
PLASMOpred: A Machine Learning-Based Web Application for Predicting Antimalarial Small Molecules Targeting the Apical Membrane Antigen 1–Rhoptry Neck Protein 2 Invasion Complex

 ,
,  , , , , , and
, , , , , and
Abstract
1. Introduction
2. Results and Discussion
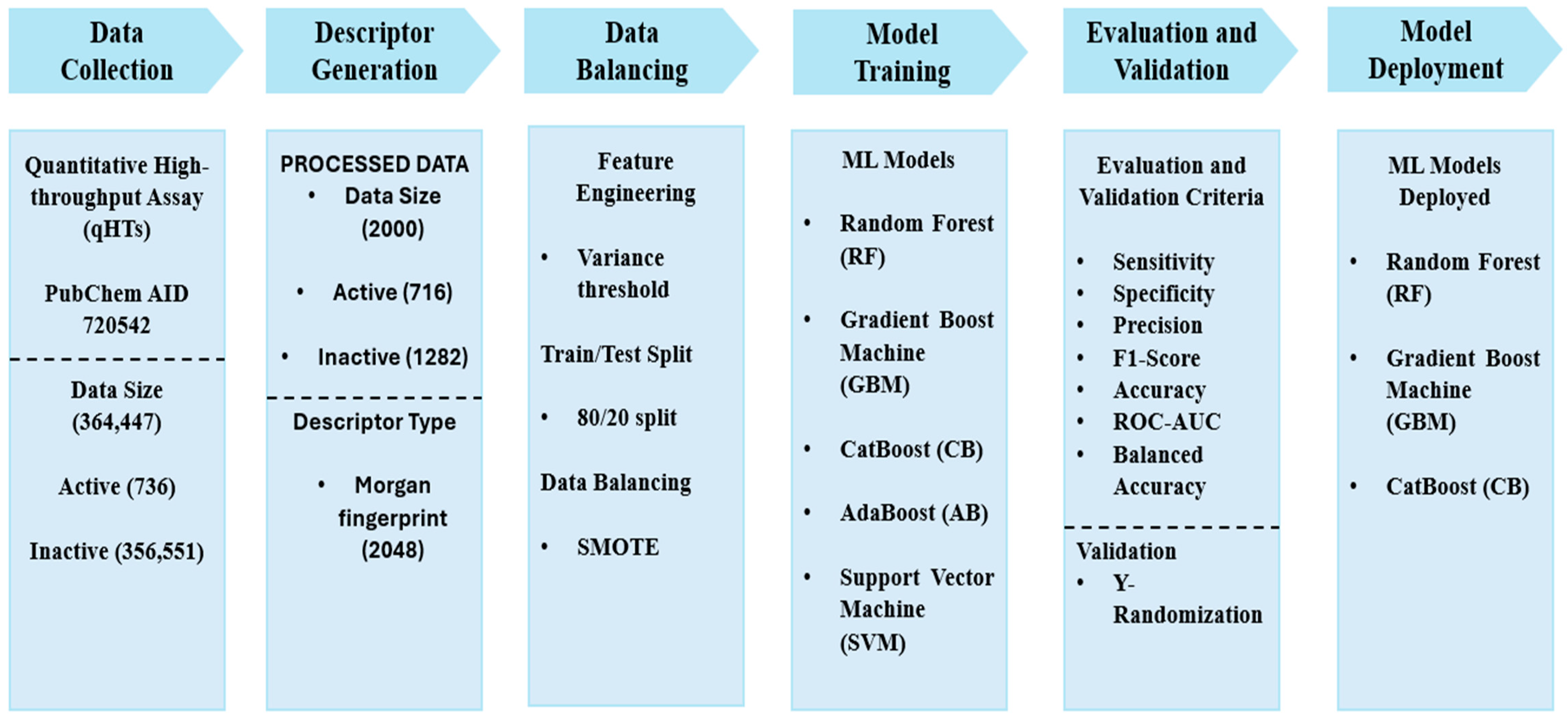
2.1. Data Collection and Pre-Processing
2.2. Descriptor Generation and Feature Engineering
2.3. Data Splitting and Data Balancing
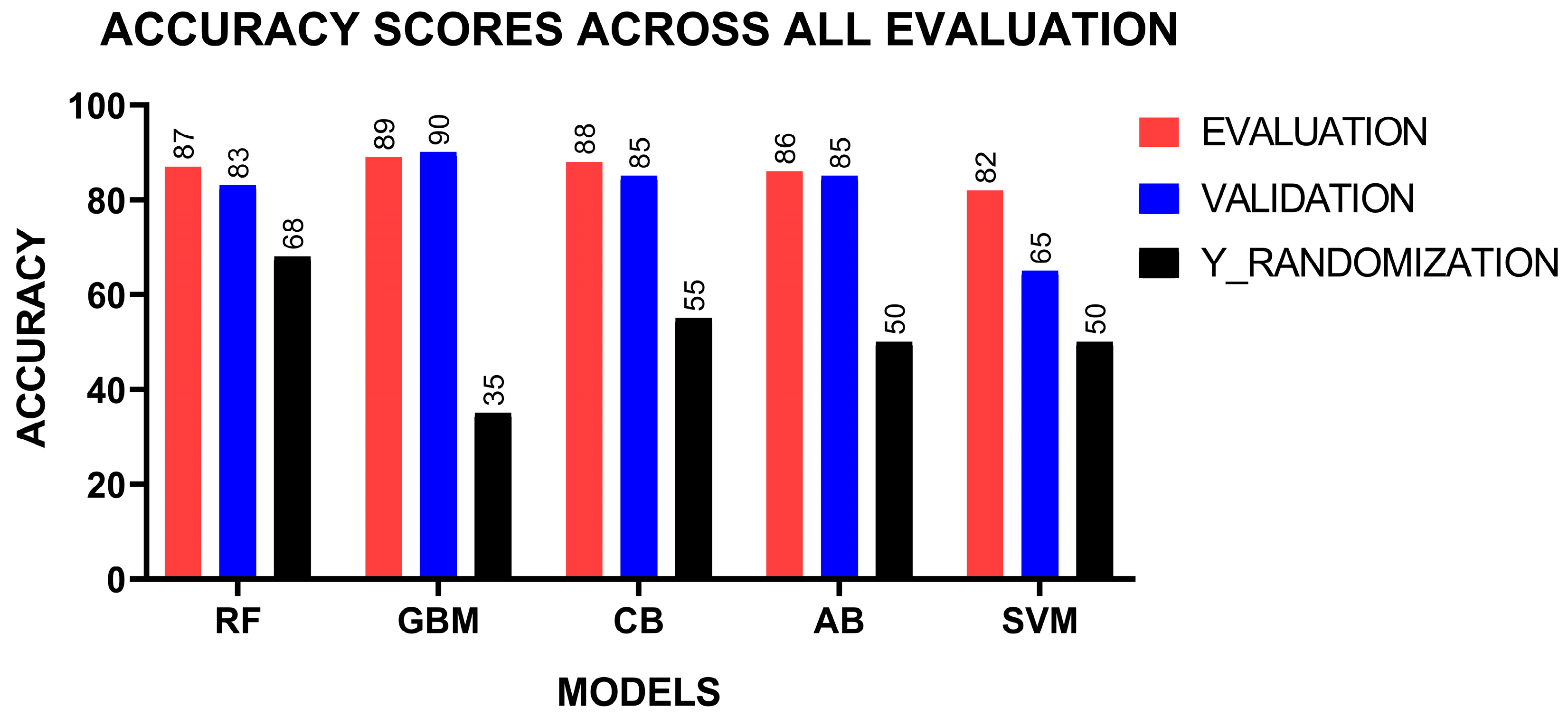
2.4. Development and Evaluation of Machine Learning Models
2.5. Validation of Machine Learning Models
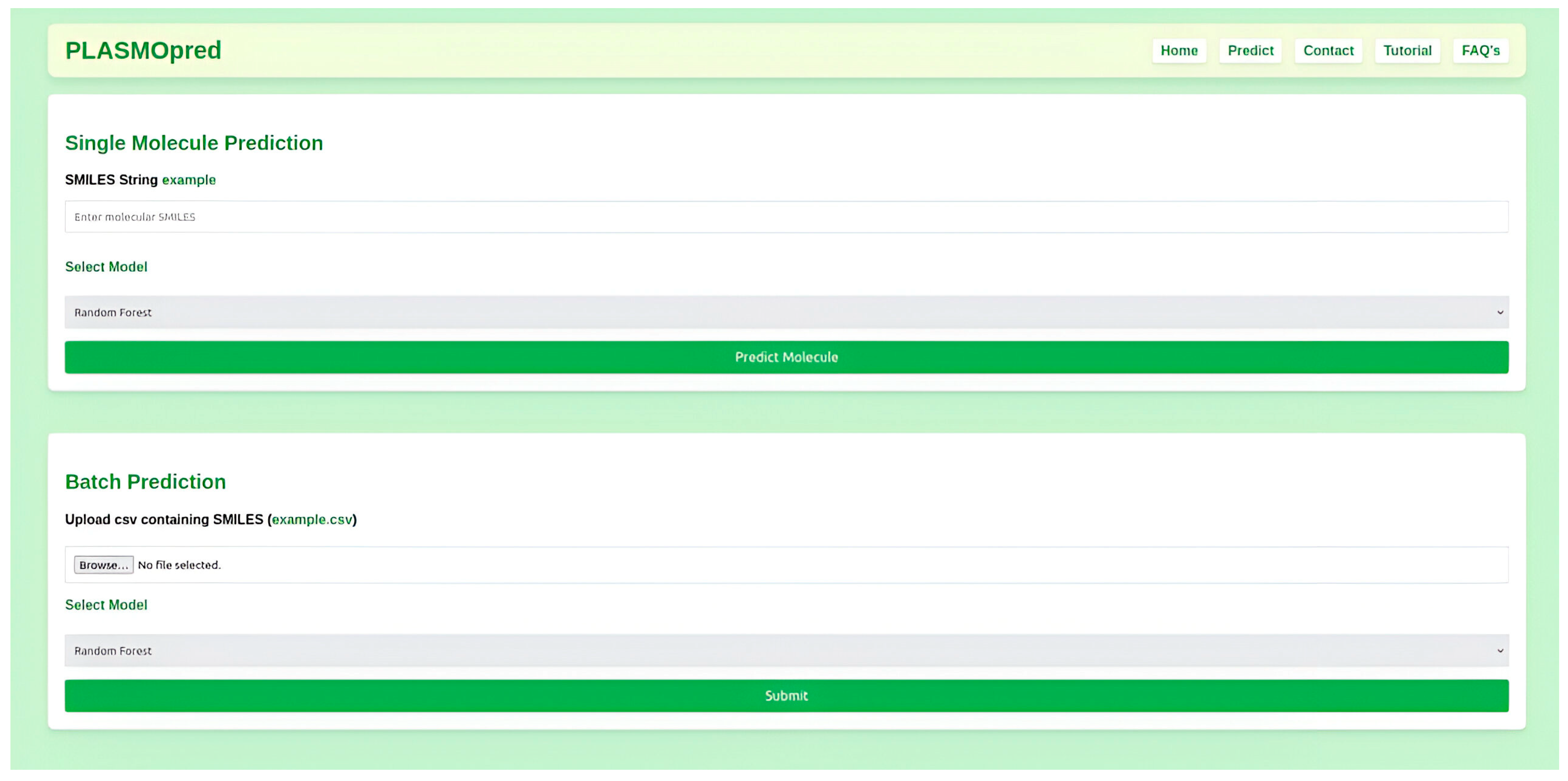
2.6. Model Deployment
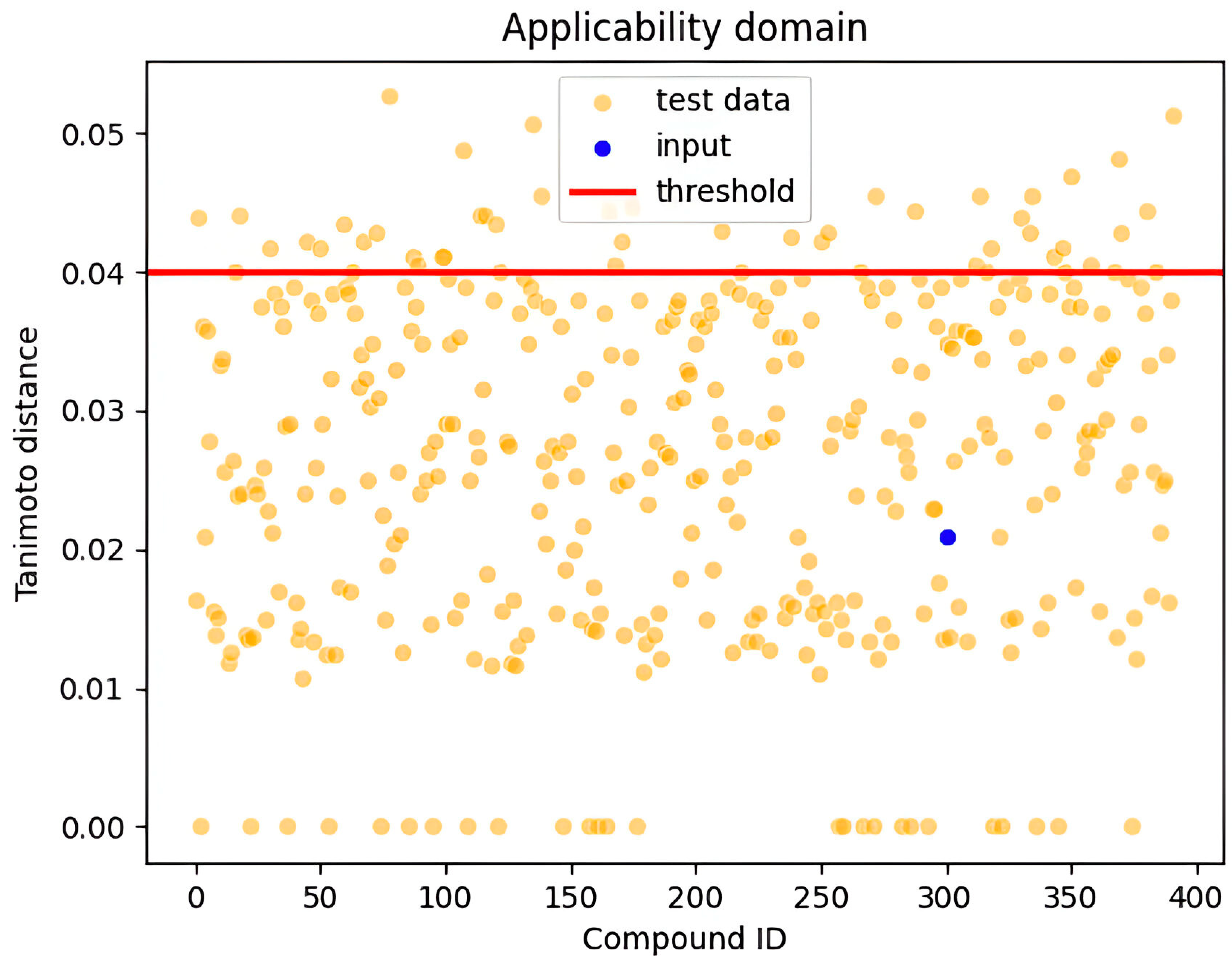
3. Applicability Domain
4. Materials and Methods
4.1. Data Collection
4.2. Descriptor Generation and Feature Engineering
4.3. Development and Evaluation of Machine Learning Models
4.4. Validation of Machine Learning Models
4.5. Model Deployment
5. Limitations
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Munisi, D.Z.; Mathania, M.M. Adult Anopheles Mosquito Distribution at a Low and High Malaria Transmission Site in Tanzania. Biomed Res. Int. 2022, 2022, 6098536. [Google Scholar] [CrossRef] [PubMed]
- Ogola, E.O.; Fillinger, U.; Ondiba, I.M.; Villinger, J.; Masiga, D.K.; Torto, B.; Tchouassi, D.P. Insights into malaria transmission among Anopheles funestus mosquitoes, Kenya. Parasites Vectors 2018, 11, 577. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, M.; Burda, P.C.; Ingmundson, A. Moving on: How malaria parasites exit the liver. Mol. Microbiol. 2024, 121, 328–340. [Google Scholar] [CrossRef] [PubMed]
- Garcia, L.S. Malaria. Clin. Lab. Med. 2010, 30, 93–129. [Google Scholar] [CrossRef]
- Maeno, Y.; Tuyen Quang, N.; Culleton, R.; Kawai, S.; Masuda, G.; Nakazawa, S.; Marchand, R.P. Humans frequently exposed to a range of non-human primate malaria parasite species through the bites of Anopheles dirus mosquitoes in South-central Vietnam. Parasites Vectors 2015, 8, 376. [Google Scholar] [CrossRef]
- Kho, S.; Anstey, N.M.; Barber, B.E.; Piera, K.; William, T.; Kenangalem, E.; McCarthy, J.S.; Jang, I.K.; Domingo, G.J.; Britton, S.; et al. Diagnostic performance of a 5-plex malaria immunoassay in regions co-endemic for Plasmodium falciparum, P. vivax, P. knowlesi, P. malariae and P. ovale. Sci. Rep. 2022, 12, 7286. [Google Scholar] [CrossRef]
- Anwar, M. Introduction: An Overview of Malaria and Plasmodium. In Drug Targets for Plasmodium Falciparum: Historic to Future Perspectives; Springer: Singapore, 2024; pp. 1–17. [Google Scholar] [CrossRef]
- Milner, D.A. Malaria Pathogenesis. Cold Spring Harb. Perspect. Med. 2018, 8, a025569. [Google Scholar] [CrossRef]
