
Warburg-like Metabolic Reprogramming in Endometriosis: From Molecular Mechanisms to Therapeutic Approaches

 , , , ,
, , , , 
Abstract
1. Introduction
2. Overview of the Warburg’s Effect
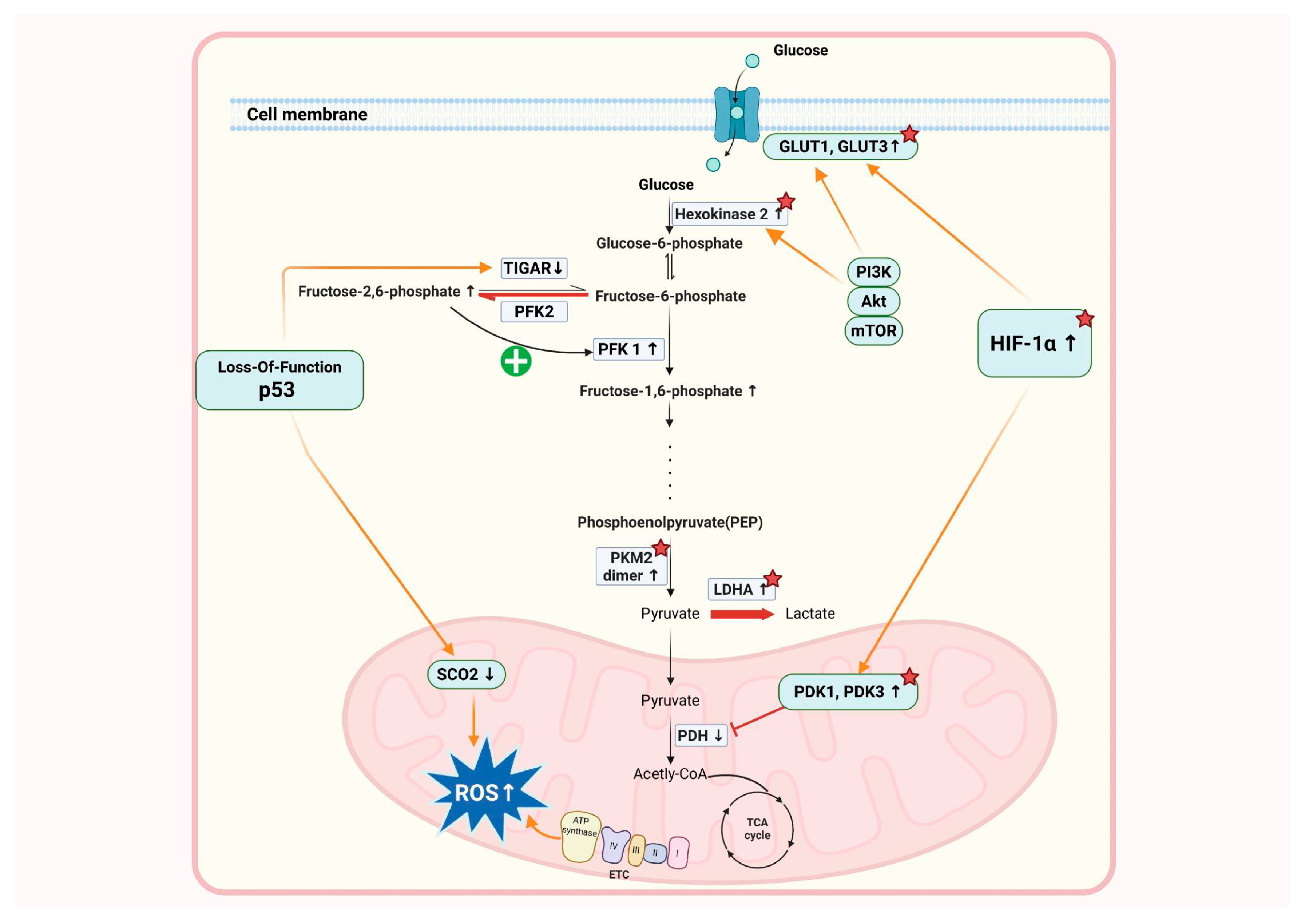
3. Metabolic Reprogramming in Endometriosis
4. Molecular Mechanisms Underlying Warburg-like Metabolic Reprogramming
4.1. Hypoxia-Inducible Factor (HIF) Signaling and Glycolysis Activation
4.2. PI3K/AKT/mTOR Pathway and Metabolic Changes
4.3. Role of Inflammatory Cytokines and Metabolic Changes
4.4. Genetic and Epigenetic Factors Contributing to Metabolic Reprogramming
4.5. The Role of Mitochondrial Dysfunction in the Progression of Endometriosis
4.6. Interaction with Angiogenesis and Matrix Remodeling
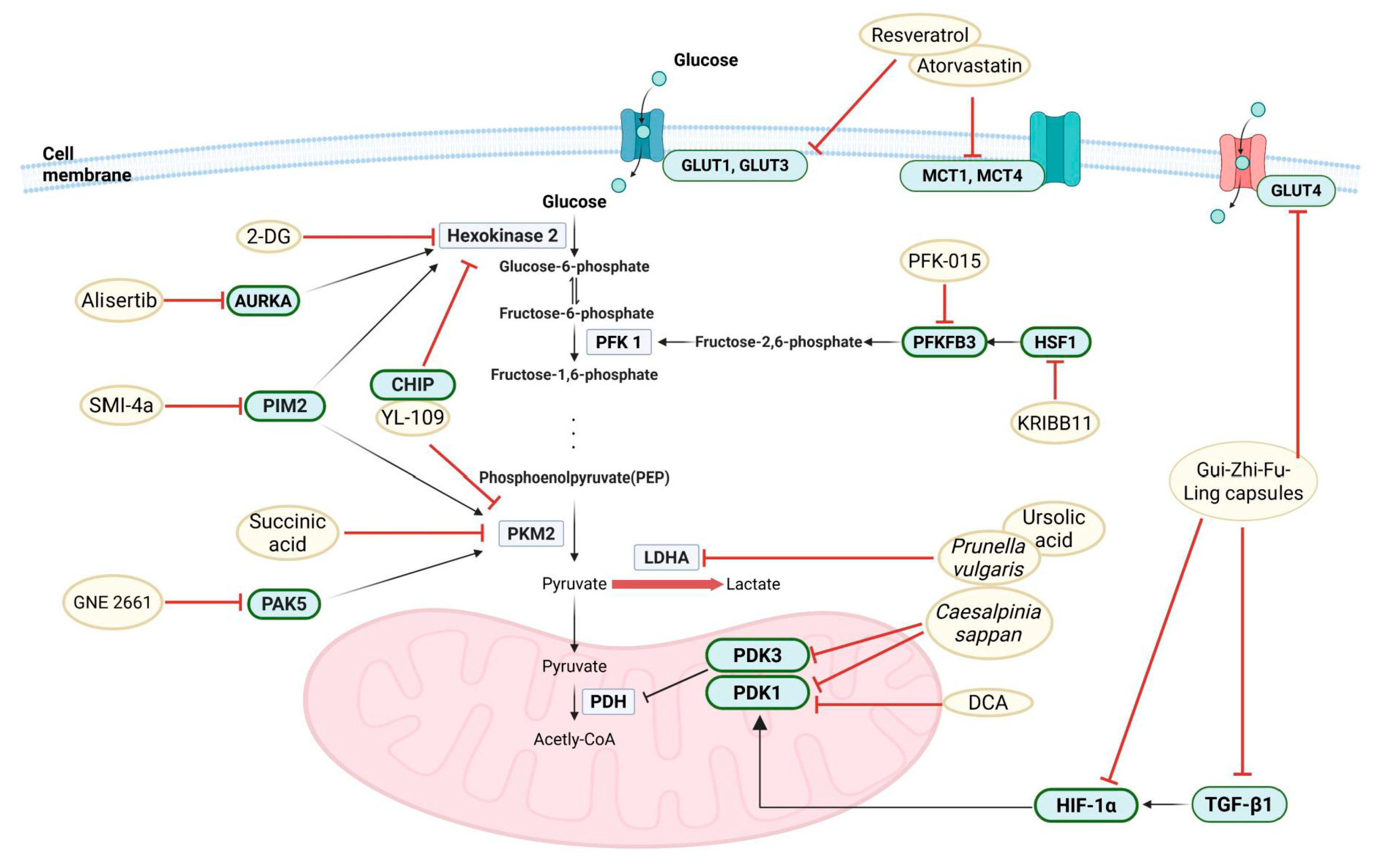
5. Current Research on Glycolysis Inhibitors in Endometriosis
6. Clinical Implications of Metabolic Reprogramming in Endometriosis
7. Conclusions and Future Perspectives
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Giudice, L.C. Clinical practice. Endometriosis. N. Engl. J. Med. 2010, 362, 2389–2398. [Google Scholar] [CrossRef] [PubMed]
- Simoens, S.; Dunselman, G.; Dirksen, C.; Hummelshoj, L.; Bokor, A.; Brandes, I.; Brodszky, V.; Canis, M.; Colombo, G.L.; DeLeire, T.; et al. The burden of endometriosis: Costs and quality of life of women with endometriosis and treated in referral centres. Hum. Reprod. 2012, 27, 1292–1299. [Google Scholar] [CrossRef]
- World Health Organization (WHO). Endometriosis. Available online: https://www.who.int/news-room/fact-sheets/detail/endometriosis (accessed on 26 May 2025).
