
Metformin-Induced Apoptosis Is Mediated Through Mitochondrial VDAC1
 , ,
, , 
Abstract
1. Introduction
2. Results
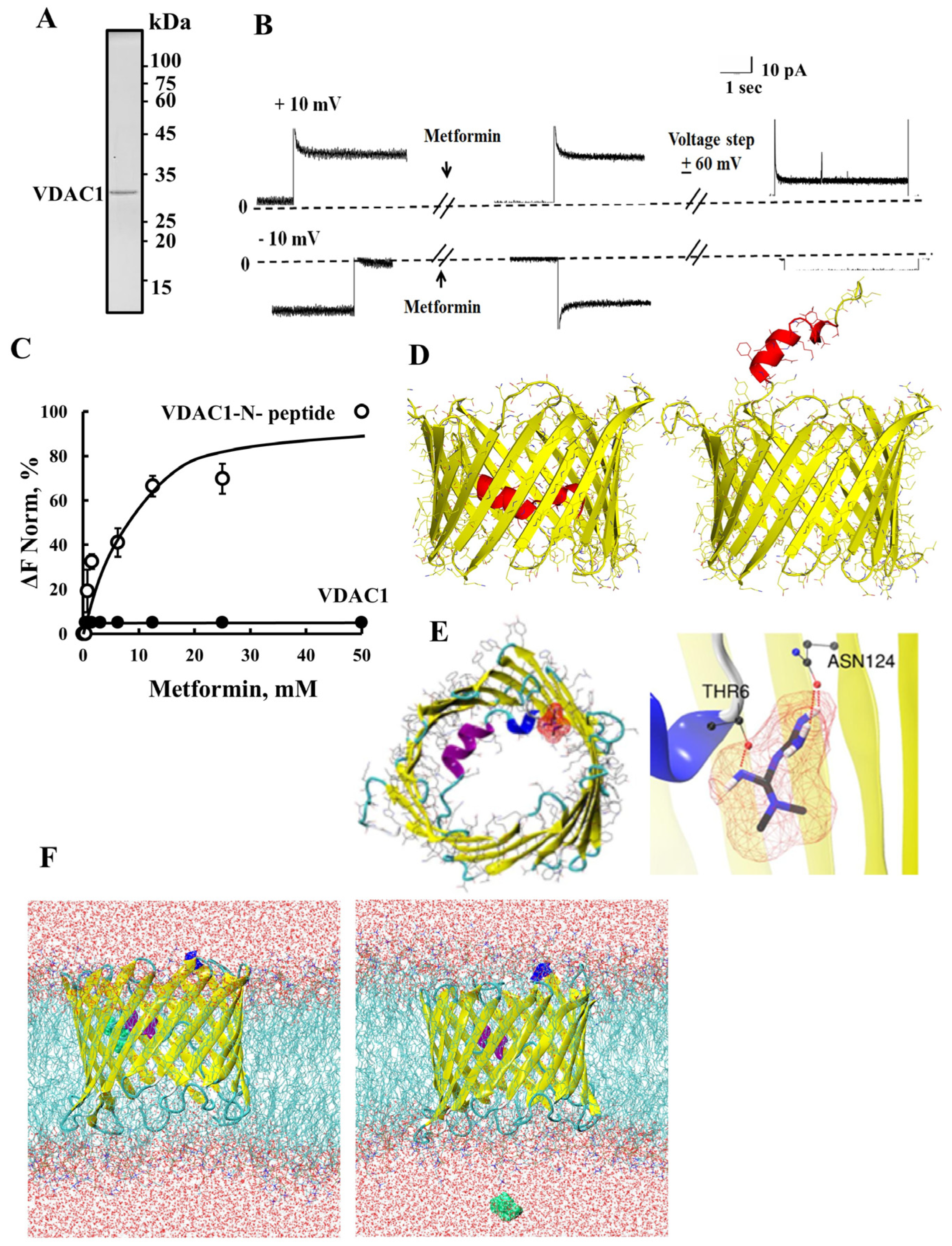
2.1. Metformin Induces VDAC1 Overexpression, Oligomerization, and Apoptosis
2.2. Metformin Binds to the VDAC1 N-Terminal Domain
2.3. Metformin Directly Interacts with Hexokinase and Detaches It from the Mitochondria
2.4. Metformin Increased NLRP3 and ASC Levels
3. Discussion
3.1. Metformin-Induced Apoptosis Can Be Mediated via VDAC1
3.2. Metformin Interaction with HK and Its Detachment from VDAC1
3.3. Metformin and Inflammation
3.4. Metformin, Buformin, and Phenformin as Potential Cancer Treatments
4. Materials and Methods
4.1. Materials
4.2. Cell Culture and Metformin, Buformin, or Phenformin Treatment
4.3. HK-I Detachment
4.4. VDAC1 Oligomerization Assay
4.5. Protein Extraction from Cells, Gel Electrophoresis, and Immunoblotting
4.6. Cell Death
4.7. Real-Time Quantitative PCR (q-RT-PCR) Analysis
4.8. Micro-Scale Thermophoresis (MST)
4.9. Intracellular Ca2+ Level Analysis
4.10. Reactive Oxygen Species (ROS) Level Analysis
4.11. HK-I and VDAC1 Purification and Channel Activity Reconstitution into a Planar Lipid Bilayer (PLB)
4.12. Immunofluorescence (IF) Staining
4.13. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AMPK | AMP-activated kinase |
| AD | Alzheimer’s disease |
| ASC | Apoptosis-associated speck-like protein containing a CARD |
| CVDs | Cardiovascular diseases |
| DPC | Diphenylamine-2-carboxylate |
| G-6-P | Glucose-6-phosphate |
| HK | Hexokinase |
| IBDs | Inflammatory bowel diseases |
| mTOR | Mammalian target of rapamycin |
| mtDNA | Mitochondria DNA |
| MST | Micro-scale thermophoresis |
| NAFLD | Non-alcoholic fatty liver disease |
| NLRP3 | NLR pyrin domain contains three |
| OCTs | Organic cation transporters |
| PLB | Planar lipid bilayer |
| ROS | Reactive oxygen species |
| T2DM | Type 2 diabetes mellitus |
| VDAC1 | Voltage-dependent anion channel |
References
- Bailey, C.J. Metformin: Historical overview. Diabetologia 2017, 60, 1566–1576. [Google Scholar] [CrossRef] [PubMed]
- Viollet, B.; Guigas, B.; Sanz Garcia, N.; Leclerc, J.; Foretz, M.; Andreelli, F. Cellular and molecular mechanisms of metformin: An overview. Clin. Sci. 2012, 122, 253–270. [Google Scholar] [CrossRef] [PubMed]
- Pawlyk, A.C.; Giacomini, K.M.; McKeon, C.; Shuldiner, A.R.; Florez, J.C. Metformin pharmacogenomics: Current status and future directions. Diabetes 2014, 63, 2590–2599. [Google Scholar] [CrossRef] [PubMed]
- Foretz, M.; Guigas, B.; Bertrand, L.; Pollak, M.; Viollet, B. Metformin: From mechanisms of action to therapies. Cell Metab. 2014, 20, 953–966. [Google Scholar] [CrossRef]
- Kasznicki, J.; Sliwinska, A.; Drzewoski, J. Metformin in cancer prevention and therapy. Ann. Transl. Med. 2014, 2, 57. [Google Scholar] [CrossRef]
- Mazza, A.; Fruci, B.; Garinis, G.A.; Giuliano, S.; Malaguarnera, R.; Belfiore, A. The role of metformin in the management of NAFLD. Exp. Diabetes Res. 2012, 2012, 716404. [Google Scholar] [CrossRef]
- Scheen, A.J.; Esser, N.; Paquot, N. Antidiabetic agents: Potential anti-inflammatory activity beyond glucose control. Diabetes Metab. 2015, 41, 183–194. [Google Scholar] [CrossRef]
- Dardano, A.; Del Prato, S. Metformin: An inexpensive and effective treatment in people with diabetes and COVID-19? Lancet Healthy Longev. 2021, 2, e6–e7. [Google Scholar] [CrossRef]
- Hitchings, A.W.; Lai, D.; Jones, P.W.; Baker, E.H. Metformin in COPD Trial Team. Metformin in severe exacerbations of chronic obstructive pulmonary disease: A randomised controlled trial. Thorax 2016, 71, 587–593. [Google Scholar] [CrossRef]
- Lv, Z.Q.; Guo, Y.J. Metformin and Its Benefits for Various Diseases. Front. Endocrinol. 2020, 11, 191. [Google Scholar] [CrossRef]
- Bost, F.; Rena, G.; Viollet, B. Editorial: Metformin: Beyond Diabetes. Front. Endocrinol. 2019, 10, 851. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.; Wang, Z.; Xu, X.; Sun, X.; Dong, X.; Luo, Y.; Sun, X. Extensive therapeutic effects, underlying molecular mechanisms and disease treatment prediction of Metformin: A systematic review. Transl. Res. 2024, 263, 73–92. [Google Scholar] [CrossRef]