- White, N.J. Anaemia and malaria. Malar. J. 2018, 17, 371. [Google Scholar] [CrossRef]
- White, N.J. Severe malaria. Malar. J. 2022, 21, 284. [Google Scholar] [CrossRef]
- Shin, H.-I.; Ku, B.; Jung, H.; Lee, S.; Lee, S.-Y.; Ju, J.-W.; Kim, J.; Lee, H.-I. 2023 World Malaria Report (Status of World Malaria in 2022). Public Heal. Wkly. Rep. 2024, 17, 1351–1377. [Google Scholar] [CrossRef]
- Tewari, S.G.; Swift, R.P.; Reifman, J.; Prigge, S.T.; Wallqvist, A. Metabolic alterations in the erythrocyte during blood-stage development of the malaria parasite. Malar. J. 2020, 19, 94. [Google Scholar] [CrossRef] [PubMed]
- Salinas, N.D.; Tang, W.K.; Tolia, N.H. Blood-Stage Malaria Parasite Antigens: Structure, Function, and Vaccine Potential. J. Mol. Biol. 2019, 431, 4259–4280. [Google Scholar] [CrossRef]
- Balaji, S.; Deshmukh, R.; Trivedi, V. Severe malaria: Biology, clinical manifestation, pathogenesis and consequences. J. Vector Borne Dis. 2020, 57, 1–13. [Google Scholar] [CrossRef]
- Vijayan, A.; Chitnis, C.E. Development of Blood Stage Malaria Vaccines. Methods Mol. Biol. 2019, 2013, 199–218. [Google Scholar] [CrossRef]
- Salamanca, D.R.; Gómez, M.; Camargo, A.; Cuy-Chaparro, L.; Molina-Franky, J.; Reyes, C.; Patarroyo, M.A.; Patarroyo, M.E. Plasmodium falciparum Blood Stage Antimalarial Vaccines: An Analysis of Ongoing Clinical Trials and New Perspectives Related to Synthetic Vaccines. Front. Microbiol. 2019, 10, 2712. [Google Scholar] [CrossRef]
- Hillringhaus, S.; Dasanna, A.K.; Gompper, G.; Fedosov, D.A. Importance of Erythrocyte Deformability for the Alignment of Malaria Parasite upon Invasion. Biophys. J. 2019, 117, 1202–1214. [Google Scholar] [CrossRef]
- Fikri Heikal, M.; Eka Putra, W.; Sustiprijatno; Hidayatullah, A.; Widiastuti, D.; Lelitawati, M.; Fikri Heikal, M.; Eka Putra, W.; Hidayatullah, A. A Molecular Docking and Dynamics Simulation Study on Prevention of Merozoite Red Blood Cell Invasion by Targeting Plasmodium vivax Duffy Binding Protein with Zingiberaceae Bioactive Compounds. Uniciencia 2024, 38, 326–340. [Google Scholar] [CrossRef]
- King, N.R.; Freire, C.M.; Touhami, J.; Sitbon, M.; Toye, A.M.; Satchwell, T.J. Basigin mediation of Plasmodium falciparum red blood cell invasion does not require its transmembrane domain or interaction with monocarboxylate transporter 1. PLOS Pathog. 2024, 20, e1011989. [Google Scholar] [CrossRef]
- Molina-Franky, J.; Patarroyo, M.E.; Kalkum, M.; Patarroyo, M.A. The Cellular and Molecular Interaction Between Erythrocytes and Plasmodium falciparum Merozoites. Front. Cell. Infect. Microbiol. 2022, 12, 816574. [Google Scholar] [CrossRef]
