

Design and Synthesis of Pyridine-Based Pyrrolo[2,3-d]pyrimidine Analogs as CSF1R Inhibitors: Molecular Hybridization and Scaffold Hopping Approach

 , , ,
, , ,  ,
,  and
and
Abstract

1. Introduction
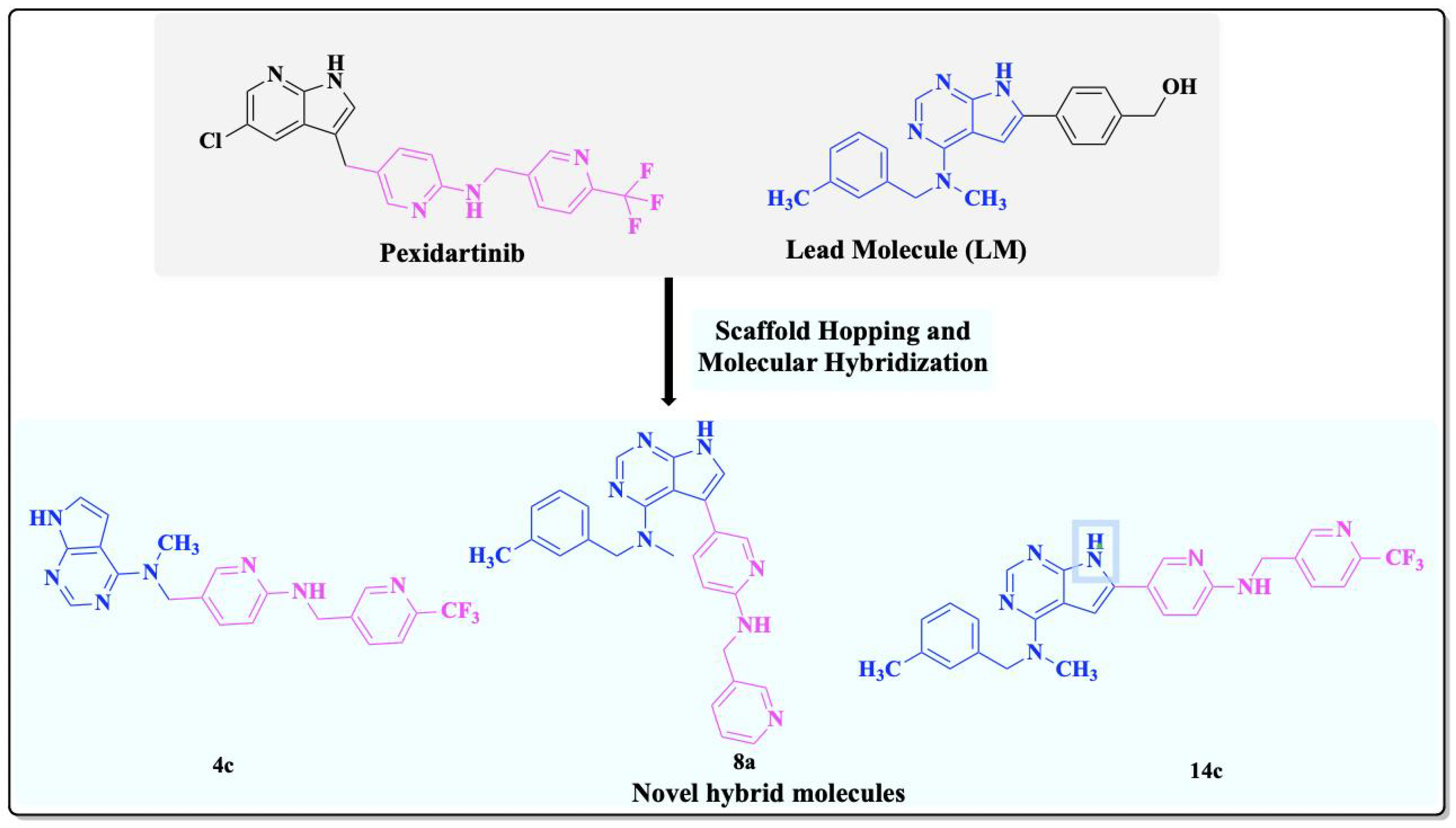
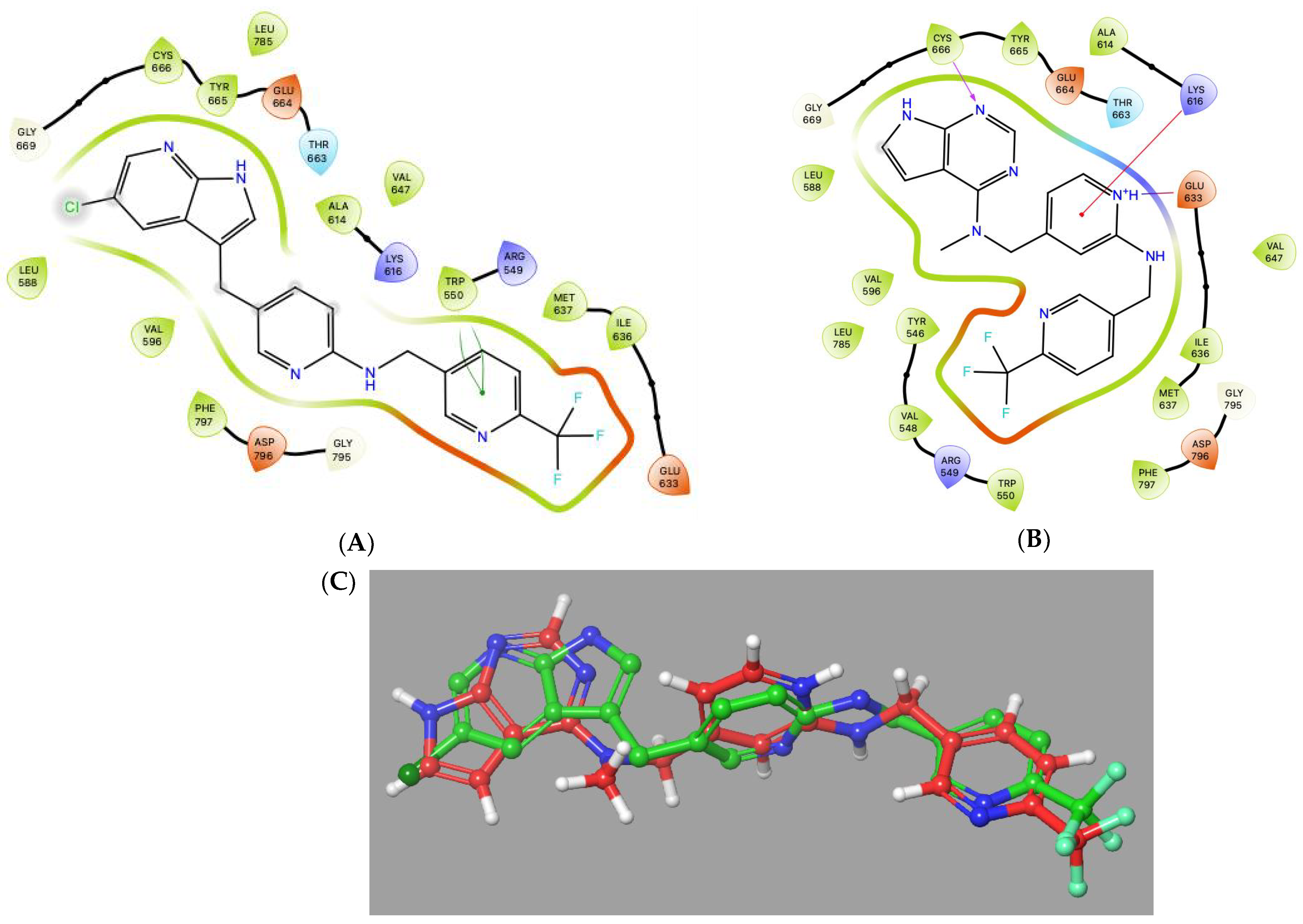
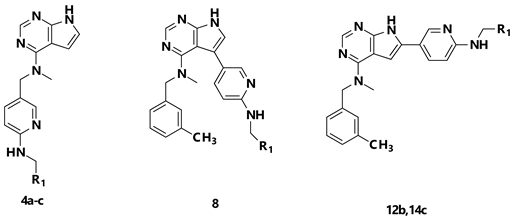
2. Results and Discussion
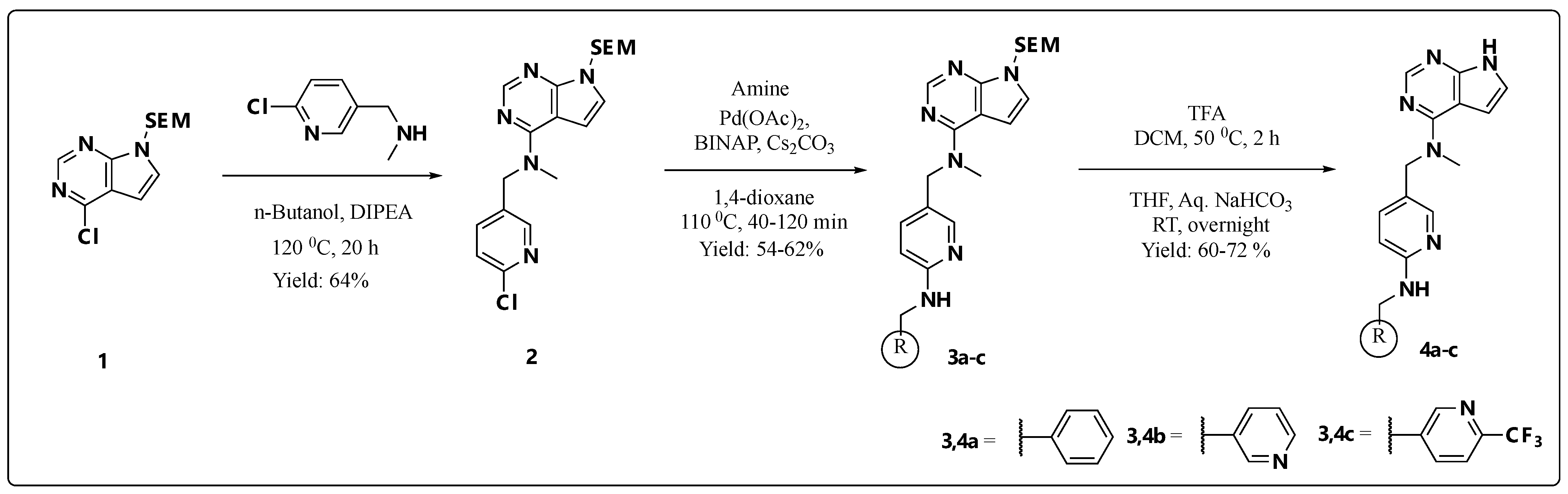
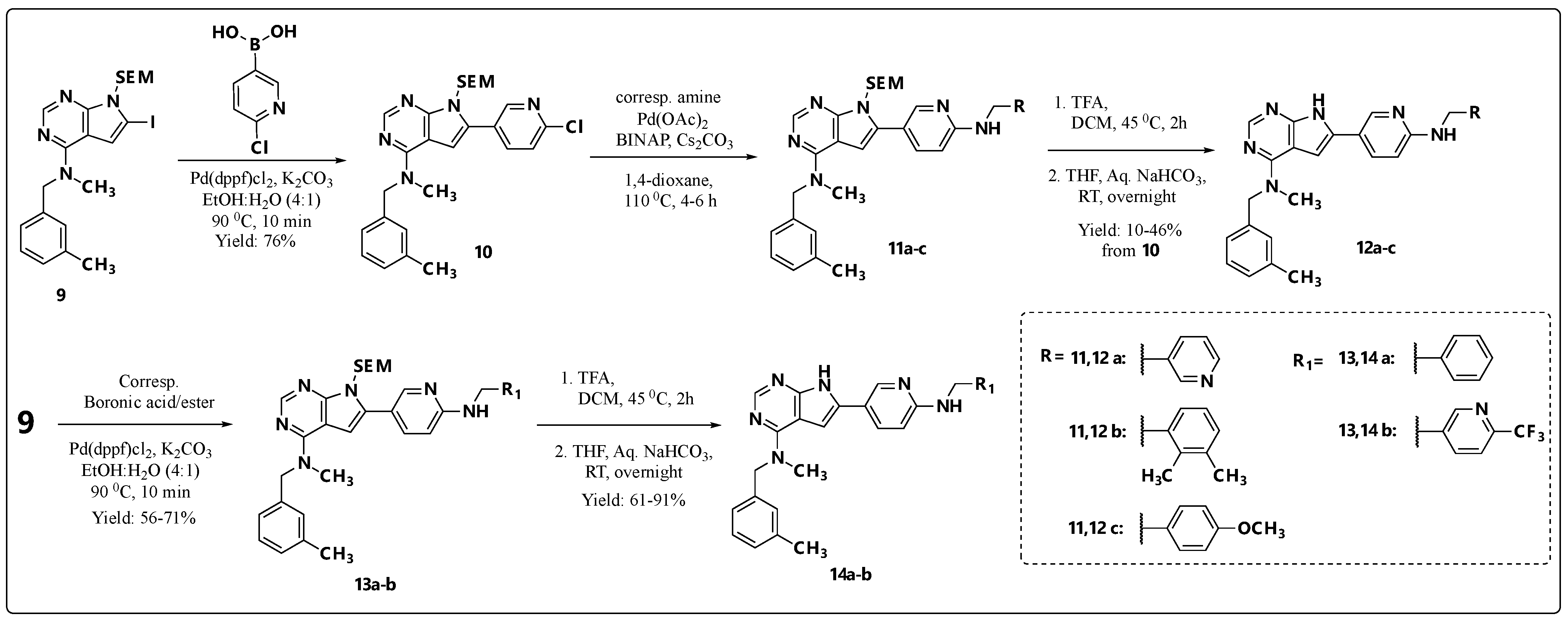
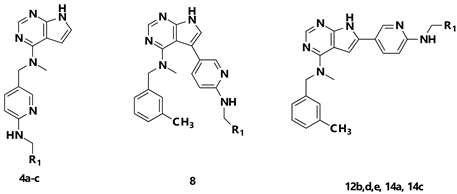
2.1. Chemistry
2.2. Enzymatic and Cellular Assays
2.3. ADME Profiling of Selected Compounds
2.4. Kinase Selectivity
3. Materials and Methods
3.1. Chemicals and Data Analysis
3.2. Synthesis of Building Blocks
3.2.1. N-((6-Chloropyridin-3-yl)methyl)-N-methyl-7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (2)
3.2.2. General Procedure A: Synthesis of 3a–c and 11a–c via Buchwald–Hartwig Amination Reaction
3.2.3. N-((6-(Benzylamino)pyridin-3-yl)methyl)-N-methyl-7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (3a)
3.2.4. N-Methyl-N-((6-((pyridin-2-ylmethyl)amino)pyridin-3-yl)methyl)-7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (3b)
3.2.5. N-Methyl-N-((6-(((6-(trifluoromethyl)pyridin-3-yl)methyl)amino)pyridin-3-yl)methyl)-7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (3c)
3.2.6. General Procedure B: Synthesis of Final Compounds 4a–c, 8a and 14a,b via SEM-Deprotection
3.2.7. N-((6-(Benzylamino)pyridin-3-yl)methyl)-N-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine (4a)
3.2.8. N-Methyl-N-((6-((pyridin-2-ylmethyl)amino)pyridin-3-yl)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (4b)
3.2.9. N-Methyl-N-((6-(((6-(trifluoromethyl)pyridin-3-yl)methyl)amino)pyridin-3-yl)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (4c)
3.2.10. 5-Iodo-N-methyl-N-(3-methylbenzyl)-7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (6)
3.2.11. 5-(6-Chloropyridin-3-yl)-N-methyl-N-(3-methylbenzyl)-7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (7)