- Wijesekara, N.; Goncalves da Silva, R.A.; De Felice, F.G.; Fraser, P.E. Impaired peripheral glucose homeostasis and Alzheimer’s disease. Neuropharmacology 2017, 136, 172–181. [Google Scholar] [CrossRef]
- Campbell, J.M.; Stephenson, M.D.; de Courten, B.; Chapman, I.; Bellman, S.M.; Aromataris, E. Metformin Use Associated with Reduced Risk of Dementia in Patients with Diabetes: A Systematic Review and Meta-Analysis. J. Alzheimer’s Dis. 2018, 65, 1225–1236. [Google Scholar] [CrossRef]
- Bridges, H.R.; Jones, A.J.; Pollak, M.N.; Hirst, J. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem. J. 2014, 462, 475–487. [Google Scholar] [CrossRef]
- Madiraju, A.K.; Erion, D.M.; Rahimi, Y.; Zhang, X.M.; Braddock, D.T.; Albright, R.A.; Prigaro, B.J.; Wood, J.L.; Bhanot, S.; MacDonald, M.J.; et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature 2014, 510, 542–546. [Google Scholar] [CrossRef]
- Sorrentino, V.; Menzies, K.J.; Auwerx, J. Repairing Mitochondrial Dysfunction in Disease. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 353–389. [Google Scholar] [CrossRef]
- Bédoui, I.; Nasr, H.B.; Ksouda, K.; Ayadi, W.; Louati, N.; Chamkha, M.; Choura, S.; Gargouri, J.; Hammami, S.; Affes, H.; et al. Phytochemical Composition, Bioavailability and Pharmacokinetics of Methanolic Extracts: Antioxidant, Anticancer, and Apoptotic Effects on MCF7 Cells. Pharmacogn. Mag. 2024, 20, 218–229. [Google Scholar] [CrossRef]
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 2000, 348, 607–614. [Google Scholar] [CrossRef]
- Luengo, A.; Sullivan, L.B.; Heiden, M.G. Understanding the complex-I-ty of metformin action: Limiting mitochondrial respiration to improve cancer therapy. BMC Biol. 2014, 12, 82. [Google Scholar] [CrossRef]
- Wang, J.; Xia, S.; Zhu, Z. Synergistic effect of phenformin in non-small cell lung cancer (NSCLC) ionizing radiation treatment. Cell Biochem. Biophys. 2015, 71, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Kwong, S.C.; Brubacher, J. Phenformin and lactic acidosis: A case report and review. J. Emerg. Med. 1998, 16, 881–886. [Google Scholar] [CrossRef] [PubMed]
- Rubiño, M.E.G.; Carrillo, E.; Alcalá, G.R.; Domínguez-Martín, A.; Marchal, J.A.; Boulaiz, H. Phenformin as an Anticancer Agent: Challenges and Prospects. Int. J. Mol. Sci. 2019, 20, 3316. [Google Scholar] [CrossRef]
- Wu, C.; Qiu, S.; Zhu, X.; Lin, H.; Li, L. OCT1-Mediated Metformin Uptake Regulates Pancreatic Stellate Cell Activity. Cell Physiol. Biochem. 2018, 47, 1711–1720. [Google Scholar] [CrossRef]
- Veiga, S.R.; Ge, X.; Mercer, C.A.; Hernandez-Alvarez, M.I.; Thomas, H.E.; Hernandez-Losa, J.; Ramon, Y.C.S.; Zorzano, A.; Thomas, G.; Kozma, S.C. Phenformin-Induced Mitochondrial Dysfunction Sensitizes Hepatocellular Carcinoma for Dual Inhibition of mTOR. Clin. Cancer Res. 2018, 24, 3767–3780. [Google Scholar] [CrossRef]
- Jackson, A.L.; Sun, W.C.; Kilgore, J.; Guo, H.; Fang, Z.W.; Yin, Y.J.; Jones, H.M.; Gilliam, T.P.; Zhou, C.X.; Bae-Jump, V.L. Phenformin has anti-tumorigenic effects in human ovarian cancer cells and in an orthotopic mouse model of serous ovarian cancer. Oncotarget 2017, 8, 100113–100127. [Google Scholar] [CrossRef]
- Miller, R.A.; Chu, Q.; Xie, J.; Foretz, M.; Viollet, B.; Birnbaum, M.J. Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature 2013, 494, 256–260. [Google Scholar] [CrossRef]
- Zhu, Z.; Jiang, W.; Thompson, M.D.; Echeverria, D.; McGinley, J.N.; Thompson, H.J. Effects of metformin, buformin, and phenformin on the post-initiation stage of chemically induced mammary carcinogenesis in the rat. Cancer Prev. Res. 2015, 8, 518–527. [Google Scholar] [CrossRef]
- Miskimins, W.K.; Ahn, H.J.; Kim, J.Y.; Ryu, S.; Jung, Y.S.; Choi, J.Y. Synergistic anti-cancer effect of phenformin and oxamate. PLoS ONE 2014, 9, e85576. [Google Scholar] [CrossRef]
- Hernández-Velázquez, E.D.; Herrera, M.D.; Alba-Betancourt, C.; Navarro-Santos, P.; Ortíz-Alvarado, R.; Solorio-Alvarado, C.R. Synthesis and Evaluation of Fluorobenzyl Metformin Derivatives as Potential Drugs in The Diabetes Treatment. Asian J. Org. Chem. 2023, 12, e202300200. [Google Scholar] [CrossRef]