- Srinivasan, P.; Yasgar, A.; Luci, D.K.; Beatty, W.L.; Hu, X.; Andersen, J.; Narum, D.L.; Moch, J.K.; Sun, H.; Haynes, J.D.; et al. Disrupting malaria parasite AMA1–RON2 interaction with a small molecule prevents erythrocyte invasion. Nat. Commun. 2013, 4, 2261. [Google Scholar] [CrossRef]
- Wang, G.; Drinkwater, N.; Drew, D.R.; MacRaild, C.A.; Chalmers, D.K.; Mohanty, B.; Lim, S.S.; Anders, R.F.; Beeson, J.G.; Thompson, P.E.; et al. Structure–Activity Studies of β-Hairpin Peptide Inhibitors of the Plasmodium falciparum AMA1–RON2 Interaction. J. Mol. Biol. 2016, 428, 3986–3998. [Google Scholar] [CrossRef] [PubMed]
- Wright, K.E.; Hjerrild, K.A.; Bartlett, J.; Douglas, A.D.; Jin, J.; Brown, R.E.; Illingworth, J.J.; Ashfield, R.; Clemmensen, S.B.; De Jongh, W.A.; et al. Structure of malaria invasion protein RH5 with erythrocyte basigin and blocking antibodies. Nature 2014, 515, 427–430. [Google Scholar] [CrossRef]
- Wong, W.; Huang, R.; Menant, S.; Hong, C.; Sandow, J.J.; Birkinshaw, R.W.; Healer, J.; Hodder, A.N.; Kanjee, U.; Tonkin, C.J.; et al. Structure of Plasmodium falciparum Rh5-CyRPA-Ripr invasion complex. Nature 2019, 565, 118–121. [Google Scholar] [CrossRef]
- Ragotte, R.J.; Higgins, M.K.; Draper, S.J. The RH5-CyRPA-Ripr Complex as a Malaria Vaccine Target. Trends Parasitol. 2020, 36, 545. [Google Scholar] [CrossRef]
- Sleebs, B.E.; Jarman, K.E.; Frolich, S.; Wong, W.; Healer, J.; Dai, W.; Lucet, I.S.; Wilson, D.W.; Cowman, A.F. Development and application of a high-throughput screening assay for identification of small molecule inhibitors of the P. falciparum reticulocyte binding-like homologue 5 protein. Int. J. Parasitol. Drugs drug Resist. 2020, 14, 188–200. [Google Scholar] [CrossRef]
- Volz, J.C.; Yap, A.; Sisquella, X.; Thompson, J.K.; Lim, N.T.Y.; Whitehead, L.W.; Chen, L.; Lampe, M.; Tham, W.H.; Wilson, D.; et al. Essential Role of the PfRh5/PfRipr/CyRPA Complex during Plasmodium falciparum Invasion of Erythrocytes. Cell Host Microbe 2016, 20, 60–71. [Google Scholar] [CrossRef]
- Wanaguru, M.; Liu, W.; Hahn, B.H.; Rayner, J.C.; Wright, G.J. RH5-Basigin interaction plays a major role in the host tropism of Plasmodium falciparum. Proc. Natl. Acad. Sci. USA. 2013, 110, 20735–20740. [Google Scholar] [CrossRef]
- Miller, L.H.; Ackerman, H.C.; Su, X.Z.; Wellems, T.E. Malaria biology and disease pathogenesis: Insights for new treatments. Nat. Med. 2013, 19, 156–167. [Google Scholar] [CrossRef]
- Lamarque, M.; Besteiro, S.; Papoin, J.; Roques, M.; Vulliez-Le Normand, B.; Morlon-Guyot, J.; Dubremetz, J.F.; Fauquenoy, S.; Tomavo, S.; Faber, B.W.; et al. The RON2-AMA1 Interaction is a Critical Step in Moving Junction-Dependent Invasion by Apicomplexan Parasites. PLOS Pathog. 2011, 7, e1001276. [Google Scholar] [CrossRef]
