
Exploring Brazilian Green Propolis Phytochemicals in the Search for Potential Inhibitors of B-Raf600E Enzyme: A Theoretical Approach
 and
and
Abstract
:1. Introduction
2. Results
2.1. Physicochemical Properties and ADME Studies
2.2. Molecular Modeling
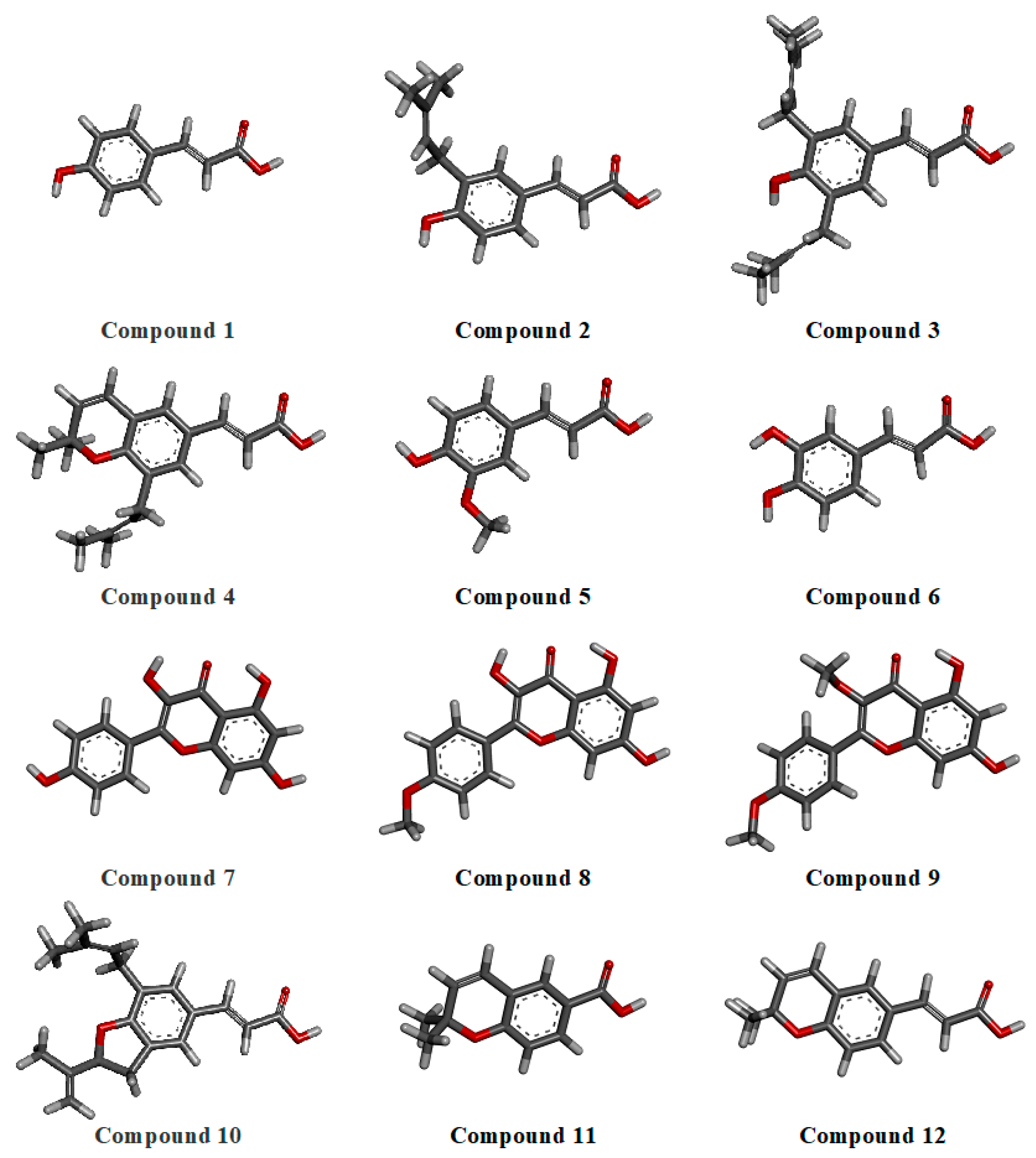
2.2.1. Geometry Optimization
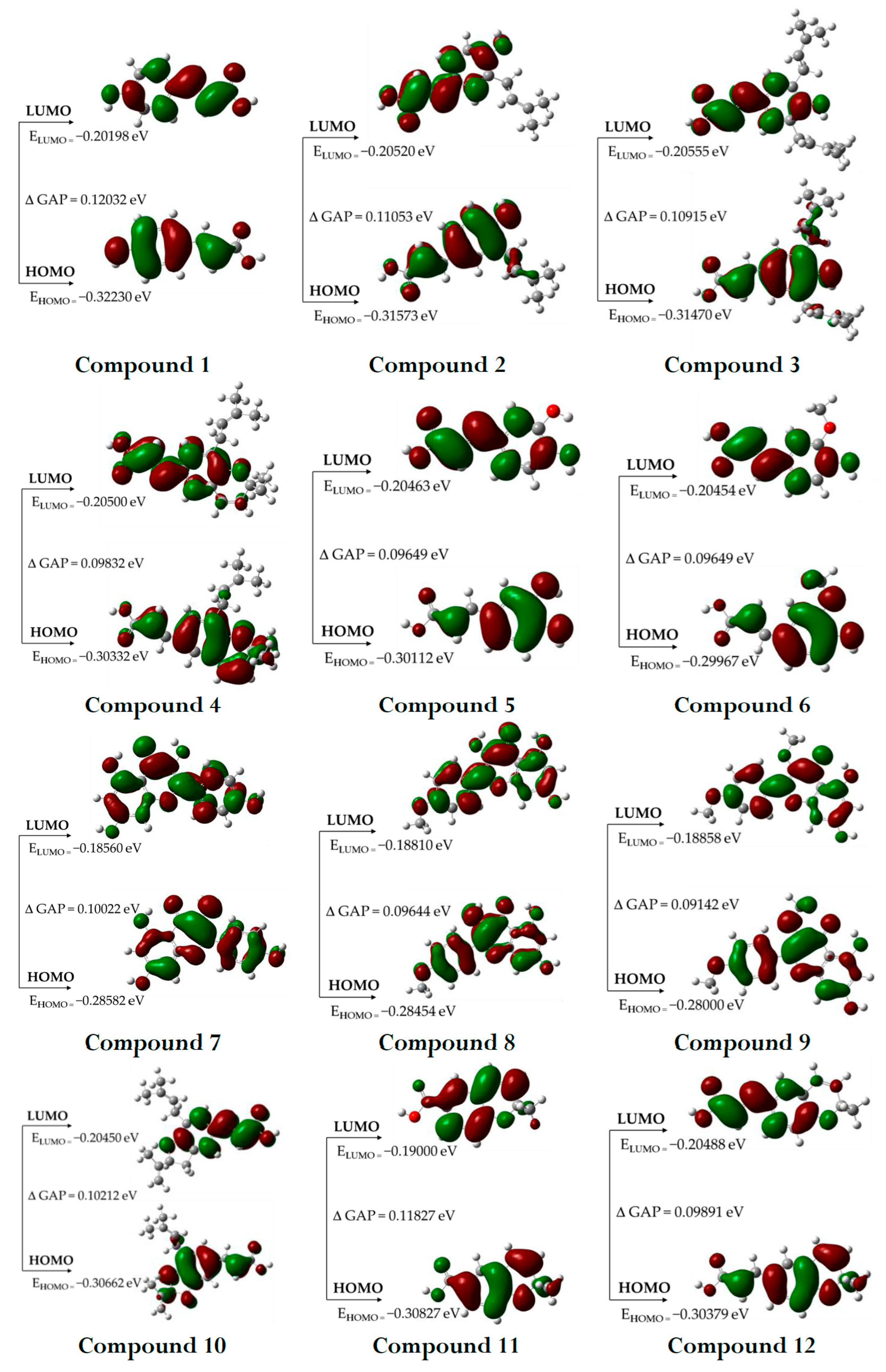
2.2.2. HOMO and LUMO Orbitals
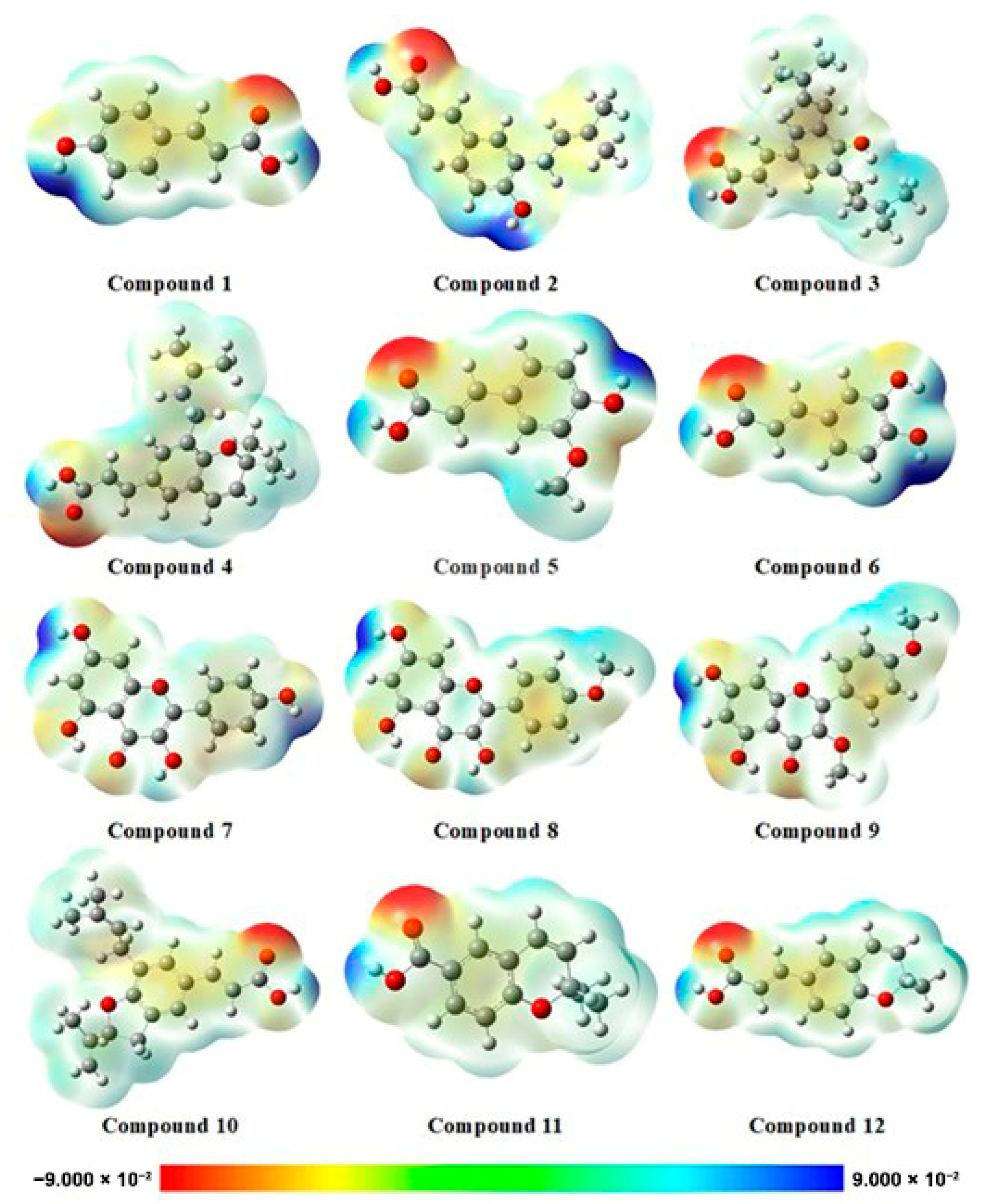
2.2.3. Electrostatic Potential Map (EPM)
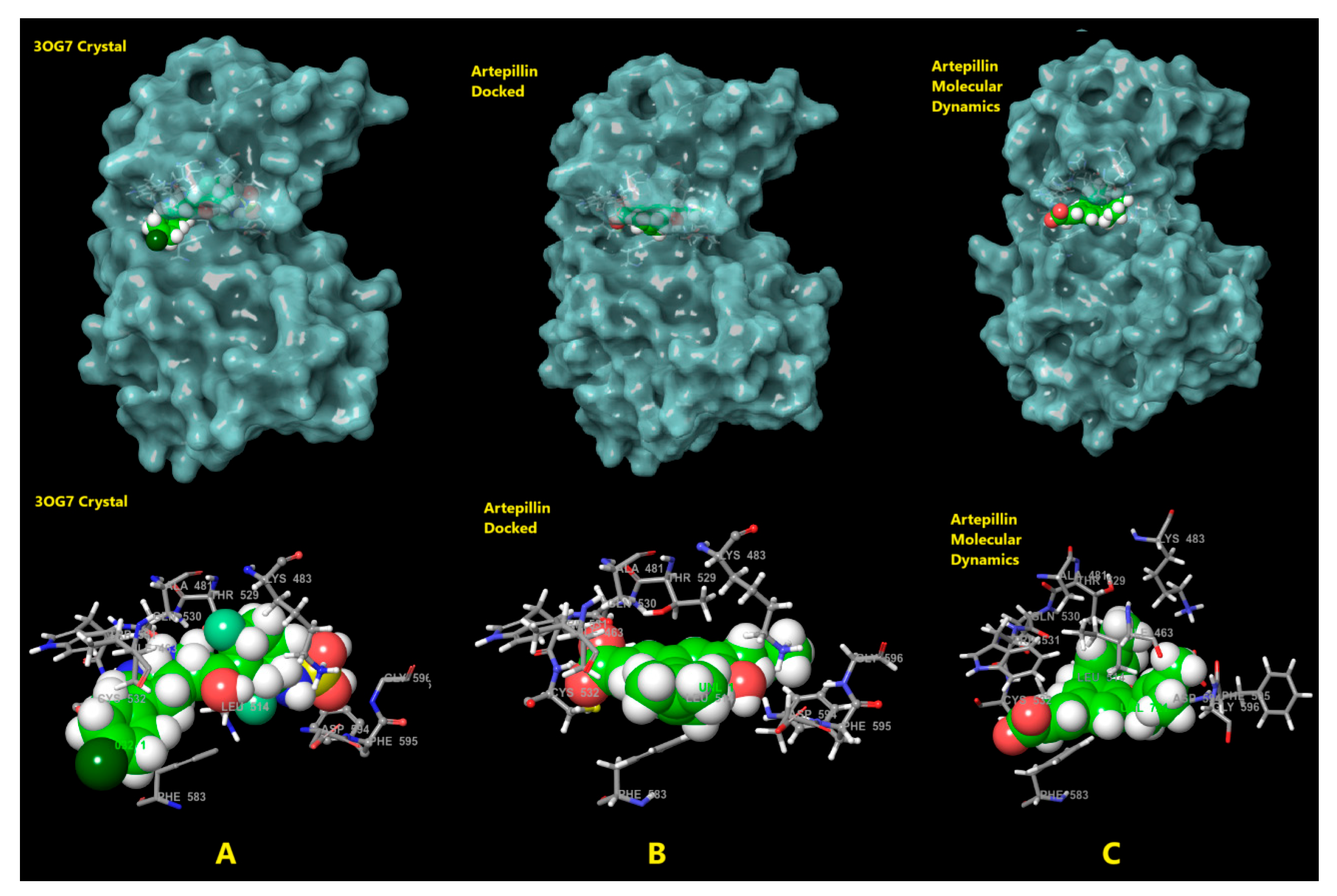
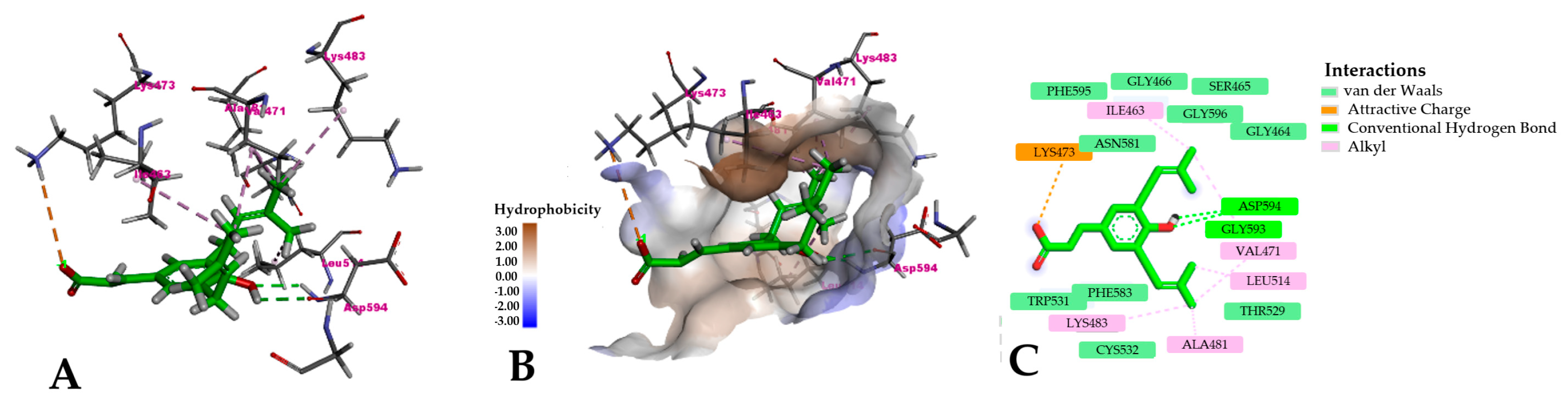
2.3. Molecular Docking
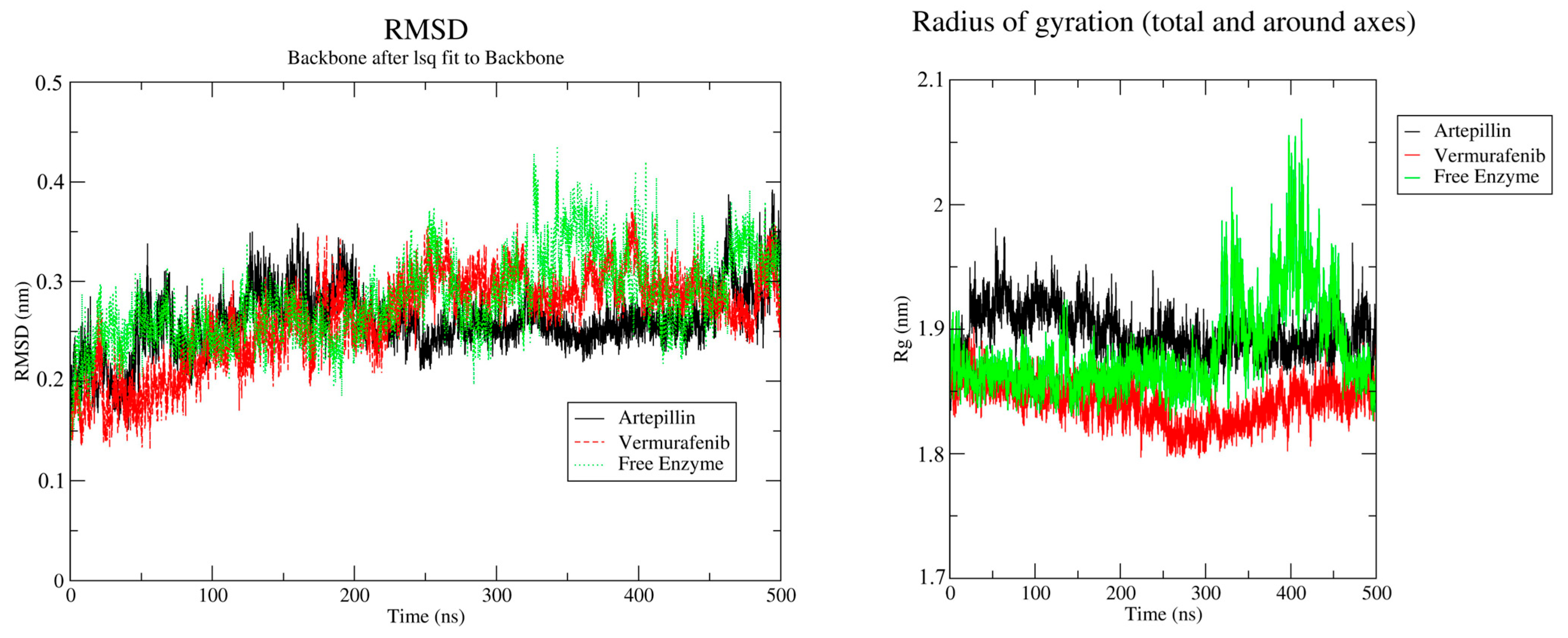
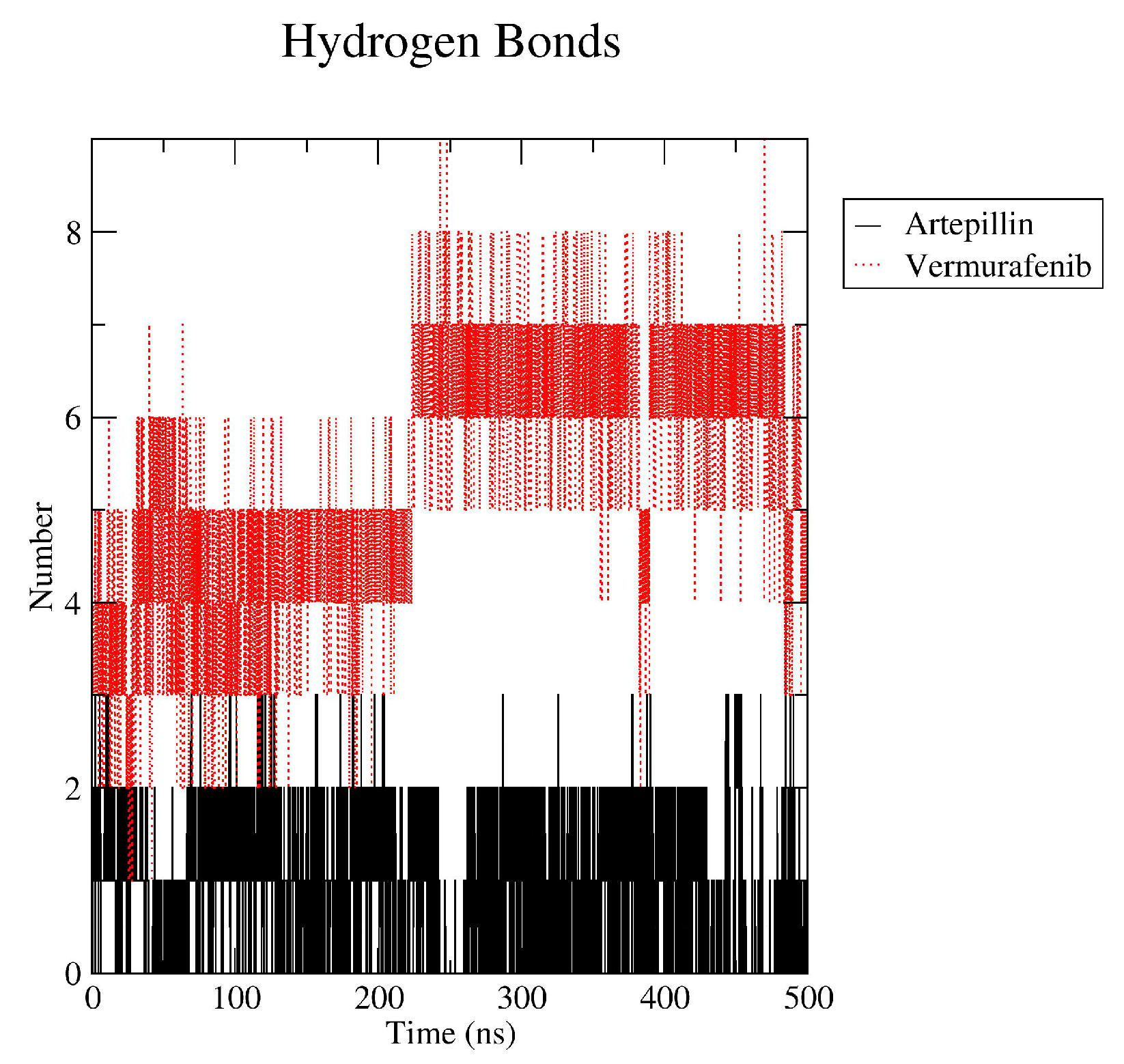
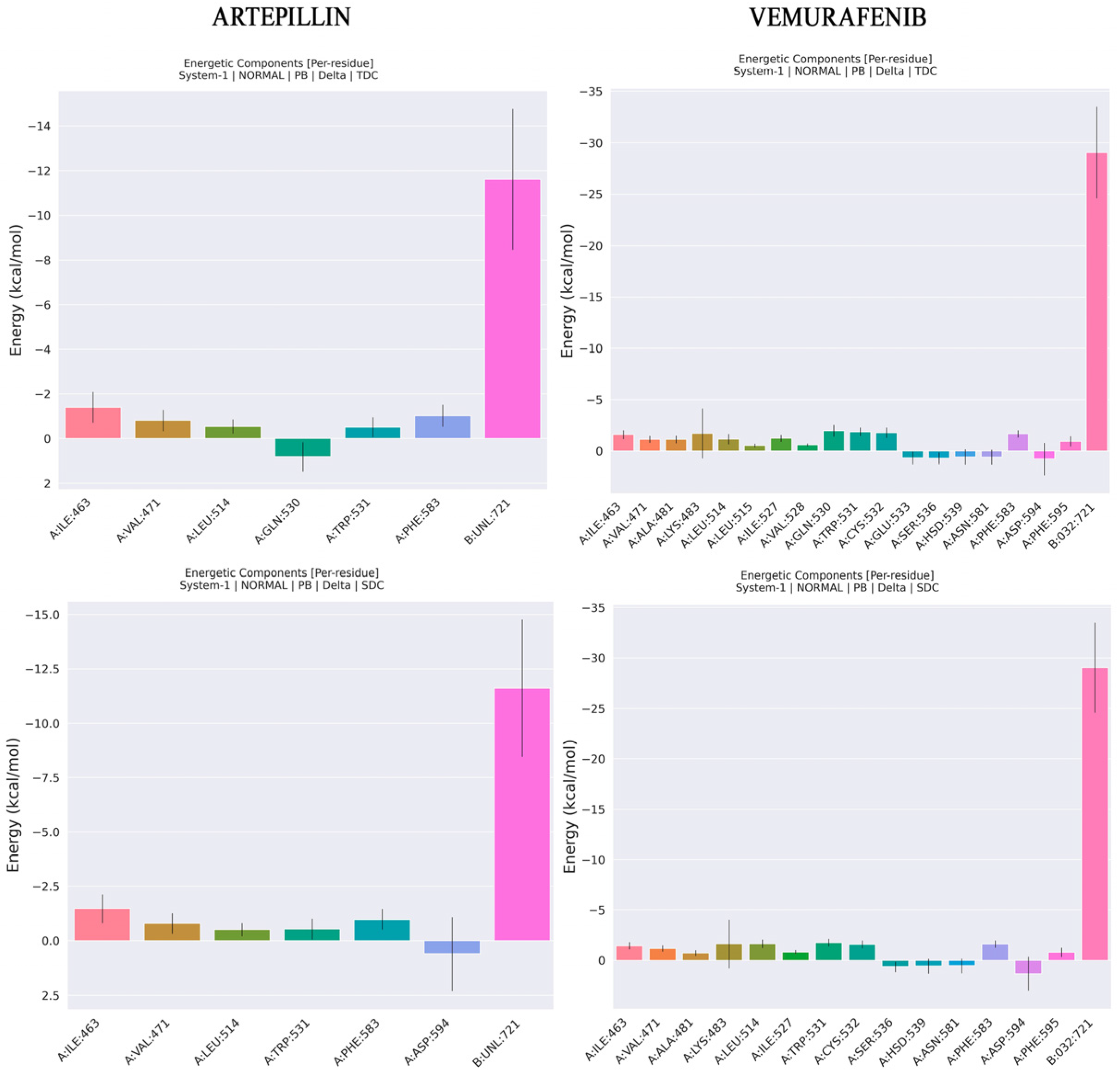
2.4. Molecular Dynamics and Free Energy of Binding
3. Discussion
3.1. Physicochemical Properties and ADME Studies
3.2. Molecular Modeling
3.2.1. Geometry Optimization
3.2.2. HOMO and LUMO Orbitals
3.2.3. Electrostatic Potential Map (EPM)
3.3. Molecular Docking
3.4. Molecular Dynamics and Free Energy
4. Materials and Methods
4.1. Physicochemical Properties and ADME Studies
4.2. Electronic Properties Calculations
4.3. Preparation of Target Protein
4.4. Molecular Docking
4.5. Molecular Dynamics
4.6. Free Energy of Binding
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Imani, S.; Roozitalab, G.; Emadi, M.; Moradi, A.; Behzadi, P.; Kaboli, P.J. The Evolution of BRAF-Targeted Therapies in Melanoma: Overcoming Hurdles and Unleashing Novel Strategies. Front. Oncol. 2024, 14, 1504142. [Google Scholar] [CrossRef] [PubMed]
- Czarnecka, A.M.; Bartnik, E.; Fiedorowicz, M.; Rutkowski, P. Targeted therapy in melanoma and mechanisms of resistance. Int. J. Mol. Sci. 2020, 21, 4576. [Google Scholar] [CrossRef]
- Śmiech, M.; Leszczyński, P.; Kono, H.; Wardell, C.; Taniguchi, H. Emerging BRAF Mutations in Cancer Progression and Their Possible Effects on Transcriptional Networks. Genes 2020, 11, 1342. [Google Scholar] [CrossRef] [PubMed]
- Amanuel, B.; Grieu, F.; Kular, J.; Millward, M.; Iacopetta, B. Incidence of BRAF p.Val600Glu and p.Val600Lys Mutations in a Consecutive Series of 183 Metastatic Melanoma Patients from a High Incidence Region. Pathology 2012, 44, 357–359. [Google Scholar] [CrossRef]