- Mehedintu, C.; Plotogea, M.N.; Ionescu, S.; Antonovici, M. Endometriosis still a challenge. J. Med. Life 2014, 7, 349–357. [Google Scholar] [PubMed]
- Ping, S.; Ma, C.; Liu, P.; Yang, L.; Yang, X.; Wu, Q.; Zhao, X.; Gong, B. Molecular mechanisms underlying endometriosis pathogenesis revealed by bioinformatics analysis of microarray data. Arch. Gynecol. Obstet. 2016, 293, 797–804. [Google Scholar] [CrossRef]
- Ferrero, S.; Evangelisti, G.; Barra, F. Current and emerging treatment options for endometriosis. Expert Opin. Pharmacother. 2018, 19, 1109–1125. [Google Scholar] [CrossRef]
- Guo, S.W. Recurrence of endometriosis and its control. Hum. Reprod. Update 2009, 15, 441–461. [Google Scholar] [CrossRef] [PubMed]
- Hughes, C.L.; Foster, W.G.; Agarwal, S.K. The Impact of Endometriosis across the Lifespan of Women: Foreseeable Research and Therapeutic Prospects. BioMed Res. Int. 2015, 2015, 158490. [Google Scholar] [CrossRef] [PubMed]
- Thomas, E.J.; Campbell, I.G. Evidence that endometriosis behaves in a malignant manner. Gynecol. Obstet. Investig. 2000, 50 (Suppl. S1), 2–10. [Google Scholar] [CrossRef]
- Young, V.J.; Brown, J.K.; Maybin, J.; Saunders, P.T.; Duncan, W.C.; Horne, A.W. Transforming growth factor-beta induced Warburg-like metabolic reprogramming may underpin the development of peritoneal endometriosis. J. Clin. Endocrinol. Metab. 2014, 99, 3450–3459. [Google Scholar] [CrossRef]
- Hou, S.; Lei, S.; Peng, H.; Weng, L.; Lv, S.; Li, M.; Zhao, D. Downregulating HK2 inhibits proliferation of endometrial stromal cells through a noncanonical pathway involving phosphorylation of signal transducer and activator of transcription 1 in endometriosis. Biol. Reprod. 2022, 107, 488–499. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The Warburg effect: How does it benefit cancer cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Mathupala, S.P.; Ko, Y.H.; Pedersen, P.L. Hexokinase-2 bound to mitochondria: Cancer’s stygian link to the “Warburg Effect” and a pivotal target for effective therapy. Semin. Cancer Biol. 2009, 19, 17–24. [Google Scholar] [CrossRef]
- Zahra, K.; Dey, T.; Ashish; Mishra, S.P.; Pandey, U. Pyruvate Kinase M2 and Cancer: The Role of PKM2 in Promoting Tumorigenesis. Front. Oncol. 2020, 10, 159. [Google Scholar] [CrossRef]
- Courtnay, R.; Ngo, D.C.; Malik, N.; Ververis, K.; Tortorella, S.M.; Karagiannis, T.C. Cancer metabolism and the Warburg effect: The role of HIF-1 and PI3K. Mol. Biol. Rep. 2015, 42, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Hoxhaj, G.; Manning, B.D. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef]
- Dong, Y.; Tu, R.; Liu, H.; Qing, G. Regulation of cancer cell metabolism: Oncogenic MYC in the driver’s seat. Signal Transduct. Target. Ther. 2020, 5, 124. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, J.; Liang, Y.; Wu, R.; Zhao, Y.; Hong, X.; Lin, M.; Yu, H.; Liu, L.; Levine, A.J.; et al. Tumour-associated mutant p53 drives the Warburg effect. Nat. Commun. 2013, 4, 2935. [Google Scholar] [CrossRef]
- Liang, Y.; Liu, J.; Feng, Z. The regulation of cellular metabolism by tumor suppressor p53. Cell Biosci. 2013, 3, 9. [Google Scholar] [CrossRef]
- Lee, P.; Vousden, K.H.; Cheung, E.C. TIGAR, TIGAR, burning bright. Cancer Metab. 2014, 2, 1. [Google Scholar] [CrossRef]
- Zhang, X.D.; Qin, Z.H.; Wang, J. The role of p53 in cell metabolism. Acta Pharmacol. Sin. 2010, 31, 1208–1212. [Google Scholar] [CrossRef] [PubMed]
- Zong, W.X.; Rabinowitz, J.D.; White, E. Mitochondria and Cancer. Mol. Cell 2016, 61, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Fendt, S.M.; Frezza, C.; Erez, A. Targeting Metabolic Plasticity and Flexibility Dynamics for Cancer Therapy. Cancer Discov. 2020, 10, 1797–1807. [Google Scholar] [CrossRef]
- Cluntun, A.A.; Lukey, M.J.; Cerione, R.A.; Locasale, J.W. Glutamine Metabolism in Cancer: Understanding the Heterogeneity. Trends Cancer 2017, 3, 169–180. [Google Scholar] [CrossRef]
- Arner, E.N.; Rathmell, J.C. Metabolic programming and immune suppression in the tumor microenvironment. Cancer Cell 2023, 41, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, K.; Morii, E. Metabolic Reprogramming of Cancer Cells during Tumor Progression and Metastasis. Metabolites 2021, 11, 28. [Google Scholar] [CrossRef]
- Schiliro, C.; Firestein, B.L. Mechanisms of Metabolic Reprogramming in Cancer Cells Supporting Enhanced Growth and Proliferation. Cells 2021, 10, 1056. [Google Scholar] [CrossRef]
- Sarsenova, M.; Lawarde, A.; Pathare, A.D.S.; Saare, M.; Modhukur, V.; Soplepmann, P.; Terasmaa, A.; Kaambre, T.; Gemzell-Danielsson, K.; Lalitkumar, P.G.L.; et al. Endometriotic lesions exhibit distinct metabolic signature compared to paired eutopic endometrium at the single-cell level. Commun. Biol. 2024, 7, 1026. [Google Scholar] [CrossRef]
- Chen, Q.; Jiao, Y.; Yin, Z.; Fu, X.; Guo, S.; Zhou, Y.; Wang, Y. Establishment of a novel glycolysis-immune-related diagnosis gene signature for endometriosis by machine learning. J. Assist. Reprod. Genet. 2023, 40, 1147–1161. [Google Scholar] [CrossRef]
- Guo, S.; Chen, Q.; Liang, J.; Wu, H.; Li, L.; Wang, Y. Correlation of Glycolysis-immune-related Genes in the Follicular Microenvironment of Endometriosis Patients with ART Outcomes. Reprod. Sci. 2024, 31, 3357–3367. [Google Scholar] [CrossRef] [PubMed]
- Toniyan, K.A.; Malkov, A.A.; Biryukov, N.S.; Gorbacheva, E.Y.; Boyarintsev, V.V.; Ogneva, I.V. The Cellular Respiration of Endometrial Biopsies from Patients with Various Forms of Endometriosis. Int. J. Mol. Sci. 2024, 25, 3680. [Google Scholar] [CrossRef] [PubMed]
- Khashchenko, E.P.; Vysokikh, M.Y.; Marey, M.V.; Sidorova, K.O.; Manukhova, L.A.; Shkavro, N.N.; Uvarova, E.V.; Chuprynin, V.D.; Fatkhudinov, T.K.; Adamyan, L.V.; et al. Altered Glycolysis, Mitochondrial Biogenesis, Autophagy and Apoptosis in Peritoneal Endometriosis in Adolescents. Int. J. Mol. Sci. 2024, 25, 4238. [Google Scholar] [CrossRef]