- Devine, S.M.; MacRaild, C.A.; Norton, R.S.; Scammells, P.J. Antimalarial drug discovery targeting apical membrane antigen 1. Medchemcomm 2016, 8, 13. [Google Scholar] [CrossRef]
- Srinivasan, P.; Beatty, W.L.; Diouf, A.; Herrera, R.; Ambroggio, X.; Moch, J.K.; Tyler, J.S.; Narum, D.L.; Pierce, S.K.; Boothroyd, J.C.; et al. Binding of Plasmodium merozoite proteins RON2 and AMA1 triggers commitment to invasion. Proc. Natl. Acad. Sci. USA 2011, 108, 13275–13280. [Google Scholar] [CrossRef] [PubMed]
- Straub, K.W.; Peng, E.D.; Hajagos, B.E.; Tyler, J.S.; Bradley, P.J. The Moving Junction Protein RON8 Facilitates Firm Attachment and Host Cell Invasion in Toxoplasma gondii. PLOS Pathog. 2011, 7, e1002007. [Google Scholar] [CrossRef]
- Su, X.Z.; Lane, K.D.; Xia, L.; Sá, J.M.; Wellems, T.E. Plasmodium Genomics and Genetics: New Insights into Malaria Pathogenesis, Drug Resistance, Epidemiology, and Evolution. Clin. Microbiol. Rev. 2019, 32. [Google Scholar] [CrossRef]
- Girgis, S.T.; Adika, E.; Nenyewodey, F.E.; Senoo Jnr, D.K.; Ngoi, J.M.; Bandoh, K.; Lorenz, O.; van de Steeg, G.; Harrott, A.J.R.; Nsoh, S.; et al. Drug resistance and vaccine target surveillance of Plasmodium falciparum using nanopore sequencing in Ghana. Nat. Microbiol. 2023, 8, 2365–2377. [Google Scholar] [CrossRef] [PubMed]
- Duffy, P.E.; Patrick Gorres, J. Malaria vaccines since 2000: Progress, priorities, products. npj Vaccines 2020, 5, 48. [Google Scholar] [CrossRef]
- de Almeida, M.E.M.; de Vasconcelos, M.G.S.; Tarragô, A.M.; Mariúba, L.A.M. Circumsporozoite Surface Protein-based malaria vaccines: A review. Rev. Inst. Med. Trop. Sao Paulo 2021, 63, e11. [Google Scholar] [CrossRef]
- Shockley, K.R. Quantitative high-throughput screening data analysis: Challenges and recent advances. Drug Discov. Today 2015, 20, 296–300. [Google Scholar] [CrossRef]
- Vamathevan, J.; Clark, D.; Czodrowski, P.; Dunham, I.; Ferran, E.; Lee, G.; Li, B.; Madabhushi, A.; Shah, P.; Spitzer, M.; et al. Applications of machine learning in drug discovery and development. Nat. Rev. Drug Discov. 2019, 18, 463–477. [Google Scholar] [CrossRef]
- Lavecchia, A. Machine-learning approaches in drug discovery: Methods and applications. Drug Discov. Today 2015, 20, 318–331. [Google Scholar] [CrossRef]
- Hochreiter, S.; Klambauer, G.; Rarey, M. Machine Learning in Drug Discovery. J. Chem. Inf. Model. 2018, 58, 1723–1724. [Google Scholar] [CrossRef]
- Dara, S.; Dhamercherla, S.; Jadav, S.S.; Babu, C.M.; Ahsan, M.J. Machine Learning in Drug Discovery: A Review. Artif. Intell. Rev. 2021, 55, 1947–1999. [Google Scholar] [CrossRef] [PubMed]
