

Innovative Formulation Strategies for Biosimilars: Trends Focused on Buffer-Free Systems, Safety, Regulatory Alignment, and Intellectual Property Challenges

Abstract
1. Introduction
2. Methodology
Keywords and Document Identification Process
3. Design, Manufacturing, and Analytical Characterization of Biosimilars
3.1. Analytical Characterization and Processing Strategies of Biosimilars
- Differences in expression systems (e.g., CHO vs. HEK293);
- Cell culture conditions, including pH, oxygen, and nutrients;
- Bioprocessing variables, such as purification methods or buffer composition.
3.2. Different Strategies and Processing Advances in the Manufacture of Biosimilars
3.3. Integrating AI and Machine Learning into the Biosimilar Development Process
3.4. Innovations in the Field of Bioprocessing
4. Biosimilar Formulations Based on Monoclonal Antibodies and Recombinant Proteins
Dissemination of Novel Ormulations
- Lipid encapsulation: This technology is being explored to improve the bioavailability and stability of therapeutic proteins, particularly in liquid formulations [176].
- Advances in nanotechnology: The use of nanocarriers offers promising strategies to deliver biologics while minimizing immunogenic responses and improving therapeutic effects through targeted delivery systems [179].
5. Advances in Biosimilar Formulation Technologies (Buffer-Free Strategies) and Excipients
5.1. Formulation and Selection of Excipients
5.2. Buffer-Free High-Concentration Formulations and the Role of Excipients in Immunogenicity Mitigation
5.3. Trends in Change: Buffer-Free Formulations
5.4. Classification and Safety Profiles of Approved Biosimilar Formulations
5.5. Formulation Trends and Considerations on Stability, Bioavailability, and Immunogenicity
6. Importance of the FDA and EMA Regulatory Frameworks in the Development of Biosimilars
6.1. FDA and EMA Regulatory Approach to Biosimilars
6.2. ICH-Guided Analytical Characterization in Biosimilar Development
7. IP Challenges Associated with the Development of Biosimilar Formulations
8. Discussion
8.1. Innovation in Formulation and Patient-Centered Strategies
8.2. Analytical and Manufacturing Advances
8.3. Regulatory Evolution and Market Access
8.4. Intellectual Property and Strategic Development
8.5. Clinical and Psychosocial Considerations
8.6. The Role of Digital Tools and Predictive Modeling
9. Conclusions
Future Research Directions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
| Aspect | Description | The Last Insinuations | Challenges | Refserences |
|---|---|---|---|---|
| Design and formulation of strategies | Molecular design and formulation optimization replicate the stability, efficacy, and immunogenicity of reference biologics through molecular engineering, innovative excipients, and stability enhancements. |
|
| [2,148,209,353,354,355] |
| Cell line and upstream engineering | Development and optimization of cell lines to ensure high similarity with the attributes of the reference product. Production of recombinant proteins using systems such as CHO cells, crucial for protein structure and PTMs. |
|
| [356,357,358,359,360,361] |
| Improvement processes and PAT | Protein purification and recovery that ensures removal of impurities, integrity of protein folding/refolding, and consistency of the product. Real-time monitoring/control through PAT, ensuring QbD. |
|
| [53,57,361,362,363] |
| Analytical Characterization and Bioassays | Structural and functional evaluation using orthogonal methods to establish biosimilarity throughout the body of evidence. |
|
| [37,188,364,365,366,367,368,369] |
| Regulations and Harmonization | Regulatory evaluations that ensure the safety, efficacy, and quality of biosimilars through comparability studies, ensuring that biosimilars comply with the strict FDA/EMA regulatory frameworks. |
|
| [5,37,168,273,370,371] |
| IP, Innovation Strategy, and Market Access | Patents related to formulation, manufacturing, and analytical techniques that affect market entry and competitiveness. |
|
| [371,372,373] |
| Integration of Digital Tools and AI | Application of digital platforms, AI, and simulations to optimize biosimilar development. |
|
| [374,375] |
References
- Bas, T.G. Biosimilars for the next decade in Latin America: A window of opportunity. Expert Opin. Biol. Ther. 2023, 23, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Bas, T.G.; Duarte, V. Biosimilars in the Era of Artificial Intelligence: International Regulations and the Use in Oncological Treatments. Pharmaceuticals 2024, 17, 925. [Google Scholar] [CrossRef] [PubMed]
- Mascarenhas-Melo, F.; Diaz, M.; Gonçalves, M.B.S.; Vieira, P.; Bell, V.; Viana, S.; Nunes, S.; Paiva-Santos, A.C.; Veiga, F. An overview of biosimilars—Development, quality, regulatory issues, and management in healthcare. Pharmaceuticals 2024, 17, 235. [Google Scholar] [CrossRef] [PubMed]
- Raposo, M.C.; Feiteira, C.; Ribeiro, M.H. Regulatory and clinical aspects in biosimilar medicines: Comparability, extrapolation, interchangeability, and safety. Drugs Ther. Perspect. 2025, 41, 111–125. [Google Scholar] [CrossRef]
- Gaylis, N.; Both, C.; Lemke, L.; Richter, O.; Yamauchi, P. ‘Totality of evidence’ approach in the development of GP2017, an approved adalimumab biosimilar. Adv. Ther. 2024, 41, 1795–1814. [Google Scholar] [CrossRef]
- Heinemann, L.; Davies, M.J.; Home, P.; Först, T.; Vilsbøll, T.; Schnell, O. Understanding biosimilar insulins—Development, manufacturing, and clinical trials. J. Diabetes Sci. Technol. 2022, 17, 1649–1661. [Google Scholar] [CrossRef]
- Sarin, D.; Krishna, K.; Nejadnik, M.; Suryanarayanan, R.; Rathore, A. Impact of excipient extraction and buffer exchange on recombinant monoclonal antibody stability. Mol. Pharm. 2024, 21, 1872–1883. [Google Scholar] [CrossRef]
- García-Beloso, N.; Altabás-González, I.; Samartín-Ucha, M.; Gayoso-Rey, M.; Castro-Parga, M.; Salgado-Barreira, Á.; Barreira, Á.S.; Badia, A.C.; Corrales, M.G.P.; Vilas, D.G.; et al. Switching between reference adalimumab and biosimilars in chronic immune-mediated inflammatory diseases: A systematic literature review. Br. J. Clin. Pharmacol. 2021, 88, 1529–1550. [Google Scholar] [CrossRef]
- Germain, M.; Caputo, F.; Metcalfe, S.; Tosi, G.; Spring, K.; Åslund, A.; Pottier, A.; Schiffelers, R.; Ceccaldi, A.; Schmid, R. Delivering the power of nanomedicine to patients today. J. Control. Release 2020, 326, 164–171. [Google Scholar] [CrossRef]
- Vulto, A.G.; Jaquez, O. The process defines the product: What really matters in biosimilar design and production? Rheumatology 2017, 56 (Suppl. S4), iv14–iv29. [Google Scholar] [CrossRef]
- Rahban, M.; Ahmad, F.; Piatyszek, M.A.; Haertlé, T.; Saso, L.; Saboury, A.A. Stabilization challenges and aggregation in protein-based therapeutics in the pharmaceutical industry. RSC Adv. 2023, 13, 35947–35963. [Google Scholar] [CrossRef] [PubMed]
- Akbarian, M.; Chen, S. Instability Challenges and Stabilization Strategies of Pharmaceutical Proteins. Pharmaceutics 2022, 14, 2533. [Google Scholar] [CrossRef] [PubMed]
- Yuanquiong, H.; Eynikel, D.; Boulet, P.; Krikorian, G. Supplementary protection certificates and their impact on access to medicines in Europe: Case studies of sofosbuvir, trastuzumab and imatinib. J. Pharm. Policy Pract. 2020, 13, 1. [Google Scholar] [CrossRef]
- Mehr, S.R.; Brook, R.A. Biosimilars in the USA: Will new efforts to spur approvals and access spur uptake and cost savings? Pharm. Med. 2019, 33, 1–8. [Google Scholar] [CrossRef]
- Dutta, B.; Huys, I.; Vulto, A.G.; Simoens, S. Identifying key benefits in European off-patent biologics and biosimilar markets: It is not only about price! BioDrugs 2019, 34, 159–170. [Google Scholar] [CrossRef]
- Geaghan-Breiner, C. The patent trap: The struggle for competition and affordability in the field of biologic drugs. Columbia J. Law Soc. Probl. 2020, 54, 589. [Google Scholar]
- Barszczewska, O.; Piechota, A. The impact of introducing successive biosimilars on changes in prices of adalimumab, infliximab, and trastuzumab—Polish experiences. Int. J. Environ. Res. Public Health 2021, 18, 6952. [Google Scholar] [CrossRef]
- Vandenplas, Y.; Simoens, S.; Wilder, P.V.; Vulto, A.G.; Huys, I. Off-patent biological and biosimilar medicines in Belgium: A market landscape analysis. Front. Pharmacol. 2021, 12, 644187. [Google Scholar] [CrossRef]
- Carrier, M.A.; Tu, S.S. Why pharmaceutical patent thickets are unique. Tex. Intellect. Prop. Law J. 2023, 32, 79–110. [Google Scholar] [CrossRef]
- Jarab, A.S.; Heshmeh, S.A.; Meslamani, A.Z.A. Bridging the gap: The future of biosimilars regulations. Hum. Vaccines Immunother. 2024, 20, 2362450. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, L.; Li, S.; Yuan, Y.; Jiang, B.; Jiang, Z.; Zhang, X.; Zhou, X.; Liu, M. Detecting biomarkers by dynamic nuclear polarization enhanced magnetic resonance. Natl. Sci. Rev. 2024, 11, nwae228. [Google Scholar] [CrossRef] [PubMed]
- Mennini, F.; Marcellusi, A.; Bini, C.; Rotundo, M.; Giunta, A.; Gasbarrini, A.; Valesini, G.; Canonico, P.L.; Novellino, E.; Orlando, V.; et al. The economic impact of biosimilars in Italy: A scenario analysis. Glob. Reg. Health Technol. Assess. 2019, 2019, 228424031985802. [Google Scholar] [CrossRef]
- Berto, P.; Bellone, M.; Sabinot, A.; Pinto, C.; Martino, M.; Generali, D.; Carriero, P.L.; Sanna, M.D. Budget saving potential of pegfilgrastim biosimilar for the treatment of chemotherapy-induced febrile neutropenia, in Italy. Farmeconomia Health Econ. Ther. Pathw. 2022, 23, 1–12. [Google Scholar] [CrossRef]
- AlRuthia, Y.; Bahari, O.; Alghnam, S.; Alrumaih, A.; Asiri, H.; Alshammari, M.; Alhowimel, M.; Al-Abdulkarim, H.A. Real-world impact of switching from insulin glargine (Lantus®) to Basaglar® and potential cost saving in a large public healthcare system in Saudi Arabia. Front. Public Health 2022, 10, 852721. [Google Scholar] [CrossRef]
- McClean, A.; Cheng, L.; Bansback, N.; Clement, F.; Tadrous, M.; Harrison, M.; Law, M.R. Uptake and spending on biosimilar infliximab and etanercept after new start and switching policies in Canada: An interrupted time series analysis. Arthritis Care Res. 2023, 75, 2011–2021. [Google Scholar] [CrossRef]
- Malakar, S.; Gontor, E.; Dugbaye, M.; Shah, K.; Sinha, S.; Sutaoney, P.; Chauhan, N.S. Cancer treatment with biosimilar drugs: A review. Cancer Innov. 2024, 3, e115. [Google Scholar] [CrossRef]
- Aladul, M.; Fitzpatrick, R.; Chapman, S. The effect of new biosimilars in rheumatology and gastroenterology specialities on UK healthcare budgets: Results of a budget impact analysis. Res. Soc. Adm. Pharm. 2019, 15, 310–317. [Google Scholar] [CrossRef]
- Sheridan, M.; Massich, M.; Ashourian, N. Biosimilars: From Production to Patient. J. Infus. Nurs. 2024, 47, 19–29. [Google Scholar] [CrossRef]
- Tenni, B.; Moir, H.; Townsend, B.; Kilic, B.; Farrell, A.; Keegel, T.; Gleeson, D. What is the impact of intellectual property rules on access to medicines? A systematic review. Glob. Health 2022, 18, 40. [Google Scholar] [CrossRef]
- Garai, P.; Chakraborty, A.; Banerjee, M.; Mukherjee, P. A comprehensive review on biosimilars: Present & future. J. Pharm. Res. Int. 2024, 36, 198–205. [Google Scholar] [CrossRef]
- Ratih, R.; Asmari, M.; Abdel-Megied, A.M.; Elbarbry, F.; Deeb, S.E. Biosimilars: Review of regulatory, manufacturing, analytical aspects and beyond. Microchem. J. 2021, 165, 106143. [Google Scholar] [CrossRef]
- Niazi, S.K. Affordable mRNA novel proteins, recombinant protein conversions, and biosimilars—Advice to developers and regulatory agencies. Biomedicines 2025, 13, 97. [Google Scholar] [CrossRef] [PubMed]
- Patil, N.; Ranjan, A.; Gaurav, G.; Mukherjee, D.; Panda, B.K.; Diksha, D.; Komal; Narang, R.K.; Singh, A. Navigating biosimilar regulatory pathways in emerging markets: Insights from BRICS nations. Appl. Drug Res. Clin. Trials Regul. Aff. 2025, 10, e26673371316219. [Google Scholar] [CrossRef]
- Bhatnagar, A.; Hazra, P.; Pai, H.V.; Vajpai, N.; Ramani, K.; Govindappa, N. Process Development for Biologics Therapeutics. In Approved: The Life Cycle of Drug Development; Springer Nature: Cham, Switzerland, 2025; pp. 249–297. [Google Scholar]
- Ranjan, R. Development of Complex Generics and Similar Biological Products: An Industrial Perspective of Reverse Engineering. AAPS PharmSciTech 2025, 26, 95. [Google Scholar] [CrossRef]
- Manwatkar, S.; Kumar, B. Biosimilars: Promising and rapidly emerging biotherapeutics. In Novel Technologies in Biosystems, Biomedical & Drug Delivery; Springer: Singapore, 2023; pp. 45–67. [Google Scholar] [CrossRef]
- Nupur, N.; Joshi, S.; Gulliarme, D.; Rathore, A. Analytical similarity assessment of biosimilars: Global regulatory landscape, recent studies and major advancements in orthogonal platforms. Front. Bioeng. Biotechnol. 2022, 10, 832059. [Google Scholar] [CrossRef]
- Naas, H.; Mehennaoui, S.; Gharbi, A. Characterization of biosimilars: Description of the analytical approach. GSC Biol. Pharm. Sci. 2023, 25, 215–221. [Google Scholar] [CrossRef]
- Pathak, M.; Pokhriyal, P.; Gandhi, I.; Khambhampaty, S. Implementation of chemometrics, design of experiments, and neural network analysis for prior process knowledge assessment, failure modes and effect analysis, scale-down model development, and process characterization for a chromatographic purification of teriparatide. Biotechnol. Prog. 2022, 38, e3252. [Google Scholar] [CrossRef]
- Wasalathanthri, D.; Rehmann, M.; Song, Y.; Gu, Y.; Luo, M.; Shao, C.; Shao, C.; Chemmalil, L.; Lee, J.; Ghose, S.; et al. Technology outlook for real-time quality attribute and process parameter monitoring in biopharmaceutical development—A review. Biotechnol. Bioeng. 2020, 117, 3182–3198. [Google Scholar] [CrossRef]
- Rahalkar, H.; Sheppard, A.; López-Morales, C.; Lobo, L.; Salek, S. Challenges faced by the biopharmaceutical industry in the development and marketing authorization of biosimilar medicines in BRICS-TM countries: An exploratory study. Pharm. Med. 2021, 35, 235–251. [Google Scholar] [CrossRef]