3.2.12. N-Methyl-N-(3-methylbenzyl)-5-(6-((pyridin-3-ylmethyl)amino)pyridin-3-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (8a)
3.2.13. 6-(6-Chloropyridin-3-yl)-N-methyl-N-(3-methylbenzyl)-7-((2-(trimethylsilyl) ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (10)
3.2.14. N-Methyl-N-(3-methylbenzyl)-6-(6-((pyridin-3-ylmethyl)amino)pyridin-3-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (12a)
3.2.15. 6-(6-((2,3-Dimethylbenzyl)amino)pyridin-3-yl)-N-methyl-N-(3-methylbenzyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (12b)
3.2.16. 6-(6-((4-Methoxybenzyl)amino)pyridin-3-yl)-N-methyl-N-(3-methylbenzyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (12c)
3.2.17. 6-(6-(Benzylamino)pyridin-3-yl)-N-methyl-N-(3-methylbenzyl)-7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (13a)
3.2.18. N-Methyl-N-(3-methylbenzyl)-6-(6-(((6-(trifluoromethyl)pyridin-3-yl)methyl)amino)pyridin-3-yl)-7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (13b)
3.2.19. 6-(6-(Benzylamino)pyridin-3-yl)-N-methyl-N-(3-methylbenzyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (14a)
3.2.20. N-Methyl-N-(3-methylbenzyl)-6-(6-(((6-(trifluoromethyl)pyridin-3-yl)methyl) amino)pyridin-3-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (14b)
3.3. Bio-Chemical Assays
3.3.1. CSF1R Enzymatic Inhibitory Assay (LANCE)
3.3.2. Kinase Panel
3.3.3. KIT-WT Lantha Assay (Kd)
3.3.4. Cell Viability Assay with Ba/F3-hCSF1R Cells
3.3.5. ADME Properties
Kinetic Solubility and Microsomal Stability Phase I
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kumari, A.; Silakari, O.; Singh, R.K. Recent advances in colony stimulating factor-1 receptor/c-FMS as an emerging target for various therapeutic implications. Biomed. Pharmacother. 2018, 103, 662–679. [Google Scholar] [CrossRef] [PubMed]