- Silva, G.d.B.; Mendes, A.P.; Macedo, M.P.d.; Pinto, C.A.L.; Gibbons, I.L.; Duprat Neto, J.P. Vemurafenib and Cutaneous Adverse Events—Report of Five Cases. An. Bras. Dermatol. 2015, 90, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Sonawane, P.; Kumar, A.; Singh, H.; Naumovich, V.; Pathak, P.; Grishina, M.; Khalilullah, H.; Jaremko, M.; Emwas, A.-H.; et al. Challenges and Opportunities in the Crusade of BRAF Inhibitors: From 2002 to 2022. ACS Omega 2023, 8, 27819–27844. [Google Scholar] [CrossRef]
- de Carvalho, F.M.d.A.; Schneider, J.K.; de Jesus, C.V.F.; de Andrade, L.N.; Amaral, R.G.; David, J.M.; Krause, L.C.; Severino, P.; Soares, C.M.F.; Caramão Bastos, E.; et al. Brazilian Red Propolis: Extracts Production, Physicochemical Characterization, and Cytotoxicity Profile for Antitumor Activity. Biomolecules 2020, 10, 726. [Google Scholar] [CrossRef]
- Pereira, F.A.N.; Barboza, J.R.; Vasconcelos, C.C.; Lopes, A.J.O.; Ribeiro, M.N.d.S. Use of Stingless Bee Propolis and Geopropolis against Cancer—A Literature Review of Preclinical Studies. Pharmaceuticals 2021, 14, 1161. [Google Scholar] [CrossRef]
- Batista, C.M.; de Queiroz, L.A.; Alves, Â.V.F.; Reis, E.C.A.; Santos, F.A.; Castro, T.N.; Lima, B.S.; Araújo, A.A.S.; Godoy, C.A.P.; Severino, P.; et al. Photoprotection and Skin Irritation Effect of Hydrogels Containing Hydroalcoholic Extract of Red Propolis: A Natural Pathway against Skin Cancer. Heliyon 2022, 8, e08893. [Google Scholar] [CrossRef]
- Saito, Y.; Tsuruma, K.; Ichihara, K.; Shimazawa, M.; Hara, H. Brazilian Green Propolis Water Extract Up-Regulates the Early Expression Level of HO-1 and Accelerates Nrf2 after UVA Irradiation. BMC Complement. Altern. Med. 2015, 15, 421. [Google Scholar] [CrossRef]
- Szliszka, E.; Helewski, K.J.; Mizgala, E.; Krol, W. Ethanolic Extract of Brazilian Green Propolis Sensitizes Prostate Cancer Cells to TRAIL-Induced Apoptosis. Int. J. Oncol. 2011, 38, 941–953. [Google Scholar] [PubMed]
- Gastaldello, G.H.; Cazeloto, A.C.V.; Ferreira, J.C.; Rodrigues, D.M.; Bastos, J.K.; Campo, V.L.; Zoccal, K.F.; Tefé-Silva, C. Green Propolis Compounds (Baccharin and p-Coumaric Acid) Show Beneficial Effects in Mice for Melanoma Induced by B16f10. Medicines 2021, 8, 20. [Google Scholar] [CrossRef]
- Quintino, R.L.; Reis, A.C.; Fernandes, C.C.; Martins, C.H.G.; Colli, A.C.; Crotti, A.E.M.; Squarisi, I.S.; Ribeiro, A.B.; Tavares, D.C.; Miranda, M.L.D. Brazilian Green Propolis: Chemical Composition of Essential Oil and Their In Vitro Antioxidant, Antibacterial and Antiproliferative Activities. Braz. Arch. Biol. Technol. 2020, 63, e20190408. [Google Scholar] [CrossRef]