- Chemmalil, L.; Prabhakar, T.; Kuang, J.; West, J.; Tan, Z.; Ehamparanathan, V.; Song, Y.; Xu, J.; Ding, J.; Li, Z. Online/at-line measurement, analysis and control of product titer and critical product quality attributes (CQAs) during process development. Biotechnol. Bioeng. 2020, 117, 3757–3765. [Google Scholar] [CrossRef]
- García-Holgado, A.; Marcos-Pablos, S.; García-Peñalvo, F.J. Guidelines for performing systematic research projects reviews. Int. J. Interact. Multimed. Artif. Intell. 2020, 6, 9. [Google Scholar] [CrossRef]
- Mengist, W.; Soromessa, T.; Legese, G. Method for conducting systematic literature review and meta-analysis for environmental science research. MethodsX 2020, 7, 100777. [Google Scholar] [CrossRef] [PubMed]
- Scells, H.; Zuccon, G.; Koopman, B.; Clark, J. Automatic Boolean query formulation for systematic review literature search. In Proceedings of the WWW ’20: The Web Conference 2020, Taipei, Taiwan, 20–24 April 2020; pp. 1071–1081. [Google Scholar]
- Scells, H.; Zuccon, G.; Koopman, B. A comparison of automatic Boolean query formulation for systematic reviews. Inf. Retr. J. 2021, 24, 3–28. [Google Scholar] [CrossRef]
- Page, M.; McKenzie, J.; Bossuyt, P.; Boutron, I.; Hoffmann, T.; Mulrow, C.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef]
- Akl, E.; Khabsa, J.; Iannizzi, C.; Piechotta, V.; Kahale, L.; Barker, J.; Skoetz, N. Extension of the PRISMA 2020 Statement for Living Systematic Reviews (PRISMA-LSR): Checklist and Explanation. BMJ 2024, 384, e079183. [Google Scholar] [CrossRef]
- Gusenbauer, M.; Haddaway, N.R. Which academic search systems are suitable for systematic reviews or meta-analyses? Evaluating retrieval qualities of Google Scholar, PubMed, and 26 other resources. Res. Synth. Methods 2020, 11, 181–217. [Google Scholar] [CrossRef]
- Duivelshof, B.; Jiskoot, W.; Beck, A.; Veuthey, J.; Guillarme, D.; D’Atri, V. Glycosylation of biosimilars: Recent advances in analytical characterization and clinical implications. Anal. Chim. Acta 2019, 1089, 1–18. [Google Scholar] [CrossRef]
- Saleem, R.; Cantin, G.; Wikström, M.; Bolton, G.; Kuhns, S.; McBride, H.; Liu, J. Analytical and functional similarity assessment of ABP 710, a biosimilar to infliximab reference product. Pharm. Res. 2020, 37, 114. [Google Scholar] [CrossRef]
- Drobnjaković, M.; Hart, R.A.; Kulvatunyou, B.; Ivezic, N.; Srinivasan, V. Current challenges and recent advances on the path towards continuous biomanufacturing. Biotechnol. Prog. 2023, 39, e3378. [Google Scholar] [CrossRef]
- Sykes, A.; Ingram, L.; Kronthaler, U.; Chevalet, L. Demonstration of physicochemical and functional similarity between Stimufend (pegfilgrastim-fpgk) and Neulasta (pegfilgrastim): A comparative analytical assessment. PLoS ONE 2024, 19, e0309480. [Google Scholar] [CrossRef]
- Nag, K.; Sarker, E.; Kumar, S.; Chakraborty, S.; Khan, M.; Chowdhury, M.; Roy, R.; Roy, R.; Biswas, B.K.; Bappi, E.H.; et al. Satisfying QTPP of erythropoietin biosimilar by QbD through DoE-derived downstream process engineering. Pharmaceutics 2023, 15, 2087. [Google Scholar] [CrossRef] [PubMed]
- Neugebauer, P.; Zettl, M.; Moser, D.; Poms, J.; Kuchler, L.; Sacher, S. Process analytical technology in downstream-processing of drug substances—A review. Int. J. Pharm. 2024, 661, 124412. [Google Scholar] [CrossRef] [PubMed]
- Gyorgypal, A.; Chundawat, S. Integrated process analytical platform for automated monitoring of monoclonal antibody N-linked glycosylation. Anal. Chem. 2022, 94, 6986–6995. [Google Scholar] [CrossRef] [PubMed]
- Puranik, A.; Dandekar, P.; Jain, R. Exploring the potential of machine learning for more efficient development and production of biopharmaceuticals. Biotechnol. Prog. 2022, 38, e3291. [Google Scholar] [CrossRef]
- Demir, F.; Albarri, R.; Ünal, D.Ö. An overview of biotechnological drug’s various techniques of downstream process, guidelines and different chromatographic analysis. Curr. Pharm. Anal. 2024, 20, 729–742. [Google Scholar] [CrossRef]
- Luca, C.; Felletti, S.; Lievore, G.; Chenet, T.; Morbidelli, M.; Sponchioni, M.; Cavazzini, A.; Catani, M. Modern trends in downstream processing of biotherapeutics through continuous chromatography: The potential of multicolumn countercurrent solvent gradient purification. TrAC Trends Anal. Chem. 2020, 132, 116051. [Google Scholar] [CrossRef]
- Kim, H.; Bang, G.; Park, Y.E.; Park, M.; Choi, J.H.; Oh, M.J.; Hwang, H. Advanced Assessment through Intact Glycopeptide Analysis of Infliximab’s Biologics and Biosimilar. Front. Mol. Biosci. 2022, 9, 1006866. [Google Scholar] [CrossRef]
- Coghlan, J.; Benet, A.; Kumaran, P.; Ford, M.; Veale, L.; Skilton, S.J.; Schwendeman, A. Streamlining the Characterization of Disulfide Bond Shuffling and Protein Degradation in IgG1 Biopharmaceuticals under Native and Stressed Conditions. Front. Bioeng. Biotechnol. 2022, 10, 862456. [Google Scholar] [CrossRef]
- Abidin, A.Z.; Snoswell, C.L.; Hanjani, L.S.; Callaghan, G.; Edmonds, M.L. Infliximab Switching from Reference Product to Biosimilar: A Review of Evidence Regarding the Clinical Efficacy, Safety Profile and Immunogenicity. J. Pharm. Pract. Res. 2021, 51, 358–373. [Google Scholar] [CrossRef]
- Bandara, S.A.T.; Raveendran, S. Current Landscape and Future Directions in Cancer Immunotherapy: Therapies, Trials, and Challenges. Cancers 2025, 17, 821. [Google Scholar] [CrossRef]
- Bouvarel, T.; Camperi, J.; Guillarme, D. Multi-dimensional technology—Recent advances and applications for biotherapeutic characterization. J. Sep. Sci. 2024, 47, 2300928. [Google Scholar] [CrossRef] [PubMed]
- Oh, M.J.; Seo, Y.; Seo, N.; An, H.J. MS-based glycome characterization of biotherapeutics with N- and O-glycosylation. Mass Spectrom. Rev. 2025. [Google Scholar] [CrossRef] [PubMed]
- Mans, J.D.; Oyugi, M.; Asmelash, B.; Sommers, C.D.; Rogstad, S. The Use of Mass Spectrometry in Therapeutic Protein Biologics License Applications: A Retrospective Review Revisited. J. Am. Soc. Mass Spectrom. 2023, 34, 2575–2584. [Google Scholar] [CrossRef] [PubMed]
- Song, N.K.; Musa, H.; Soriano, M.; Batger, M.; Hawkins, B.A.; Ramzan, I.; Ong, J. Safety and Efficacy Comparisons of Intravenous Trastuzumab Biosimilars to the Reference Product Medicine in Treatment-Naïve and Switch-Over Patients with Breast Cancer: A Systematic and Meta-Analysis. J. Pharm. Pract. Res. 2023, 54, 1–32. [Google Scholar] [CrossRef]
- Atik, A.E. Comparative Analysis of Glycoform Profiles between Biosimilar and Originator Monoclonal Antibodies by Liquid Chromatography–Mass Spectrometry. J. Turk. Chem. Soc. Sect. A Chem. 2024, 11, 365–376. [Google Scholar] [CrossRef]
- Gupta, S.; Shah, B.; Fung, C.S.; Chan, P.K.; Wakefield, D.L.; Kuhns, S.; Piret, J.M. Engineering Protein Glycosylation in CHO Cells to Be Highly Similar to Murine Host Cells. Front. Bioeng. Biotechnol. 2023, 11, 1113994. [Google Scholar] [CrossRef]
- Rathore, A.S.; Malani, H. Need for a Risk-Based Control Strategy for Managing Glycosylation Profile for Biosimilar Products. Expert Opin. Biol. Ther. 2021, 22, 123–131. [Google Scholar] [CrossRef]
- Coliat, P.; Erb, S.; Diemer, H.; Karouby, D.; Martin, T.; Banerjee, M.; Pivot, X. Influence of Pneumatic Transportation on the Stability of Monoclonal Antibodies. Sci. Rep. 2023, 13, 21875. [Google Scholar] [CrossRef]
- Castel, J.; Delaux, S.; Hernandez-Alba, O.; Cianférani, S. Recent Advances in Structural Mass Spectrometry Methods in the Context of Biosimilarity Assessment: From Sequence Heterogeneities to Higher Order Structures. J. Pharm. Biomed. Anal. 2023, 236, 115696. [Google Scholar] [CrossRef]
- Rocca, D.; Redis, R.S.; Meyer, P.; Rohleff, I.; Oswald, E.; Martin, S.; Bryans, J.S. Abstract 3794: Developing an mRNA Nanomedicine Platform to Democratise Therapeutic Antibodies. Cancer Res. 2025, 85 (Suppl. S1), 3794. [Google Scholar] [CrossRef]
- McGonigle, P. How Biologics Have Changed the Drug Discovery Landscape. Annu. Rev. Pharmacol. Toxicol. 2025, 65, 29–46. [Google Scholar] [CrossRef] [PubMed]
- Sagi, S.; Kalsekar, S.; Kottke, A.; Cohen, H.P. Long-Term Real-World Post-Approval Safety Data of Multiple Biosimilars from One Marketing-Authorization Holder after More Than 18 Years since Their First Biosimilar Launch. Drug Saf. 2023, 46, 1391–1404. [Google Scholar] [CrossRef] [PubMed]
- Böttinger, K.; Regl, C.; Schäpertöns, V.; Rapp, E.; Wohlschlager, T.; Huber, C.G. “Small Is Beautiful”—Examining Reliable Determination of Low-Abundant Therapeutic Antibody Glycovariants. J. Pharm. Anal. 2024, 14, 100982. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Xiang, Y.; Zhang, X.; Sun, Y.; Li, Y.; Wang, L.; Li, J. Analysis Strategy for Identifying the O-Linked Glycan Profile and O-Glycosylation Sites on Recombinant Human Follicle Stimulating Hormone-C-Terminal Peptide (rhFSH-CTP). Molecules 2025, 30, 2141. [Google Scholar] [CrossRef]
- Reddy, J.; Leibiger, T.; Singh, S.; Lee, K.; Papoutsakis, E.; Ierapetritou, M. A novel, site-specific N-linked glycosylation model provides mechanistic insights into the process-condition dependent distinct Fab and Fc glycosylation of an IgG1 monoclonal antibody produced by CHO VRC01 cells. Biotechnol. Bioeng. 2024, 122, 761–778. [Google Scholar] [CrossRef]
- Mesonzhnik, N.V.; Belushenko, A.; Novikova, P.V.; Kukharenko, A.B.; Afonin, M. Enhanced N-Glycan Profiling of Therapeutic Monoclonal Antibodies through the Application of Upper-Hinge Middle-Up Level LC-HRMS Analysis. Antibodies 2024, 13, 66. [Google Scholar] [CrossRef]
- Hudnik, D.; Bohanec, N.; Drobnak, I.; Ernst, P.B.; Hanke, A.T.; Horvat, M.; Omladič, M. Automatic Peak Annotation and Area Estimation of Glycan Map Peaks Directly from Chromatograms. J. Chemom. 2023, 37, e3521. [Google Scholar] [CrossRef]
- David, P.O. Abstract A001: Navigating the Future of Therapeutic Development: Innovations in Safety and Efficacy through Advanced Research Methodologies and Nanotechnology. Mol. Cancer Ther. 2024, 23 (Suppl. S12), A001. [Google Scholar] [CrossRef]
- Eidenberger, L.; Kogelmann, B.; Steinkellner, H. Plant-Based Biopharmaceutical Engineering. Nat. Rev. Bioeng. 2023, 1, 426–439. [Google Scholar] [CrossRef]
- Saunders, M.; Woods, R.J.; Yang, L. Simplifying the Detection and Monitoring of Protein Glycosylation during In Vitro Glycoengineering. Sci. Rep. 2023, 13, 567. [Google Scholar] [CrossRef]
- Barron, A.; Chung, J.; Ferner, R.E.; Leandro, M.; Maru, S.; Scourfield, A.; Sofat, R. Effectiveness of Biosimilar Adoption within a UK Tertiary Hospital: 6-Year Follow-Up. Br. J. Clin. Pharmacol. 2023, 89, 2944–2949. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Chen, Y.; Li, X.; Gu, X.; Dong, W.; Shi, J.; Ji, S. Protein Succinylation: Regulating Metabolism and Beyond. Front. Nutr. 2024, 11, 1336057. [Google Scholar] [CrossRef] [PubMed]
- Sharma, C.; Hamza, A.; Boyle, E.; Donu, D.; Cen, Y. Post-Translational Modifications and Diabetes. Biomolecules 2024, 14, 310. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Liu, H.; Bai, B.; Wang, H. Quantification of Epigenetic DNA and RNA Modifications by UHPLC–MS/MS Technologies: New Concepts and New Improvements for the Special Collections. J. Sep. Sci. 2025, 48, e70159. [Google Scholar] [CrossRef]
- Popova, L.; Carr, R.A.; Carabetta, V.J. Recent Contributions of Proteomics to Our Understanding of Reversible Nε-Lysine Acylation in Bacteria. J. Proteome Res. 2024, 23, 2733–2749. [Google Scholar] [CrossRef]
- McClellan, J.; Ómarsdóttir, S.; Roy, N.; Berger, V.; Michel, C.; Berti, F. The totality of evidence approach in the development of AVT02 (adalimumab), a biosimilar to Humira. Ther. Adv. Chronic Dis. 2024, 15, 20406223231223286. [Google Scholar] [CrossRef]
- Lerch, T.; Sharpe, P.; Mayclin, S.; Edwards, T.; Polleck, S.; Rouse, J.; Zou, Q.; Conlon, H.D. Crystal structures of PF-06438179/GP1111, an infliximab biosimilar. BioDrugs 2019, 34, 77–87. [Google Scholar] [CrossRef]
- Tamara, S.; Boer, M.; Heck, A. High-resolution native mass spectrometry. Chem. Rev. 2021, 122, 7269–7326. [Google Scholar] [CrossRef]
- Camperi, J.; Goyon, A.; Guillarme, D.; Zhang, K.; Stella, C. Multi-dimensional LC-MS: The next generation characterization of antibody-based therapeutics by unified online bottom-up, middle-up and intact approaches. Analyst 2021, 146, 747–769. [Google Scholar] [CrossRef]
- Zahel, T. A novel bootstrapping test for analytical biosimilarity. AAPS J. 2022, 24, 112. [Google Scholar] [CrossRef]
- Wen, J.; Razick, A.; How-Volkman, C.; Bernstein, E.; Nadora, D.; Truong, A.; Razick, D.; Akhtar, M.; Karabala, M.; Frezza, E. An exploratory analysis of glucagon-like peptide-1 (GLP-1) agonists and biosimilars: A literature review. Diabetes Obes. Metab. 2024, 27, 1113–1122. [Google Scholar] [CrossRef] [PubMed]
- Pedro, M.; Klijn, M.; Eppink, M.; Ottens, M. Process analytical technique (PAT) miniaturization for monoclonal antibody aggregate detection in continuous downstream processing. J. Chem. Technol. Biotechnol. 2021, 97, 2347–2364. [Google Scholar] [CrossRef]
- Kim, E.; Kim, J.; Kim, M.; Jeong, S.; Choi, D. Process analytical technology tools for monitoring pharmaceutical unit operations: A control strategy for continuous process verification. Pharmaceutics 2021, 13, 919. [Google Scholar] [CrossRef] [PubMed]
- Esmonde-White, K.; Cuellar, M.; Lewis, I. The role of Raman spectroscopy in biopharmaceuticals from development to manufacturing. Anal. Bioanal. Chem. 2021, 414, 969–991. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, C.; Wasalathanthri, D.; Ritz, D.; Zhou, G.; Davis, K.; Wucherpfennig, T.; Hazelwood, N. Systematic assessment of process analytical technologies for biologics. Biotechnol. Bioeng. 2021, 119, 423–434. [Google Scholar] [CrossRef]
- Klein, K.; Gencoglu, M.; Heisterberg, J.; Acha, V.; Stolk, P. The Global Landscape of Manufacturers of Follow-On Biologics: An Overview of Five Major Biosimilar Markets and 15 Countries. BioDrugs 2022, 37, 235–245. [Google Scholar] [CrossRef]
