776198 | Precision Therapy Based on Immune Microenvironment by Transcriptome Sequencing of Osteosarcoma, a Prospective, Multi-cohort Exploratory Clinical Study | Osteosarcoma transcriptome tumor microenvironment | Peking University People’s Hospital | Observational |
| NCT06922825 | Pilot Study Evaluating Tumor Microenvironment Interaction in Solid Tumor Patients | Solid tumor cancer; | Eben Rosenthal | Interventional |
| NCT02042781 | Study of the Safety and Tolerability of IV Infused PG545 (Pixatimod) in Patients With Advanced Solid Tumours | Advanced solid tumours | Zucero Pty Ltd. | Interventional |
| NCT03844763 | Targeting the Tumor Microenvironment in R/M SCCHN (Avelumab) | Head and neck cancer | Gruppo Oncologico del Nord-Ovest | Interventional |
| NCT04225390 | Preconditioning of Tumor, Tumor Microenvironment and the Immune System to Immunotherapy (Dacarbazine) | Immunotherapy | University of Erlangen-Nürnberg Medical School | Interventional |
| NCT06439173 | Study of the Hematopoietic Niche and the Role of Inflammation in the Pathophysiology of Cytopenias After CAR-T Cell Therapy: Potential of Therapies Directed to Repair the Bone Marrow Microenvironment | Diffuse large B cell lymphoma. Biological: CAR-T cell therapy | Instituto de Investigación Biomédica de Salamanca | Observational |
| NCT03647839 | Modulation Of The Tumour Microenvironment Using Either Vascular Disrupting Agents or STAT3 Inhibition in Order to Synergise With PD1 Inhibition in Microsatellite Stable, Refractory Colorectal Cancer | Colorectal cancer metastatic | Australasian Gastro-Intestinal Trials Group | Interventional |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ravera, M.; Gabano, E.; Tonello, S.; Colangelo, D. Tumor Niche Influences the Activity and Delivery of Anticancer Drugs: Pharmacology Meets Chemistry. Pharmaceuticals 2025, 18, 1047. https://doi.org/10.3390/ph18071047
Ravera M, Gabano E, Tonello S, Colangelo D. Tumor Niche Influences the Activity and Delivery of Anticancer Drugs: Pharmacology Meets Chemistry. Pharmaceuticals. 2025; 18(7):1047. https://doi.org/10.3390/ph18071047
Chicago/Turabian StyleRavera, Mauro, Elisabetta Gabano, Stelvio Tonello, and Donato Colangelo. 2025. "Tumor Niche Influences the Activity and Delivery of Anticancer Drugs: Pharmacology Meets Chemistry" Pharmaceuticals 18, no. 7: 1047. https://doi.org/10.3390/ph18071047
APA StyleRavera, M., Gabano, E., Tonello, S., & Colangelo, D. (2025). Tumor Niche Influences the Activity and Delivery of Anticancer Drugs: Pharmacology Meets Chemistry. Pharmaceuticals, 18(7), 1047. https://doi.org/10.3390/ph18071047