- Markowicz-Piasecka, M.; Sadkowska, A.; Sikora, J.; Broncel, M.; Huttunen, K.M. Novel Sulfonamide-Based Analogs of Metformin Exert Promising Anti-Coagulant Effects without Compromising Glucose-Lowering Activity. Pharmaceuticals 2020, 13, 323. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Zielonka, J.; Ouari, O.; Lopez, M.; McAllister, D.; Boyle, K.; Barrios, C.S.; Weber, J.J.; Johnson, B.D.; Hardy, M.; et al. Mitochondria-Targeted Analogues of Metformin Exhibit Enhanced Antiproliferative and Radiosensitizing Effects in Pancreatic Cancer Cells. Cancer Res. 2016, 76, 3904–3915. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Anand, U.; Nahon-Crystal, E.; Di Carlo, M.; Shteinfer-Kuzmine, A. Adverse Effects of Metformin from Diabetes to COVID-19, Cancer, Neurodegenerative Diseases, and Aging: Is VDAC1 a Common Target? Front. Physiol. 2021, 12, 730048. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Maldonado, E.N.; Krelin, Y. VDAC1 at the crossroads of cell metabolism, apoptosis and cell stress. Cell Stress 2017, 1, 11–36. [Google Scholar] [CrossRef]
- Magri, A.; Reina, S.; De Pinto, V. VDAC1 as Pharmacological Target in Cancer and Neurodegeneration: Focus on Its Role in Apoptosis. Front. Chem. 2018, 6, 108. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Shteinfer-Kuzmine, A.; Verma, A. VDAC1 at the Intersection of Cell Metabolism, Apoptosis, and Diseases. Biomolecules 2020, 10, 1485. [Google Scholar] [CrossRef]
- Koushi, M.; Aoyama, Y.; Kamei, Y.; Asakai, R. Bisindolylpyrrole triggers transient mitochondrial permeability transitions to cause apoptosis in a VDAC1/2 and cyclophilin D-dependent manner via the ANT-associated pore. Sci. Rep. 2020, 10, 16751. [Google Scholar] [CrossRef]
- Liang, Y.; Chu, P.H.; Tian, L.; Ho, K.F.; Ip, M.S.M.; Mak, J.C.W. Targeting mitochondrial permeability transition pore ameliorates PM(2.5)-induced mitochondrial dysfunction in airway epithelial cells. Environ. Pollut. 2022, 295, 118720. [Google Scholar] [CrossRef]
- Messina, A.; Reina, S.; Guarino, F.; De Pinto, V. VDAC isoforms in mammals. Biochim. Biophys. Acta 2012, 1818, 1466–1476. [Google Scholar] [CrossRef]
- Geula, S.; Ben-Hail, D.; Shoshan-Barmatz, V. Structure-based analysis of VDAC1: N-terminus location, translocation, channel gating and association with anti-apoptotic proteins. Biochem. J. 2012, 444, 475–485. [Google Scholar] [CrossRef]
- Abu-Hamad, S.; Zaid, H.; Israelson, A.; Nahon, E.; Shoshan-Barmatz, V. Hexokinase-I protection against apoptotic cell death is mediated via interaction with the voltage-dependent anion channel-1: Mapping the site of binding. J. Biol. Chem. 2008, 283, 13482–13490. [Google Scholar] [CrossRef] [PubMed]
- Azoulay-Zohar, H.; Israelson, A.; Abu-Hamad, S.; Shoshan-Barmatz, V. In self-defence: Hexokinase promotes voltage-dependent anion channel closure and prevents mitochondria-mediated apoptotic cell death. Biochem. J. 2004, 377, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Arbel, N.; Ben-Hail, D.; Shoshan-Barmatz, V. Mediation of the antiapoptotic activity of Bcl-xL protein upon interaction with VDAC1 protein. J. Biol. Chem. 2012, 287, 23152–23161. [Google Scholar] [CrossRef] [PubMed]
- Abu-Hamad, S.; Arbel, N.; Calo, D.; Arzoine, L.; Israelson, A.; Keinan, N.; Ben-Romano, R.; Friedman, O.; Shoshan-Barmatz, V. The VDAC1 N-terminus is essential both for apoptosis and the protective effect of anti-apoptotic proteins. J. Cell Sci. 2009, 122, 1906–1916. [Google Scholar] [CrossRef]
- Arbel, N.; Shoshan-Barmatz, V. Voltage-dependent anion channel 1-based peptides interact with Bcl-2 to prevent antiapoptotic activity. J. Biol. Chem. 2010, 285, 6053–6062. [Google Scholar] [CrossRef]
- Smilansky, A.; Dangoor, L.; Nakdimon, I.; Ben-Hail, D.; Mizrachi, D.; Shoshan-Barmatz, V. The Voltage-dependent Anion Channel 1 Mediates Amyloid beta Toxicity and Represents a Potential Target for Alzheimer Disease Therapy. J. Biol. Chem. 2015, 290, 30670–30683. [Google Scholar] [CrossRef]
- Betaneli, V.; Petrov, E.P.; Schwille, P. The role of lipids in VDAC oligomerization. Biophys. J. 2012, 102, 523–531. [Google Scholar] [CrossRef]
- Keinan, N.; Tyomkin, D.; Shoshan-Barmatz, V. Oligomerization of the mitochondrial protein voltage-dependent anion channel is coupled to the induction of apoptosis. Mol. Cell Biol. 2010, 30, 5698–5709. [Google Scholar] [CrossRef]