- Xu, X.; Yang, B.; Wang, D.; Zhu, Y.; Miao, X.; Yang, W. The Chemical Composition of Brazilian Green Propolis and Its Protective Effects on Mouse Aortic Endothelial Cells against Inflammatory Injury. Molecules 2020, 25, 4612. [Google Scholar] [CrossRef] [PubMed]
- de Moura, S.A.L.; Negri, G.; Salatino, A.; Lima, L.D.d.C.; Dourado, L.P.A.; Mendes, J.B.; Andrade, S.P.; Ferreira, M.A.N.D.; Cara, D.C. Aqueous Extract of Brazilian Green Propolis: Primary Components, Evaluation of Inflammation and Wound Healing by Using Subcutaneous Implanted Sponges. Evid.-Based Complem Altern. Med. 2011, 1, 748283. [Google Scholar] [CrossRef]
- Yamaga, M.; Nohara, M.; Hata, A.; Ito, T.; Furumoto, K.; Ohta, N.; Miyamae, J.; Tani, H.; Yamaki, A.; Fujitani, N. Tissue Distribution and Accumulation of Cinnamic Acid Derivatives from Brazilian Green Propolis in Mice. NFS J. 2025, 38, 100222. [Google Scholar] [CrossRef]
- Celińska-Janowicz, K.; Zaręba, I.; Lazarek, U.; Teul, J.; Tomczyk, M.; Pałka, J.; Miltyk, W. Constituents of Propolis: Chrysin, Caffeic Acid, p-Coumaric Acid, and Ferulic Acid Induce PRODH/POX-Dependent Apoptosis in Human Tongue Squamous Cell Carcinoma Cell (CAL-27). Front. Pharmacol. 2018, 9, 336. [Google Scholar] [CrossRef]
- Ueda, T.; Inden, M.; Shirai, K.; Sekine, S.; Masaki, Y.; Kurita, H.; Ichihara, K.; Inuzuka, T.; Hozumi, I. The Effects of Brazilian Green Propolis That Contains Flavonols against Mutant Copper-Zinc Superoxide Dismutase-Mediated Toxicity. Sci. Rep. 2017, 7, 2882. [Google Scholar] [CrossRef]
- Son, N.T.; Ribeiro, V.P.; Bastos, J.K. Flavonoids from Green Propolis of the Northeastern Brazilian Caatinga Mimosa tenuiflora (Willd.) Poir.: A Chemotaxonomic Aspect. Biochem. Syst. Ecol. 2022, 104, 104473. [Google Scholar] [CrossRef]
- Bankova, V.S.; de Castro, S.L.; Marcucci, M.C. Propolis: Recent Advances in Chemistry and Plant Origin. Apidologie 2000, 31, 3–15. [Google Scholar] [CrossRef]
- Huang, Y.; Rong, C.; Zhang, R.; Liu, S. Evaluating Frontier Orbital Energy and HOMO/LUMO Gap with Descriptors from Density Functional Reactivity Theory. J. Mol. Model. 2017, 23, 3. [Google Scholar] [CrossRef] [PubMed]
- Rathi, P.C.; Ludlow, R.F.; Verdonk, M.L. Practical High-Quality Electrostatic Potential Surfaces for Drug Discovery Using a Graph-Convolutional Deep Neural Network. J. Med. Chem. 2020, 63, 8778–8790. [Google Scholar] [CrossRef]