- Xiang, C.G.; Li, H.; Tang, W. Targeting CSF-1R represents an effective strategy in modulating inflammatory diseases. Pharmacol. Res. 2023, 187, 106566. [Google Scholar] [CrossRef]
- Barca, C.; Foray, C.; Hermann, S.; Herrlinger, U.; Remory, I.; Laoui, D.; Schaefers, M.; Grauer, O.M.; Zinnhardt, B.; Jacobs, A.H. The Colony Stimulating Factor-1 Receptor (CSF-1R)-Mediated Regulation of Microglia/Macrophages as a Target for Neurological Disorders (Glioma, Stroke). Front. Immunol. 2021, 12, 787307. [Google Scholar] [CrossRef]
- Abu-Duhier, F.M.; Goodeve, A.C.; Care, R.S.; Gari, M.; Wilson, G.A.; Peake, I.R.; Reilly, J.T. Mutational analysis of class III receptor tyrosine kinases (C-KIT, C-FMS, FLT3) in idiopathic myelofibrosis. Br. J. Haematol. 2003, 120, 464–470. [Google Scholar] [CrossRef]
- Tarale, P.; Alam, M.M. Colony-stimulating factor 1 receptor signaling in the central nervous system and the potential of its pharmacological inhibitors to halt the progression of neurological disorders. Inflammopharmacology 2022, 30, 821–842. [Google Scholar] [CrossRef] [PubMed]
- Denny, W.A.; Flanagan, J.U. Small-molecule CSF1R kinase inhibitors; review of patents 2015-present. Expert Opin. Ther. Pat. 2021, 31, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Tap, W.D.; Wainberg, Z.A.; Anthony, S.P.; Ibrahim, P.N.; Zhang, C.; Healey, J.H.; Chmielowski, B.; Staddon, A.P.; Cohn, A.L.; Shapiro, G.I.; et al. Structure-Guided Blockade of CSF1R Kinase in Tenosynovial Giant-Cell Tumor. N. Engl. J. Med. 2015, 373, 428–437. [Google Scholar] [CrossRef]
- Smith, B.D.; Kaufman, M.D.; Wise, S.C.; Ahn, Y.M.; Caldwell, T.M.; Leary, C.B.; Lu, W.P.; Tan, G.G.; Vogeti, L.; Vogeti, S.; et al. Vimseltinib: A Precision CSF1R Therapy for Tenosynovial Giant Cell Tumors and Diseases Promoted by Macrophages. Mol. Cancer Ther. 2021, 20, 2098–2109. [Google Scholar] [CrossRef]