- Geula, S.; Naveed, H.; Liang, J.; Shoshan-Barmatz, V. Structure-based analysis of VDAC1 protein: Defining oligomer contact sites. J. Biol. Chem. 2012, 287, 2179–2190. [Google Scholar] [CrossRef]
- Ben-Hail, D.; Begas-Shvartz, R.; Shalev, M.; Shteinfer-Kuzmine, A.; Gruzman, A.; Reina, S.; De Pinto, V.; Shoshan-Barmatz, V. Novel Compounds Targeting the Mitochondrial Protein VDAC1 Inhibit Apoptosis and Protect against Mitochondrial Dysfunction. J. Biol. Chem. 2016, 291, 24986–25003. [Google Scholar] [CrossRef]
- Weisthal, S.; Keinan, N.; Ben-Hail, D.; Arif, T.; Shoshan-Barmatz, V. Ca2+-mediated regulation of VDAC1 expression levels is associated with cell death induction. Biochim. Biophys. Acta 2014, 1843, 2270–2281. [Google Scholar] [CrossRef] [PubMed]
- Ben-Hail, D.; Shoshan-Barmatz, V. VDAC1-interacting anion transport inhibitors inhibit VDAC1 oligomerization and apoptosis. Biochim. Biophys. Acta 2016, 1863, 1612–1623. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Gupta, R.; Blanco, L.P.; Yang, S.T.; Shteinfer-Kuzmine, A.; Wang, K.N.; Zhu, J.; Yoon, H.E.; Wang, X.H.; Kerkhofs, M.; et al. VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease. Science 2019, 366, 1531. [Google Scholar] [CrossRef]
- Riley, J.S.; Tait, S.W. Mitochondrial DNA in inflammation and immunity. EMBO Rep. 2020, 21, e49799. [Google Scholar] [CrossRef]
- Baik, S.H.; Ramanujan, V.K.; Becker, C.; Fett, S.; Underhill, D.M.; Wolf, A.J. Hexokinase dissociation from mitochondria promotes oligomerization of VDAC that facilitates NLRP3 inflammasome assembly and activation. Sci. Immunol. 2023, 8, eade7652. [Google Scholar] [CrossRef]
- Pittala, S.; Krelin, Y.; Shoshan-Barmatz, V. Targeting Liver Cancer and Associated Pathologies in Mice with a Mitochondrial VDAC1-Based Peptide. Neoplasia 2018, 20, 594–609. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Krelin, Y.; Shteinfer-Kuzmine, A.; Arif, T. Voltage-Dependent Anion Channel 1 As an Emerging Drug Target for Novel Anti-Cancer Therapeutics. Front. Oncol. 2017, 7, 154. [Google Scholar] [CrossRef]
- Manczak, M.; Reddy, P.H. Abnormal interaction of VDAC1 with amyloid beta and phosphorylated tau causes mitochondrial dysfunction in Alzheimer’s disease. Hum. Mol. Genet. 2012, 21, 5131–5146. [Google Scholar] [CrossRef]
- Cuadrado-Tejedor, M.; Vilarino, M.; Cabodevilla, F.; Del Rio, J.; Frechilla, D.; Perez-Mediavilla, A. Enhanced expression of the voltage-dependent anion channel 1 (VDAC1) in Alzheimer’s disease transgenic mice: An insight into the pathogenic effects of amyloid-beta. J. Alzheimer’s Dis. 2011, 23, 195–206. [Google Scholar] [CrossRef]
- Verma, A.; Shteinfer-Kuzmine, A.; Kamenetsky, N.; Pittala, S.; Paul, A.; Crystal, E.N.; Ouro, A.; Chalifa-Caspi, V.; Pandey, S.K.; Monsengo, A.; et al. Targeting the overexpressed mitochondrial protein VDAC1 in a mouse model of Alzheimer’s disease protects against mitochondrial dysfunction and mitigates brain pathology. Transl. Neurodegener. 2022, 11, 58. [Google Scholar] [CrossRef]
- Ahmed, M.; Muhammed, S.J.; Kessler, B.; Salehi, A. Mitochondrial proteome analysis reveals altered expression of voltage dependent anion channels in pancreatic beta-cells exposed to high glucose. Islets 2010, 2, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Donthamsetty, R.; Heldak, M.; Cho, Y.E.; Scott, B.T.; Makino, A. VDAC: Old protein with new roles in diabetes. Am. J. Physiol. Cell Physiol. 2012, 303, C1055–C1060. [Google Scholar] [CrossRef]
- Zhang, E.; Mohammed Al-Amily, I.; Mohammed, S.; Luan, C.; Asplund, O.; Ahmed, M.; Ye, Y.; Ben-Hail, D.; Soni, A.; Vishnu, N.; et al. Preserving Insulin Secretion in Diabetes by Inhibiting VDAC1 Overexpression and Surface Translocation in beta Cells. Cell Metab. 2019, 29, 64–77.e6. [Google Scholar] [CrossRef]
- Jiang, L.; Wang, H.; Chen, G.; Feng, Y.; Zou, J.; Liu, M.; Liu, K.; Wang, N.; Zhang, H.; Wang, K.; et al. WDR26/MIP2 interacts with VDAC1 and regulates VDAC1 expression levels in H9c2 cells. Free Radic. Biol. Med. 2018, 117, 58–65. [Google Scholar] [CrossRef]