- Tu, J.L.; Fang, R.X. Identification of fatty acid metabolism hub genes in endometriosis using integrative bioinformatics analysis. Front. Med. 2025, 12, 1529074. [Google Scholar] [CrossRef]
- Wang, H.; Cao, Y.; Gou, Y.; Wang, H.; Liang, Z.; Wu, Q.; Tan, J.; Liu, J.; Li, Z.; Cui, J.; et al. IGF2BP3 promotes glutamine metabolism of endometriosis by interacting with UCA1 to enhances the mRNA stability of GLS1. Mol. Med. 2024, 30, 64. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, K.-Y.; Lin, S.-C.; Wu, M.-H.; Tsai, S.-J. Pathological functions of hypoxia in endometriosis. Front. Biosci. (Elite Ed.) 2015, 7, 309–321. [Google Scholar] [CrossRef]
- Li, W.N.; Wu, M.H.; Tsai, S.J. HYPOXIA AND REPRODUCTIVE HEALTH: The role of hypoxia in the development and progression of endometriosis. Reproduction 2021, 161, F19–F31. [Google Scholar] [CrossRef] [PubMed]
- Rytkonen, K.T.; Heinosalo, T.; Mahmoudian, M.; Ma, X.; Perheentupa, A.; Elo, L.L.; Poutanen, M.; Wagner, G.P. Transcriptomic responses to hypoxia in endometrial and decidual stromal cells. Reproduction 2020, 160, 39–51. [Google Scholar] [CrossRef]
- Lac, V.; Nazeran, T.M.; Tessier-Cloutier, B.; Aguirre-Hernandez, R.; Albert, A.; Lum, A.; Khattra, J.; Praetorius, T.; Mason, M.; Chiu, D.; et al. Oncogenic mutations in histologically normal endometrium: The new normal? J. Pathol. 2019, 249, 173–181. [Google Scholar] [CrossRef]
- Suda, K.; Nakaoka, H.; Yoshihara, K.; Ishiguro, T.; Tamura, R.; Mori, Y.; Yamawaki, K.; Adachi, S.; Takahashi, T.; Kase, H.; et al. Clonal Expansion and Diversification of Cancer-Associated Mutations in Endometriosis and Normal Endometrium. Cell Rep. 2018, 24, 1777–1789. [Google Scholar] [CrossRef]
- Pocate-Cheriet, K.; Santulli, P.; Kateb, F.; Bourdon, M.; Maignien, C.; Batteux, F.; Chouzenoux, S.; Patrat, C.; Wolf, J.P.; Bertho, G.; et al. The follicular fluid metabolome differs according to the endometriosis phenotype. Reprod. Biomed. Online 2020, 41, 1023–1037. [Google Scholar] [CrossRef]
- McKinnon, B.; Bertschi, D.; Wotzkow, C.; Bersinger, N.A.; Evers, J.; Mueller, M.D. Glucose transporter expression in eutopic endometrial tissue and ectopic endometriotic lesions. J. Mol. Endocrinol. 2014, 52, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Kierans, S.J.; Taylor, C.T. Regulation of glycolysis by the hypoxia-inducible factor (HIF): Implications for cellular physiology. J. Physiol. 2021, 599, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Wang, X.; Ran, L.; Song, J.; Luo, R.; Wang, Y. Hypoxia-Inducible Factor 1alpha Regulates the Transforming Growth Factor beta1/SMAD Family Member 3 Pathway to Promote Breast Cancer Progression. J. Breast Cancer 2018, 21, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liu, L.; Fan, R. The PKM2/HIF-1alpha Axis is Involved in the Pathogenesis of Endometriosis via TGF-beta1 under Endometrial Polyps. Front. Biosci. (Landmark Ed.) 2024, 29, 417. [Google Scholar] [CrossRef]
- Sarsenova, M.; Boggavarapu, N.R.; Kask, K.; Modhukur, V.; Samuel, K.; Karro, H.; Gemzell-Danielsson, K.; Lalitkumar, P.G.L.; Salumets, A.; Peters, M.; et al. Hypoxic conditions affect transcriptome of endometrial stromal cells in endometriosis and promote TGFBI axis. Front. Endocrinol. 2024, 15, 1465393. [Google Scholar] [CrossRef]
- Skuli, N.; Majmundar, A.J.; Krock, B.L.; Mesquita, R.C.; Mathew, L.K.; Quinn, Z.L.; Runge, A.; Liu, L.; Kim, M.N.; Liang, J.; et al. Endothelial HIF-2alpha regulates murine pathological angiogenesis and revascularization processes. J. Clin. Investig. 2012, 122, 1427–1443. [Google Scholar] [CrossRef]
- Rankin, E.B.; Rha, J.; Unger, T.L.; Wu, C.H.; Shutt, H.P.; Johnson, R.S.; Simon, M.C.; Keith, B.; Haase, V.H. Hypoxia-inducible factor-2 regulates vascular tumorigenesis in mice. Oncogene 2008, 27, 5354–5358. [Google Scholar] [CrossRef]
- Iommarini, L.; Porcelli, A.M.; Gasparre, G.; Kurelac, I. Non-Canonical Mechanisms Regulating Hypoxia-Inducible Factor 1 Alpha in Cancer. Front. Oncol. 2017, 7, 286. [Google Scholar] [CrossRef]
- Ngo, C.; Chereau, C.; Nicco, C.; Weill, B.; Chapron, C.; Batteux, F. Reactive oxygen species controls endometriosis progression. Am. J. Pathol. 2009, 175, 225–234. [Google Scholar] [CrossRef]
- Dai, W.; Guo, R.; Na, X.; Jiang, S.; Liang, J.; Guo, C.; Fang, Y.; Na, Z.; Li, D. Hypoxia and the endometrium: An indispensable role for HIF-1α as therapeutic strategies. Redox Biol. 2024, 73, 103205. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.J.; Isaacs, J.S.; Lee, S.; Trepel, J.; Neckers, L. IL-1beta-mediated up-regulation of HIF-1alpha via an NFkappaB/COX-2 pathway identifies HIF-1 as a critical link between inflammation and oncogenesis. FASEB J. 2003, 17, 2115–2117. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Dong, S.; Liu, W.; Wang, M.; Tian, S.; Ai, Y.; Wang, H. Accumulated ROS Activates HIF-1alpha-Induced Glycolysis and Exerts a Protective Effect on Sensory Hair Cells Against Noise-Induced Damage. Front. Mol. Biosci. 2021, 8, 806650. [Google Scholar] [CrossRef]
- Lingappan, K. NF-kappaB in Oxidative Stress. Curr. Opin. Toxicol. 2018, 7, 81–86. [Google Scholar] [CrossRef]
- Gao, J.; Zhao, Y.; Li, T.; Gan, X.; Yu, H. The Role of PKM2 in the Regulation of Mitochondrial Function: Focus on Mitochondrial Metabolism, Oxidative Stress, Dynamic, and Apoptosis. PKM2 in Mitochondrial Function. Oxid. Med. Cell. Longev. 2022, 2022, 7702681. [Google Scholar] [CrossRef]
- Barra, F.; Ferro Desideri, L.; Ferrero, S. Inhibition of PI3K/AKT/mTOR pathway for the treatment of endometriosis. Br. J. Pharmacol. 2018, 175, 3626–3627. [Google Scholar] [CrossRef] [PubMed]
- Makker, A.; Goel, M.M.; Das, V.; Agarwal, A. PI3K-Akt-mTOR and MAPK signaling pathways in polycystic ovarian syndrome, uterine leiomyomas and endometriosis: An update. Gynecol. Endocrinol. 2012, 28, 175–181. [Google Scholar] [CrossRef]