- Kwofie, S.K.; Adams, J.; Broni, E.; Enninful, K.S.; Agoni, C.; Soliman, M.E.S.; Wilson, M.D. Artificial Intelligence, Machine Learning, and Big Data for Ebola Virus Drug Discovery. Pharmaceuticals 2023, 16, 332. [Google Scholar] [CrossRef]
- Agyapong, O.; Miller, W.A.; Wilson, M.D.; Kwofie, S.K. Development of a proteochemometric-based support vector machine model for predicting bioactive molecules of tubulin receptors. Mol. Divers. 2022, 26, 2231–2242. [Google Scholar] [CrossRef]
- Adams, J.; Agyenkwa-Mawuli, K.; Agyapong, O.; Wilson, M.D.; Kwofie, S.K. EBOLApred: A Machine Learning-Based Web Application for Predicting Cell Entry Inhibitors of the Ebola Virus. Comput. Biol. Chem. 2022, 101, 107766. [Google Scholar] [CrossRef]
- Lee, Y.W.; Choi, J.W.; Shin, E.H. Machine learning model for predicting malaria using clinical information. Comput. Biol. Med. 2021, 129, 104151. [Google Scholar] [CrossRef]
- Maindola, P.; Jamal, S.; Grover, A. Cheminformatics Based Machine Learning Models for AMA1-RON2 Abrogators for Inhibiting Plasmodium falciparum Erythrocyte Invasion. Mol. Inform. 2015, 34, 655–664. [Google Scholar] [CrossRef]
- Vetrivel, U.; Muralikumar, S.; Mahalakshmi, B.; Lily Therese, K.; Madhavan, H.; Alameen, M.; Thirumudi, I. Multilevel Precision-Based Rational Design of Chemical Inhibitors Targeting the Hydrophobic Cleft of Toxoplasma gondii Apical Membrane Antigen 1 (AMA1). Genomics Inform. 2016, 14, 53. [Google Scholar] [CrossRef]
- Bharti, D.R.; Lynn, A.M. QSAR based predictive modeling for anti-malarial molecules. Bioinformation 2017, 13, 154. [Google Scholar] [CrossRef]
- Inglese, J.; Auld, D.S.; Jadhav, A.; Johnson, R.L.; Simeonov, A.; Yasgar, A.; Zheng, W.; Austin, C.P. Quantitative high-throughput screening: A titration-based approach that efficiently identifies biological activities in large chemical libraries. Proc. Natl. Acad. Sci. USA 2006, 103, 11473–11478. [Google Scholar] [CrossRef]
- Yasgar, A.; Shinn, P.; Jadhav, A.; Auld, D.; Michael, S.; Zheng, W.; Austin, C.P.; Inglese, J.; Simeonov, A. Compound Management for Quantitative High-Throughput Screening. J. Lab. Autom. 2008, 13, 79–89. [Google Scholar] [CrossRef]
- Huang, R. A quantitative high-throughput screening data analysis pipeline for activity profiling. Methods Mol. Biol. 2016, 1473, 111–122. [Google Scholar] [CrossRef] [PubMed]
- AID 720542—qHTS for Inhibitors of AMA1-RON.; Towards Development of Antimalarial Drug Lead: Primary Screen—PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/bioassay/720542 (accessed on 16 February 2025).