- Xu, J.M.; Shen, L.; Zhou, Z.W.; Li, J.; Bai, C.M.; Chi, Y.H.B.L.; Li, Z.P.; Xu, N.; Li, E.X.; Liu, T.S.; et al. Surufatinib in advanced extrapancreatic neuroendocrine tumours (SANET-ep): A randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 2020, 21, 1500–1512. [Google Scholar] [CrossRef]
- Wen, J.C.; Wang, S.Y.; Guo, R.X.; Liu, D. CSF1R inhibitors are emerging immunotherapeutic drugs for cancer treatment. Eur. J. Med. Chem. 2023, 245, 114884. [Google Scholar] [CrossRef]
- Benner, B.; Good, L.; Quiroga, D.; Schultz, T.E.; Kassem, M.; Carson, W.E.; Cherian, M.A.; Sardesai, S.; Wesolowski, R. Pexidartinib, a Novel Small Molecule CSF-1R Inhibitor in Use for Tenosynovial Giant Cell Tumor: A Systematic Review of Pre-Clinical and Clinical Development. Drug Des. Dev. Ther. 2020, 14, 1693–1704. [Google Scholar] [CrossRef] [PubMed]
- Wesolowski, R.; Sharma, N.; Reebel, L.; Rodal, M.B.; Peck, A.; West, B.L.; Marimuthu, A.; Severson, P.; Karlin, D.A.; Dowlati, A.; et al. Phase Ib study of the combination of pexidartinib (PLX3397), a CSF-1R inhibitor, and paclitaxel in patients with advanced solid tumors. Ther. Adv. Med. Oncol. 2019, 11, 1758835919854238. [Google Scholar] [CrossRef]
- Rosenbaum, E.; Kelly, C.; D’Angelo, S.P.; Dickson, M.A.; Gounder, M.; Keohan, M.L.; Movva, S.; Condy, M.; Adamson, T.; Mcfadyen, C.R.; et al. A Phase I Study of Binimetinib (MEK162) Combined with Pexidartinib (PLX3397) in Patients with Advanced Gastrointestinal Stromal Tumor. Oncologist 2019, 24, 1309. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.M.; Shen, L.; Bai, C.M.; Wang, W.; Li, J.; Yu, X.J.; Li, Z.P.; Li, E.X.; Yuan, X.L.; Chi, Y.H.B.L.; et al. Surufatinib in advanced pancreatic neuroendocrine tumours (SANET-p): A randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 2020, 21, 1489–1499. [Google Scholar] [CrossRef]
- Movva, S.; Spira, A.I.; Hamilton, E.P.; Wang, J.S.; Cohn, A.L.; Strauss, J.F.; Stacchiotti, S.; Tucci, C.; Kauh, J.; Nanda, S.; et al. Surufatinib in US patients with soft tissue sarcoma. J. Clin. Oncol. 2022, 40, 11557. [Google Scholar] [CrossRef]