- Gandhi, S.; Patankar, D.; Kashiramka, S.; Rathore, A.S. The Economics of Translating a Biosimilar from Lab to Market in India. Ann. N. Y. Acad. Sci. 2024, 1541, 219–229. [Google Scholar] [CrossRef]
- Lee, J.J.; Lee, N.; Seo, Y.J.; Kim, I. Consistency of Product Quality for SB5, an Adalimumab Biosimilar. BioDrugs 2023, 37, 271–277. [Google Scholar] [CrossRef]
- Lees, J.; Dias, J.; Han, S. Applications of cryo-EM in small molecule and biologics drug design. Biochem. Soc. Trans. 2021, 49, 2627–2638. [Google Scholar] [CrossRef]
- Deshmukh, A.; Goyal, R.; Sundaram, K.; Dange, K.; Lakhote, T.; Niranjan, S.; Bharucha, J.; Mishra, A.; Vats, B.; Tiwari, S.; et al. Analytical sameness methodology for the evaluation of structural, physicochemical, and biological characteristics of ARMLUPEG: A pegfilgrastim biosimilar case study. PLoS ONE 2023, 18, e0289745. [Google Scholar] [CrossRef]
- Stüber, J.C.; Uhland, K.; Reiter, A.; Jakob, S.; Wolschin, F. Comparative Analytical Evaluation of the Proposed Biosimilar FYB206 and Its Reference Medicinal Product Keytruda®. Drugs RD 2024, 24, 447–464. [Google Scholar] [CrossRef] [PubMed]
- Shukla, V.; Niveria, K.; Shashidhar, P.; Verma, A. Dynamic Light Scattering (DLS) Particle Size Analysis for Biomedical Nanotechnology. In Analytical Techniques for Biomedical Nanotechnology; IOP Publishing: Bristol, UK, 2023; pp. 16-1–16-25. [Google Scholar] [CrossRef]
- Kang, H.; Wadhwa, M.; Knežević, I.; Ondari, C.; Simão, M. WHO Guidelines on Biosimilars: Toward Improved Access to Safe and Effective Products. Ann. N. Y. Acad. Sci. 2023, 1521, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Chari, A.; Stark, H. Prospects and limitations of high-resolution single-particle cryo-electron microscopy. Annu. Rev. Biophys. 2023, 52, 391–411. [Google Scholar] [CrossRef] [PubMed]
- Kryshtafovych, A.; Moult, J.; Albrecht, R.; Chang, G.; Chao, K.; Fraser, A.; Greenfield, J.; Hartmann, M.D.; Herzberg, O.; Josts, I.; et al. Computational models in the service of X-ray and cryo-electron microscopy structure determination. Proteins 2021, 89, 1633–1646. [Google Scholar] [CrossRef]
- Tercan, H.; Meisen, T. Machine learning and deep learning based predictive quality in manufacturing: A systematic review. J. Intell. Manuf. 2022, 33, 1879–1905. [Google Scholar] [CrossRef]
- Gangwar, N.; Balraj, K.; Rathore, A. Explainable AI for CHO cell culture media optimization and prediction of critical quality attribute. Appl. Microbiol. Biotechnol. 2024, 108, 308. [Google Scholar] [CrossRef]
- Kumar, N.; Pakalapati, A.; Chandrasekhar, K.; Durthi, C. Comparison of analytical method validation guidelines used for release, stability in biosimilar manufacturing process. Curr. Trends Biotechnol. Pharm. 2024, 18, 1798–1812. [Google Scholar] [CrossRef]
- Borza, B.; Hajba, L.; Guttman, A. N-glycan analysis in molecular medicine: Innovator and biosimilar protein therapeutics. Curr. Mol. Med. 2021, 20, 828–839. [Google Scholar] [CrossRef]
- Kodumuru, R.; Sarkar, S.; Parepally, V.; Chandarana, J. Artificial Intelligence and Internet of Things Integration in Pharmaceutical Manufacturing: A Smart Synergy. Pharmaceutics 2025, 17, 290. [Google Scholar] [CrossRef]
- Wu, C.; Chan, B.; Sarich, Z.; Duan, Y.; Chen, J.; Song, J.; Goudar, C.T. Accelerating Attribute-Focused Process and Product Development through the Development and Deployment of Autonomous Process Analytical Technology Platform System. Biotechnol. Bioeng. 2024, 121, 1256–1269. [Google Scholar] [CrossRef]
- Feng, X.; Ma, Z.; Yu, C.; Xin, R. MRNDR: Multihead Attention-Based Recommendation Network for Drug Repurposing. J. Chem. Inf. Model. 2024, 64, 2654–2669. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Chao, B.; Zhang, C.; Li, Y.; Jing-liang, L. Real-Time Tool Wear Monitoring and Multi-Step Forward Prediction Based on Multi-Information Fusion. Meas. Sci. Technol. 2025, 36, 025104. [Google Scholar] [CrossRef]
- Basaure, V. Models in medicine: The digital twin for health. SerieST 2025, 10, 3–16. [Google Scholar] [CrossRef]
- Haq, M.A.U.; Rehman, S.U.; Alhulayyil, H.A.; Alzahrani, T.; AlSagri, H.S.; Faheem, M. Wireless Antenna Sensors for Biosimilar Monitoring toward Cyber-Physical Systems: A Review of Current Trends and Future Prospects. IEEE Access 2023, 11, 132037–132054. [Google Scholar] [CrossRef]
- Kuang, X. Application and Difficulties of Deep Learning in Drug Target Discovery. MedScien 2024, 1, 1–4. [Google Scholar] [CrossRef]
- Kalakoti, Y.; Yadav, S.; Sundar, D. TransDTI: Transformer-based language models for estimating DTIs and building a drug recommendation workflow. ACS Omega 2022, 7, 2706–2717. [Google Scholar] [CrossRef]
- Lee, S.; Cho, J.; Lee, B.; Hwang, D.; Park, J. Design and prediction of aptamers assisted by in silico methods. Biomedicines 2023, 11, 356. [Google Scholar] [CrossRef]
- Saleh, D.; Hess, R.; Ahlers-Hesse, M.; Rischawy, F.; Wang, G.; Grosch, J.; Schwab, T.; Kluters, S.; Studts, J.; Hubbuch, J. A multiscale modeling method for therapeutic antibodies in ion exchange chromatography. Biotechnol. Bioeng. 2022, 120, 125–138. [Google Scholar] [CrossRef]
- Wittkopp, F.; Welsh, J.P.; Todd, R.; Staby, A.; Roush, D.J.; Lyall, J.Y.; Babi, D.K. Current State of Implementation of In Silico Tools in the Biopharmaceutical Industry—Proceedings of the 5th Modeling Workshop. Biotechnol. Bioeng. 2024, 121, 2952–2973. [Google Scholar] [CrossRef]
- Wang, X.; Mohsin, A.; Sun, Y.; Li, C.; Zhuang, Y.; Wang, G. From Spatial-Temporal Multiscale Modeling to Application: Bridging the Valley of Death in Industrial Biotechnology. Bioengineering 2023, 10, 744. [Google Scholar] [CrossRef]
- Dutta, D.J.; Kumar, P.; Singh, A.; Khade, S. Bioprocess Strategies for Enhanced Performance in Single-Use Bioreactors for Biomolecule Synthesis: A Biokinetic Approach. Food Bioeng. 2024, 3, 337–351. [Google Scholar] [CrossRef]
- Islam, A.; Jena, D.; Mondal, N.; Teli, A.; Mondal, S.; Gautam, M. In-silico approaches for drug designing technology: Bridging discovery and development. Curr. Drug Discov. Technol. 2025, 22, e15701638326869. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Pan, S.; Chen, F.; Long, G.; Zhang, C.; Yu, P. A comprehensive survey on graph neural networks. IEEE Trans. Neural Netw. Learn. Syst. 2021, 32, 4–24. [Google Scholar] [CrossRef] [PubMed]
- Ye, W.; Li, J.; Cai, X. MF-GNN: Multi-scale feature-attentive graph neural networks for molecular property prediction. J. Comput. Chem. 2025, 46, e70011. [Google Scholar] [CrossRef]
- Shi, X.; Zhou, L.; Huang, Y.; Wu, Y.; Hong, Z. A review on the applications of graph neural networks in materials science at the atomic scale. Mater. Genome Eng. Adv. 2024, 2, e50. [Google Scholar] [CrossRef]
- Satheeskumar, R. Enhancing drug discovery with AI: Predictive modeling of pharmacokinetics using graph neural networks and ensemble learning. Intell. Pharm. 2025, 3, 127–140. [Google Scholar] [CrossRef]
- Shi, Z.; Ma, M.; Ning, H.; Yang, B.; Dang, J. A multiscale molecular structural neural network for molecular property prediction. Mol. Divers. 2025. [Google Scholar] [CrossRef]
- Pancino, N.; Gallegati, C.; Romagnoli, F.; Bongini, P.; Bianchini, M. Protein–protein interfaces: A graph neural network approach. Int. J. Mol. Sci. 2024, 25, 5870. [Google Scholar] [CrossRef]
- Dong, L.; Shi, S.; Qu, X.; Ding, L.; Wang, B. Ligand binding affinity prediction with fusion of graph neural networks and 3D structure-based complex graph. Phys. Chem. Chem. Phys. 2023, 25, 24110–24120. [Google Scholar] [CrossRef]
- Shi, W.; Singha, M.; Srivastava, G.; Pu, L.; Ramanujam, J.; Bryliński, M. Pocket2drug: An encoder-decoder deep neural network for the target-based drug design. Front. Pharmacol. 2022, 13, 837715. [Google Scholar] [CrossRef]
- Davis, D.; Jackson, K.; Hoppe, C. Evaluation of Cost and Infusion-Related Reactions among Intravenous Trastuzumab, Subcutaneous Trastuzumab, and Trastuzumab Biosimilars at an Academic Medical Center. J. Oncol. Pharm. Pract. 2024, 31, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Du, K.; Zhang, W.; Mao, J. Empowering Digital Transformation: The Roles of Platforms. J. Inf. Technol. 2023, 39, 650–667. [Google Scholar] [CrossRef]
- Chkarat, H.; Abid, T.; Sauvée, L. Conditions for a Convergence between Digital Platforms and Sustainability in Agriculture. Sustainability 2023, 15, 14195. [Google Scholar] [CrossRef]
- Junaid, M.A.L. Artificial Intelligence Driven Innovations in Biochemistry: A Review of Emerging Research Frontiers. Biomol. Biomed. 2025, 25, 739–750. [Google Scholar] [CrossRef]
- Langer, E. Single-use technology in biopharmaceutical manufacture and beyond. Chem. Ing. Tech. 2022, 94, 1892–1901. [Google Scholar] [CrossRef]
- Ding, L.; Bazaz, S.; Fardjahromi, M.; Mckinnirey, F.; Saputro, B.; Banerjee, B.; Banerjee, B.; Vesey, G.; Warkiani, M.E. A modular 3D printed microfluidic system: A potential solution for continuous cell harvesting in large-scale bioprocessing. Bioresour. Bioprocess. 2022, 9, 64. [Google Scholar] [CrossRef] [PubMed]
- Saha, R.; Sarkar, M.; Choudhury, S.S.; Kumar, H.; Bhatt, G.; Bhattacharya, S. Evolution of 3D Printing Technology in Fabrication of Microfluidic Devices and Biological Applications: A Comprehensive Review. J. Micromanuf. 2024, 7, 110–140. [Google Scholar] [CrossRef]
- Brunner, V.; Siegl, M.; Geier, D.; Becker, T. Challenges in the Development of Soft Sensors for Bioprocesses: A Critical Review. Front. Bioeng. Biotechnol. 2021, 9, 722202. [Google Scholar] [CrossRef]
- Rivera, E.C.; Yamakawa, C.K.; Rossell, C.E.; Nolasco, J.; Kwon, H.J. Prediction of Intensified Ethanol Fermentation of Sugarcane Using a Deep Learning Soft Sensor and Process Analytical Technology. J. Chem. Technol. Biotechnol. 2023, 99, 207–216. [Google Scholar] [CrossRef]
- Dürauer, A.; Jungbauer, A.; Scharl, T. Sensors and Chemometrics in Downstream Processing. Biotechnol. Bioeng. 2023, 121, 2347–2364. [Google Scholar] [CrossRef]
- Akram, M.; Jabeen, F.; Daniyal, M.; Zainab, R.; Haq, U.u.; Adetunji, C.O.; Egbuna, C.; Ephraim-Emmanuel, B.C.; Patrick-Iwuanyanwu, K.C.; Ogbo, A.B. Genetic engineering of novel products of health significance: Recombinant DNA technology. In Functional Foods and Nutraceuticals; Springer: Cham, Switzerland, 2020; pp. 595–611. [Google Scholar] [CrossRef]
- Bellinvia, S.; Edwards, C.J. Explaining biosimilars and how reverse engineering plays a critical role in their development. Expert Opin. Drug Discov. 2020, 15, 1283–1289. [Google Scholar] [CrossRef] [PubMed]
- Svilenov, H.; Kulakova, A.; Zalar, M.; Golovanov, A.; Harris, P.; Winter, G. Orthogonal techniques to study the effect of pH, sucrose, and arginine salts on monoclonal antibody physical stability and aggregation during long-term storage. J. Pharm. Sci. 2020, 109, 584–594. [Google Scholar] [CrossRef] [PubMed]
- Castañeda Ruiz, A.J.; Shetab Boushehri, M.A.; Phan, T.; Carle, S.; Garidel, P.; Buske, J.; Lamprecht, A. Alternative excipients for protein stabilization in protein therapeutics: Overcoming the limitations of polysorbates. Pharmaceutics 2022, 14, 2575. [Google Scholar] [CrossRef] [PubMed]
- Panda, C.; Kumar, S.; Gupta, S.; Pandey, L.M. Insulin fibrillation under physicochemical parameters of bioprocessing and intervention by peptides and surface-active agents. Crit. Rev. Biotechnol. 2024, 45, 643–664. [Google Scholar] [CrossRef]
- Abitbol, V.; Benkhalifa, S.; Habauzit, C.; Marotte, H. Navigating Adalimumab Biosimilars: An Expert Opinion. J. Comp. Eff. Res. 2023, 12, e230117. [Google Scholar] [CrossRef]
- Wu, Y.; Gardner, R.; Schöneich, C. Near UV and visible light-induced degradation of bovine serum albumin and a monoclonal antibody mediated by citrate buffer and Fe(III): Reduction vs oxidation pathways. Mol. Pharm. 2024, 21, 4060–4073. [Google Scholar] [CrossRef]
- Abdulkareem, R.; Doekhie, A.; Fotaki, N.; Koumanov, F.; Dodson, C.; Sartbaeva, A. Thermal Stabilisation of Lysozyme through Ensilication. Molecules 2024, 29, 4207. [Google Scholar] [CrossRef] [PubMed]
- Akbarian, M.; Farjadian, F. Chitosan-Coated Glass Beads as Stabilizer of Insulin; A Novel Strategy for the Storage of Pharmaceutical Proteins. ChemistrySelect 2024, 9, e202402813. [Google Scholar] [CrossRef]
- Koynov, A.; Lin, W.; Bothe, J.; Schenck, L.; Parajuli, B.; Li, Z.; Ruzanski, R.; Hoffman, N.; Frank, D.; VanAernum, Z. A precipitation-based process to generate a solid formulation of a therapeutic monoclonal antibody: An alternative to lyophilization. JPBI 2025, 2, 2. [Google Scholar] [CrossRef]
- Liu, X.; Dong, Y.; Wang, C.; Guo, Z. Application of Chitosan as Nano Carrier in the Treatment of Inflammatory Bowel Disease. Int. J. Biol. Macromol. 2024, 278, 134899. [Google Scholar] [CrossRef]
- Arcos Rosero, W.A.; Bueno Barbezan, A.; Daruich de Souza, C.; Chuery Martins Rostelato, M.E. Review of Advances in Coating and Functionalization of Gold Nanoparticles: From Theory to Biomedical Application. Pharmaceutics 2024, 16, 255. [Google Scholar] [CrossRef] [PubMed]
- Aparna, M.; Giuggioli, N.R. Chitosan Based Edible Coatings: Enhancing Shelf Life and Quality in Fruits and Vegetables. J. Adv. Biol. Biotechnol. 2024, 27, 178–191. [Google Scholar] [CrossRef]
- Ghafoor, K.; Juhaimi, F.A.; Babiker, E.E.; Ahmed, I.A.M.; Shahzad, S.A.; Alsawmahi, O.N. Quality Attributes of Refrigerated Barhi Dates Coated with Edible Chitosan Containing Natural Functional Ingredients. Foods 2022, 11, 1584. [Google Scholar] [CrossRef] [PubMed]
- Skerritt, J.H. Considerations for mRNA Product Development, Regulation and Deployment Across the Lifecycle. Vaccines 2025, 13, 473. [Google Scholar] [CrossRef]
- Zárate-Moreno, J.C.; Escobar-Sierra, D.M.; Ríos-Estepa, R. Development and Evaluation of Chitosan-Based Food Coatings for Exotic Fruit Preservation. BioTech 2023, 12, 20. [Google Scholar] [CrossRef]
- Aguiar, A.C.d.; Bianchi, J.R.O.; Lopes, J.H.; Ferreira, F.V.; Lona, L.M.F. Nanocellulose-Based Capsules with pH Responsiveness for Colon-Targeted Curcumin Delivery. ACS Appl. Nano Mater. 2025, 8, 2033–2045. [Google Scholar] [CrossRef]