- Assaf, L.; Eid, A.A.; Nassif, J. Role of AMPK/mTOR, mitochondria, and ROS in the pathogenesis of endometriosis. Life Sci. 2022, 306, 120805. [Google Scholar] [CrossRef]
- Zhang, J.; Kim, S.; Li, L.; Kemp, C.J.; Jiang, C.; Lu, J. Proteomic and transcriptomic profiling of Pten gene-knockout mouse model of prostate cancer. Prostate 2020, 80, 588–605. [Google Scholar] [CrossRef]
- Xi, Y.; Qi, Z.; Ma, J.; Chen, Y. PTEN loss activates a functional AKT/CXCR4 signaling axis to potentiate tumor growth and lung metastasis in human osteosarcoma cells. Clin. Exp. Metastasis 2020, 37, 173–185. [Google Scholar] [CrossRef]
- Dai, F.; Li, J.; Liu, Y. Phosphatase and tensin homolog deficiency induces M2 macrophage polarization by promoting glycolytic activity in endometrial stromal cells. Biol. Reprod. 2025, 112, 640–650. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; He, T.; Liu, S.; Zheng, Y.; Xiang, L.; Pei, X.; Wang, Z.; Yang, H. The PIK3CA E542K and E545K mutations promote glycolysis and proliferation via induction of the beta-catenin/SIRT3 signaling pathway in cervical cancer. J. Hematol. Oncol. 2018, 11, 139. [Google Scholar] [CrossRef] [PubMed]
- Young, V.J.; Ahmad, S.F.; Brown, J.K.; Duncan, W.C.; Horne, A.W. ID2 mediates the transforming growth factor-beta1-induced Warburg-like effect seen in the peritoneum of women with endometriosis. Mol. Hum. Reprod. 2016, 22, 648–654. [Google Scholar] [CrossRef]
- Remels, A.; Gosker, H.; Verhees, K.; Langen, R.; Schols, A. TNF-α-induced NF-κB activation stimulates skeletal muscle glycolytic metabolism through activation of HIF-1α. Endocrinology 2015, 156, 1770–1781. [Google Scholar] [CrossRef] [PubMed]
- Camporeale, A.; Demaria, M.; Monteleone, E.; Giorgi, C.; Wieckowski, M.R.; Pinton, P.; Poli, V. STAT3 Activities and Energy Metabolism: Dangerous Liaisons. Cancers 2014, 6, 1579–1596. [Google Scholar] [CrossRef]
- Luckow Invitti, A.; Schor, E.; Martins Parreira, R.; Kopelman, A.; Kamergorodsky, G.; Gonçalves, G.A.; Batista Castello Girão, M.J. Inflammatory cytokine profile of co-cultivated primary cells from the endometrium of women with and without endometriosis. Mol. Med. Rep. 2018, 18, 1287–1296. [Google Scholar] [CrossRef]
- Taylor, D.J.; Faragher, E.B.; Evanson, J.M. Inflammatory cytokines stimulate glucose uptake and glycolysis but reduce glucose oxidation in human dermal fibroblasts in vitro. Circ. Shock 1992, 37, 105–110. [Google Scholar]
- Pak, H.K.; Nam, B.; Lee, Y.K.; Kim, Y.W.; Roh, J.; Son, J.; Chung, Y.S.; Choe, J.; Park, C.S. Human Plasmablast Migration Toward CXCL12 Requires Glucose Oxidation by Enhanced Pyruvate Dehydrogenase Activity via AKT. Front. Immunol. 2018, 9, 1742. [Google Scholar] [CrossRef]
- Luker, K.E.; Luker, G.D. The CXCL12/CXCR4/ACKR3 Signaling Axis Regulates PKM2 and Glycolysis. Cells 2022, 11, 1775. [Google Scholar] [CrossRef]
- de Azambuja Rodrigues, P.M.; Valente, R.H.; Brunoro, G.V.F.; Nakaya, H.T.I.; Araujo-Pereira, M.; Bozza, P.T.; Bozza, F.A.; Trugilho, M.R.O. Proteomics reveals disturbances in the immune response and energy metabolism of monocytes from patients with septic shock. Sci. Rep. 2021, 11, 15149. [Google Scholar] [CrossRef]
- Luo, Y.; Jiang, Q.; Zhu, Z.; Sattar, H.; Wu, J.; Huang, W.; Su, S.; Liang, Y.; Wang, P.; Meng, X. Phosphoproteomics and Proteomics Reveal Metabolism as a Key Node in LPS-Induced Acute Inflammation in RAW264.7. Inflammation 2020, 43, 1667–1679. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Yang, X.; Yuan, Z.; Wang, H. Metabolic Reprogramming in Immune Response and Tissue Inflammation. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 1990–2001. [Google Scholar] [CrossRef] [PubMed]
- Penariol, L.B.; Thomé, C.H.; Tozetti, P.A.; Paier, C.R.; Buono, F.O.; Peronni, K.C.; Orellana, M.D.; Covas, D.T.; Moraes, M.E.; Silva, W.A., Jr. What do the transcriptome and proteome of menstrual blood-derived mesenchymal stem cells tell us about endometriosis? Int. J. Mol. Sci. 2022, 23, 11515. [Google Scholar] [CrossRef] [PubMed]
- Kasvandik, S.; Samuel, K.; Peters, M.; Eimre, M.; Peet, N.; Roost, A.M.; Padrik, L.; Paju, K.; Peil, L.; Salumets, A. Deep Quantitative Proteomics Reveals Extensive Metabolic Reprogramming and Cancer-Like Changes of Ectopic Endometriotic Stromal Cells. J. Proteome Res. 2016, 15, 572–584. [Google Scholar] [CrossRef]
- Sapkota, Y.; Steinthorsdottir, V.; Morris, A.P.; Fassbender, A.; Rahmioglu, N.; De Vivo, I.; Buring, J.E.; Zhang, F.; Edwards, T.L.; Jones, S.; et al. Meta-analysis identifies five novel loci associated with endometriosis highlighting key genes involved in hormone metabolism. Nat. Commun. 2017, 8, 15539. [Google Scholar] [CrossRef]
- Ducreux, B.; Patrat, C.; Firmin, J.; Ferreux, L.; Chapron, C.; Marcellin, L.; Parpex, G.; Bourdon, M.; Vaiman, D.; Santulli, P.; et al. Systematic review on the DNA methylation role in endometriosis: Current evidence and perspectives. Clin. Epigenet. 2025, 17, 32. [Google Scholar] [CrossRef]
- Zubrzycka, A.; Zubrzycki, M.; Perdas, E.; Zubrzycka, M. Genetic, Epigenetic, and Steroidogenic Modulation Mechanisms in Endometriosis. J. Clin. Med. 2020, 9, 1309. [Google Scholar] [CrossRef]
- Adammek, M.; Greve, B.; Kassens, N.; Schneider, C.; Bruggemann, K.; Schuring, A.N.; Starzinski-Powitz, A.; Kiesel, L.; Gotte, M. MicroRNA miR-145 inhibits proliferation, invasiveness, and stem cell phenotype of an in vitro endometriosis model by targeting multiple cytoskeletal elements and pluripotency factors. Fertil. Steril. 2013, 99, 1346–1355.e1345. [Google Scholar] [CrossRef]
- Bjorkman, S.; Taylor, H.S. MicroRNAs in endometriosis: Biological function and emerging biomarker candidatesdagger. Biol. Reprod. 2019, 100, 1135–1146. [Google Scholar] [CrossRef]