- Son, Y.; Jeong, Y.J.; Shin, N.R.; Oh, S.J.; Nam, K.R.; Choi, H.D.; Choi, J.Y.; Lee, H.J. Inhibition of Colony-Stimulating Factor 1 Receptor by PLX3397 Prevents Amyloid Beta Pathology and Rescues Dopaminergic Signaling in Aging 5xFAD Mice. Int. J. Mol. Sci. 2020, 21, 5553. [Google Scholar] [CrossRef]
- Neal, M.L.; Fleming, S.M.; Budge, K.M.; Boyle, A.M.; Kim, C.; Alam, G.; Beier, E.E.; Wu, L.J.; Richardson, J.R. Pharmacological inhibition of CSF1R by GW2580 reduces microglial proliferation and is protective against neuroinflammation and dopaminergic neurodegeneration. FASEB J. 2020, 34, 1679–1694. [Google Scholar] [CrossRef]
- Mancuso, R.; Fryatt, G.; Cleal, M.; Obst, J.; Pipi, E.; Monzon-Sandoval, J.; Ribe, E.; Winchester, L.; Webber, C.; Nevado, A.; et al. CSF1R inhibitor JNJ-40346527 attenuates microglial proliferation and neurodegeneration in P301S mice. Brain 2019, 142, 3243–3264. [Google Scholar] [CrossRef] [PubMed]
- Spiteri, A.G.; Ni, D.; Ling, Z.L.; Macia, L.; Campbell, I.L.; Hofer, M.J.; King, N.J.C. PLX5622 Reduces Disease Severity in Lethal CNS Infection by Off-Target Inhibition of Peripheral Inflammatory Monocyte Production. Front. Immunol. 2022, 13, 851556. [Google Scholar] [CrossRef]
- Albert, D.H.; Tapang, P.; Magoc, T.J.; Pease, L.J.; Reuter, D.R.; Wei, R.-Q.; Li, J.; Guo, J.; Bousquet, P.F.; Ghoreishi-Haack, N.S.; et al. Preclinical activity of ABT-869, a multitargeted receptor tyrosine kinase inhibitor. Mol. Cancer Ther. 2006, 5, 995–1006. [Google Scholar] [CrossRef]
- Zhou, Y.; Shan, S.; Li, Z.B.; Xin, L.J.; Pan, D.S.; Yang, Q.J.; Liu, Y.P.; Yue, X.P.; Liu, X.R.; Gao, J.Z.; et al. CS2164, a novel multi-target inhibitor against tumor angiogenesis, mitosis and chronic inflammation with anti-tumor potency. Cancer Sci. 2017, 108, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.Q.; Sheng, X.N.; Tang, B.X.; Chi, Z.H.; Cui, C.L.; Si, L.; Mao, L.L.; Lian, B.; Li, S.M.; Zhou, L.; et al. Anti-VEGFR, PDGFR, and CSF1R tyrosine kinase inhibitor CM082 (X-82) in combination with everolimus for treatment of metastatic renal cell carcinoma: A phase 1 clinical trial. Lancet Oncol. 2017, 18, S8. [Google Scholar] [CrossRef]