- Hemmingsen, L.M.; Panchai, P.; Julin, K.; Basnet, P.; Nystad, M.; Johannessen, M.; Škalko-Basnet, N. Chitosan-Based Delivery System Enhances Antimicrobial Activity of Chlorhexidine. Front. Microbiol. 2022, 13, 1023083. [Google Scholar] [CrossRef]
- Jia, Y.; Cao, J.; Zhou, J.; Ping, Z. Methyl Chitosan Coating for Glycoform Analysis of Glycoproteins by Capillary Electrophoresis. Electrophoresis 2020, 41, 729–734. [Google Scholar] [CrossRef]
- Cao, X.; Islam, M.N.; Chitrakar, B.; Duan, Z.; Xu, W.; Zhong, S. Effect of Combined Chlorogenic Acid and Chitosan Coating on Antioxidant, Antimicrobial, and Sensory Properties of Snakehead Fish in Cold Storage. Food Sci. Nutr. 2020, 8, 973–981. [Google Scholar] [CrossRef]
- Fan, S.; Wang, H.; Yu, Y.; Cao, S.; Lin, Y. Advancements in the Utilization of Chitosan and Its Derivatives for Metal Corrosion Inhibition: A Comprehensive Review. Acad. J. Sci. Technol. 2024, 11, 81–84. [Google Scholar] [CrossRef]
- Banks, D.; Kempf, J.; Du, Y.; Reichert, P.; Narasimhan, C.; Fang, R.; Kwon, S.; Ling, J.; Lay-Fortenbery, A.; Zhang, Y.; et al. Investigation of protein therapeutics in frozen conditions using DNP MAS NMR: A study on pembrolizumab. Mol. Pharm. 2024, 21, 6363–6375. [Google Scholar] [CrossRef] [PubMed]
- Fox, G.; Bernauer, M.; Stephens, J.; Jackson, B.; Roth, J.; Shelbaya, A. Clinical and economic value of a biosimilar portfolio to stakeholders: An integrative literature review. Clin. Outcomes Res. 2024, 16, 247–416. [Google Scholar] [CrossRef] [PubMed]
- Almaaytah, A. Technical guidance on the physicochemical and functional comparability exercise for trastuzumab biosimilars. Int. J. Appl. Pharm. 2022, 14, 71–76. [Google Scholar] [CrossRef]
- Yang, S. Aspects and implementation of pharmaceutical quality by design from conceptual frameworks to industrial applications. Pharmaceutics 2025, 17, 623. [Google Scholar] [CrossRef] [PubMed]
- Huizinga, T.; Torii, Y.; Muniz, R. Adalimumab biosimilars in the treatment of rheumatoid arthritis: A systematic review of the evidence for biosimilarity. Rheumatol. Ther. 2020, 8, 41–61. [Google Scholar] [CrossRef]
- Cook, J.; McGrath, M.; Dixon, M.; Switchenko, J.; Harvey, R.; Pentz, R. Academic oncology clinicians’ understanding of biosimilars and information needed before prescribing. Ther. Adv. Med. Oncol. 2019, 11, 1758835918818335. [Google Scholar] [CrossRef]
- Derikx, L.; Dolby, H.; Plevris, N.; Lucaciu, L.; Rees, C.; Lyons, M.; Siakavellas, S.I.; Constantine-Cooke, N.; Jenkinson, P.; Su, S.; et al. Effectiveness and safety of adalimumab biosimilar SB5 in inflammatory bowel disease: Outcomes in originator to SB5 switch, double biosimilar switch and bio-naïve SB5 observational cohorts. J. Crohn’s Colitis 2021, 15, 2011–2021. [Google Scholar] [CrossRef]
- Fadhil, H.; Al-Jumaili, A.; Al-Ani, N. Cost-effectiveness analysis of reference infliximab (Remicade) compared to its biosimilar (Remsima) in Iraqi patients with rheumatoid arthritis (conference paper). Iraqi J. Pharm. Sci. 2023, 31, 100–110. [Google Scholar] [CrossRef]
- Smolen, J.; Gonçalves, J.; Quinn, M.; Benedetti, F.; Lee, J. Era of biosimilars in rheumatology: Reshaping the healthcare environment. RMD Open 2019, 5, e000900. [Google Scholar] [CrossRef]
- Blevins, T.; Barve, A.; Raiter, Y.; Aubonnet, P.; Athalye, S.; Sun, B.; Muniz, R. Efficacy and safety of MYL-1501D versus insulin glargine in people with type 1 diabetes mellitus: Results of the INSTRIDE 3 phase 3 switch study. Diabetes Obes. Metab. 2019, 22, 365–372. [Google Scholar] [CrossRef]
- Shuptrine, C.; Chen, Y.; Miriyala, J.; Lenz, K.; Moffett, D.; Nguyen, T.; Michaux, J.; Campbell, K.; Smith, C.; Morra, M.; et al. Lipid-encapsulated mRNAs encoding complex fusion proteins potentiate antitumor immune responses. Cancer Res. 2024, 84, 1550–1559. [Google Scholar] [CrossRef] [PubMed]
- Strickley, R.; Lambert, W. A review of formulations of commercially available antibodies. J. Pharm. Sci. 2021, 110, 2590–2608.e56. [Google Scholar] [CrossRef] [PubMed]
- Kurki, P.; Barry, S.; Bourges, I.; Tsantili, P.; Wolff-Holz, E. Safety, immunogenicity and interchangeability of biosimilar monoclonal antibodies and fusion proteins: A regulatory perspective. Drugs 2021, 81, 1881–1896. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Chen, C. Carcinomembrane-camouflaged perfluorochemical dual-layer nanopolymersomes bearing indocyanine green and camptothecin effectuate targeting photochemotherapy of cancer. ACS Biomater. Sci. Eng. 2024, 10, 6332–6343. [Google Scholar] [CrossRef]
- Ionova, Y.; Wilson, L. Biologic excipients: Importance of clinical awareness of inactive ingredients. PLoS ONE 2020, 15, e0235076. [Google Scholar] [CrossRef]
- Krishna, M. Product-related factors and immunogenicity of biotherapeutics. J. Pharm. Innov. 2019, 15, 219–231. [Google Scholar] [CrossRef]
- Grudzinska-Goebel, J.; Benstein, K.; Bloem, K.; Cowan, K.J.; Gorovits, B.; Jadhav, M.; Janssen, M.; Jawa, V.; Kiessling, A.; Kramer, D. Immunogenicity risk assessment for tailored mitigation and monitoring of biotherapeutics during development: Recommendations from the European immunogenicity platform. Front. Immunol. 2025, 16, 1581153. [Google Scholar] [CrossRef]
- Previ, T.; Rodrigues, A. Relationship between innovative and biosimilar trastuzumab used in the treatment of breast cancer: Cross-sectional study of financial impact. J. Cancer Prev. Curr. Res. 2021, 12, 123–126. [Google Scholar] [CrossRef]
- Xin, L.; Lan, L.; Mellal, M.; McChesney, N.; Vaughan, R.; Berdugo, C.; Li, Y.; Zhang, J. Leveraging high-throughput analytics and automation to rapidly develop high-concentration mAb formulations: Integrated excipient compatibility and viscosity screening. Antib. Ther. 2024, 7, 335–350. [Google Scholar] [CrossRef]
- Goli, V.A.R.; Butreddy, A. Biosimilar monoclonal antibodies: Challenges and approaches towards formulation. Chem. -Biol. Interact. 2022, 366, 110116. [Google Scholar] [CrossRef]
- Ingle, R.G.; Fang, W. An Overview of the Stability and Delivery Challenges of Commercial Nucleic Acid Therapeutics. Pharmaceutics 2023, 15, 1158. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.; Kundu, A.; Hageman, M.; Lou, H.; Boisvert, D. Monoclonal Antibody and Protein Therapeutic Formulations for Subcutaneous Delivery: High-Concentration, Low-Volume vs. Low-Concentration, High-Volume. mAbs 2023, 15, 2285277. [Google Scholar] [CrossRef] [PubMed]
- Papamichael, K.; Stocco, G.; Agua, A.R.d. Challenges in Therapeutic Drug Monitoring: Optimizing Biological Treatments in Patients with Inflammatory Bowel Disease and Other Immune-Mediated Inflammatory Diseases. Ther. Drug Monit. 2023, 45, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.; Lee, M.; Baek, Y.; Lee, S. A Randomized Pharmacokinetic Study in Healthy Male Subjects Comparing a High-Concentration, Citrate-Free SB5 Formulation (40 mg/0.4 mL) and Prior SB5 (Adalimumab Biosimilar). Rheumatol. Ther. 2022, 9, 1157–1169. [Google Scholar] [CrossRef]
- D’Abbundo, G.; Nachury, M.; Wartski, A.; Blondeaux, A.; Hambli, S.; Gérard, R.; Wils, P. Switch Acceptance and Persistence of Adalimumab Biosimilars in IBD Patients: A Prospective Observational Study. Ther. Adv. Gastroenterol. 2025, 18, 17562848251332025. [Google Scholar] [CrossRef]
- Lv, J.; Ingle, R.G.; Wu, H.; Liu, C.; Fang, W. Histidine as a Versatile Excipient in the Protein-Based Biopharmaceutical Formulations. Int. J. Pharm. 2024, 662, 124472. [Google Scholar] [CrossRef]
- Zarzar, J.; Khan, T.A.; Bhagawati, M.; Weiche, B.; Sydow-Andersen, J.; Sreedhara, A. High concentration formulation developability approaches and considerations. mAbs 2023, 15, 2211185. [Google Scholar] [CrossRef]
- Ghosh, I.; Gutka, H.J.; Krause, M.E.; Clemens, R.; Kashi, R.S. A systematic review of commercial high concentration antibody drug products approved in the US: Formulation composition, dosage form design and primary packaging considerations. mAbs 2023, 15, 2205540. [Google Scholar] [CrossRef]
- Lebar, B.; Zidar, M.; Mravljak, J.; Šink, R.; Žula, A.; Pajk, S. Alternative buffer systems in biopharmaceutical formulations and their effect on protein stability. Pharm. Acta 2024, 74, 479–493. [Google Scholar] [CrossRef]
- Pardeshi, S.R.; Deshmukh, N.S.; Telange, D.R.; Nangare, S.; Sonar, Y.Y.; Lakade, S.; Harde, M.T.; Pardeshi, C.V.; Gholap, A.; Deshmukh, P.K.; et al. Process development and quality attributes for the freeze-drying process in pharmaceuticals, biopharmaceuticals and nanomedicine delivery: A state-of-the-art review. Future J. Pharm. Sci. 2023, 9, 99. [Google Scholar] [CrossRef]
- Upadhya, R.; Yarger, T.; Blanco, M.; Bothe, J.; Pabit, S.; XI, H. Leveraging SAXS for biologics formulation development in the pharmaceutical industry. Struct. Dyn. 2025, 12 (Suppl. S2), A189. [Google Scholar] [CrossRef]
- Mieczkowski, C. The evolution of commercial antibody formulations. J. Pharm. Sci. 2023, 112, 1801–1810. [Google Scholar] [CrossRef] [PubMed]
- Erfani, A.; Schieferstein, J.; Reichert, P.; Narasimhan, C.; Pastuskovas, C.; Parab, V.; Simmons, D.; Yang, X.; Shanker, A.; Hammond, P.; et al. Crystalline antibody-laden alginate particles: A platform for enabling high concentration subcutaneous delivery of antibodies. Adv. Healthc. Mater. 2023, 12, 2202370. [Google Scholar] [CrossRef] [PubMed]
- Saurabh, S.; Li, Z.; Hollowell, P.; Waigh, T.; Li, P.; Webster, J.; Seddon, J.M.; Kalonia, C.; Lu, J.R.; Bresme, F. Structure and interaction of therapeutic proteins in solution: A combined simulation and experimental study. Mol. Phys. 2023, 121, e2236248. [Google Scholar] [CrossRef] [PubMed]
- Mosca, I.; Pounot, K.; Beck, C.; Colin, L.; Matsarskaia, O.; Grapentin, C.; Seydel, T.; Schreiber, F. Biophysical determinants for the viscosity of concentrated monoclonal antibody solutions. Mol. Pharm. 2023, 20, 4698–4713. [Google Scholar] [CrossRef] [PubMed]
- Zürcher, D.; Wuchner, K.; Arosio, P. Real-Time Observation of Protein Aggregation at Liquid-Liquid Interfaces in a Microfluidic Device. Small 2024, 20, 2401085. [Google Scholar] [CrossRef]
- Meza, N.P.; Hardy, C.A.; Morin, K.H.; Huang, C.; Raghava, S.; Song, J.; Wang, Y. Predicting Colloidal Stability of High-Concentration Monoclonal Antibody Formulations in Common Pharmaceutical Buffers Using Improved Polyethylene Glycol Induced Protein Precipitation Assay. Mol. Pharm. 2023, 20, 5842–5855. [Google Scholar] [CrossRef]
- Kramar, H.I.; Kryvoviaz, O.V.; Tomashevska, Y.O.; Toziuk, O.I.; Kudria, V.V.; Koval, V.M.; Alchuk, O.I. Analysis of the use of excipients in medicines of biological origin. Rep. Vinnytsia Natl. Med. Univ. 2024, 28, 145–150. [Google Scholar] [CrossRef]
- Xiao, E.; Mirabel, C.; Clénet, D.; Zhu, S.; James, A.; Ettorre, L.; Williams, T.; Szeto, J.; Rahman, N.; Ausar, S.F. Formulation development of a COVID-19 recombinant spike protein-based vaccine. Vaccines 2024, 12, 830. [Google Scholar] [CrossRef]
- Rospiccio, M.; Casucci, P.; Arsiccio, A.; Udrescu, C.; Pisano, R. Mechanistic investigation of tert-butanol’s impact on biopharmaceutical formulations: When experiments meet molecular dynamics. Mol. Pharm. 2023, 20, 3975–3986. [Google Scholar] [CrossRef]
- Park, J.; Liu, R.; Kim, A.; Cyr, N.; Boehlein, S.; Resende, M.; Savin, D.A.; Bailey, L.S.; Sumerlin, B.S.; Hudalla, G.A. Sweet corn phytoglycogen dendrimers as a lyoprotectant for dry-state protein storage. J. Biomed. Mater. Res. A 2024, 112, 2026–2041. [Google Scholar] [CrossRef] [PubMed]
- Lebar, B.; Lekić, T.; Košir, P.; Kastelic, M.; Zidar, M.; Mravljak, J.; Pajk, S. Polysorbate stability: Effects of packaging materials, buffers, counterions, and ph. Int. J. Pharm. 2024, 665, 124598. [Google Scholar] [CrossRef] [PubMed]
- Žiberna, M.; Grabnar, P.; Gašperlin, M.; Matjaž, M. Lyophilised protein formulations as a patient-centric dosage form: A contribution toward sustainability paradigm. Acta Pharm. 2024, 74, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Niazi, S. No two classes of biosimilars: Urgent advice to the US Congress and the FDA. J. Clin. Pharm. Ther. 2022, 47, 1352–1361. [Google Scholar] [CrossRef]
- Gianoncelli, A.; Bertuzzi, M.; Guarienti, M.; Vezzoli, S.; Bonini, S.A.; Mastinu, A.; Sigala, S.; Memo, M. Parallelism of chemicostructural properties between filgrastim originator and three of its biosimilar drugs. J. Chem. 2019, 2019, 2751461. [Google Scholar] [CrossRef]
- Ghasriani, H.; Frahm, G.; Johnston, M.; Aubin, Y. Effects of excipients on the structure and dynamics of filgrastim monitored by thermal unfolding studies by CD and NMR spectroscopy. ACS Omega 2020, 5, 31845–31857. [Google Scholar] [CrossRef]
- Arvinte, T.; Poirier, E.; Cudd, A.; Ersayin, N.; Darpin, G.; Dowd, J.; Brokx, S. Aggregation of human plasma and of human blood induced in vitro by filgrastim originator product; effect of PEGylation. Eur. J. Pharm. Biopharm. 2024, 194, 148–158. [Google Scholar] [CrossRef]
- Lonshakov, D.V.; Sheremet’ev, S.V.; Khursevich, E.M.; Bobrysheva, I.V.; Grebennikova, O.A.; Korovkin, S.A. Study of pH-dependent kinetics of filgrastim pegylation with succinimide derivatives of poly(ethylene glycol) and comparison of the properties of pegylated forms obtained under various conditions. Pharm. Chem. J. 2024, 58, 306–319. [Google Scholar] [CrossRef]
- Allegretti, J.; Brady, J.; Wicker, A.; Latymer, M.; Wells, A. Relevance of adalimumab product attributes to patient experience in the biosimilar era: A narrative review. Adv. Ther. 2024, 41, 1775–1794. [Google Scholar] [CrossRef]
- Chiu, I.; Tsai, T. Risk factors of ixekizumab-induced injection site reactions in patients with psoriatic diseases: Report from a single medical center. Biomedicines 2023, 11, 1718. [Google Scholar] [CrossRef]
- Patel, A.; Luu, P. Changes in patient reported pain measures with the citrate-free adalimumab formulation in pediatric inflammatory bowel disease patients. JPGN Rep. 2020, 1, e016. [Google Scholar] [CrossRef] [PubMed]