- Suresh, C.H.; Remya, G.S.; Anjalikrishna, P.K. Molecular Electrostatic Potential Analysis: A Powerful Tool to Interpret and Predict Chemical Reactivity. WIREs Comp. Mol. Sci. 2022, 12, e1601. [Google Scholar] [CrossRef]
- Dugave, C.; Demange, L. Cis–Trans Isomerization of Organic Molecules and Biomolecules: Implications and Applications. Chem. Rev. 2003, 103, 2475–2532. [Google Scholar] [CrossRef]
- Aakeröy, C.B.; Panikkattu, S.; Chopade, P.D.; Desper, J. Competing Hydrogen-Bond and Halogen-Bond Donors in Crystal Engineering. Cryst. Eng. Comm. 2013, 15, 3125–3136. [Google Scholar] [CrossRef]
- Nishio, M. The CH/π Hydrogen Bond in Chemistry. Conformation, Supramolecules, Optical Resolution and Interactions Involving Carbohydrates. Phys. Chem. Chem. Phys. 2011, 13, 13873. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Muegge, I.; Heald, S.L.; Brittelli, D. Simple Selection Criteria for Drug-like Chemical Matter. J. Med. Chem. 2001, 44, 1841–1846. [Google Scholar] [CrossRef]
- Beserra, F.P.; Gushiken, L.F.S.; Hussni, M.F.; Ribeiro, V.P.; Bonamin, F.; Jackson, C.J.; Pellizzon, C.H.; Bastos, J.K. Artepillin C as an Outstanding Phenolic Compound of Brazilian Green Propolis for Disease Treatment: A Review on Pharmacological Aspects. Phytother. Res. 2021, 35, 2274–2286. [Google Scholar] [CrossRef]
- de Freitas Meirelles, L.E.; de Assis Carvalho, A.R.B.; Ferreira Damke, G.M.Z.; Souza, R.P.; Damke, E.; de Souza Bonfim-Mendonça, P.; de Oliveira Dembogurski, D.S.; da Silva, D.B.; Consolaro, M.E.L.; da Silva, V.R.S. Antitumoral Potential of Artepillin C, a Compound Derived from Brazilian Propolis, against Breast Cancer Cell Lines. Anticancer. Agents Med. Chem. 2024, 24, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Valivand, N.; Aravand, S.; Lotfi, H.; Esfahani, A.J.; Ahmadpour-Yazdi, H.; Gheibi, N. Propolis: A Natural Compound with Potential as an Adjuvant in Cancer Therapy—A Review of Signaling Pathways. Mol. Biol. Rep. 2024, 51, 931. [Google Scholar] [CrossRef]
- Shahinozzaman, M.; Basak, B.; Emran, R.; Rozario, P.; Obanda, D.N. Artepillin C: A Comprehensive Review of Its Chemistry, Bioavailability, and Pharmacological Properties. Fitoterapia 2020, 147, 104775. [Google Scholar] [CrossRef] [PubMed]
- Hirata, Y.; Motoyama, M.; Kimura, S.; Takashima, M.; Ikawa, T.; Oh-Hashi, K.; Kamatari, Y.O. Artepillin C, a major component of Brazilian green propolis, inhibits endoplasmic reticulum stress and protein aggregation. Eur. J. Pharmacol. 2021, 5, 174572. [Google Scholar] [CrossRef] [PubMed]