- Okamoto, M.; Nasu, K.; Abe, W.; Aoyagi, Y.; Kawano, Y.; Kai, K.; Moriyama, M.; Narahara, H. Enhanced miR-210 expression promotes the pathogenesis of endometriosis through activation of signal transducer and activator of transcription 3. Hum. Reprod. 2015, 30, 632–641. [Google Scholar] [CrossRef]
- Huang, P.; Zhu, S.; Liang, X.; Zhang, Q.; Luo, X.; Liu, C.; Song, L. Regulatory Mechanisms of LncRNAs in Cancer Glycolysis: Facts and Perspectives. Cancer Manag. Res. 2021, 13, 5317–5336. [Google Scholar] [CrossRef] [PubMed]
- Malakar, P.; Stein, I.; Saragovi, A.; Winkler, R.; Stern-Ginossar, N.; Berger, M.; Pikarsky, E.; Karni, R. Long Noncoding RNA MALAT1 Regulates Cancer Glucose Metabolism by Enhancing mTOR-Mediated Translation of TCF7L2. Cancer Res. 2019, 79, 2480–2493. [Google Scholar] [CrossRef]
- Ji, X.; Sun, W.; Lv, C.; Huang, J.; Zhang, H. Circular RNAs Regulate Glucose Metabolism in Cancer Cells. Onco Targets Ther. 2021, 14, 4005–4021. [Google Scholar] [CrossRef]
- Li, L.; Sun, B.; Sun, Y. Identification of functional TF-miRNA-hub gene regulatory network associated with ovarian endometriosis. Front. Genet. 2022, 13, 998417. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Zhang, J.; Xu, Z.; Li, M.; Dong, X.; Du, Y.; Xu, Z.; Yan, L. Highly expressed lncRNA H19 in endometriosis promotes aerobic glycolysis and histone lactylation. Reproduction 2024, 168, e240018. [Google Scholar] [CrossRef]
- Qi, X.; Zhang, Y.; Ji, H.; Wu, X.; Wang, F.; Xie, M.; Shu, L.; Jiang, S.; Mao, Y.; Cui, Y.; et al. Knockdown of prohibitin expression promotes glucose metabolism in eutopic endometrial stromal cells from women with endometriosis. Reprod. Biomed. Online 2014, 29, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.; Zhang, J.; Cai, L.; Cui, Y.; Liu, J.; Mao, Y. Elevated prohibitin 1 expression mitigates glucose metabolism defects in granulosa cells of infertile patients with endometriosis. Mol. Hum. Reprod. 2022, 28, gaac018. [Google Scholar] [CrossRef]
- Signorile, A.; Sgaramella, G.; Bellomo, F.; De Rasmo, D. Prohibitins: A Critical Role in Mitochondrial Functions and Implication in Diseases. Cells 2019, 8, 71. [Google Scholar] [CrossRef]
- Zong, Y.; Li, H.; Liao, P.; Chen, L.; Pan, Y.; Zheng, Y.; Zhang, C.; Liu, D.; Zheng, M.; Gao, J. Mitochondrial dysfunction: Mechanisms and advances in therapy. Signal Transduct. Target. Ther. 2024, 9, 124. [Google Scholar] [CrossRef]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef]
- Kuhl, I.; Miranda, M.; Atanassov, I.; Kuznetsova, I.; Hinze, Y.; Mourier, A.; Filipovska, A.; Larsson, N.G. Transcriptomic and proteomic landscape of mitochondrial dysfunction reveals secondary coenzyme Q deficiency in mammals. eLife 2017, 6, e30952. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Matsubara, S.; Yoshimoto, C.; Shigetomi, H.; Imanaka, S. The role of mitochondrial dynamics in the pathophysiology of endometriosis. J. Obstet. Gynaecol. Res. 2023, 49, 2783–2791. [Google Scholar] [CrossRef]
- Zhan, L.; Wang, W.; Zhang, Y.; Song, E.; Fan, Y.; Wei, B. Hypoxia-inducible factor-1alpha: A promising therapeutic target in endometriosis. Biochimie 2016, 123, 130–137. [Google Scholar] [CrossRef]
- Hirschhaeuser, F.; Sattler, U.G.; Mueller-Klieser, W. Lactate: A metabolic key player in cancer. Cancer Res. 2011, 71, 6921–6925. [Google Scholar] [CrossRef]
- Marianna, S.; Alessia, P.; Susan, C.; Francesca, C.; Angela, S.; Francesca, C.; Antonella, N.; Patrizia, I.; Nicola, C.; Emilio, C. Metabolomic profiling and biochemical evaluation of the follicular fluid of endometriosis patients. Mol. Biosyst. 2017, 13, 1213–1222. [Google Scholar] [CrossRef] [PubMed]
- Ke, J.; Ye, J.; Li, M.; Zhu, Z. The Role of Matrix Metalloproteinases in Endometriosis: A Potential Target. Biomolecules 2021, 11, 1739. [Google Scholar] [CrossRef]
- Niland, S.; Riscanevo, A.X.; Eble, J.A. Matrix Metalloproteinases Shape the Tumor Microenvironment in Cancer Progression. Int. J. Mol. Sci. 2021, 23, 146. [Google Scholar] [CrossRef]
- Lee, H.C.; Lin, S.C.; Wu, M.H.; Tsai, S.J. Induction of Pyruvate Dehydrogenase Kinase 1 by Hypoxia Alters Cellular Metabolism and Inhibits Apoptosis in Endometriotic Stromal Cells. Reprod. Sci. 2019, 26, 734–744. [Google Scholar] [CrossRef] [PubMed]
- Horne, A.W.; Ahmad, S.F.; Carter, R.; Simitsidellis, I.; Greaves, E.; Hogg, C.; Morton, N.M.; Saunders, P.T.K. Repurposing dichloroacetate for the treatment of women with endometriosis. Proc. Natl. Acad. Sci. USA 2019, 116, 25389–25391. [Google Scholar] [CrossRef]
- Leow, H.W.; Koscielniak, M.; Williams, L.; Saunders, P.T.K.; Daniels, J.; Doust, A.M.; Jones, M.C.; Ferguson, G.D.; Bagger, Y.; Horne, A.W.; et al. Dichloroacetate as a possible treatment for endometriosis-associated pain: A single-arm open-label exploratory clinical trial (EPiC). Pilot Feasibility Stud. 2021, 7, 67. [Google Scholar] [CrossRef]
- Kim, B.S.; Chung, T.W.; Choi, H.J.; Bae, S.J.; Cho, H.R.; Lee, S.O.; Choi, J.H.; Joo, J.K.; Ha, K.T. Caesalpinia sappan induces apoptotic cell death in ectopic endometrial 12Z cells through suppressing pyruvate dehydrogenase kinase 1 expression. Exp. Ther. Med. 2021, 21, 357. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liang, Z.; Gou, Y.; Li, Z.; Cao, Y.; Jiao, N.; Tan, J.; Yu, Y.; Zhang, Z. FTO-dependent N(6)-Methyladenosine regulates the progression of endometriosis via the ATG5/PKM2 Axis. Cell Signal. 2022, 98, 110406. [Google Scholar] [CrossRef]
- Zheng, J.; Dai, Y.; Lin, X.; Huang, Q.; Shi, L.; Jin, X.; Liu, N.; Zhou, F.; Zhang, S. Hypoxia-induced lactate dehydrogenase A protects cells from apoptosis in endometriosis. Mol. Med. Rep. 2021, 24, 637. [Google Scholar] [CrossRef]
- Cho, M.K.; Jin, L.; Han, J.H.; Jin, J.S.; Cheon, S.Y.; Shin, S.; Bae, S.J.; Park, J.K.; Ha, K.T. Water-Extracted Prunella vulgaris Alleviates Endometriosis by Reducing Aerobic Glycolysis. Front. Pharmacol. 2022, 13, 872810. [Google Scholar] [CrossRef]