- Severa, W.; Vineyard, C.M.; Dellana, R.; Verzi, S.J.; Aimone, J.B. Training deep neural networks for binary communication with the Whetstone method. Nat. Mach. Intell. 2019, 1, 86–94. [Google Scholar] [CrossRef]
- Korshunova, M.; Huang, N.; Capuzzi, S.; Radchenko, D.S.; Savych, O.; Moroz, Y.S.; Wells, C.I.; Willson, T.M.; Tropsha, A.; Isayev, O. Generative and reinforcement learning approaches for the automated de novo design of bioactive compounds. Commun. Chem. 2022, 5, 129. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Skolnick, J. Utility of the Morgan Fingerprint in Structure-Based Virtual Ligand Screening. J. Phys. Chem. B 2024, 128, 5363–5370. [Google Scholar] [CrossRef]
- Zhong, S.; Guan, X. Count-Based Morgan Fingerprint: A More Efficient and Interpretable Molecular Representation in Developing Machine Learning-Based Predictive Regression Models for Water Contaminants’ Activities and Properties. Environ. Sci. Technol. 2023, 57, 18193–18202. [Google Scholar] [CrossRef]
- Morgan, L.O.; Johnson, M.; Cornelison, J.B.; Isaac, C.V.; deJong, J.L.; Prahlow, J.A. Autopsy Fingerprint Technique Using Fingerprint Powder. J. Forensic Sci. 2018, 63, 262–265. [Google Scholar] [CrossRef]
- Ehiro, T. Descriptor generation from Morgan fingerprint using persistent homology. SAR QSAR Environ. Res. 2024, 35, 31–51. [Google Scholar] [CrossRef]
- Cherrington, M.; Thabtah, F.; Lu, J.; Xu, Q. Feature selection: Filter methods performance challenges. In Proceedings of the 2019 International Conference on Computer and Information Sciences ICCIS, Sakaka, Saudi Arabia, 3–4 April 2019. [Google Scholar] [CrossRef]
- Blagus, R.; Lusa, L. SMOTE for high-dimensional class-imbalanced data. BMC Bioinformatics 2013, 14, 106. [Google Scholar] [CrossRef]
- Dablain, D.; Krawczyk, B.; Chawla, N.V. DeepSMOTE: Fusing Deep Learning and SMOTE for Imbalanced Data. IEEE Trans. Neural Networks Learn. Syst. 2023, 34, 6390–6404. [Google Scholar] [CrossRef]
- Pradipta, G.A.; Wardoyo, R.; Musdholifah, A.; Sanjaya, I.N.H.; Ismail, M. SMOTE for Handling Imbalanced Data Problem: A Review. In Proceedings of the 2021 6th International Conference on Informatics and Computing ICIC, Jakarta, Indonesia, 3–4 November 2021. [Google Scholar] [CrossRef]
- Liu, X.Y.; Zhou, Z.H. Ensemble Methods for Class Imbalance Learning. In Imbalanced Learning: Foundations, Algorithms, and Applications; Wiley-IEEE Press: Hoboken, NJ, USA, 2013; pp. 61–82. [Google Scholar] [CrossRef]
- Huda, S.; Liu, K.; Abdelrazek, M.; Ibrahim, A.; Alyahya, S.; Al-Dossari, H.; Ahmad, S. An Ensemble Oversampling Model for Class Imbalance Problem in Software Defect Prediction. IEEE Access 2018, 6, 24184–24195. [Google Scholar] [CrossRef]
- Galar, M.; Fernandez, A.; Barrenechea, E.; Bustince, H.; Herrera, F. A review on ensembles for the class imbalance problem: Bagging-, boosting-, and hybrid-based approaches. IEEE Trans. Syst. Man Cybern. Part C Appl. Rev. 2012, 42, 463–484. [Google Scholar] [CrossRef]
- Ogunsanya, M.; Isichei, J.; Desai, S. Grid search hyperparameter tuning in additive manufacturing processes. Manuf. Lett. 2023, 35, 1031–1042. [Google Scholar] [CrossRef]
- Belete, D.M.; Huchaiah, M.D. Grid search in hyperparameter optimization of machine learning models for prediction of HIV/AIDS test results. Int. J. Comput. Appl. 2022, 44, 875–886. [Google Scholar] [CrossRef]
- Shekar, B.H.; Dagnew, G. Grid search-based hyperparameter tuning and classification of microarray cancer data. In Proceedings of the 2019 Second International Conference on Advanced Computational and Communication Paradigms ICACCP, Gangtok, India, 25–28 February 2019. [Google Scholar] [CrossRef]
- Alireza, B.; Mostafa, H.; Ahmed, N.; Gehad, E.A. Part 1: Simple Definition and Calculation of Accuracy, Sensitivity and Specificity. Arch. Acad. Emerg. Med. 2015, 3, 48–49. [Google Scholar]
- Kelly, H.; Bull, A.; Russo, P.; McBryde, E.S. Estimating sensitivity and specificity from positive predictive value, negative predictive value and prevalence: Application to surveillance systems for hospital-acquired infections. J. Hosp. Infect. 2008, 69, 164–168. [Google Scholar] [CrossRef]
- Hawass, N.E.D. Comparing the sensitivities and specificities of two diagnostic procedures performed on the same group of patients. Br. J. Radiol. 1997, 70, 360–366. [Google Scholar] [CrossRef]
- Goutte, C.; Gaussier, E. A Probabilistic Interpretation of Precision, Recall and F-Score, with Implication for Evaluation. Lect. Notes Comput. Sci. 2005, 3408, 345–359. [Google Scholar] [CrossRef]
- Wardhani, N.W.S.; Rochayani, M.Y.; Iriany, A.; Sulistyono, A.D.; Lestantyo, P. Cross-validation Metrics for Evaluating Classification Performance on Imbalanced Data. In Proceedings of the 2019 International Conference on Computer, Control, Informatics and its Applications IC3INA, Tangerang, Indonesia, 23–24 October 2019; pp. 14–18. [Google Scholar] [CrossRef]
- Haque, M.R.; Islam, M.M.; Iqbal, H.; Reza, M.S.; Hasan, M.K. Performance Evaluation of Random Forests and Artificial Neural Networks for the Classification of Liver Disorder. In Proceedings of the 2018 International Conference on Computer, Communication, Chemical, Material and Electronic Engineering IC4ME2, Rajshahi, Bangladesh, 8–9 February 2018. [Google Scholar] [CrossRef]