- Arruda, C.; Pena Ribeiro, V.; Oliveira Almeida, M.; Aldana Mejía, J.A.; Casoti, R.; Kenupp Bastos, J. Effect of Light, Oxygen and Temperature on the Stability of Artepillin C and p-Coumaric Acid from Brazilian Green Propolis. J. Pharm. Biomed. Anal. 2020, 178, 112922. [Google Scholar] [CrossRef]
- Oteiza, P.I.; Fraga, C.G.; Mills, D.A.; Taft, D.H. Flavonoids and the Gastrointestinal Tract: Local and Systemic Effects. Mol. Aspects Med. 2018, 61, 41–49. [Google Scholar] [CrossRef]
- Panche, A.N.; Diwan, A.D.; Chandra, S.R. Flavonoids: An Overview. J. Nutr. Sci. 2016, 5, e47. [Google Scholar] [CrossRef]
- de Souza Farias, S.A.; da Costa, K.S.; Martins, J.B.L. Analysis of Conformational, Structural, Magnetic, and Electronic Properties Related to Antioxidant Activity: Revisiting Flavan, Anthocyanidin, Flavanone, Flavonol, Isoflavone, Flavone, and Flavan-3-Ol. ACS Omega 2021, 6, 8908–8918. [Google Scholar] [CrossRef]
- Jan, R.; Khan, M.; Asaf, S.; Lubna; Asif, S.; Kim, K.-M. Bioactivity and Therapeutic Potential of Kaempferol and Quercetin: New Insights for Plant and Human Health. Plants 2022, 11, 2623. [Google Scholar] [CrossRef]
- Echeverri, F.; Cardona, G.; Torres, F.; Pelaez, C.; Quiñones, W.; Renteria, E. Ermanin: An Insect Deterrent Flavonoid from Passiflora Foetida Resin. Phytochemistry 1991, 30, 153–155. [Google Scholar] [CrossRef]
- Lu, L.; Luo, K.; Luan, Y.; Zhao, M.; Wang, R.; Zhao, X.; Wu, S. Effect of Caffeic Acid Esters on Antioxidant Activity and Oxidative Stability of Sunflower Oil: Molecular Simulation and Experiments. Food Res. Int. 2022, 160, 111760. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Biswas, S.; Al-Dayan, N.; Alhegaili, A.S.; Sarwat, M. Antioxidant Role of Kaempferol in Prevention of Hepatocellular Carcinoma. Antioxidants 2021, 10, 1419. [Google Scholar] [CrossRef] [PubMed]
- Okińczyc, P.; Widelski, J.; Szperlik, J.; Żuk, M.; Mroczek, T.; Skalicka-Woźniak, K.; Sakipova, Z.; Widelska, G.; Kuś, P.M. Impact of Plant Origin on Eurasian Propolis on Phenolic Profile and Classical Antioxidant Activity. Biomolecules 2021, 11, 68. [Google Scholar] [CrossRef]
- Wang, Y.; Li, C.; Li, Z.; Moalin, M.; Hartog, G.J.M.d.; Zhang, M. Computational Chemistry Strategies to Investigate the Antioxidant Activity of Flavonoids—An Overview. Molecules 2024, 29, 2627. [Google Scholar] [CrossRef]