- Klapper-Goldstein, H.; Verma, A.; Elyagon, S.; Gillis, R.; Murninkas, M.; Pittala, S.; Paul, A.; Shoshan-Barmatz, V.A.E.Y. VDAC1 in the diseased myocardium and the effect of VDAC1-interacting compound on atrial fibrosis induced by hyperaldosteronism. Sci. Rep. 2020, 10, 22101. [Google Scholar] [CrossRef]
- Liao, Z.; Liu, D.; Tang, L.; Yin, D.; Yin, S.; Lai, S.; Yao, J.; He, M. Long-term oral resveratrol intake provides nutritional preconditioning against myocardial ischemia/reperfusion injury: Involvement of VDAC1 downregulation. Mol. Nutr. Food Res. 2015, 59, 454–464. [Google Scholar] [CrossRef]
- Schwertz, H.; Carter, J.M.; Abdudureheman, M.; Russ, M.; Buerke, U.; Schlitt, A.; Muller-Werdan, U.; Prondzinsky, R.; Werdan, K.; Buerke, M. Myocardial ischemia/reperfusion causes VDAC phosphorylation which is reduced by cardioprotection with a p38 MAP kinase inhibitor. Proteomics 2007, 7, 4579–4588. [Google Scholar] [CrossRef]
- Tian, M.; Xie, Y.; Meng, Y.; Ma, W.; Tong, Z.; Yang, X.; Lai, S.; Zhou, Y.; He, M.; Liao, Z. Resveratrol protects cardiomyocytes against anoxia/reoxygenation via dephosphorylation of VDAC1 by Akt-GSK3 beta pathway. Eur. J. Pharmacol. 2019, 843, 80–87. [Google Scholar] [CrossRef]
- Tong, Z.H.; Xie, Y.Y.; He, M.; Ma, W.; Zhou, Y.; Lai, S.Q.; Meng, Y.; Liao, Z.P. VDAC1 deacetylation is involved in the protective effects of resveratrol against mitochondria-mediated apoptosis in cardiomyocytes subjected to anoxia/reoxygenation injury. Biomed. Pharmacother. 2017, 95, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Pittala, S.; Alhozeel, B.; Shteinfer-Kuzmine, A.; Ohana, E.; Gupta, R.; Chung, J.H.; Shoshan-Barmatz, V. The role of the mitochondrial protein VDAC1 in inflammatory bowel disease: A potential therapeutic target. Mol. Ther. 2021, 30, 726–744. [Google Scholar] [CrossRef]
- Pittala, S.; Krelin, Y.; Kuperman, Y.; Shoshan-Barmatz, V. A Mitochondrial VDAC1-Based Peptide Greatly Suppresses Steatosis and NASH-Associated Pathologies in a Mouse Model. Mol. Ther. 2019, 27, 1848–1862. [Google Scholar] [CrossRef] [PubMed]
- Thompson, E.A.; Cascino, K.; Ordonez, A.A.; Zhou, W.; Vaghasia, A.; Hamacher-Brady, A.; Brady, N.R.; Sun, I.H.; Wang, R.; Rosenberg, A.Z.; et al. Metabolic programs define dysfunctional immune responses in severe COVID-19 patients. Cell Rep. 2021, 34, 108863. [Google Scholar] [CrossRef] [PubMed]
- Di Domizio, J.; Gulen, M.F.; Saidoune, F.; Thacker, V.V.; Yatim, A.; Sharma, K.; Nass, T.; Guenova, E.; Schaller, M.; Conrad, C.; et al. The cGAS-STING pathway drives type I IFN immunopathology in COVID-19. Nature 2022, 603, 145–151. [Google Scholar] [CrossRef]
- Gao, Z.Y.; Liu, Z.; Bi, M.H.; Zhang, J.J.; Han, Z.Q.; Han, X.; Wang, H.Y.; Sun, G.P.; Liu, H. Metformin induces apoptosis via a mitochondria-mediated pathway in human breast cancer cells in vitro. Exp. Ther. Med. 2016, 11, 1700–1706. [Google Scholar] [CrossRef]
- Arzoine, L.; Zilberberg, N.; Ben-Romano, R.; Shoshan-Barmatz, V. Voltage-dependent anion channel 1-based peptides interact with hexokinase to prevent its anti-apoptotic activity. J. Biol. Chem. 2009, 284, 3946–3955. [Google Scholar] [CrossRef]
- Shi, Y.; Chen, J.; Weng, C.; Chen, R.; Zheng, Y.; Chen, Q.; Tang, H. Identification of the protein-protein contact site and interaction mode of human VDAC1 with Bcl-2 family proteins. Biochem. Biophys. Res. Commun. 2003, 305, 989–996. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Mondal, S.; Samajdar, R.N.; Mukherjee, S.; Bhattacharyya, A.J.; Bagchi, B. Unique Features of Metformin: A Combined Experimental, Theoretical, and Simulation Study of Its Structure, Dynamics, and Interaction Energetics with DNA Grooves. J. Phys. Chem. B 2018, 122, 2227–2242. [Google Scholar] [CrossRef]
- Briones, R.; Weichbrodt, C.; Paltrinieri, L.; Mey, I.; Villinger, S.; Giller, K.; Lange, A.; Zweckstetter, M.; Griesinger, C.; Becker, S.; et al. Voltage Dependence of Conformational Dynamics and Subconducting States of VDAC-1. Biophys. J. 2016, 111, 1223–1234. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmuller, H.; MacKerell, A.D., Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Salani, B.; Marini, C.; Rio, A.D.; Ravera, S.; Massollo, M.; Orengo, A.M.; Amaro, A.; Passalacqua, M.; Maffioli, S.; Pfeffer, U.; et al. Metformin impairs glucose consumption and survival in Calu-1 cells by direct inhibition of hexokinase-II. Sci. Rep. 2013, 3, 2070. [Google Scholar] [CrossRef]