- Maxwell, A.E.; Warner, T.A.; Guillén, L.A. Accuracy Assessment in Convolutional Neural Network-Based Deep Learning Remote Sensing Studies—Part 1: Literature Review. Remote Sens. 2021, 13, 2450. [Google Scholar] [CrossRef]
- Eusebi, P. Diagnostic Accuracy Measures. Cerebrovasc. Dis. 2013, 36, 267–272. [Google Scholar] [CrossRef]
- Varoquaux, G.; Colliot, O. Evaluating Machine Learning Models and Their Diagnostic Value. Neuromethods 2023, 197, 601–630. [Google Scholar] [CrossRef]
- Ferris, M.H.; McLaughlin, M.; Grieggs, S.; Ezekiel, S.; Blasch, E.; Alford, M.; Cornacchia, M.; Bubalo, A. Using ROC curves and AUC to evaluate performance of no-reference image fusion metrics. In Proceedings of the 2015 National Aerospace and Electronics Conference (NAECON), Dayton, OH, USA, 15–19 June 2015; pp. 27–34. [Google Scholar] [CrossRef]
- Rücker, C.; Rücker, G.; Meringer, M. Y-randomization and its variants in QSPR/QSAR. J. Chem. Inf. Model. 2007, 47, 2345–2357. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, H. Estimation of predictive performance for test data in applicability domains using y-randomization. J. Chemom. 2019, 33, e3171. [Google Scholar] [CrossRef]
- Chung, N.C.; Miasojedow, B.Z.; Startek, M.; Gambin, A. Jaccard/Tanimoto similarity test and estimation methods for biological presence-absence data. BMC Bioinformatics 2019, 20, 644. [Google Scholar] [CrossRef]
- Mathea, M.; Klingspohn, W.; Baumann, K. Chemoinformatic Classification Methods and their Applicability Domain. Mol. Inform. 2016, 35, 160–180. [Google Scholar] [CrossRef]
- Bichri, H.; Chergui, A.; Hain, M. Investigating the Impact of Train / Test Split Ratio on the Performance of Pre-Trained Models with Custom Datasets. Int. J. Adv. Comput. Sci. Appl. 2024, 15, 331. [Google Scholar] [CrossRef]
- Rácz, A.; Bajusz, D.; Héberger, K. Effect of Dataset Size and Train/Test Split Ratios in QSAR/QSPR Multiclass Classification. Mol. 2021, 26, 1111. [Google Scholar] [CrossRef]
- Han, H.; Wang, W.Y.; Mao, B.H. Borderline-SMOTE: A new over-sampling method in imbalanced data sets learning. In Advances in Intelligent Computing; Lecture Notes in Computer Science (PART I); Springer: Berlin/Heidelberg, Germany, 2005; Volume 3644, pp. 878–887. [Google Scholar] [CrossRef]
- Nagassou, M.; Mwangi, R.W.; Nyarige, E.; Nagassou, M.; Mwangi, R.W.; Nyarige, E. A Hybrid Ensemble Learning Approach Utilizing Light Gradient Boosting Machine and Category Boosting Model for Lifestyle-Based Prediction of Type-II Diabetes Mellitus. J. Data Anal. Inf. Process. 2023, 11, 480–511. [Google Scholar] [CrossRef]
- Kumar, V.; Kedam, N.; Sharma, K.V.; Khedher, K.M.; Alluqmani, A.E. A Comparison of Machine Learning Models for Predicting Rainfall in Urban Metropolitan Cities. Sustainability 2023, 15, 13724. [Google Scholar] [CrossRef]
- Joshi, A.; Saggar, P.; Jain, R.; Sharma, M.; Gupta, D.; Khanna, A. CatBoost—An Ensemble Machine Learning Model for Prediction and Classification of Student Academic Performance. Adv. Data Sci. Adapt. Anal. 2021, 13, 2141002. [Google Scholar] [CrossRef]
- Contreras, P.; Orellana-Alvear, J.; Muñoz, P.; Bendix, J.; Célleri, R. Influence of Random Forest Hyperparameterization on Short-Term Runoff Forecasting in an Andean Mountain Catchment. Atmos 2021, 12, 238. [Google Scholar] [CrossRef]
- Fan, M.; Xiao, K.; Sun, L.; Zhang, S.; Xu, Y. Automated Hyperparameter Optimization of Gradient Boosting Decision Tree Approach for Gold Mineral Prospectivity Mapping in the Xiong’ershan Area. Miner 2022, 12, 1621. [Google Scholar] [CrossRef]
- Kiatkarun, K.; Phunchongharn, P. Automatic Hyper-Parameter Tuning for Gradient Boosting Machine. In Proceedings of the 2020 1st International Conference on Big Data Analytics and Practices IBDAP, Bangkok, Thailand, 25–26 September 2020. [Google Scholar] [CrossRef]
- Kavzoglu, T.; Teke, A. Advanced hyperparameter optimization for improved spatial prediction of shallow landslides using extreme gradient boosting (XGBoost). Bull. Eng. Geol. Environ. 2022, 81, 201. [Google Scholar] [CrossRef]
- Hancock, J.T.; Khoshgoftaar, T.M. CatBoost for big data: An interdisciplinary review. J. Big Data 2020, 7, 94. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Zhang, S.; Xue, D.; Xiao, F.; Liu, C. Improved estimation of coalbed methane content using the revised estimate of depth and CatBoost algorithm: A case study from southern Sichuan Basin, China. Comput. Geosci. 2022, 158, 104973. [Google Scholar] [CrossRef]
- Krithiga, R.; Ilavarasan, E. Hyperparameter tuning of AdaBoost algorithm for social spammer identification. Int. J. Pervasive Comput. Commun. 2020, 17, 462–482. [Google Scholar] [CrossRef]
- Gao, R.; Liu, Z. An Improved AdaBoost Algorithm for Hyperparameter Optimization. J. Phys. Conf. Ser. 2020, 1631, 012048. [Google Scholar] [CrossRef]
- Fahrezi, S.F.; Nugraha, A.; Luthfiarta, A.; Primadya, N.D. Optimizing Performance of AdaBoost Algorithm through Undersampling and Hyperparameter Tuning on CICIoT 2023 Dataset. Techné J. Ilm. Elektrotek. 2024, 10. [Google Scholar] [CrossRef]