- Feldman, S.; Reznichenko, N.; Pulka, G.; Kingo, K.; Galdava, G.; Berti, F.; Sobierska, J.; Dias, R.; Guenzi, E.; Otto, H.; et al. Efficacy, safety and immunogenicity of AVT02 versus originator adalimumab in subjects with moderate to severe chronic plaque psoriasis: A multicentre, double-blind, randomised, parallel group, active control, phase III study. BioDrugs 2021, 35, 735–748. [Google Scholar] [CrossRef] [PubMed]
- Uricoli, B.; Birnbaum, L.A.; Do, P.; Kelvin, J.; Jain, J.; Costanza, E.; Chyong, A.; Porter, C.C.; Rafiq, S.; Dreaden, E.C. Engineered cytokines for cancer and autoimmune disease immunotherapy. Adv. Healthc. Mater. 2021, 10, 2002214. [Google Scholar] [CrossRef] [PubMed]
- Barbier, L.; Ebbers, H.C.; Declerck, P.; Simoens, S.; Vulto, A.G.; Huys, I. The efficacy, safety, and immunogenicity of switching between reference biopharmaceuticals and biosimilars: A systematic review. Clin. Pharmacol. Ther. 2020, 108, 734–755. [Google Scholar] [CrossRef] [PubMed]
- Vinukonda, A.; Rapolu, K.; Jadi, R.K.; Devadasu, V.R. Complex peptide injectables: Development and challenges. Int. J. Pept. Res. Ther. 2025, 31, 51. [Google Scholar] [CrossRef]
- Ismail, S.; Esba, L.; Khan, M.; Alabdulkarim, H.; Modimagh, H.; Yousef, C. An institutional guide for formulary decisions of biosimilars. Hosp. Pharm. 2022, 58, 38–48. [Google Scholar] [CrossRef]
- Li, J.; Krause, M.; Chen, X.; Cheng, Y.; Dai, W.; Hill, J.; Huang, M.; Jordan, S.; LaCasse, D.; Narhi, L.; et al. Interfacial stress in the development of biologics: Fundamental understanding, current practice, and future perspective. AAPS J. 2019, 21, 44. [Google Scholar] [CrossRef]
- Velankar, K.; Gawalt, E.; Wen, Y.; Meng, W. Pharmaceutical proteins at the interfaces and the role of albumin. Biotechnol. Prog. 2024, 40, e3474. [Google Scholar] [CrossRef]
- Tosstorff, A.; Menzen, T.; Winter, G. Exploring chemical space for new substances to stabilize a therapeutic monoclonal antibody. J. Pharm. Sci. 2020, 109, 301–307. [Google Scholar] [CrossRef]
- Bramham, J.; Davies, S.; Podmore, A.; Golovanov, A. Stability of a high-concentration monoclonal antibody solution produced by liquid–liquid phase separation. mAbs 2021, 13, 1940666. [Google Scholar] [CrossRef]
- Stolzke, T.; Krieg, F.; Peng, T.; Zhang, H.; Häusler, O.; Brandenbusch, C. Hydroxylpropyl-β-cyclodextrin as potential excipient to prevent stress-induced aggregation in liquid protein formulations. Molecules 2022, 27, 5094. [Google Scholar] [CrossRef] [PubMed]
- Bianco, S.; Hasan, M.; Ahmad, A.; Richards, S.; Dietrich, B.; Wallace, M.; Tang, Q.; Smith, A.J.; Gibson, M.I.; Adams, D.J. Mechanical release of homogenous proteins from supramolecular gels. Nature 2024, 631, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Escobar, E.; Vaclaw, M.; Lozenski, J.; Dhar, P. Using passive microrheology to measure the evolution of the rheological properties of NIST mAb formulations during adsorption to the air–water interface. Langmuir 2024, 40, 4789–4800. [Google Scholar] [CrossRef] [PubMed]
- Arsiccio, A.; McCarty, J.; Pisano, R.; Shea, J. Effect of Surfactants on Surface-Induced Denaturation of Proteins: Evidence of an Orientation-Dependent Mechanism. J. Phys. Chem. B 2018, 122, 11390–11399. [Google Scholar] [CrossRef]
- Buckland, B.C.; Sanyal, G.; Ranheim, T.; Pollard, D.; Searles, J.; Behrens, S.; Pluschkell, S.; Josefsberg, J.; Roberts, C.J. Vaccine process technology—A decade of progress. Biotechnol. Bioeng. 2024, 121, 2604–2635. [Google Scholar] [CrossRef]
- Crommelin, D.J.; Hawe, A.; Jiskoot, W. Formulation of biologics including biopharmaceutical considerations. In Pharmaceutical Biotechnology: Fundamentals and Applications; Springer International Publishing: Cham, Switzerland, 2024. [Google Scholar]
- Nongkhlaw, R.; Patra, P.; Chavrasiya, A.; Nirmal, J.; Dubey, S. Biologics: Delivery options and formulation strategies. In Drug Delivery Aspects; 2020; pp. 115–155. [Google Scholar] [CrossRef]
- Mathias, N.; Huille, S.; Picci, M.; Mahoney, R.P.; Pettis, R.J.; Case, B.; Helk, B.; Kang, D.; Shah, R.; Ma, J.; et al. Towards more tolerable subcutaneous administration: Review of contributing factors for improving combination product design. Adv. Drug Deliv. Rev. 2024, 209, 115301. [Google Scholar] [CrossRef]
- Knowles, S.; Printz, M.; Kang, D.; LaBarre, M.; Tannenbaum, R. Safety of recombinant human hyaluronidase PH20 for subcutaneous drug delivery. Expert Opin. Drug Deliv. 2021, 18, 1673–1685. [Google Scholar] [CrossRef]
- Gracia, G.; Cao, E.; Yuen, D.; Ferreira, V.; Chen, M.; Senyschyn, D.; Mintern, J.D.; Johnston, A.P.; Kang, D.W.; Feeney, O.M.; et al. Abstract B001: Removal of interstitial hyaluronan facilitates subcutaneous administration and lymphatic delivery of anti-CTLA4 antibody and improves anti-tumor efficacy. Cancer Immunol. Res. 2024, 12, B001. [Google Scholar] [CrossRef]
- Nolan, R.; Printz, M. Modeling the subcutaneous pharmacokinetics of antibodies co-administered with rHuPH20. Clin. Transl. Sci. 2024, 17, e13788. [Google Scholar] [CrossRef]
- Shibata, H.; Saitoh, S.; Kiyoshi, M.; Hayashi, Y.; Inaba, K.; Katsura, S.; Sakurai, M.; Komine, Y.; Okabe, S.; Ohbayashi, N.; et al. Survey and establishment of points to consider for application of analytical techniques to evaluate protein aggregates and insoluble particles in biopharmaceuticals: Experiences in japan biopharmaceutical consortium. AAPS J. 2025, 27, 3. [Google Scholar] [CrossRef]
- Fayed, B.; Luo, S.; Yassin, A.E.B. Challenges and recent advances in erythropoietin stability. Pharm. Dev. Technol. 2024, 29, 930–944. [Google Scholar] [CrossRef] [PubMed]
- Bohlius, J.; Bohlke, K.; Castelli, R.; Djulbegović, B.; Lustberg, M.; Martino, M.; Mountzios, G.; Peswani, N.; Porter, L.; Tanaka, T.N.; et al. Management of cancer-associated anemia with erythropoiesis-stimulating agents: ASCO/ASH clinical practice guideline update. Blood Adv. 2019, 3, 1197–1210. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Mendívil, C.; Kinsella, N.; Ebbers, H. A retrospective analysis of the potential impact of differences in aggregates on clinical immunogenicity of biosimilars and their reference products. Clin. Pharmacol. Ther. 2024, 115, 1122–1131. [Google Scholar] [CrossRef] [PubMed]
- Gascón, P.; Goldsmith, D.; Aapro, M.; Dellanna, F.; Esmael, A.; Zabransky, M. Epoetin alfa biosimilar (HX575): A review of 15 years’ post-approval clinical experience. Crit. Rev. Oncol./Hematol. 2023, 181, 103894. [Google Scholar] [CrossRef]
- Venetsanopoulou, A.I.; Voulgari, P.V.; Drosos, A.A. Optimizing withdrawal strategies for anti-TNF-α therapies in rheumatoid arthritis. Expert Opin. Biol. Ther. 2024, 24, 815–825. [Google Scholar] [CrossRef]
- Jyssum, I.; Gehin, J.; Sexton, J.; Kristianslund, E.; Hu, Y.; Warren, D.; Kvien, T.K.; Haavardsholm, E.A.; Syversen, S.W.; Bolstad, N.; et al. Adalimumab serum levels and anti-drug antibodies: Associations to treatment response and drug survival in inflammatory joint diseases. Rheumatology 2023, 63, 1746–1755. [Google Scholar] [CrossRef]
- Zagalo, D.M.; Simões, S.; Sousa, J. Regulatory science approach in pharmaceutical development of follow-on versions of non-biological complex drug products. J. Pharm. Sci. 2022, 111, 2687–2713. [Google Scholar] [CrossRef]
- Monga, A.; Gagan; Jamwal, P.; Sharma, S.; Kaur, A. Biosimilars: A Critical Review of Development, Regulatory Landscape, and Clinical Implications. AAPS PharmSciTech 2025, 26, 46. [Google Scholar] [CrossRef]
- D’Amico, F.; Peyrin-Biroulet, L.; Danese, S. Benefits of biosimilars in the management of patients with inflammatory bowel disease: An international survey. J. Clin. Med. 2024, 13, 3069. [Google Scholar] [CrossRef]
- Seo, N.; Guan, X.; Wang, T.; Chung, H.S.H.; Wikström, M.; Padaki, R.; Kalenian, K.; Kuhns, S.; Matthies, K.; Crouse-Zeineddini, J.; et al. Analytical and functional similarity of aflibercept biosimilar ABP 938 with aflibercept reference product. Ophthalmol. Ther. 2024, 13, 1303–1320. [Google Scholar] [CrossRef]
- Feagan, B.; Marabani, M.; Wu, J.; Faccin, F.; Spronk, C.; Castañeda-Hernández, G. The challenges of switching therapies in an evolving multiple biosimilars landscape: A narrative review of current evidence. Adv. Ther. 2020, 37, 4491–4518. [Google Scholar] [CrossRef] [PubMed]
- Abu-Zaid, M.; Adebajo, A.; Miedany, Y. Potential for Biosimilars in Rheumatology in Africa. Ann. Rheum. Dis. 2023, 82, 1508–1510. [Google Scholar] [CrossRef] [PubMed]
- Joppi, R.; Bertelè, V.; Vannini, T.; Garattini, S.; Banzi, R. Food and Drug Administration vs European Medicines Agency: Review times and clinical evidence on novel drugs at the time of approval. Br. J. Clin. Pharmacol. 2019, 86, 170–174. [Google Scholar] [CrossRef] [PubMed]
- Greene, L.; Singh, R.; Carden, M.; Pardo, C.; Lichtenstein, G. Strategies for overcoming barriers to adopting biosimilars and achieving goals of the Biologics Price Competition and Innovation Act: A survey of managed care and specialty pharmacy professionals. J. Manag. Care Spec. Pharm. 2019, 25, 904–912. [Google Scholar] [CrossRef]
- Shan, H.; Wang, M.; Huang, S.; Liu, H.; Liu, J.; Du, Q. Efficacy and safety of bevacizumab biosimilar (encoda) compared with reference bevacizumab (Avastin) in patients with metastatic colorectal cancer: A multicenter, real-world study. Clin. Med. Insights Oncol. 2024, 18, 11795549241303726. [Google Scholar] [CrossRef]
- Niazi, S. A critical analysis of the FDA’s omics-driven pharmacodynamic biomarkers to establish biosimilarity. Pharmaceuticals 2023, 16, 1556. [Google Scholar] [CrossRef]
- Patel, A.; Bhatt, N.; Prakash, S.S.; Biswas, G.; Nagarkar, R.; Roy, B.; Doshi, M. Rituximab Biosimilar for the Treatment of Diffuse Large B-Cell Lymphoma: A Phase 3 Randomized Study in India. Cancer Chemother. Pharmacol. 2023, 91, 457–468. [Google Scholar] [CrossRef]
- Hariprasad, S.; Gale, R.; Weng, C.; Ebbers, H.; Rezk, M.; Tadayoni, R. An introduction to biosimilars for the treatment of retinal diseases: A narrative review. Ophthalmol. Ther. 2022, 11, 959–982. [Google Scholar] [CrossRef]
- Lin, S.; Lou, Y.; Hao, R.; Shao, Y.; Yü, J.; Lü, F.; Zhang, Y. A Single-Dose, Randomized, Open-Label, Four-Period, Crossover Equivalence Trial Comparing the Clinical Similarity of the Proposed Biosimilar Rupatadine Fumarate to Reference Wystamm® in Healthy Chinese Subjects. Front. Pharmacol. 2024, 15, 1328142. [Google Scholar] [CrossRef]
- Bhatt, M.; Tank, S.; Shah, J.S.; Maheshwari, D. Regulatory Framework and Disparities of Complex Generics in United States, European Union & Latin America. J. Generic Med. 2023, 19, 130–140. [Google Scholar] [CrossRef]
- Sadda, S.R.; Bradvica, M.; Vajas, A.; Sagong, M.; Ernest, J.; Studnička, J.; Woo, S.J. Biosimilar SB15 versus Reference Aflibercept in Neovascular Age-Related Macular Degeneration: 1-Year and Switching Results of a Phase 3 Clinical Trial. BMJ Open Ophthalmol. 2023, 8, e001561. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Delate, T.; Hui, R.L.; Le, K.; Pham, C. Real-World Noninferiority Assessment of Two Filgrastim Biosimilars in Patients Receiving Myelosuppressive Chemotherapy. JCO Oncol. Pract. 2025, 21, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Gherghescu, I.A.; Delgado-Charro, M.B. The Biosimilar Landscape: An Overview of Regulatory Approvals by the EMA and FDA. Pharmaceutics 2020, 13, 48. [Google Scholar] [CrossRef] [PubMed]
- Wouters, O.J.; Vogel, M.; Feldman, W.B.; Beall, R.F.; Kesselheim, A.S.; Tu, S.S. Differential Legal Protections for Biologics vs Small-Molecule Drugs in the US. JAMA 2024, 332, 2101. [Google Scholar] [CrossRef]
- Available online: http://www.ich.org (accessed on 7 June 2025).
- Yamaci, M. Comparative Forced Degradation Study of Anticomplement C5 Biosimilar and Originator Monoclonal Antibodies. Pharmaceuticals 2025, 18, 579. [Google Scholar] [CrossRef]
- Gaggero, A.; Jeremic, D.; Sattler, R.; Paudel, A.; Urich, J. Evaluation of Analytical Quality by Design Approaches on the Development and Optimization of Size Exclusion Chromatography Analytical Procedures for Bovine Serum Albumin. J. Sep. Sci. 2025, 48, e70168. [Google Scholar] [CrossRef]
- Low, K.; Kok, Y.; Tate, S.; Bi, X. Multilevel—Intact, Subunits, and Peptides—Characterization of Antibody-Based Therapeutics by a Single-Column LC–MS Setup. Anal. Chem. 2025, 97, 5118–5125. [Google Scholar] [CrossRef]
- Wu, G.; Yu, C.; Wang, W.; Du, J.; Fu, Z.; Xu, G.; Wang, L. Mass Spectrometry-Based Charge Heterogeneity Characterization of Therapeutic mAbs with Imaged Capillary Isoelectric Focusing and Ion-Exchange Chromatography as Separation Techniques. Anal. Chem. 2023, 95, 2548–2560. [Google Scholar] [CrossRef]
- Kwok, T.; Chan, S.; Shi, J.; Zhou, M.; Schaefer, A.; Tao, B.; Chen, T. Imaged Capillary Isoelectric Focusing Employing Fluorocarbon and Methylcellulose Coated Fused Silica Capillary for Characterization of Charge Heterogeneity of Protein Biopharmaceuticals. Sep. Sci. Plus 2023, 6, 2200160. [Google Scholar] [CrossRef]
- Dawdy, A.; Trujillo, E.; Liu, Z.; Crone, C.; Kraegenbring, J.; Scheffler, K. Improved Mass Accuracy and Precision for Multi-Attribute Methods Using a New Internally Calibrated High Resolution Orbitrap Mass Detector. Anal. Chem. 2024, 96, 6528–6533. [Google Scholar] [CrossRef]
- Waldenmaier, H.; Gorre, E.; Poltash, M.; Gunawardena, H.; Zhai, X.; Li, J.; Nanda, H. “Lab of the Future”—Today: Fully Automated System for High-Throughput Mass Spectrometry Analysis of Biotherapeutics. J. Am. Soc. Mass Spectrom. 2023, 34, 1073–1085. [Google Scholar] [CrossRef] [PubMed]
- Feldman, R. Paucity of intellectual property rights information in the us biologics system a decade after passage of the biosimilars act. PLoS Med. 2024, 21, e1004381. [Google Scholar] [CrossRef] [PubMed]
- FDA. Guidance for Industry: Q8(R2) Pharmaceutical Development. U.S. Food and Drug Administration; 2009. Available online: https://www.fda.gov/media/71535/download (accessed on 15 May 2025).