- Salani, B.; Del Rio, A.; Marini, C.; Sambuceti, G.; Cordera, R.; Maggi, D. Metformin, cancer and glucose metabolism. Endocr. Relat. Cancer 2014, 21, R461–R471. [Google Scholar] [CrossRef]
- Allam, R.; Lawlor, K.E.; Yu, E.C.; Mildenhall, A.L.; Moujalled, D.M.; Lewis, R.S.; Ke, F.; Mason, K.D.; White, M.J.; Stacey, K.J.; et al. Mitochondrial apoptosis is dispensable for NLRP3 inflammasome activation but non-apoptotic caspase-8 is required for inflammasome priming. EMBO Rep. 2014, 15, 982–990. [Google Scholar] [CrossRef]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef]
- Vajjhala, P.R.; Mirams, R.E.; Hill, J.M. Multiple binding sites on the pyrin domain of ASC protein allow self-association and interaction with NLRP3 protein. J. Biol. Chem. 2012, 287, 41732–41743. [Google Scholar] [CrossRef]
- Pollak, M. Potential applications for biguanides in oncology. J. Clin. Investig. 2013, 123, 3693–3700. [Google Scholar] [CrossRef]
- Lord, S.R.; Harris, A.L. Is it still worth pursuing the repurposing of metformin as a cancer therapeutic? Br. J. Cancer 2023, 128, 958–966. [Google Scholar] [CrossRef]
- Romero, R.; Erez, O.; Hüttemann, M.; Maymon, E.; Panaitescu, B.; Conde-Agudelo, A.; Pacora, P.; Yoon, B.H.; Grossman, L.I. Metformin, the aspirin of the 21st century: Its role in gestational diabetes mellitus, prevention of preeclampsia and cancer, and the promotion of longevity. Am. J. Obstet. Gynecol. 2017, 217, 282–302. [Google Scholar] [CrossRef]
- Anisimov, V.N. Metformin for cancer and aging prevention: Is it a time to make the long story short? Oncotarget 2015, 6, 39398. [Google Scholar] [CrossRef]
- Triggle, C.R.; Mohammed, I.; Bshesh, K.; Marei, I.; Ye, K.; Ding, H.; MacDonald, R.; Hollenberg, M.D.; Hill, M.A. Metformin: Is it a drug for all reasons and diseases? Metabolism 2022, 133, 155223. [Google Scholar] [CrossRef] [PubMed]
- Malki, A.; Youssef, A. Antidiabetic drug metformin induces apoptosis in human MCF breast cancer via targeting ERK signaling. Oncol. Res. 2011, 19, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hu, M.; Meng, F.; Sun, X.; Xu, H.; Zhang, J.; Cui, P.; Morina, N.; Li, X.; Li, W.; et al. Metformin Ameliorates Uterine Defects in a Rat Model of Polycystic Ovary Syndrome. eBioMedicine 2017, 18, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Ko, M.; Kim, J.; Lazim, R.; Lee, J.Y.; Kim, J.Y.; Gosu, V.; Lee, Y.; Choi, S.; Kwon, H.J. The anticancer effect of metformin targets VDAC1 via ER-mitochondria interactions-mediated autophagy in HCC. Exp. Mol. Med. 2024, 56, 2714–2725. [Google Scholar] [CrossRef]
- Zhang, J.; Guo, Y.; Ge, W.; Zhou, X.; Pan, M. High glucose induces the apoptosis of HUVECs in mitochondria dependent manner by enhancing VDAC1 expression. Pharmazie 2018, 73, 725–728. [Google Scholar] [CrossRef]
- Mathupala, S.P.; Ko, Y.H.; Pedersen, P.L. Hexokinase II: Cancer’s double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene 2006, 25, 4777–4786. [Google Scholar] [CrossRef]
- Bryson, J.M.; Coy, P.E.; Gottlob, K.; Hay, N.; Robey, R.B. Increased hexokinase activity, of either ectopic or endogenous origin, protects renal epithelial cells against acute oxidant-induced cell death. J. Biol. Chem. 2002, 277, 11392–11400. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kepp, O.; Tajeddine, N.; Kroemer, G. Disruption of the hexokinase–VDAC complex for tumor therapy. Oncogene 2008, 27, 4633–4635. [Google Scholar] [CrossRef]
- Gelb, B.D.; Adams, V.; Jones, S.N.; Griffin, L.D.; MacGregor, G.R.; McCabe, E.R. Targeting of hexokinase 1 to liver and hepatoma mitochondria. Proc. Natl. Acad. Sci. USA 1992, 89, 202–206. [Google Scholar] [CrossRef]
- Goldin, N.; Arzoine, L.; Heyfets, A.; Israelson, A.; Zaslavsky, Z.; Bravman, T.; Bronner, V.; Notcovich, A.; Shoshan-Barmatz, V.; Flescher, E. Methyl jasmonate binds to and detaches mitochondria-bound hexokinase. Oncogene 2008, 27, 4636–4643. [Google Scholar] [CrossRef]