- Han, J.H.; Lee, E.J.; Park, W.; Ha, K.T.; Chung, H.S. Natural compounds as lactate dehydrogenase inhibitors: Potential therapeutics for lactate dehydrogenase inhibitors-related diseases. Front. Pharmacol. 2023, 14, 1275000. [Google Scholar] [CrossRef] [PubMed]
- Yao, Q.; Jing, G.; Zhang, X.; Li, M.; Yao, Q.; Wang, L. Cinnamic acid inhibits cell viability, invasion, and glycolysis in primary endometrial stromal cells by suppressing NF-kappaB-induced transcription of PKM2. Biosci. Rep. 2021, 9, BSR20211828. [Google Scholar] [CrossRef]
- Wang, M.; Fan, R.; Jiang, J.; Sun, F.; Sun, Y.; Wang, Q.; Jiang, A.; Yu, Z.; Yang, T. PIM2 Promotes the Development of Ovarian Endometriosis by Enhancing Glycolysis and Fibrosis. Reprod. Sci. 2023, 30, 2692–2702. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Qiao, P.; Fu, R.; Wang, Y.; Lu, J.; Ling, X.; Liu, L.; Sun, Y.; Ren, C.; Yu, Z. Phosphorylation of PFKFB4 by PIM2 promotes anaerobic glycolysis and cell proliferation in endometriosis. Cell Death Dis. 2022, 13, 790. [Google Scholar] [CrossRef]
- Lu, J.; Wang, X.; Shi, X.; Jiang, J.; Liu, L.; Liu, L.; Ren, C.; Lu, C.; Yu, Z. PAK5-mediated PKM2 phosphorylation is critical for anaerobic glycolysis in endometriosis. Front. Med. 2024, 18, 1054–1067. [Google Scholar] [CrossRef]
- Zhou, J.; Ding, Z.M.; Hardiman, P.J. Understanding the Role of Gui-Zhi-Fu-Ling-Capsules (Chinese Medicine) for Treatment of Endometriosis in the Rat Model: Using NMR Based Metabolomics. Evid. Based Complement. Altern. Med. 2018, 2018, 9864963. [Google Scholar] [CrossRef]
- Bahrami, A.; Ayen, E.; Razi, M.; Behfar, M. Effects of atorvastatin and resveratrol against the experimental endometriosis; evidence for glucose and monocarboxylate transporters, neoangiogenesis. Life Sci. 2021, 272, 119230. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xiu, J.; Yang, T.; Ren, C.; Yu, Z. HSF1 promotes endometriosis development and glycolysis by up-regulating PFKFB3 expression. Reprod. Biol. Endocrinol. 2021, 19, 86. [Google Scholar] [CrossRef]
- Ling, X.; Lu, J.; Wang, X.; Liu, L.; Liu, L.; Wang, Y.; Sun, Y.; Ren, C.; Lu, C.; Yu, Z. Ovarian tumorB1-mediated heat shock transcription factor 1 deubiquitination is critical for glycolysis and development of endometriosis. iScience 2022, 25, 105363. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Wang, Q.; Wang, M.; Sun, F.; Qiao, P.; Jiang, A.; Ren, C.; Yu, Z.; Yang, T. CHIP induces ubiquitination and degradation of HMGB1 to regulate glycolysis in ovarian endometriosis. Cell Mol. Life Sci. 2022, 80, 13. [Google Scholar] [CrossRef] [PubMed]
- Gou, Y.; Wang, H.; Wang, T.; Wang, H.; Wang, B.; Jiao, N.; Yu, Y.; Cao, Y.; Wang, H.; Zhang, Z. Ectopic endometriotic stromal cells-derived lactate induces M2 macrophage polarization via Mettl3/Trib1/ERK/STAT3 signalling pathway in endometriosis. Immunology 2023, 168, 389–402. [Google Scholar] [CrossRef]
- Ling, X.; Liu, L.; Jiang, A.; Shi, X.; Liu, L.; Wang, X.; Lu, C.; Ren, C.; Yu, Z. PFKFB3 promotes endometriosis cell proliferation via enhancing the protein stability of beta-catenin. Mol. Cell. Endocrinol. 2024, 579, 112083. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, S.; Zhang, X.; Li, G.; Sun, F.; Wang, M.; Ren, C.; Jiang, A.; Yang, T. AURKA Enhances the Glycolysis and Development of Ovarian Endometriosis Through ERbeta. Endocrinology 2024, 165, bqae018. [Google Scholar] [CrossRef]
- Huang, Z.X.; Lin, D.C.; Zhang, H.Y.; Yang, M.J.; Chen, J.H.; Ding, X.Y.; Dai, S.J.; Hong, Y.H.; Liang, G.S.; Li, Q.Y.; et al. The dysfunction of CD8(+) T cells triggered by endometriotic stromal cells promotes the immune survival of endometriosis. Immunology 2024, 172, 469–485. [Google Scholar] [CrossRef]
- Gao, X.; Shao, W.; Wang, J.; Gao, H.; Zhang, X.; Xia, C.; Li, M.; Liu, S. Integrin beta3 enhances glycolysis and increases lactate production in endometriosis. J. Reprod. Immunol. 2024, 165, 104312. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, R.; Gu, H.; Zhang, E.; Qu, J.; Cao, W.; Huang, X.; Yan, H.; He, J.; Cai, Z. Metabolic reprogramming in macrophage responses. Biomark. Res. 2021, 9, 1. [Google Scholar] [CrossRef]
- Yee, J.L.; Huang, C.Y.; Yu, Y.C.; Huang, S.J. Potential Mechanisms of Guizhi Fuling Wan in Treating Endometriosis: An Analysis Based on TCMSP and DisGeNET Databases. J. Ethnopharmacol. 2024, 329, 118190. [Google Scholar] [CrossRef]
- Medina, M.G.; Lebovic, D.I. Endometriosis-associated nerve fibers and pain. Acta Obstet. Gynecol. Scand. 2009, 88, 968–975. [Google Scholar] [CrossRef] [PubMed]
- Mohammed Rasheed, H.A.; Hamid, P. Inflammation to Infertility: Panoramic View on Endometriosis. Cureus 2020, 12, e11516. [Google Scholar] [CrossRef]
- Fan, W.; Yuan, Z.; Li, M.; Zhang, Y.; Nan, F. Decreased oocyte quality in patients with endometriosis is closely related to abnormal granulosa cells. Front. Endocrinol. 2023, 14, 1226687. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Kimura, M.; Maruyama, S.; Nagayasu, M.; Imanaka, S. Revisiting estrogen-dependent signaling pathways in endometriosis: Potential targets for non-hormonal therapeutics. Eur. J. Obstet. Gynecol. Reprod. Biol. 2021, 258, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Scutiero, G.; Iannone, P.; Bernardi, G.; Bonaccorsi, G.; Spadaro, S.; Volta, C.A.; Greco, P.; Nappi, L. Oxidative Stress and Endometriosis: A Systematic Review of the Literature. Oxid. Med. Cell. Longev. 2017, 2017, 7265238. [Google Scholar] [CrossRef]
- Song, S.Y.; Jung, Y.W.; Shin, W.; Park, M.; Lee, G.W.; Jeong, S.; An, S.; Kim, K.; Ko, Y.B.; Lee, K.H.; et al. Endometriosis-Related Chronic Pelvic Pain. Biomedicines 2023, 11, 2868. [Google Scholar] [CrossRef]
- Kalaitzopoulos, D.R.; Samartzis, N.; Kolovos, G.N.; Mareti, E.; Samartzis, E.P.; Eberhard, M.; Dinas, K.; Daniilidis, A. Treatment of endometriosis: A review with comparison of 8 guidelines. BMC Womens Health 2021, 21, 397. [Google Scholar] [CrossRef]