- Aarhus, T.I.; Bjørnstad, F.; Wolowczyk, C.; Larsen, K.U.; Rognstad, L.; Leithaug, T.; Unger, A.; Habenberger, P.; Wolf, A.; Bjørkøy, G.; et al. Synthesis and Development of Highly Selective Pyrrolo[2,3- d]pyrimidine CSF1R Inhibitors Targeting the Autoinhibited Form. J. Med. Chem. 2023, 66, 6959–6980. [Google Scholar] [CrossRef] [PubMed]
- Aarhus, T.I.; Eickhoff, J.; Klebl, B.; Unger, A.; Boros, J.; Choidas, A.; Zischinsky, M.L.; Wolowczyk, C.; Bjørkøy, G.; Sundby, E.; et al. A highly selective purine-based inhibitor of CSF1R potently inhibits osteoclast differentiation. Eur. J. Med. Chem. 2023, 255, 115344. [Google Scholar] [CrossRef] [PubMed]
- Aarhus, T.I.; Teksum, V.; Unger, A.; Habenberger, P.; Wolf, A.; Eickhoff, J.; Klebl, B.; Wolowczyk, C.; Bjørkøy, G.; Sundby, E.; et al. Negishi Cross-Coupling in the Preparation of Benzyl Substituted Pyrrolo[2,3-d]pyrimidine Based CSF1R Inhibitors. Eur. J. Org. Chem. 2023, 26, e202300052. [Google Scholar] [CrossRef]
- Bjørnstad, F.; Havik, S.; Aarhus, T.I.; Mahdi, I.; Unger, A.; Habenberger, P.; Degenhart, C.; Eickhoff, J.; Klebl, B.M.; Sundby, E.; et al. Pyrrolopyrimidine based CSF1R inhibitors: Attempted departure from Flatland. Eur. J. Med. Chem. 2024, 265, 116053. [Google Scholar] [CrossRef]
- Bérubé, G. An overview of molecular hybrids in drug discovery. Expert Opin. Drug Discov. 2016, 11, 281–305. [Google Scholar] [CrossRef]
- Soltan, O.M.; Shoman, M.E.; Abdel-Aziz, S.A.; Narumi, A.; Konno, H.; Abdel-Aziz, M. Molecular hybrids: A five-year survey on structures of multiple targeted hybrids of protein kinase inhibitors for cancer therapy. Eur. J. Med. Chem. 2021, 225, 113768. [Google Scholar] [CrossRef]