- Sumayya, P.C.; Mujeeb, V.M.A.; Muraleedharan, K. Radical Scavenging Capacity, UV Activity, and Molecular Docking Studies of 2′,5′,3,4-Tetrahydroxychalcone: An Insight into the Photoprotection. Chem. Phys. Imp. 2022, 5, 100126. [Google Scholar] [CrossRef]
- Rana, J.N.; Mumtaz, S. Prunin: An Emerging Anticancer Flavonoid. Int. J. Mol. Sci. 2025, 26, 2678. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. J. Cheminformatics 2012, 4, 17. [Google Scholar] [CrossRef]
- Dewar, M.J.S.; Thiel, W. Ground States of Molecules. 38. The MNDO Method. Approximations and Parameters. J. Am. Chem. Soc. 1977, 99, 4899–4907. [Google Scholar] [CrossRef]
- Boys, S.F. Electronic Wave Functions. I. A General Method of Calculation for the Stationary States of any Molecular System. Proc. R. Soc. 1950, 200, 542–554. [Google Scholar]
- Neese, F. The ORCA Program System. WIREs Comp. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Berman, H.M. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Goodsell, D.S.; Olson, A.J. Automated Docking of Substrates to Proteins by Simulated Annealing. Proteins 1990, 8, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A. PDBsum: Summaries and Analyses of PDB Structures. Nucleic Acids Res. 2001, 29, 221–222. [Google Scholar] [CrossRef]
- Brooks, B.R.; Brooks, C.L.; Mackerell, A.D.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The Biomolecular Simulation Program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef]
- Bekker, H.; Berendsen, H.; Dijkstra, E. Physics Computing; DeGroot, R., Nadrchal, J., Eds.; World Scientific Publishing: Praha, Czech Republic, 1993; pp. 252–256. [Google Scholar]
- Bill, R.; Miller, I.; McGee, D., Jr.; Swails, J.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comp. 2012, 8, 3314–3321. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | MF | MW (g/mol) | nHA | nAHA | F.Csp3 | nRB | nHBA | nHBD | MR | TPSA (Å2) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | C9H8O3 | 164.16 | 12 | 6 | 0 | 2 | 3 | 2 | 45.13 | 57.53 |
| 2 | C14H16O3 | 232.28 | 17 | 6 | 0.21 | 4 | 3 | 2 | 68.85 | 57.53 |
| 3 | C19H24O3 | 300.39 | 22 | 6 | 0.32 | 6 | 3 | 2 | 92.57 | 57.53 |
| 4 | C19H22O3 | 298.38 | 22 | 6 | 0.32 | 4 | 3 | 1 | 90.95 | 46.53 |
| 5 | C10H10O4 | 194.18 | 14 | 6 | 0.1 | 3 | 4 | 2 | 51.63 | 66.76 |
| 6 | C9H8O4 | 180.16 | 13 | 6 | 0 | 2 | 4 | 3 | 47.16 | 77.76 |
| 7 | C15H10O6 | 286.24 | 21 | 16 | 0 | 1 | 6 | 4 | 76.01 | 111.13 |