- Capezzuoli, T.; Rossi, M.; La Torre, F.; Vannuccini, S.; Petraglia, F. Hormonal drugs for the treatment of endometriosis. Curr. Opin. Pharmacol. 2022, 67, 102311. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| First Author | Key Mechanism | Molecular Target | Drug | Experiments | Publication Year | Ref |
|---|---|---|---|---|---|---|
| McKinnon B | Altered glucose metabolism is mediated by glucose transporter | GLUT4 | - | In vitro, In vivo | 2014 | [43] |
| Young VJ | TGF-β induces a Warburg-like effect | HIF1A, PDK1, LDHA, GLUT1 | - | In vitro, Human sample | 2014 | [10] |
| Qi X | Enhanced glycolysis is due to prohibitin (PHB) downregulation | PHB | - | In vitro, Human sample | 2014 | [87] |
| Kasvandik S | Warburg effect leads to enhanced invasiveness, adhesion, and immune evasion. | - | - | Human sample, In silico | 2016 | [75] |
| Young VJ | TGF-β1-induced Warburg effect via suppression of ID2, which in turn upregulates HIF-1α expression. | ID2 | - | In vitro, Human sample | 2016 | [64] |
| Zhou J | Regulation of glycolysis and gluconeogenesis leads to metabolic changes and modulation of TGF-β1, GLUT-4, and VEGF expression | TGFB1, GLUT4, VEGF | Gui-Zhi-Fu-Ling Capsules | In vivo, In silico | 2018 | [111] |
| Lee HC | Hypoxia upregulates PDK1 to enhance glycolysis | PDK1 | Dichloroacetate | In vitro | 2019 | [99] |
| Horne AW | TGF-β1 drives increased glycolysis, reduced mitochondrial respiration, and enhanced lactate production. | PDK1 | Dichloroacetate | In vitro, In vivo, Human sample | 2019 | [100] |
| Rytkönen KT | Hypoxia-driven transcription factors Jun/Fos and CEBP, alters glycolysis, epithelial-mesenchymal transition, and inflammatory pathways. | Jun, Fos, CEBP | - | In vitro, Human sample | 2020 | [39] |
| Pocate-Cheriet K | Metabolic reprogramming in follicular fluid is characterized by altered glycolysis, beta-oxidation, and mitochondrial dysfunction. | - | - | Human sample, In silico | 2020 | [42] |
| Bahrami A | Inhibition of glycolysis and neovascularization suppresses endometriosis development. | GLUT1, GLUT3, MCT1, MCT4 | Atorvastatin and Resveratrol | In vivo | 2021 | [112] |
| Leow HW | Metabolic reprogramming correction reduces endometriosis-associated pain. | PDK1 | Dichloroacetate | Human clinical trials | 2021 | [101] |
| Kim BS | Inhibition of aerobic glycolysis and induction of ROS-mitochondria-mediated apoptosis. | PDK1, PDK3 | Caesalpinia sappan | In vitro | 2021 | [102] |
| Wang Y | HSF1 promotes glycolysis via upregulating PFKFB3 facilitates endometriosis progression. | HSF1, PFKFB3 | KRIBB11 (HSF1 inhibitor) | in vitro, In vivo | 2021 | [113] |
| Zheng J | LDHA promotes glycolysis and inhibits mitochondrial function. | LDHA | shLDHA | In vitro, Human sample | 2021 | [104] |
| Yao Q | PKM2 inhibition and may serve as a potential treatment for endometriosis. | PKM2 | Cinnamic acid | In vitro, Human sample | 2021 | [107] |
| Cho MK | Inhibition of aerobic glycolysis prevents lesion growth and metabolic adaptation. | LDHA, PDK1, PDK3 | Prunella vulgaris | In vitro, In vivo | 2022 | [105] |
| Hou S | Downregulation of HK2 reduces migration, invasion, and proliferation of endometrial stromal cells. | HK2, STAT1 | - | In vitro | 2022 | [11] |
| Mao J | Upregulation of PHB1 enhances glucose metabolism, ATP synthesis, and ROS production. | PHB1 | - | In vitro, Human sample | 2022 | [88] |
| Wang H | FTO regulates ATG5 expression through m6A methylation and in turn, suppresses the expression of PKM2. | FTO, ATG5, PKM2 | - | In vitro, Human sample | 2022 | [103] |
| Lu C | The PIM2-PFKFB4 axis drives glycolysis and cell growth, contributing to EM progression. | PIM2, PFKFB4 | - | In vitro, In vivo, Human sample | 2022 | [109] |
| Li L | Three key feedback loops were discovered in the TF-miRNA-hub gene network | MYC, YY1, HIF1A, LDHA, RELA, miR-34a-5p, miR-155-5p, miR-93-5p | - | Human sample, In silico | 2022 | [85] |
| Ling X | OTUB1 increase the HSF1 stability and enhances glycolysis, EMT, and progression of endometriosis. | OTUB1, HSF1 | - | In vitro, In vivo | 2022 | [114] |
| Sun Y | CHIP activation reduces HMGB1 expression, limiting the energy supply for endometriotic cell growth. | CHIP, HMGB1 | YL-109 (CHIP agonist) | In vitro, In vivo, Human sample | 2022 | [115] |
| Gou Y | Glycolysis-driven lactate accumulation promotes M2 macrophage polarization, enhancing endometriotic lesion invasion via the Mettl3/Trib1/ERK-STAT3 pathway. | Mettl3, Trib1, ERK, STAT3 | 2-Deoxy-D-glucose (2-DG) | In vitro, In vivo, Human sample | 2023 | [116] |
| Chen Q | Glycolysis-related genes influence immune cell infiltration, contributing to endometriosis progression. | CHPF, CITED2, GPC3, PDK3, ADH6 | - | Human sample, In silico | 2023 | [31] |
| Wang M | PIM2 enhances glycolysis and fibrosis in endometriotic cells by upregulating PKM2, promoting endometriosis progression. | PIM2, PKM2, HK2, SMH, Desmin, α-SMA | SMI-4a (PIM2 inhibitor) | In vitro, In vivo, Human sample | 2023 | [108] |
| Ling X | PFKFB3 enhances glycolysis, proliferation, migration, and invasion of endometriosis cells by stabilizing β-catenin, promoting epithelial-mesenchymal transition (EMT). | PFKFB3, β-catenin | PFK-015 (PFKFB3 inhibitor) | In vitro, In vivo, Human sample | 2024 | [117] |
| Sun Y | URKA promotes proliferation, migration, invasion, and glycolysis in ovarian endometriosis by upregulating ERβ. | AURKA, ERβ | Alisertib (AURKA inhibitor) | In vitro, In vivo, Human sample | 2024 | [118] |
| Huang ZX | Endometriosis involves dysfunctional CD8+ T cells, where glycolysis and the STAT1/PDCD1 pathway reduce T-cell cytotoxicity and promote lesion growth. | CDK1, CCNB1, STAT1, PDCD1 | - | In vitro, In vivo, Human sample, In silico | 2024 | [119] |