- Hu, Y.; Stumpfe, D.; Bajorath, J. Recent Advances in Scaffold Hopping. J. Med. Chem. 2017, 60, 1238–1246. [Google Scholar] [CrossRef]
- Azhar, Z.; Grose, R.P.; Raza, A.; Raza, Z. In silico targeting of colony-stimulating factor-1 receptor: Delineating immunotherapy in cancer. Explor. Target. Anti-Tumor Ther. 2023, 4, 727–742. [Google Scholar] [CrossRef]
- Schrödinger_Release_2025-1. Glide; Schrödinger, LLC: New York, NY, USA, 2025. [Google Scholar]
- Reiersølmoen, A.C.; Aarhus, T.I.; Eckelt, S.; Nørsett, K.G.; Sundby, E.; Hoff, B.H. Potent and selective EGFR inhibitors based on 5-aryl-7H-pyrrolopyrimidin-4-amines. Bioorg. Chem. 2019, 88, 102918. [Google Scholar] [CrossRef] [PubMed]
- Karaman, M.W.; Herrgard, S.; Treiber, D.K.; Gallant, P.; Atteridge, C.E.; Campbell, B.T.; Chan, K.W.; Ciceri, P.; Davis, M.I.; Edeen, P.T.; et al. A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2008, 26, 127–132. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Compound | R1 | Enzymatic Assay | Cellular Assay | ||
| CSF1R (IC50, nM) 1 | KIT (Kd, nM) 2 | IC50 (μM), Ba/F3 CSF1R 3 |
IC50 (μM), Ba/F3 IL-3 4 | ||
| 4a | 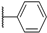 | 857 (0.96) | >1000 (0.94) | 1.16 ± 0.22 | 1.39 ± 0.40 |
| 4b |  | >1000 (0.86) | >1000 (0.97) | >10 | >10 |
| 4c |  | >1000 (0.81) | >1000 (0.99) | 9.96 ± 0.02 | >10 |
| 8 | 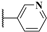 | >1000 (0.81) | 397 (0.98) | 8.46 ± 0.89 | >10 |
| 12b |  | 1.54 (0.97) | 375 (0.98) | 1.15 ± 0.36 | >10 |
| 12d |  | >1000 (0.47) | >1000 (-) | ND | ND 5 |
| 12e |  | 10.50 (0.91) | >1000 (-) | 6.24 5 | 9.25 5 |
| 14a | 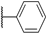 | 6.00 (0.92) | >1000 (0.84) | ND | ND |
| 14c | 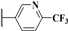 | 7.18 (0.97) | >1000 (0.82) | >10 | >10 |
| Pexidartinib | 18.10 (0.97) | 0.036 (1.0) | 0.064 ± 0.04 | 6.54 ± 0.61 | |
 | |||||||
|---|---|---|---|---|---|---|---|