- Marini, C.; Salani, B.; Massollo, M.; Amaro, A.; Esposito, A.I.; Maria Orengo, A.; Capitanio, S.; Emionite, L.; Riondato, M.; Bottoni, G. Direct inhibition of hexokinase activity by metformin at least partially impairs glucose metabolism and tumor growth in experimental breast cancer. Cell Cycle 2013, 12, 3490–3499. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Guilbaud, E.; Tait, S.W.G.; Yamazaki, T.; Galluzzi, L. Mitochondrial control of inflammation. Nat. Rev. Immunol. 2022, 23, 159–173. [Google Scholar] [CrossRef] [PubMed]
- Elliott, E.I.; Miller, A.N.; Banoth, B.; Iyer, S.S.; Stotland, A.; Weiss, J.P.; Gottlieb, R.A.; Sutterwala, F.S.; Cassel, S.L. Cutting Edge: Mitochondrial Assembly of the NLRP3 Inflammasome Complex Is Initiated at Priming. J. Immunol. 2018, 200, 3047–3052. [Google Scholar] [CrossRef]
- Mills, E.L.; Kelly, B.; O’Neill, L.A.J. Mitochondria are the powerhouses of immunity. Nat. Immunol. 2017, 18, 488–498. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, D.; Hu, D.; Zhou, X.; Zhou, Y. The role of mitochondria in NLRP3 inflammasome activation. Mol. Immunol. 2018, 103, 115–124. [Google Scholar] [CrossRef]
- Xian, H.; Watari, K.; Sanchez-Lopez, E.; Offenberger, J.; Onyuru, J.; Sampath, H.; Ying, W.; Hoffman, H.M.; Shadel, G.S.; Karin, M. Oxidized DNA fragments exit mitochondria via mPTP- and VDAC-dependent channels to activate NLRP3 inflammasome and interferon signaling. Immunity 2022, 55, 1370–1385.e1378. [Google Scholar] [CrossRef]
- Hu, H.; Guo, L.; Overholser, J.; Wang, X. Mitochondrial VDAC1: A Potential Therapeutic Target of Inflammation-Related Diseases and Clinical Opportunities. Cells 2022, 11, 3174. [Google Scholar] [CrossRef]
- Xie, J.; Cui, Y.; Chen, X.; Yu, H.; Chen, J.; Huang, T.; He, M.; Zhang, L.; Meng, Q. VDAC1 regulates mitophagy in NLRP3 inflammasome activation in retinal capillary endothelial cells under high-glucose conditions. Exp. Eye Res. 2021, 209, 108640. [Google Scholar] [CrossRef]
- Lu, A.; Magupalli, V.G.; Ruan, J.; Yin, Q.; Atianand, M.K.; Vos, M.R.; Schroder, G.F.; Fitzgerald, K.A.; Wu, H.; Egelman, E.H. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 2014, 156, 1193–1206. [Google Scholar] [CrossRef]
- Hochheiser, I.V.; Behrmann, H.; Hagelueken, G.; Rodriguez-Alcazar, J.F.; Kopp, A.; Latz, E.; Behrmann, E.; Geyer, M. Directionality of PYD filament growth determined by the transition of NLRP3 nucleation seeds to ASC elongation. Sci. Adv. 2022, 8, eabn7583. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Wu, H. Structural Mechanisms of NLRP3 Inflammasome Assembly and Activation. Annu. Rev. Immunol. 2023, 41, 301–316. [Google Scholar] [CrossRef]
- Andrzejewski, S.; Siegel, P.M.; St-Pierre, J. Metabolic Profiles Associated With Metformin Efficacy in Cancer. Front. Endocrinol. 2018, 9, 372. [Google Scholar] [CrossRef]
- Biondani, G.; Peyron, J.F. Metformin, an Anti-Diabetic Drug to Target Leukemia. Front. Endocrinol. 2018, 9, 446. [Google Scholar] [CrossRef]
- Chen, K.; Qian, W.; Jiang, Z.; Cheng, L.; Li, J.; Sun, L.; Zhou, C.; Gao, L.; Lei, M.; Yan, B.; et al. Metformin suppresses cancer initiation and progression in genetic mouse models of pancreatic cancer. Mol. Cancer 2017, 16, 131. [Google Scholar] [CrossRef]
- Xie, J.; Xia, L.; Xiang, W.; He, W.; Yin, H.; Wang, F.; Gao, T.; Qi, W.; Yang, Z.; Yang, X.; et al. Metformin selectively inhibits metastatic colorectal cancer with the KRAS mutation by intracellular accumulation through silencing MATE1. Proc. Natl. Acad. Sci. USA 2020, 117, 13012–13022. [Google Scholar] [CrossRef]
- Ben Sahra, I.; Laurent, K.; Giuliano, S.; Larbret, F.; Ponzio, G.; Gounon, P.; Le Marchand-Brustel, Y.; Giorgetti-Peraldi, S.; Cormont, M.; Bertolotto, C.; et al. Targeting cancer cell metabolism: The combination of metformin and 2-deoxyglucose induces p53-dependent apoptosis in prostate cancer cells. Cancer Res. 2010, 70, 2465–2475. [Google Scholar] [CrossRef]
- Zhang, W.B.; Wang, Z.; Shu, F.; Jin, Y.H.; Liu, H.Y.; Wang, Q.J.; Yang, Y. Activation of AMP-activated protein kinase by temozolomide contributes to apoptosis in glioblastoma cells via p53 activation and mTORC1 inhibition. J. Biol. Chem. 2010, 285, 40461–40471. [Google Scholar] [CrossRef]