| 8 | C16H12O6 | 300.26 | 22 | 16 | 0.06 | 2 | 6 | 3 | 80.48 | 100.13 |
| 9 | C17H14O6 | 314.29 | 23 | 16 | 0.12 | 3 | 6 | 2 | 84.95 | 89.13 |
| 10 | C19H22O3 | 298.38 | 22 | 6 | 0.32 | 6 | 3 | 1 | 90.12 | 46.53 |
| 11 | C12H12O3 | 204.22 | 15 | 6 | 0.25 | 1 | 3 | 1 | 57.52 | 46.53 |
| 12 | C14H14O3 | 230.26 | 17 | 6 | 0.21 | 2 | 3 | 1 | 67.23 | 46.53 |
| Ligand | LogP | GI | BBB | P-gp Substrate | Cytochrome Inhibitor P450 | ||||
|---|---|---|---|---|---|---|---|---|---|
| CYP1A2 | CYP2C19 | CYP2C9 | CYP2D6 | CYP3A4 | |||||
| 1 | 1.26 | High | Yes | No | No | No | No | No | No |
| 2 | 2.83 | High | Yes | No | No | No | No | No | No |
| 3 | 4.27 | High | Yes | No | No | Yes | Yes | No | No |
| 4 | 4.02 | High | Yes | No | No | Yes | Yes | Yes | No |
| 5 | 1.36 | High | Yes | No | No | No | No | No | No |
| 6 | 0.93 | High | No | No | No | No | No | No | No |
| 7 | 1.58 | High | No | Yes | No | No | Yes | Yes | No |
| 8 | 2.00 | High | No | Yes | No | No | Yes | Yes | No |
| 9 | 2.35 | High | No | Yes | No | Yes | Yes | Yes | No |
| 10 | 4.11 | High | Yes | Yes | Yes | Yes | No | Yes | Yes |
| 11 | 2.33 | High | Yes | Yes | No | No | No | Yes | No |
| 12 | 2.67 | High | Yes | Yes | Yes | No | No | Yes | Yes |
| Ligand | Binding Energy (kcal/mol) | Hydrogen Interacting Residue of Target Along with Their Bond Length | Hydrophobic Interacting Residue of Target Along with Their Bond Length |
|---|---|---|---|
| 1 | −5.11 | CYS532 (2.01), GLY534 (1.88), GLN530 (1.72) | TRP531, CYS532 |
| 2 | −7.02 | CYS532 (1.76), ILE527 (2.62), GLN530 (2.05) | THR529, PHE595, LEU505, LEU505, LEU514, ALA481, LYS483 |
| 3 | −8.17 | LYS483 (2.09), CYS532 (2.07), GLN530 (2.02) | VAL471, ILE463, VAL471, LEU505, LEU505, LEU514, PHE468, PHE595, VAL471 |
| 4 | −7.57 | CYS532 (1.85) | CYS532, PHE583, VAL471, ALA481, ALA481, LEU514, VAL471, LYS483, VAL471, LYS483, VAL471, PHE468, PHE583, PHE583, VAL471, ALA481 |
| 5 | −5.51 | PHE595 (2.94) | GLY596, ALA481, VAL471, VAL471, ALA481, LYS483 |
| 6 | −5.41 | LYS483 (2.62), PHE595 (2.59), GLY596 (2.13), ALA481 (2.21), ILE527 (2.04) | VAL471, ALA481, LYS483 |
| 7 | −7.15 | THR529 (2.59), CYS532 (1.77), ILE527 (2.04), GLN530 (2.03), CYS532 (2.29) | TRP531, TRP531, PHE583, PHE583, VAL471, ALA481, CYS532, ILE463, VAL471, VAL471, ALA481, LYS483 |
| 8 | −7.38 | LYS483 (2.34), CYS532 (1.69), GLN530 (1.98), CYS532 (2.23) | TRP531, TRP531, PHE583, PHE583, VAL471, ALA481, CYS532, ILE463, LYS483, LEU514 |