- Chen, Y.C.; Su, C.T. Distance-based margin support vector machine for classification. Appl. Math. Comput. 2016, 283, 141–152. [Google Scholar] [CrossRef]
- Aoudi, W.; Barbar, A.M. Support vector machines: A distance-based approach to multi-class classification. In Proceedings of the 2016 IEEE International Multidisciplinary Conference on Engineering Technology IMCET, Beirut, Lebanon, 2–4 November 2016; pp. 75–80. [Google Scholar] [CrossRef]
- Gaspar, P.; Carbonell, J.; Oliveira, J.L. On the parameter optimization of Support Vector Machines for binary classification. J. Integr. Bioinform. 2012, 9, 201. [Google Scholar] [CrossRef]
- Min, J.H.; Lee, Y.C. Bankruptcy prediction using support vector machine with optimal choice of kernel function parameters. Expert Syst. Appl. 2005, 28, 603–614. [Google Scholar] [CrossRef]
- Dioşan, L.; Rogozan, A.; Pecuchet, J.P. Improving classification performance of Support Vector Machine by genetically optimising kernel shape and hyper-parameters. Appl. Intell. 2012, 36, 280–294. [Google Scholar] [CrossRef]
- Naidu, G.; Zuva, T.; Sibanda, E.M. A Review of Evaluation Metrics in Machine Learning Algorithms. In Artificial Intelligence Application in Networks and Systems; Lecture Notes in Networks and Systems (LNNS); Springer: Cham, Switzerland, 2023; Volume 724, pp. 15–25. [Google Scholar] [CrossRef]
- Rainio, O.; Teuho, J.; Klén, R. Evaluation metrics and statistical tests for machine learning. Sci. Rep. 2024, 14, 6086. [Google Scholar] [CrossRef]
- Erickson, B.J.; Kitamura, F. Magician’s corner: 9. performance metrics for machine learning models. Radiol. Artif. Intell. 2021, 7. [Google Scholar] [CrossRef]
- Taneja, S.; Gupta, P.R. Python as a tool for web server application development. JIMS 8i-Int’l J. Inf. Comm. Comput. Technol. 2014, 2, 77–83. [Google Scholar]
- Mufid, M.R.; Basofi, A.; Al Rasyid, M.U.H.; Rochimansyah, I.F.; Rokhim, A. Design an MVC Model using Python for Flask Framework Development. In Proceedings of the 2019 International Electronics Symposium (IES), Surabaya, Indonesia, 27–28 September 2019; pp. 214–219. [Google Scholar] [CrossRef]
- Uzayr, S.B.; Cloud, N.; Ambler, T. JavaScript Frameworks for Modern Web Development; Apress: Berkeley, CA, USA, 2019. [Google Scholar]
- Prusa, J.; Khoshgoftaar, T.M.; DIttman, D.J.; Napolitano, A. Using Random Undersampling to Alleviate Class Imbalance on Tweet Sentiment Data. In Proceedings of the 2015 IEEE International Conference on Information Reuse and Integration, San Francisco, CA, USA, 13–15 August 2015; pp. 197–202. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| METRICS | MODELS | ||||
|---|---|---|---|---|---|
| RF | GBMs | CB | AB | SVM | |
| Sensitivity | 0.85 | 0.85 | 0.87 | 0.77 | 0.86 |
| Specificity | 0.88 | 0.90 | 0.88 | 0.92 | 0.80 |
| Precision | 0.81 | 0.84 | 0.80 | 0.85 | 0.71 |
| F1-score | 0.83 | 0.84 | 0.84 | 0.81 | 0.78 |
| Accuracy | 0.87 | 0.89 | 0.88 | 0.86 | 0.82 |
| ROC-AUC | 0.91 | 0.92 | 0.93 | 0.93 | 0.90 |
| METRICS | MODELS | ||||
|---|---|---|---|---|---|
| RF | GBMs | CB | AB | SVM | |
| Sensitivity | 0.80 | 0.85 | 0.85 | 0.80 | 0.50 |
| Specificity | 0.85 | 0.95 | 0.85 | 0.90 | 0.80 |
| Precision | 0.84 | 0.94 | 0.85 | 0.88 | 0.71 |
| F1-score | 0.82 | 0.89 | 0.85 | 0.84 | 0.59 |
| Accuracy | 0.83 | 0.90 | 0.85 | 0.85 | 0.65 |
| ROC-AUC | 0.80 | 0.85 | 0.85 | 0.80 | 0.50 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lamptey, E.; Oparebea, J.; Anyaele, G.; Ofosu, B.; Hanson, G.; Sakyi, P.O.; Agyapong, O.; Amuzu, D.S.Y.; Miller, W.A., III; Kwofie, S.K.; et al. PLASMOpred: A Machine Learning-Based Web Application for Predicting Antimalarial Small Molecules Targeting the Apical Membrane Antigen 1–Rhoptry Neck Protein 2 Invasion Complex. Pharmaceuticals 2025, 18, 776. https://doi.org/10.3390/ph18060776
Lamptey E, Oparebea J, Anyaele G, Ofosu B, Hanson G, Sakyi PO, Agyapong O, Amuzu DSY, Miller WA III, Kwofie SK, et al. PLASMOpred: A Machine Learning-Based Web Application for Predicting Antimalarial Small Molecules Targeting the Apical Membrane Antigen 1–Rhoptry Neck Protein 2 Invasion Complex. Pharmaceuticals. 2025; 18(6):776. https://doi.org/10.3390/ph18060776
Chicago/Turabian StyleLamptey, Eugene, Jessica Oparebea, Gabriel Anyaele, Belinda Ofosu, George Hanson, Patrick O. Sakyi, Odame Agyapong, Dominic S. Y. Amuzu, Whelton A. Miller, III, Samuel K. Kwofie, and et al. 2025. "PLASMOpred: A Machine Learning-Based Web Application for Predicting Antimalarial Small Molecules Targeting the Apical Membrane Antigen 1–Rhoptry Neck Protein 2 Invasion Complex" Pharmaceuticals 18, no. 6: 776. https://doi.org/10.3390/ph18060776
APA StyleLamptey, E., Oparebea, J., Anyaele, G., Ofosu, B., Hanson, G., Sakyi, P. O., Agyapong, O., Amuzu, D. S. Y., Miller, W. A., III, Kwofie, S. K., & Mensah-Brown, H. E. (2025). PLASMOpred: A Machine Learning-Based Web Application for Predicting Antimalarial Small Molecules Targeting the Apical Membrane Antigen 1–Rhoptry Neck Protein 2 Invasion Complex. Pharmaceuticals, 18(6), 776. https://doi.org/10.3390/ph18060776