- Kourelis, T.V.; Siegel, R.D. Metformin and cancer: New applications for an old drug. Med. Oncol. 2012, 29, 1314–1327. [Google Scholar] [CrossRef]
- Cossor, F.I.; Adams-Campbell, L.L.; Chlebowski, R.T.; Gunter, M.J.; Johnson, K.; Martell, R.E.; McTiernan, A.; Simon, M.S.; Rohan, T.; Wallace, R.B.; et al. Diabetes, metformin use, and colorectal cancer survival in women: A retrospective cohort study. J. Clin. Oncol. 2012, 30, e14005. [Google Scholar] [CrossRef]
- Mc Menamin, U.C.; Murray, L.J.; Hughes, C.M.; Cardwell, C.R. Metformin use and survival after colorectal cancer: A population-based cohort study. Int. J. Cancer 2016, 138, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Shackelford, D.B.; Abt, E.; Gerken, L.; Vasquez, D.S.; Seki, A.; Leblanc, M.; Wei, L.; Fishbein, M.C.; Czernin, J.; Mischel, P.S.; et al. LKB1 Inactivation Dictates Therapeutic Response of Non-Small Cell Lung Cancer to the Metabolism Drug Phenformin. Cancer Cell 2013, 23, 143–158. [Google Scholar] [CrossRef] [PubMed]
- Bayat, S.; Shekari Khaniani, M.; Choupani, J.; Alivand, M.R.; Mansoori Derakhshan, S. HDACis (class I), cancer stem cell, and phytochemicals: Cancer therapy and prevention implications. Biomed. Pharmacother. 2018, 97, 1445–1453. [Google Scholar] [CrossRef]
- Kilgore, J.; Jackson, A.L.; Clark, L.H.; Guo, H.; Zhang, L.; Jones, H.M.; Gilliam, T.P.; Gehrig, P.A.; Zhou, C.; Bae-Jump, V.L. Buformin exhibits anti-proliferative and anti-invasive effects in endometrial cancer cells. Am. J. Transl. Res. 2016, 8, 2705–2715. [Google Scholar]
- Yakisich, J.S.; Azad, N.; Kaushik, V.; Iyer, A.K.V. The Biguanides Metformin and Buformin in Combination with 2-Deoxy-glucose or WZB-117 Inhibit the Viability of Highly Resistant Human Lung Cancer Cells. Stem Cells Int. 2019, 2019, 6254269. [Google Scholar] [CrossRef]
- Liu, J.; Li, J.; Chen, H.; Wang, R.; Li, P.; Miao, Y.; Liu, P. Metformin suppresses proliferation and invasion of drug-resistant breast cancer cells by activation of the Hippo pathway. J. Cell. Mol. Med. 2020, 24, 5786–5796. [Google Scholar] [CrossRef]
- Wienken, C.J.; Baaske, P.; Rothbauer, U.; Braun, D.; Duhr, S. Protein-binding assays in biological liquids using microscale thermophoresis. Nat. Commun. 2010, 1, 100. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Source and Catalog Number | Dilution | |
|---|---|---|---|
| WB | IF | ||
| Mouse monoclonal anti-β-Actin | Millipore, Billerica, MA, USA, MAB1501 | 1:40,000 | - |
| Goat anti-rabbit—HRP | Promega, Madison, WI, USA, W4011 | 1:15,000 | - |
| Goat anti-rabbit—Alexa Fluor 555 | Abcam, Cambridge, UK, ab150078 | - | 1:750 |
| Goat anti-mouse H and L—Alexa Fluor 488 | Abcam, Cambridge, UK, ab150114 | - | 1:750 |
| Rabbit polyclonal anti-VDAC1 | Abcam, Cambridge, UK, ab15895 | 1:5000 | 1:500 |
| Mouse monoclonal anti-ASC | Santa Cruz Biotechnology, Santa Cruz, TX, USA, sc-514414 | 1:500 | |
| Rabbit polyclonal anti-NLRP3 | Novus Biologicals, Centennial, CO, USA, NBP1-77080 | 1:500 | |
| Rabbit polyclonal anti cleaved caspase-3 | Cell signaling, Danvers, MS, USA, 9661S | 1:400 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shteinfer-Kuzmine, A.; Moyal, M.M.; Karunanithi Nivedita, A.; Trishna, S.; Nadir, A.; Tripathi, S.; Shoshan-Barmatz, V. Metformin-Induced Apoptosis Is Mediated Through Mitochondrial VDAC1. Pharmaceuticals 2025, 18, 757. https://doi.org/10.3390/ph18050757
Shteinfer-Kuzmine A, Moyal MM, Karunanithi Nivedita A, Trishna S, Nadir A, Tripathi S, Shoshan-Barmatz V. Metformin-Induced Apoptosis Is Mediated Through Mitochondrial VDAC1. Pharmaceuticals. 2025; 18(5):757. https://doi.org/10.3390/ph18050757
Chicago/Turabian StyleShteinfer-Kuzmine, Anna, Meital M. Moyal, Aditya Karunanithi Nivedita, Sweta Trishna, Almog Nadir, Shubhandra Tripathi, and Varda Shoshan-Barmatz. 2025. "Metformin-Induced Apoptosis Is Mediated Through Mitochondrial VDAC1" Pharmaceuticals 18, no. 5: 757. https://doi.org/10.3390/ph18050757
APA StyleShteinfer-Kuzmine, A., Moyal, M. M., Karunanithi Nivedita, A., Trishna, S., Nadir, A., Tripathi, S., & Shoshan-Barmatz, V. (2025). Metformin-Induced Apoptosis Is Mediated Through Mitochondrial VDAC1. Pharmaceuticals, 18(5), 757. https://doi.org/10.3390/ph18050757