| 9 | −7.29 | CYS532 (1.71), GLN530 (2.04) | ASP594, LYS483, TRP531, LEU505, VAL471, ALA481, VAL471, ALA481, CYS532, LYS483 |
| 10 | −7.74 | LYS483 (1.73), PHE595 (2.99), GLY596 (1.96) | VAL471, ALA481, ALA481, LEU514, CYS532, VAL471, TRP531, TRP531, TRP531, PHE583, VAL471, ALA481, LYS483, LEU514 |
| 11 | −6.74 | LYS483 (1.80), ASP594 (2.22), PHE595 (2.99), GLY596 (2.24) | LYS483, ALA481, ALA481, ALA481, LEU514, LEU514, VAL471, PHE583, LYS483, LEU514 |
| 12 | −8.49 | LYS483 (1.70), ASP594 (2.29), PHE595 (2.99), GLY596 (2.21), PHE583 (2.85) | VAL471, ALA481, ALA481, LEU514, CYS532, VAL471, VAL471, ILE463, VAL471, PHE468, TRP531, TRP531, PHE583, PHE583, VAL471, LEU514 |
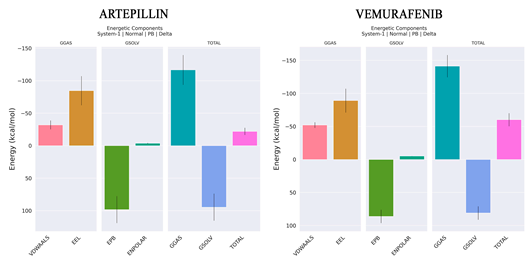 | ||
| ΔE (VDW) | −32.03 | −52.13 |
| ΔE (EEL) | −84.62 | −89.14 |
| ΔE (EPB) | 98.26 | 86.12 |
| ΔE (ENPOLAR) | −3.65 | −5.1 |
| GGAS | −116.65 | −141.27 |
| GSOLV | 94.6 | 81.02 |
| ΔH (total) | −22.05 | −60.25 |
| −TΔS | 1.28 | 42.70 |
| ΔG | −20.77 | −17.55 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Souza, G.F.; Santana, A.F.; Antunes, F.S.K.; Cogo, R.M.; Pereira, M.D.; Rando, D.G.G.; Gonçalves, C.P. Exploring Brazilian Green Propolis Phytochemicals in the Search for Potential Inhibitors of B-Raf600E Enzyme: A Theoretical Approach. Pharmaceuticals 2025, 18, 902. https://doi.org/10.3390/ph18060902
de Souza GF, Santana AF, Antunes FSK, Cogo RM, Pereira MD, Rando DGG, Gonçalves CP. Exploring Brazilian Green Propolis Phytochemicals in the Search for Potential Inhibitors of B-Raf600E Enzyme: A Theoretical Approach. Pharmaceuticals. 2025; 18(6):902. https://doi.org/10.3390/ph18060902
Chicago/Turabian Stylede Souza, Garcia Ferreira, Airis Farias Santana, Fernanda Sanches Kuhl Antunes, Ramon Martins Cogo, Matheus Dornellas Pereira, Daniela Gonçales Galasse Rando, and Carolina Passarelli Gonçalves. 2025. "Exploring Brazilian Green Propolis Phytochemicals in the Search for Potential Inhibitors of B-Raf600E Enzyme: A Theoretical Approach" Pharmaceuticals 18, no. 6: 902. https://doi.org/10.3390/ph18060902
APA Stylede Souza, G. F., Santana, A. F., Antunes, F. S. K., Cogo, R. M., Pereira, M. D., Rando, D. G. G., & Gonçalves, C. P. (2025). Exploring Brazilian Green Propolis Phytochemicals in the Search for Potential Inhibitors of B-Raf600E Enzyme: A Theoretical Approach. Pharmaceuticals, 18(6), 902. https://doi.org/10.3390/ph18060902