- Iqbal, Z.; Sadaf, S. Biosimilars: A comparative study of regulatory, safety and pharmacovigilance monograph in the developed and developing economies. J. Pharm. Pharm. Sci. 2022, 25, 149–182. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Luo, X.; Guo, Q.; Jiang, X.; Su, Z.; Zhou, W.; Zhang, Y. Assessment of Clinical Benefit, Cost and Uptake of Biosimilars versus Reference Biologics in Immune-Mediated Inflammatory Diseases in China. Front. Public Health 2024, 12, 1476213. [Google Scholar] [CrossRef]
- Amaral, C.; Rodrigues, A.R.; Veiga, F.; Bell, V. Biosimilar medicines: From development process to marketing authorization by the EMA and the FDA. Appl. Sci. 2024, 14, 7529. [Google Scholar] [CrossRef]
- Loche, S.; Kanumakala, S.; Backeljauw, P.; Schwab, K.O.; Lechuga-Sancho, A.M.; Esmael, A.; Urosevic, D.; Boldea, A.; Zabransky, M. Safety and effectiveness of a biosimilar recombinant human growth hormone in children requiring growth hormone treatment: Analysis of final data from PATRO Children, an international, post-marketing surveillance study. Drug Des. Dev. Ther. 2024, 18, 667–684. [Google Scholar] [CrossRef]
- Feldman, W. Patent thickets and product hops: Challenges and opportunities for legislative reform. J. Law Med. Ethics 2025, 1–6. [Google Scholar] [CrossRef]
- Kantaros, A.; Ganetsos, T.; Petrescu, F.; Alysandratou, E. Bioprinting and intellectual property: Challenges, opportunities, and the road ahead. Bioengineering 2025, 12, 76. [Google Scholar] [CrossRef]
- Asija, A.; Moreira, S.; Ringov, D.; Soares, T. Fragmentation of technology ownership and acquisition strategy of firms. Br. J. Manag. 2023, 35, 1392–1407. [Google Scholar] [CrossRef]
- Druedahl, L.; Almarsdóttir, A.; Sporrong, S.; Bruin, M.; Hoogland, H.; Minssen, T.; van de Weert, M.; Kesselheim, A.S.; Sarpatwari, A. A qualitative study of biosimilar manufacturing and regulatory perceptions on intellectual property and abbreviated approval pathways. Nat. Biotechnol. 2020, 38, 1253–1256. [Google Scholar] [CrossRef]
- Moorkens, E.; Vulto, A.; Huys, I. An overview of patents on therapeutic monoclonal antibodies in Europe: Are they a hurdle to biosimilar market entry? mAbs 2020, 12, 1743517. [Google Scholar] [CrossRef] [PubMed]
- Bettanti, A.; Lanati, A.; Missoni, A. Biopharmaceutical innovation ecosystems: A stakeholder model and the case of Lombardy. J. Technol. Transf. 2021, 47, 1948–1973. [Google Scholar] [CrossRef] [PubMed]
- Camperos, M.; Almodóvar, P. Tracking the literature on strategic alliances in the biotechnology industry: Insights from a bibliometric approach over the last 30 years. Eur. J. Manag. Bus. Econ. 2023, 34, 245–262. [Google Scholar] [CrossRef]
- Allmendinger, A. Opportunities in an evolving pharmaceutical development landscape: Product differentiation of biopharmaceutical drug products. Pharm. Res. 2021, 38, 739–757. [Google Scholar] [CrossRef]
- Edwards, C.; Chyrok, V.; Monnet, J.; Ullmann, M.; Vlachos, P. Fri0088 safety, immunogenicity and efficacy of the proposed biosimilar MSB11022 (modified formulation) compared with adalimumab reference product in patients with moderately to severely active rheumatoid arthritis: AURIEL-RA, a randomised, double-blind, phase III study. Ann. Rheum. Dis. 2019, 78, 707. [Google Scholar] [CrossRef]
- Alvarez, D.; Wolbink, G.; Cronenberger, C.; Orazem, J.; Kay, J. Interchangeability of biosimilars: What level of clinical evidence is needed to support the interchangeability designation in the United States? BioDrugs 2020, 34, 723–732. [Google Scholar] [CrossRef]
- Liu, L.; Wang, L.; Zonderman, J.; Rouse, J.; Kim, H. Automated, high-throughput infrared spectroscopy for secondary structure analysis of protein biopharmaceuticals. J. Pharm. Sci. 2020, 109, 3223–3230. [Google Scholar] [CrossRef]
- Montero-Vílchez, T.; Cuenca-Barrales, C.; Tejero, A.; Martínez-López, A.; Arias-Santiago, S.; Molina-Leyva, A. Switching from adalimumab originator to biosimilar: Clinical experience in patients with hidradenitis suppurativa. J. Clin. Med. 2022, 11, 1007. [Google Scholar] [CrossRef]
- Cavazzoni, P.; Yim, S. The science of biosimilars—Updating interchangeability. JAMA 2024, 332, 1235. [Google Scholar] [CrossRef]
- Horrow, C.; Gabriele, S.; Tu, S.; Sarpatwari, A.; Kesselheim, A. Patent portfolios protecting 10 top-selling prescription drugs. JAMA Intern. Med. 2024, 184, 810. [Google Scholar] [CrossRef]
- Tu, S.; Goode, R.; Feldman, W. Biologic patent thickets and terminal disclaimers. JAMA 2024, 331, 355. [Google Scholar] [CrossRef] [PubMed]
- Tu, S. The long con: An empirical analysis of pharmaceutical patent thickets. Univ. Pitt. Law Rev. 2024, 86, 213. [Google Scholar] [CrossRef]
- Singh, P.; Desai, P.N.; Dutta, V. Rising biosimilars in the Indian biopharmaceutical industry: Emerging challenges and way forward. Technol. Anal. Strateg. Manag. 2021, 35, 1145–1160. [Google Scholar] [CrossRef]
- Balaprabha, G.; Kondapalli, S.; Pochampally, L.; Reddy, T.; Rao, T. Interchangeable biosimilars: A new era in healthcare system. Int. J. Sci. Res. Arch. 2024, 13, 3763–3773. [Google Scholar] [CrossRef]
- Olsen, T.; Beall, R.; Knox, R.; Tu, S.; Kesselheim, A.; Feldman, W. Patents and regulatory exclusivities on FDA-approved insulin products: A longitudinal database study, 1986–2019. PLoS Med. 2023, 20, e1004309. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, R. Economics of the Pharmaceutical and Medical Device Industry: Supply Chain, Trade and Innovation; Taylor & Francis: London, UK, 2024. [Google Scholar]
- Franco, P.; Haefliger, S. Competition of regulatory ecosystems in approving medicines: Policy implications in the case of Europe. Drug Discov. Today 2025, 30, 104295. [Google Scholar] [CrossRef]
- Tourdot, S.; Bloem, K.; Champion, L.; Groot, A.S.D.; Ducret, A.; Garidel, P.; Grudzinska-Goebel, J.; Gutknecht, M.; Hickling, T.; Horling, F.; et al. Proceedings of the 15th European immunogenicity platform open symposium on immunogenicity of biopharmaceuticals. mAbs 2025, 17, 2487604. [Google Scholar] [CrossRef]
- Larid, G.; Baudens, G.; Dandurand, A.; Coquerelle, P.; Goëb, V.; Guyot, M.H.; Gervais, E. Differential Retention of Adalimumab and Etanercept Biosimilars Compared to Originator Treatments: Results of a Retrospective French Multicenter Study. Front. Med. 2022, 9, 989514. [Google Scholar] [CrossRef]
- Jacquot, G.; Navarro, P.L.; Grange, C.; Boudali, L.; Harlepp, S.; Pivot, X.; Detappe, A. Landscape of subcutaneous administration strategies for monoclonal antibodies in oncology. Adv. Mater. 2024, 36, 2406604. [Google Scholar] [CrossRef]
- Chabra, S.; Gill, B.; Gallo, G.; Zhu, D.; Pitou, C.; Payne, C.; Accioly, A.; Puig, L. Ixekizumab citrate-free formulation: Results from two clinical trials. Adv. Ther. 2022, 39, 2862–2872. [Google Scholar] [CrossRef]
- Schreiber, S.; Ben-Horin, S.; Leszczyszyn, J.; Dudkowiak, R.; Lahat, A.; Gawdis-Wojnarska, B.; Pukitis, A.; Horynski, M.; Farkas, K.; Kierkus, J.; et al. Randomized controlled trial: Subcutaneous vs intravenous infliximab CT-P13 maintenance in inflammatory bowel disease. Gastroenterology 2021, 160, 2340–2353. [Google Scholar] [CrossRef] [PubMed]
- Pujahari, S.R.; Engla, S.; Soni, R.; Patra, S.; Hanawal, M.K.; Kumar, A. Structural similarity of biological drugs using statistical signal processing and nuclear magnetic resonance spectral pattern analysis. Mol. Pharm. 2025, 22, 2684–2693. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Strauss, D. Advancing innovations in biosimilars. Clin. Pharmacol. Ther. 2022, 113, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Hengelbrock, A.; Strube, J. Continuous biomanufacturing in upstream and downstream processing. Phys. Sci. Rev. 2023, 9, 3167–3222. [Google Scholar] [CrossRef]
- Fautrel, B.; Bouhnik, Y.; Dieudé, P.; Richette, P.; Dougados, M.; Freudensprung, U.; Addison, J. Real-World Evidence of the Use of the Infliximab Biosimilar SB2: Data from the PERFUSE Study. Rheumatol. Adv. Pract. 2023, 7, rkad031. [Google Scholar] [CrossRef]
- Nikitina, V.; Laurini, G.S.; Montanaro, N.; Motola, D. Comparative Safety Profiles of Biosimilars vs. Originators Used in Rheumatology: A Pharmacovigilance Analysis of the EudraVigilance Database. J. Clin. Med. 2025, 14, 1644. [Google Scholar] [CrossRef]
- Borrás-Blasco, J.; Valcuende-Rosique, A.; Cornejo, S.; Aparicio-Rubio, C.; Aguilar-Zamora, M.; Garijo-Bufort, M.; Arévalo-Ruales, K.R. Persistence after Switching from Adalimumab Biosimilar MSB11022 to Adalimumab Biosimilar GP2017 in Patients with Chronic Inflammatory Rheumatic Diseases. J. Pharm. Technol. 2024, 41, 90–94. [Google Scholar] [CrossRef]
- Ntais, C.; Kontodimopoulos, N.; Fanourgiakis, J.; Talias, Μ.A. Fostering Healthcare System Sustainability through Efficient Practices: Can Adopting Biosimilars Ease the Financial Burden of Rheumatoid Arthritis? F1000Research 2025, 13, 1128. [Google Scholar] [CrossRef]
- Fiorino, G.; Ananthakrishnan, A.N.; Cohen, R.D.; Cross, R.K.; Deepak, P.; Farraye, F.A.; Steinhart, A. Accelerating Earlier Access to Anti-TNF-α Agents with Biosimilar Medicines in the Management of Inflammatory Bowel Disease. J. Clin. Med. 2025, 14, 1561. [Google Scholar] [CrossRef]
- Teixeira, F.V.; Peyrin-Biroulet, L.; Danese, S. Are We Ready for Multiple Switches between Reference Products and Biosimilars? Arq. Gastroenterol. 2024, 61, e24046. [Google Scholar] [CrossRef]
- Niazi, S.; Ómarsdóttir, S. Lectin-based fluorescent comparison of glycan profile—FDA validation to expedite approval of biosimilars. Int. J. Mol. Sci. 2024, 25, 9240. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Yan, Y.; Ho, K. US FDA-approved therapeutic antibodies with high-concentration formulation: Summaries and perspectives. Antib. Ther. 2021, 4, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Foss-Solbrekk, K. The divisional game: Using procedural rights to impede generic/biosimilar market entry. IIC—Int. Rev. Intellect. Prop. Compet. Law 2022, 53, 1007–1037. [Google Scholar] [CrossRef]
- Organon. FDA Grants Interchangeability Designation to Samsung Bioepis and Organon HADLIMA™ (Adalimumab-BWWD) Injection. Organon Newsroom, 27 May 2025. Available online: https://www.organon.com/news/us-food-and-drug-administration-fda-grants-interchangeability-designation-to-samsung-bioepis-and-organon-hadlima-adalimumab-bwwd-injection/ (accessed on 3 June 2025).
- Rome, B.N.; Kesselheim, A.S. Biosimilar Competition for Humira Is Here: Signs of Hope Despite Early Hiccups. Arthritis Rheumatol. 2023, 75, 1325–1327. [Google Scholar] [CrossRef] [PubMed]
- Junker, S.; Ebert, O.; Bartsch, R. A Systematic Literature Review of Injection Site Pain Perception in Adult Patients Treated with Citrate-Free and Citrate-Containing Biologic Agents. Curr. Rheumatol. Rev. 2023, 19, 303–313. [Google Scholar] [CrossRef]
- Bouguen, G.; Gossec, L.; Abitbol, V.; Senbel, É.; Bonnaud, G.; Roblin, X.; Marotte, H. Patient Satisfaction and Experience with CT-P17 Following Transition from Reference Adalimumab or Another Adalimumab Biosimilar: Results from the Real-World YU-MATTER Study. BioDrugs 2024, 38, 867–878. [Google Scholar] [CrossRef]
- Cheung, S.; Heidari, P.; Truong, D. A208 Real World Evidence Analysis with Adalimumab Biosimilar, MSB 11022, Reported with Patient Care Program. J. Can. Assoc. Gastroenterol. 2023, 6 (Suppl. S1), 46–47. [Google Scholar] [CrossRef]
- Tu, S.S.; Goode, R.; Turner, M.; Van de Wiele, V. Accelerating biosimilar market access: The case for allowing earlier standing. J. Law Biosci. 2025, 12, lsae030. [Google Scholar] [CrossRef]
- Goode, R.W.; Chao, B. Biological Patent Thickets and Delayed Access to Biosimilars, an American Problem. J. Law Biosci. 2022, 9, lsac022. [Google Scholar] [CrossRef]
- Halawa, M.R.; ElSayed, R.M.R.; Aderibigbe, T.; Newman, P.M.; Reid, B.E.; Carabetta, V.J. Biosimilars Targeting Pathogens: A Comprehensive Review of Their Role in Bacterial, Fungal, Parasitic, and Viral Infections. Pharmaceutics 2025, 17, 581. [Google Scholar] [CrossRef]
- Alnaqbi, K.A.; Bellanger, A.; Brill, A.; Castañeda-Hernández, G.; Estela, A.C.; Sánchez, O.D.; Simoens, S. An International Comparative Analysis and Roadmap to Sustainable Biosimilar Markets. Front. Pharmacol. 2023, 14, 1188368. [Google Scholar] [CrossRef] [PubMed]
- Reddanna, P.; Shaik, J.; Gandla, J. Innovations in the Pharma and Biopharma Industries: Role of Academia–Industry Interactions. In Biotechnology in India—Reworking A Strategy; John, G., Panda, A.K., Eds.; Springer: Singapore, 2024. [Google Scholar] [CrossRef]
- Stosch, M.v. Digital process development and manufacturing of biopharmaceuticals: Is it a revolution? In Management for Professionals; Springer: Cham, Switzerland, 2024; pp. 61–75. [Google Scholar] [CrossRef]
- Baumgart, D.C.; Misery, L.; Naeyaert, S.; Taylor, P.C. Biological therapies in immune-mediated inflammatory diseases: Can biosimilars reduce access inequities? Front. Pharmacol. 2019, 10, 279. [Google Scholar] [CrossRef] [PubMed]
- Rezk, M.F.; Pieper, B. Unlocking the value of anti-TNF biosimilars: Reducing disease burden and improving outcomes in chronic immune-mediated inflammatory diseases: A narrative review. Adv. Ther. 2020, 37, 3732–3745. [Google Scholar] [CrossRef] [PubMed]
- Vandenplas, Y.; Simoens, S.; Wilder, P.; Vulto, A.; Huys, I. The impact of policy interventions to promote the uptake of biosimilar medicines in Belgium: A nationwide interrupted time series analysis. Health Res. Policy Syst. 2023, 21, 68. [Google Scholar] [CrossRef] [PubMed]