| Compound | Solubility 3 | MLM 1 | HLM 2 | Papp, A-B 4 | Papp, B-A 4 | MDCK Ratio 5 | |
| 4a | 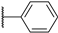 | 20.9 | 277.3 | 20.9 | 34.0 | 24.7 | 0.72 |
| 4b | 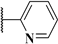 | 440.8 | 101.9 | 440.8 | 42.0 | 28.8 | 0.69 |
| 4c | 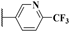 | 20.9 | 150.7 | 20.9 | 18.3 | 10.0 | 0.55 |
| 8 |  | 70.4 | 1540.3 | 70.4 | 15.9 | 4.4 | 0.27 |
| 12b |  | 4.4 | 128.4 | 115.5 | 0.3 | 0.1 | 0.19 |
| 14c |  | 0.7 | 60.5 | 77 | 5.4 | 0.8 | 0.14 |
| Pexidartinib | 20.5 | 41.4 | 11.8 | 4.5 | 1.5 | 0.33 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cherukupalli, S.; Degenhart, C.; Habenberger, P.; Unger, A.; Eickhoff, J.; Hoff, B.H.; Sundby, E. Design and Synthesis of Pyridine-Based Pyrrolo[2,3-d]pyrimidine Analogs as CSF1R Inhibitors: Molecular Hybridization and Scaffold Hopping Approach. Pharmaceuticals 2025, 18, 814. https://doi.org/10.3390/ph18060814
Cherukupalli S, Degenhart C, Habenberger P, Unger A, Eickhoff J, Hoff BH, Sundby E. Design and Synthesis of Pyridine-Based Pyrrolo[2,3-d]pyrimidine Analogs as CSF1R Inhibitors: Molecular Hybridization and Scaffold Hopping Approach. Pharmaceuticals. 2025; 18(6):814. https://doi.org/10.3390/ph18060814
Chicago/Turabian StyleCherukupalli, Srinivasulu, Carsten Degenhart, Peter Habenberger, Anke Unger, Jan Eickhoff, Bård Helge Hoff, and Eirik Sundby. 2025. "Design and Synthesis of Pyridine-Based Pyrrolo[2,3-d]pyrimidine Analogs as CSF1R Inhibitors: Molecular Hybridization and Scaffold Hopping Approach" Pharmaceuticals 18, no. 6: 814. https://doi.org/10.3390/ph18060814
APA StyleCherukupalli, S., Degenhart, C., Habenberger, P., Unger, A., Eickhoff, J., Hoff, B. H., & Sundby, E. (2025). Design and Synthesis of Pyridine-Based Pyrrolo[2,3-d]pyrimidine Analogs as CSF1R Inhibitors: Molecular Hybridization and Scaffold Hopping Approach. Pharmaceuticals, 18(6), 814. https://doi.org/10.3390/ph18060814