| Guo S | increased PDK3 and GPC3 levels and decreased ADH6 in EMT patients, which correlated with lower oocyte quality and pregnancy rates. | PDK3, GPC3, ADH6 | - | Human sample | 2024 | [32] |
| Toniyan KA | endometriomas and pelvic peritoneum lesions, oxygen absorption is significantly reduced, and there is a shift towards succinate utilization, suggesting a Warburg effect with increased glycolysis. | HK2, PK | - | Human sample | 2024 | [33] |
| Khashchenko EP | glycolysis reprogramming, mitochondrial biogenesis, and apoptosis suppression drive peritoneal endometriosis | MCT2, PDK1, GLUT1, OPA1, DRP1, Beclin1, Bnip3, ERβ | - | Human sample | 2024 | [34] |
| Wen X | H19 expression promotes abnormal glucose metabolism and histone lactylation, driving endometriosis progression. | H19 | - | In vitro, In vivo, Human sample | 2024 | [86] |
| Gao X | Macrophage-induced ITGB3 upregulation promotes glycolysis and enhances proliferation, migration, and invasion in endometriosis. | ITGB3 | - | In vitro, In vivo, In silico | 2024 | [120] |
| Sarsenova M | Metabolic reprogramming, including glutathione metabolism, oxidative phosphorylation, and glycolysis, occurs in response to AMPK signaling and HIF-1 signaling perivascular cells of endometriotic lesions. | AMPK, HIF1 | - | Human sample, In silico | 2024 | [30] |
| Lu J | PAK5 is upregulated in endometriosis, promoting glycolysis by stabilizing PKM2. | PAK5, PKM2 | GNE 2861 (PAK5 inhibitor) | In vitro, In vivo, Human sample | 2024 | [110] |
| Li J | PKM2/HIF-1α axis regulates glycolysis and plays a key role in endometriosis pathogenesis. | TGFB1, HIF1A, PKM2 | - | In vitro, Human sample | 2024 | [46] |
| Sarsenova M | Hypoxia-induced TGFBI is involved in fibrosis and angiogenesis and is more highly expressed in ectopic endometrial tissue during the secretory phase. | TGFB1 | - | In vitro, Human sample | 2024 | [47] |
| Dai F | PTEN deficiency in ectopic endometrial stromal cells (EESCs) promotes glycolytic activity, which enhances M2 macrophage polarization and increases CCL2 production. | PTEN, CCL2 | - | In vitro | 2025 | [62] |
| Drug Name | Mechanism | Development Stage | Efficacy | Safety |
|---|---|---|---|---|
| Gui-Zhi-Fu-Ling Capsules | Promotes blood circulation and removes blood stasis | Approved for clinical use (in China) | Anti-inflammatory effects, alleviates abdominal pain, regulates menstruation | Generally safe; lacks standardized clinical data |
| Dichloroacetate (DCA) | Inhibits pyruvate dehydrogenase kinase (PDK) to normalize glucose metabolism | Preclinical/Phase 2 | Reduces peritoneal lactate levels and lesion size (animal models) | Potential neurotoxicity with prolonged use |
| Atorvastatin + Resveratrol | Inhibits GLUT-1/3 and MCT-1/4 to suppress glycolysis and angiogenesis | Preclinical | 40% reduction in lesion size and angiogenesis density | Muscle toxicity risk (statin-related) |
| Caesalpinia sappan Extract | PDK1 inhibition induces mitochondrial ROS and apoptosis | Preclinical | >50% apoptosis in 12Z cells | Limited human toxicity data |
| KRIBB11 | HSF1 inhibition reduces PFKFB3 and glycolysis | Preclinical | 60% lesion weight reduction in animal models | Potential heat shock protein dysregulation |
| shLDHA | LDHA gene silencing to inhibit lactate production | Early research stage | 70% inhibition of 12Z cell migration | Unconfirmed long-term safety of gene therapy |
| Cinnamic Acid | Suppresses NF-κB/PKM2 signaling to reduce invasiveness | Preclinical | 55% reduction in stromal cell invasion | Mucosal irritation at high doses |
| Prunella vulgaris Extract | LDHA/PDK1 inhibition disrupts mitochondrial function | Preclinical | 40% decrease in oxygen consumption rate in 12Z cells | Traditional use history but lacking modern toxicity data |
| YL-109 | CHIP activation promotes HMGB1 ubiquitination and degradation | Preclinical | 65% lesion size reduction in animal models | Unclear immune system impacts |
| 2-Deoxy-D-glucose (2-DG) | Glycolysis blockade via glucose analog | Preclinical | 75% disease progression inhibition in mice | Hypoglycemia risk |
| SMI-4a | PIM2 inhibition reduces PKM2 expression and fibrosis | Preclinical | 50% reduction in lesion fibrosis in animal models | Potential hematological side effects |
| PFK-015 | PFKFB3 inhibition destabilizes β-catenin | Preclinical | 60% inhibition of endometrial cell migration | Risk of erythrocyte dysfunction |
| Alisertib | AURKA inhibition suppresses ERβ expression and glycolysis | Preclinical | 40% reduction in peritoneal lactate levels in mice | Potential mitotic disruption |
| GNE 2861 | PAK5 inhibition blocks PKM2 phosphorylation | Preclinical | 70% lesion size reduction in PAK5-KO mice | Unconfirmed neurological effects |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, B.-S.; Kim, B.; Yoon, S.; Park, W.; Bae, S.-J.; Joo, J.; Kim, W.; Ha, K.-T. Warburg-like Metabolic Reprogramming in Endometriosis: From Molecular Mechanisms to Therapeutic Approaches. Pharmaceuticals 2025, 18, 813. https://doi.org/10.3390/ph18060813
Kim B-S, Kim B, Yoon S, Park W, Bae S-J, Joo J, Kim W, Ha K-T. Warburg-like Metabolic Reprogramming in Endometriosis: From Molecular Mechanisms to Therapeutic Approaches. Pharmaceuticals. 2025; 18(6):813. https://doi.org/10.3390/ph18060813
Chicago/Turabian StyleKim, Bo-Sung, Bosung Kim, Seyeong Yoon, Wonyoung Park, Sung-Jin Bae, Jongkil Joo, Wonnam Kim, and Ki-Tae Ha. 2025. "Warburg-like Metabolic Reprogramming in Endometriosis: From Molecular Mechanisms to Therapeutic Approaches" Pharmaceuticals 18, no. 6: 813. https://doi.org/10.3390/ph18060813
APA StyleKim, B.-S., Kim, B., Yoon, S., Park, W., Bae, S.-J., Joo, J., Kim, W., & Ha, K.-T. (2025). Warburg-like Metabolic Reprogramming in Endometriosis: From Molecular Mechanisms to Therapeutic Approaches. Pharmaceuticals, 18(6), 813. https://doi.org/10.3390/ph18060813