- Kravvariti, E.; Kitas, G.D. The role of the nocebo effect in the use of biosimilars in routine rheumatology clinical practice. Mediterr. J. Rheumatol. 2019, 30 (Suppl. S1), 63. [Google Scholar] [CrossRef]
- Colloca, L.; Panaccione, R.; Murphy, T. The clinical implications of nocebo effects for biosimilar therapy. Front. Pharmacol. 2019, 10, 1372. [Google Scholar] [CrossRef]
- Car, E.; Vandenplas, Y.; Lacosta, T.B.; Simoens, S.; Huys, I.; Vulto, A.G.; Barbier, L. Mitigating the nocebo effect in biosimilar use and switching: A systematic review. Pharm. Med. 2024, 38, 429–455. [Google Scholar] [CrossRef]
- Pouillon, L.; Danese, S.; Hart, A.; Fiorino, G.; Argollo, M.; Selmi, C.; Carlo-Stella, C.; Loeuille, D.; Costanzo, A.; Lopez, A.; et al. Consensus report: Clinical recommendations for the prevention and management of the nocebo effect in biosimilar-treated IBD patients. Aliment. Pharmacol. Ther. 2019, 49, 1181–1187. [Google Scholar] [CrossRef]
- Dipasquale, V.; Romano, C. Biosimilar infliximab in paediatric inflammatory bowel disease: Efficacy, immunogenicity and safety. J. Clin. Pharm. Ther. 2020, 45, 1228–1234. [Google Scholar] [CrossRef]
- Afzali, A.; Furtner, D.; Melsheimer, R.; Molloy, P. The Automatic Substitution of Biosimilars: Definitions of Interchangeability Are Not Interchangeable. Adv. Ther. 2021, 38, 2077–2093. [Google Scholar] [CrossRef]
- Guyader, G.; Vieillard, V.; Paul, M. Physicochemical stability study of MYL-1401O, a biosimilar of trastuzumab, following a transient temperature excursion. J. Oncol. Pharm. Pract. 2020, 27, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Chew, C.; Aguiar, M.; Bansback, N.; Law, M.; Harrison, M. Patient perspectives on the British Columbia biosimilars initiative: A qualitative descriptive study. Rheumatol. Int. 2021, 42, 1831–1842. [Google Scholar] [CrossRef] [PubMed]
- Simoens, S.; Vulto, A.; Dylst, P. Simulating costs of intravenous biosimilar trastuzumab vs. subcutaneous reference trastuzumab in adjuvant HER2-positive breast cancer: A Belgian case study. Pharmaceuticals 2021, 14, 450. [Google Scholar] [CrossRef] [PubMed]
- Oskouei, S.; Kusmierczyk, A. Biosimilar uptake: The importance of healthcare provider education. Pharm. Med. 2021, 35, 215–224. [Google Scholar] [CrossRef]
- Bucci, P.L.; Cardama, G.A. In Silico Tools to Leverage Rational Drug Design and Development in LMICs. Front. Comput. Chem. 2024, 7, 1. [Google Scholar]
- Lohmann, F.; Allenspach, S.; Atz, K.; Schiebroek, C.; Hiss, J.; Schneider, G. Protein binding site representation in latent space. Mol. Inform. 2024, 44, e202400205. [Google Scholar] [CrossRef]
- Musuamba, F.; Bursi, R.; Manolis, E.; Karlsson, K.; Kulesza, A.; Courcelles, E.; Boissel, J.; Lesage, R.; Crozatier, C.; Voisin, E.M.; et al. Verifying and validating quantitative systems pharmacology and in silico models in drug development: Current needs, gaps, and challenges. CPT Pharmacomet. Syst. Pharmacol. 2020, 9, 195–197. [Google Scholar] [CrossRef]
- Hu, C.; Yi, Q.; Li, C.; Luo, Y.; Yi, S. Digital twin–based dynamic prediction and simulation model of carbon efficiency in gear hobbing process. Int. J. Adv. Manuf. Technol. 2023, 126, 3959–3980. [Google Scholar] [CrossRef]
- Jahangir, M.; Schultz, C.; Kamari, A. A review of drivers and barriers of digital twin adoption in building project development processes. J. Inf. Technol. Constr. 2024, 29, 141–178. [Google Scholar] [CrossRef]
- Botton, A.; Barberi, G.; Facco, P. Data augmentation to support biopharmaceutical process development through digital models—A proof of concept. Processes 2022, 10, 1796. [Google Scholar] [CrossRef]
- Pérez-Benito, Á.; García-Aznar, J.; Gómez-Benito, M.; Pérez, M. Patient-specific prostate tumour growth simulation: A first step towards the digital twin. Front. Physiol. 2024, 15, 1421591. [Google Scholar] [CrossRef] [PubMed]
- VT, I.; Asireddy, S.; Vallarapu, N.; Madhira, J.; Rao, T. Digital twins in drug discovery: A paradigm shift shaping pharmaceutical innovation. Int. J. Pharm. Sci. Nanotechnol. 2024, 17, 7628–7637. [Google Scholar] [CrossRef]
- Hodel, K.V.S.; Fiuza, B.S.D.; Conceição, R.S.; Aleluia, A.C.M.; Pitanga, T.N.; Fonseca, L.M.d.S.; Valente, C.O.; Minafra-Rezende, C.S.; Machado, B.A.S. Pharmacovigilance in vaccines: Importance, main aspects, perspectives, and challenges—A narrative review. Pharmaceuticals 2024, 17, 807. [Google Scholar] [CrossRef] [PubMed]
- Huanbutta, K.; Burapapadh, K.; Kraisit, P.; Sriamornsak, P.; Ganokratanaa, T.; Suwanpitak, K.; Sangnim, T. Artificial intelligence-driven pharmaceutical industry: A paradigm shift in drug discovery, formulation development, manufacturing, quality control, and post-market surveillance. Eur. J. Pharm. Sci. 2024, 203, 106938. [Google Scholar] [CrossRef]
- Pontes, M.A.; Ribeiro, A.A.; Albuquerque, F.C.; Cotenzini, S.N.L. Comparative Price Analysis of Biological Medicines: Disparities Generated by Different Pricing Policies. Front. Pharmacol. 2024, 14, 1256542. [Google Scholar] [CrossRef]
- Cohen, H.; Turner, M.; McCabe, D.; Woollett, G. Future evolution of biosimilar development by application of current science and available evidence: The developer’s perspective. BioDrugs 2023, 37, 583–593. [Google Scholar] [CrossRef]
- Chen, H.; Yemeke, T.T.; Ozawa, S. Reduction of Biologic Pricing Following Biosimilar Introduction: Analysis across 57 Countries and Regions, 2012–2019. PLoS ONE 2024, 19, e0304851. [Google Scholar] [CrossRef]
- Ascef, B.d.O.; Almeida, M.O.d.; Ribeiro, A.C.d.M.; Andrade, D.; Oliveira, H.A.d.; Soárez, P.C.d. Therapeutic Equivalence of Biosimilar and Reference Biologic Drugs in Rheumatoid Arthritis. JAMA Netw. Open 2023, 6, e2315872. [Google Scholar] [CrossRef]
- Strand, M.; Watanabe, J.H. Examining the Impact of the World Health Organization 2022 Guidelines on Evaluation of Biosimilars for Non-Local Comparators in Biosimilar Studies on Middle East and North Africa Member States. Pharmacy 2024, 12, 94. [Google Scholar] [CrossRef]
- Zhou, X.; Chen, Z.; Bi, X. An update review of biosimilars of adalimumab in psoriasis—Bioequivalence and interchangeability. Drug Des. Dev. Ther. 2021, 15, 2987–2998. [Google Scholar] [CrossRef]
- Alska, E.; Łaszczych, D.; Napiórkowska-Baran, K.; Szymczak, B.; Rajewska, A.; Rubisz, A.E.; Bartuzi, Z. Advances in Biologic Therapies for Allergic Diseases: Current Trends, Emerging Agents, and Future Perspectives. J. Clin. Med. 2025, 14, 1079. [Google Scholar] [CrossRef] [PubMed]
- Ren, S. Effects of arginine in therapeutic protein formulations: A decade review and perspectives. Antib. Ther. 2023, 6, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Lin, M.; Nguyen, B.; Chang, C.; Shih, J.; Nguyen, M.; Chang, Y.-H.; Hu, Y.-C. CRISPR-Cas13d for gene knockdown and engineering of CHO cells. ACS Synth. Biol. 2020, 9, 2808–2818. [Google Scholar] [CrossRef] [PubMed]
- Ehret, J.; Zimmermann, M.; Eichhorn, T.; Zimmer, A. Impact of cell culture media additives on IgG glycosylation produced in Chinese hamster ovary cells. Biotechnol. Bioeng. 2019, 116, 816–830. [Google Scholar] [CrossRef]
- Siew, Y.; Zhang, W. Downstream processing of recombinant human insulin and its analogues production from E. coli inclusion bodies. Bioresour. Bioprocess. 2021, 8, 65. [Google Scholar] [CrossRef]
- Glinšek, K.; Bozovičar, K.; Bratkovič, T. CRISPR Technologies in Chinese Hamster Ovary Cell Line Engineering. Int. J. Mol. Sci. 2023, 24, 8144. [Google Scholar] [CrossRef]
- Sembiring, E.; Fuad, A.; Suryadi, H. Expression and purification of recombinant human granulocyte colony-stimulating factor (rG-CSF) from Pichia pastoris. Indones. J. Biotechnol. 2024, 29, 205. [Google Scholar] [CrossRef]
- Swaminathan, N.; Priyanka, P.; Rathore, A.; Sivaprakasam, S.; Subbiah, S. Multiobjective optimization for enhanced production of therapeutic proteins in Escherichia coli: Application of real-time dielectric spectroscopy. Ind. Eng. Chem. Res. 2020, 59, 21841–21853. [Google Scholar] [CrossRef]
- Zybin, D.I.; Klishin, A.A.; Orlova, N.V.; Seregin, Y.A.; Sorokina, T.S.; Kapustin, D.V. A simple rapid HPLC-UV method for assessing C-terminal arginine content in darbepoetin alfa. Biomed. Chromatogr. 2025, 39, e70018. [Google Scholar] [CrossRef]
- Capozzi, L.C.; Trout, B.L.; Pisano, R. From Batch to Continuous: Freeze-Drying of Suspended Vials for Pharmaceuticals in Unit-Doses. Ind. Eng. Chem. Res. 2019, 58, 1635–1649. [Google Scholar] [CrossRef]
- Sahebnasagh, R.; Deli, H.; Shadboorestan, A.; Vakili-Ghartavol, Z.; Salehi, N.; Komeili–Movahhed, T.; Ghahremani, M.H. Identification of Key lncRNAs Associated with Oxaliplatin Resistance in Colorectal Cancer Cells and Isolated Exosomes: From In-Silico Prediction to In-Vitro Validation. PLoS ONE 2024, 19, e0311680. [Google Scholar] [CrossRef] [PubMed]
- Forest-Nault, C.; Gaudreault, J.; Henry, O.; Durocher, Y.; Crescenzo, G. On the use of surface plasmon resonance biosensing to understand IgG-FcγR interactions. Int. J. Mol. Sci. 2021, 22, 6616. [Google Scholar] [CrossRef] [PubMed]
- Bharali, P.; Chand, S.; Chander, H. A Cost-Effective and Robust Cell-Based Bioassay Method for Evaluating the Bioactivity of Trastuzumab-like Antibodies. Biomedicines 2025, 13, 23. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Xie, L.; Zhang, E.; Gao, W.; Wang, L.; Cao, Y.; Jiang, W.; Liu, S. Physicochemical and functional assessments demonstrating analytical similarity between rituximab biosimilar HLX01 and the MabThera®. mAbs 2019, 11, 606–620. [Google Scholar] [CrossRef]
- Kurki, P.; Kang, H.; Ekman, N.; Knežević, I.; Weise, M.; Wolff-Holz, E. Regulatory Evaluation of Biosimilars: Refinement of Principles Based on the Scientific Evidence and Clinical Experience. BioDrugs 2022, 36, 359–371. [Google Scholar] [CrossRef]
- Nair, R.; Krishnan, N.; Shenoy, V.; Seetharam, R.N. Evaluation of methods employed in establishing preclinical similarity of adalimumab biosimilars. Adv. Pharmacol. Pharm. Sci. 2025, 2025, 8816591. [Google Scholar] [CrossRef]
- Nagar, L.; Saini, A.; Gulati, N.; Solanki, N.; Dureja, H. Regulatory considerations of biosimilars in cancer. In Biosimilars for Cancer Treatment; Springer: Singapore, 2024; pp. 265–285. [Google Scholar] [CrossRef]
- Florian, J.; Gershuny, V.; Sun, Q.; Schrieber, S.; Matta, M.; Hazel, A.; Sheikhy, M.; Weaver, J.L.; Hyland, P.L.; Hsiao, C.; et al. Considerations for use of pharmacodynamic biomarkers to support biosimilar development—(III) a randomized trial with interferon beta-1a products. Clin. Pharmacol. Ther. 2022, 113, 339–348. [Google Scholar] [CrossRef]
- Jofre-Bonet, M.; McGuire, A.; Dayer, V.; Roth, J.A.; Sullivan, S.D. The price effects of biosimilars in the united states. Value Health 2025, 28, 742–750. [Google Scholar] [CrossRef]
- Ma, X.; Wang, J.; Ni, X.; Shi, J. Machine Learning Approaches for Enhancing Customer Retention and Sales Forecasting in the Biopharmaceutical Industry: A Case Study. J. Adv. Comput. Syst. 2024, 4, 10–29. [Google Scholar] [CrossRef]
- Askari, S.; Ghofrani, A.; Taherdoost, H. Transforming drug design: Innovations in computer-aided discovery for biosimilar agents. Biomedinformatics 2023, 3, 1178–1196. [Google Scholar] [CrossRef]
- Petrov, A.; Makarenko, I.; Belova, B.; Melikyan, A.; Saparova, V.; Peskov, K.; Kudryashova, N.; Kovalik, V.; Gefen, M.; Khokhlov, A.; et al. Optimization of romiplostim biosimilar efficacy trial using in silico clinical trial approach for patients with immune thrombocytopenia. Clin. Pharmacol. Drug Dev. 2024, 14, 116–126. [Google Scholar] [CrossRef]
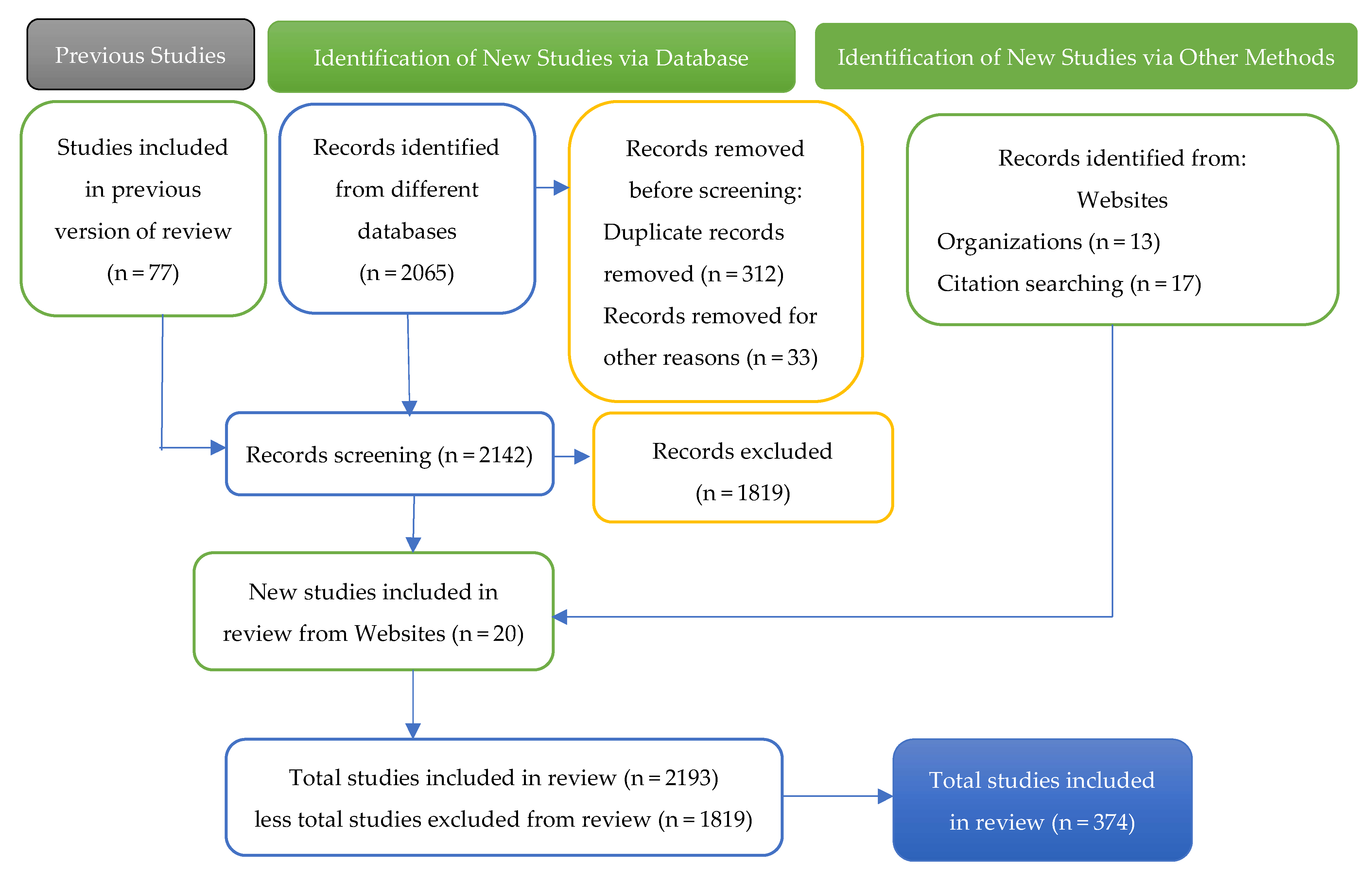
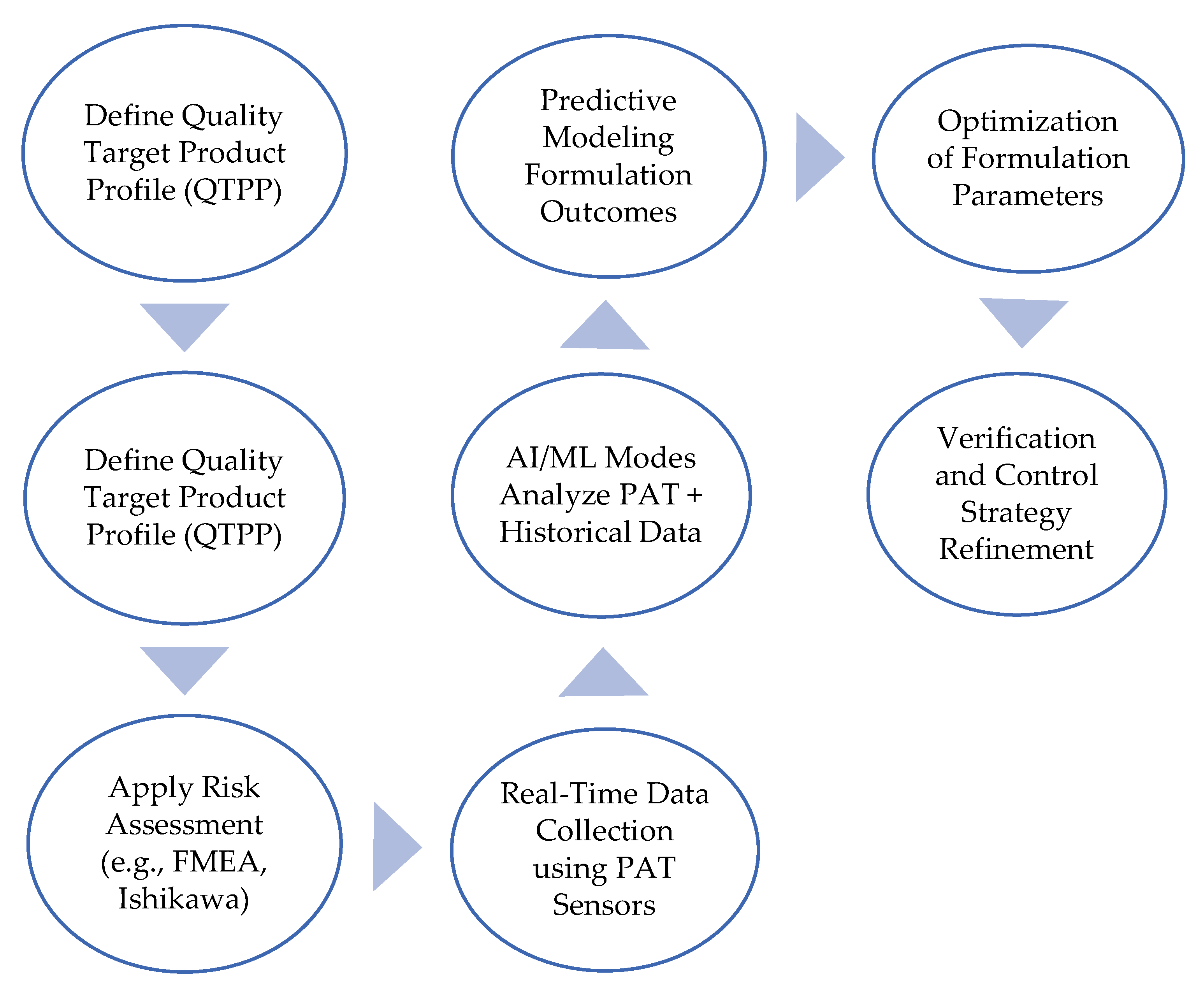
| Parameter | Chemical Drug | Generic Drug | Biological Drug | Biosimilar Drug |
|---|---|---|---|---|
| Synthesis | Production of original chemical formula | Copy of the original chemical formula | Insertion of a gene into a cell clone | Development derived from the original biological molecule |
| Size | 100–1000 Da | 100–1000 Da | 10,000–300,000 Da | 10,000–300,000 Da |
| Glycosylation process | Zero | Zero | Several | Several |
| Molecular structure | Simple | Simple | Complex | Complex |
| Ability to generate immunity | Low | Low | Medium-high | Medium-high |
| Drug development time | 7–10 years | 1–3 years | 10–15 years | 6–9 years |
| Type of Inclusion Criteria | Details |
| Relevance of the topic | Studies in biopharmaceutical and biosimilar formulation strategies in the industry, with an emphasis on buffer-free systems. |
| Formulation technologies, biosimilar design, and trends focusing on FDA and EMA regulations | Specific articles on the formulation of biosimilars to improve their accessibility, efficacy, and safety, highlighting buffer-free systems, safe excipients, intellectual property challenges, and the use of QbD, PAT, and artificial intelligence to optimize the regulated (FDA and EMA) and competitive development of biologic therapies. |
| Scope of the study | To provide an integrative review of biosimilar formulation strategies, with emphasis on buffer-free systems, safety classification, regulatory alignment, and intellectual property considerations to guide future development and market access. |
| Type of publication | Articles in peer-reviewed journals, systematic reviews, and reports from reputable institutions and websites. |
| Period of time | Publications primarily from 2018 to 2025. This range of years was specifically selected to include recent events related to biosimilar formulation. strategies, with an emphasis on buffer-free systems. Older studies will only be included to understand specific topics. |
| Exclusion Criteria | Details |
| Irrelevant topics | Studies did not directly focus on the selected keywords. |
| Limited crisis context | Articles that do not analyze the formulation of biosimilars, their accessibility, efficacy, and safety, bufferless systems, safe excipients, intellectual property challenges, and the use of QbD, PAT, and artificial intelligence to optimize the regulated (FDA and EMA) and competitive development of biological therapies. |
| Lack of regional and global focus | Research that focuses solely on localized issues without connecting them to the scope of this research. |
| Low credibility | Non-peer-reviewed articles, opinion pieces, or studies with unverifiable data. |
| Duplicate or overlapped content | Studies with existing duplicate results are not considered valid unless they offer new insights or updates. |
| Language/accessibility | Exclude studies that are not available in English unless accessible translations are provided or accessible for systematic review. |
| Step | Explanation |
|---|---|
| Definition of the Quality Target Product Profile (QTPP) | High-level product goals: route, dosage, stability, etc. |
| CQAs | Measurable attributes that affect safety and efficacy (e.g., aggregation, viscosity) |
| Apply Risk Assessment | Use FMEA or Pareto to identify high-risk parameters |
| Design of Experiments (DoE) using QbD tools | Use MODDE®, JMP®, or Design-Expert® to systematically vary inputs |
| Real-Time Data Collection Using PAT sensors | Real-time sensing: Raman, NIR, and FTIR for critical variables like pH and osmolality |
| AI/ML models analyze PAT + historical data | Platforms like BioPharma Finder™, TensorFlow, or DeepChem predict trends |
| Predictive Modeling of Formulation Outcomes | Forecast viscosity, stability, and bioequivalence from design space |
| Optimization of formulation parameters | Fine-tune concentrations, pH, excipients |
| Refinement of the verification and control strategy | Based on process capability and design space robustness |
| Regulatory submission with integrated QbD-AI evidence | Aligned with ICH Q8–Q11 and regulatory AI guidance (FDA/EMA evolving). |
| Biosimilar Name | Reference Biologic | Disease/ Condition | FDA Approval Year | Key Findings/Insights | References |
|---|---|---|---|---|---|
| Zarxio (filgrastim-sndz) | Neupogen (filgrastim) | Neutropenia | 2015 | The first biosimilar was approved in the US. Comparable efficacy and safety to Neupogen. | [167] |
| Ontruzant (trastuzumab-dttb) | Herceptin (trastuzumab) | Breast Cancer | 2019 | Demonstrated non-inferiority to Herceptin in terms of efficacy. | [51,168] |
| Amsparity (bevacizumab-maly) | Avastin (bevacizumab) | Various Cancers | 2017 | Similar safety and efficacy profile as Avastin, improving the accessibility of treatment. | [169] |
| Amgen’s Amjevita (adalimumab-atto) | Humira (adalimumab) | Rheumatoid Arthritis | 2016 | The first biosimilar approved, adalimumab, reported comparable safety and efficacy. | [170,171] |
| Kanjinti (trastuzumab-anns) | Herceptin (trastuzumab) | Breast Cancer | 2019 | Validated efficacy through phase III trials, expanding treatment options. | [168] |
| Breztri (bococizumab) | Repatha (evolocumab) | Hyperlipidemia | 2021 | Efficacy and safety profile similar to those of Repatha, although more data could be beneficial. | [167] |
| SB5 (adalimumab) | Humira (adalimumab) | Inflammatory Bowel Disease | 2021 | High similarity in clinical effectiveness observed in real-world IBD cohorts. | [172] |
| Remsima (infliximab) | Remicade (infliximab) | Rheumatoid Arthritis | 2013 | Known for cost-effectiveness; Comparable efficacy in patients transitioning from Remicade. | [173,174] |
| Neulasta Onpro (pegfilgrastim-jmdb) | Neulasta (pegfilgrastim) | Neutropenia | 2018 | Shows equivalent safety and efficacy to Neulasta, supporting oncology protocols. | [167] |
| MYL-1501D (insulin glargine) | Lantus (insulin glargine) | Diabetes | 2019 | Indicated for type 1 diabetes; demonstrated comparable safety profiles. | [175] |
| Excipient Class | Function in the Formulation | Examples |
|---|---|---|
| Buffers | Maintain a stable pH environment | Histidine, phosphate, acetate, citrate, etc. |
| Sugars/polyols | Stabilize protein conformation (lyoprotectant or bulking agent) | Sucrose, trehalose, mannitol |
| Amino acids | They are stabilized by charge or hydrophobic interactions; sometimes they act as weak buffers or tonicity agents. | Arginine, glycine, proline, histidine |
| Surfactants | Prevent surface-induced aggregation (e.g., agitation, interface) | Polysorbate 80, Polysorbate 20 |
| Chelating agents | Preventing metal ion-induced degradation | EDTA, DTPA |
| Antioxidants | Protect against protein oxidation. | Methionine, Glutathione |
| Tonicity modifiers | Adjust osmolality to the physiological range | Sodium chloride, glycerol |
| Aspect | FDA Approach | EMA Approach | Representative References |
|---|---|---|---|
| General Stance on Formulation Differences | Allows formulation differences, particularly in clinically inactive components (excipients), as long as safety, purity, and potency remain equivalent to the reference product. | Permits formulation differences if justified scientifically and clinically. Acceptable if no detrimental effects are introduced. | [2,66,171,271,272,273,274] |
| Use of Different Excipients (Clinically Inactive Components) | The differences allowed must be justified through analytical data and often clinical studies. Caution is advised if the excipients interfere with the analytical comparability. | Minor differences are acceptable; must be justified. The avoidance of new excipients is preferred unless safety is demonstrated. | [2,3,271,275,276,277] |
| Use of New Excipients or Novel Routes | Additional safety/toxicological data are expected for new excipients or excipients used via a new route. | New excipients not previously used in biology are discouraged. If included, prior safety data or similar precedent is required. | [3,244,278] |
| Justification Requirements | Sponsors must demonstrate that they have no impact on safety, purity, or potency using validated methods. | A complete scientific rationale and risk mitigation must be submitted for formulation differences. | [2,117,171,178] |
| Encouragement of Formulation Innovation | Encourage the use of advanced, cutting-edge formulation technologies if biosimilarity is preserved. Avoid imposing outdated reference formulations. | Supports formulation updates aligned with patient needs (e.g., improved tolerability, stability), but changes must be scientifically justified. | [2,53,271,272,273,274] |
| Analytical Comparability of Interference Risk | Formulations should allow robust analytical comparability. Excipients that hinder testing are discouraged. Requires assurance that excipients do not interfere with analytical comparability assays. | Demands evidence that changes in excipients do not affect comparability studies. | [209,275,276] |
| Changes in Dosage Form (e.g., liquid vs. lyophilized) | Permitted if the safety, purity, and potency remain consistent and the differences are justified. | Allowed if justified. The route of administration must remain the same; any change in dosage form must not affect product performance. | [273,274] |
| Regulatory Expectation for Documentation | Differences must be clearly documented and justified in the application. | All formulation differences are reviewed and discussed in EMA assessment reports, with a clear justification. | [272,275] |
| Clinical Testing Requirement | It is necessary to see if formulation changes could affect PK/PD or clinical response. | Clinical studies are required when formulation changes could affect bioavailability or tolerability. | [178,257] |
| New Dosage Forms | Permits with evidence of unchanged safety and efficacy. | Permits if the route is unchanged and product performance is validated. | [53,178] |
| Documentation Practices | Comprehensive submission required with full rationale and evidence for any formulation changes. | Extensive assessment reports document and justify all formulation differences. | [171,275] |
| Examples of Acceptable Changes | Omission of antimicrobial preservatives in single-dose vials; inclusion of newer and safer excipients with full justification. | Similar cases accepted; removal of preservatives in single-use formats or omission of unnecessary excipients. Emphasis on safety, patient convenience, and technological justification. | [271,275] |
| Aspect | Challenge/Strategy | Description and Examples |
|---|---|---|
| Patent Thickets | Freedom-to-operate (FTO) analysis | Biosimilar developers map reference product patents and analyze expiry timelines and claim breadth. May delay launch if critical patents expire soon. Use of inter-patent review IPR (US) or opposition (EU) to challenge weak patents. |
| Designing Around Patents | Formulation modifications | Launch with legacy formulations to avoid infringement (e.g., citrate buffered adalimumab). Switch to citrate-free after the expiration of the patent. Biosimilars of insulin glargine are used in vials instead of pen devices due to device patents. |
| Patent Multiplicity | Continuations and layered patents | Developers exploit continuation strategies (e.g., in the US) to file overlapping claims. Litigation during Phase 3 can help resolve disputes before launch. |
| Trade Secrets | Reverse engineering | Some reference formulations remain partly undisclosed (e.g., additives, processes). Biosimilar makers reverse-engineer composition and verify using analytical techniques, but some process steps (e.g., lyophilization) may remain protected. |
| Formulation Optimization | Use cutting-edge science | Prefer a ready-to-use liquid over lyophilized if stable. Use stabilizers such as trehalose or histidine. Support high-concentration subcutaneous injections if safe. The improvements must be justified with comparability data. |
| Excipient Risk Minimization | Select safe and known ingredients | Avoid new excipients unless essential. Use animal-free, pharmacopoeial-grade excipients. Minimize concentration and number of excipients to reduce the risks of immunogenicity and toxicity. |
| Pharmaceutical Equivalence | Match reference where possible | Maintain the dosage form, route, and protein concentration. Modify excipients (e.g., citrate replaced with acetate) to reduce pain but preserve administration parameters. Justify changes with data. |
| Stability and Compatibility | Comprehensive testing required | Evaluate under stress conditions (e.g., freeze-thaw, light, agitation). Ensure compatibility with containers and infusion hardware. Prevent protein aggregation or crystallization. |
| Patient-centered Design | Optimize for Comfort and Usability | Reduce injection pain (e.g., avoid citrate/glutamate), improve convenience (e.g., tolerate room temperature). Consider auto-injectors, thinner needles, or reduced injection frequency. |
| IP-Informed Development | Align Formulation with IP Landscape | Identify and avoid violating excipient combinations. If infringement is unavoidable but preferable, plan for post-expiration launch or license negotiation. Legal safety should not compromise patient safety. |
| Regulatory Engagement | Early Agency Dialog | Proactively discuss formulation deviations with FDA/EMA. Use scientific advice or type IV meetings. Emphasize patient safety and similarity to gain support for changes. |
| Formulation Challenge | Strategic Innovation | Tools/Technologies | Regulatory Relevance |
|---|---|---|---|
| Protein Aggregation and Viscosity | Buffer-free high-concentration systems | QbD, AI simulation, Ensilication | Reduces immunogenicity risk |
| Post-Translational Modifications (PTMs) | AI-assisted PTM mapping and Deep Learning prediction | GlycoAnalytics™, Mass Spectrometry, AI | Ensures biosimilarity of functional domains |
| Regulatory Equivalence | Totality of evidence approach | QbD–PAT–AI integrated systems | EMA/FDA stepwise biosimilarity validation |
| Legal Barriers (IP Thickets) | Early Freedom-to-Operate (FTO) and Formulation Design | Patent Landscape, AI-FTO Analysis Tools | Minimizes litigation, secures market access |
| Patient Tolerability | Citrate-free excipient-optimized subcutaneous delivery | AI-enhanced excipient selection | Enhances compliance and real-world outcomes |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bas, T.G. Innovative Formulation Strategies for Biosimilars: Trends Focused on Buffer-Free Systems, Safety, Regulatory Alignment, and Intellectual Property Challenges. Pharmaceuticals 2025, 18, 908. https://doi.org/10.3390/ph18060908
Bas TG. Innovative Formulation Strategies for Biosimilars: Trends Focused on Buffer-Free Systems, Safety, Regulatory Alignment, and Intellectual Property Challenges. Pharmaceuticals. 2025; 18(6):908. https://doi.org/10.3390/ph18060908
Chicago/Turabian StyleBas, Tomas Gabriel. 2025. "Innovative Formulation Strategies for Biosimilars: Trends Focused on Buffer-Free Systems, Safety, Regulatory Alignment, and Intellectual Property Challenges" Pharmaceuticals 18, no. 6: 908. https://doi.org/10.3390/ph18060908
APA StyleBas, T. G. (2025). Innovative Formulation Strategies for Biosimilars: Trends Focused on Buffer-Free Systems, Safety, Regulatory Alignment, and Intellectual Property Challenges. Pharmaceuticals, 18(6), 908. https://doi.org/10.3390/ph18060908