
Machine Learning-Driven Consensus Modeling for Activity Ranking and Chemical Landscape Analysis of HIV-1 Inhibitors

 , ,
, ,  ,
, 
Abstract
1. Introduction
2. Results
2.1. Selection of Molecular Descriptors and Chemical Space Analysis
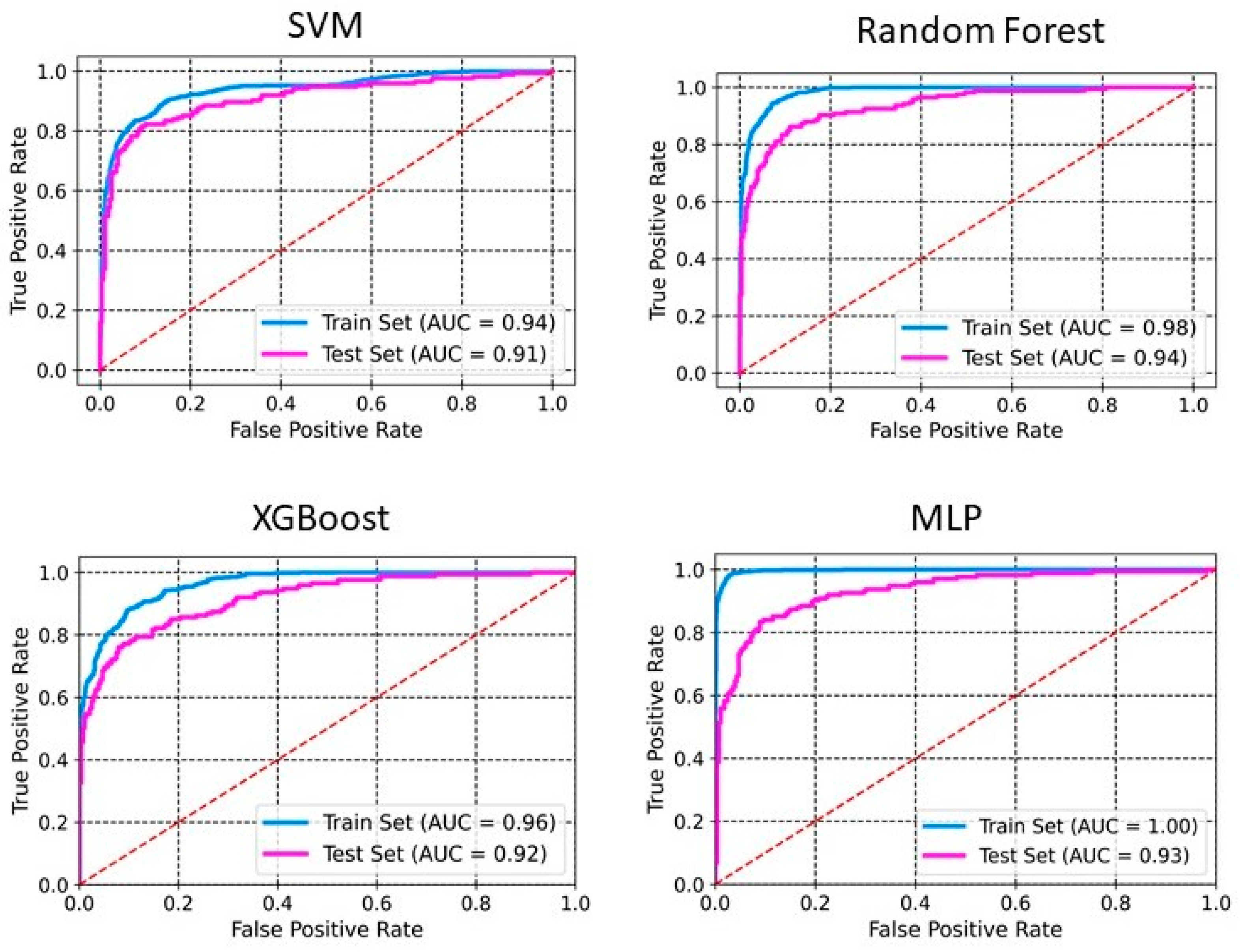
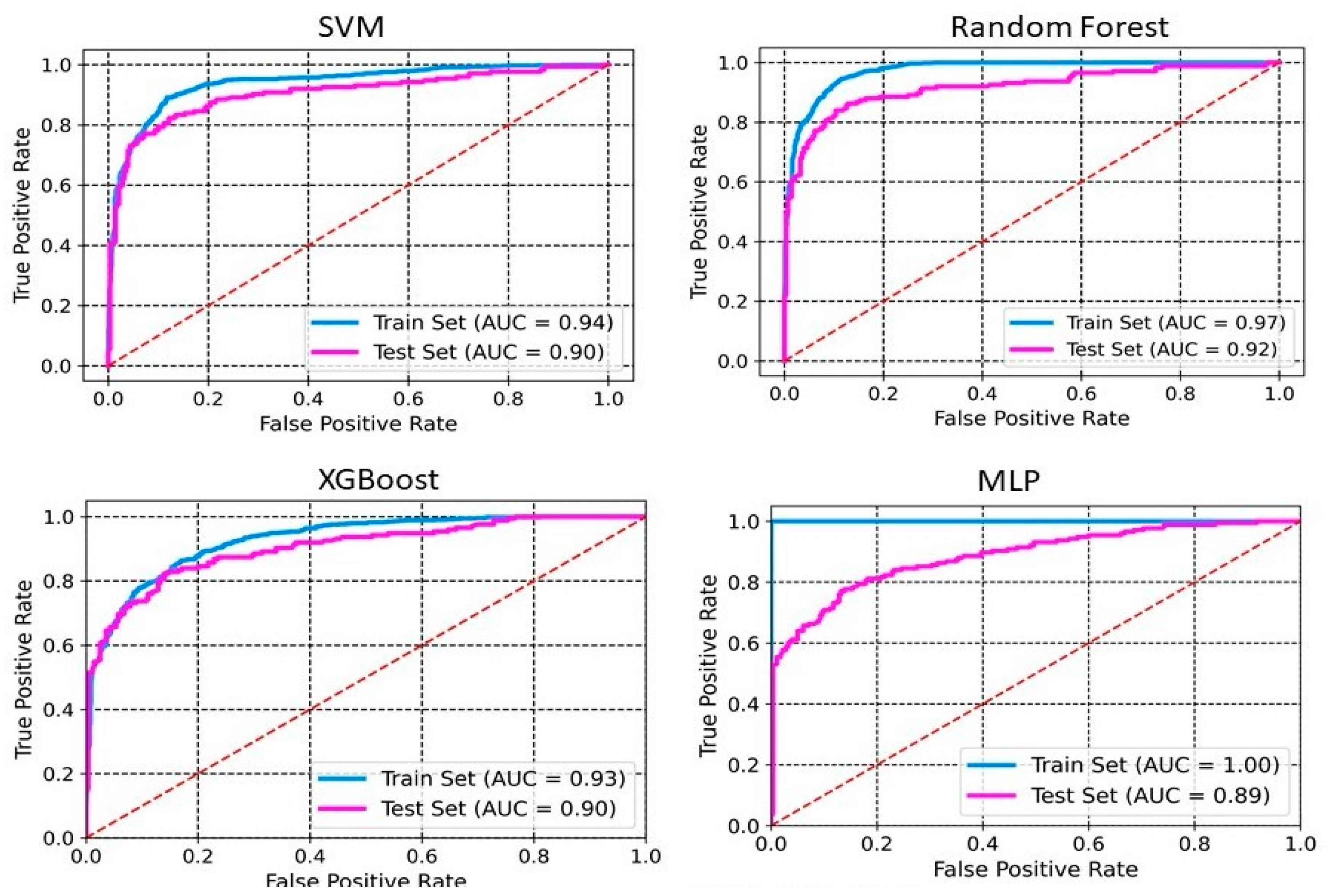
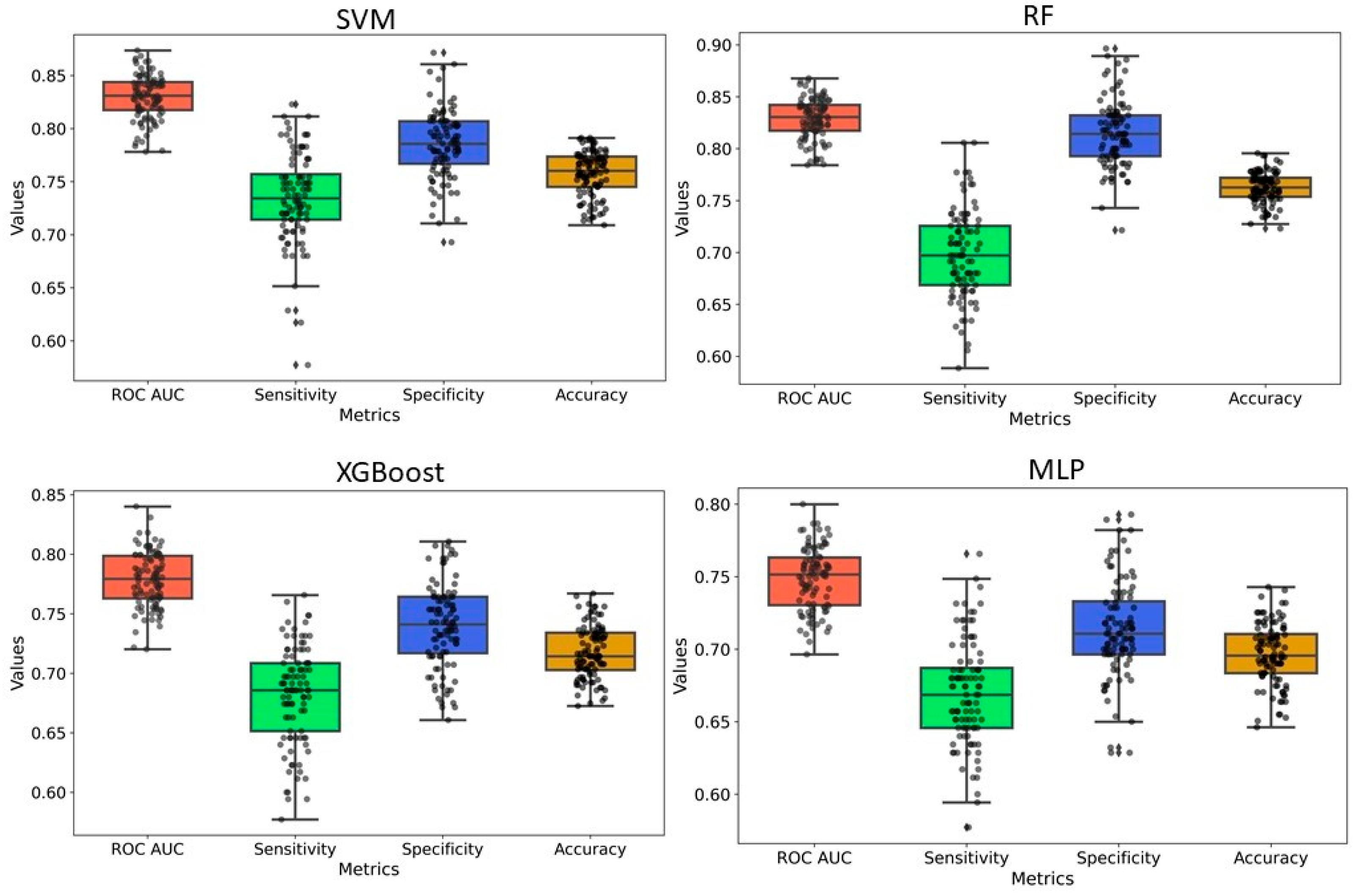
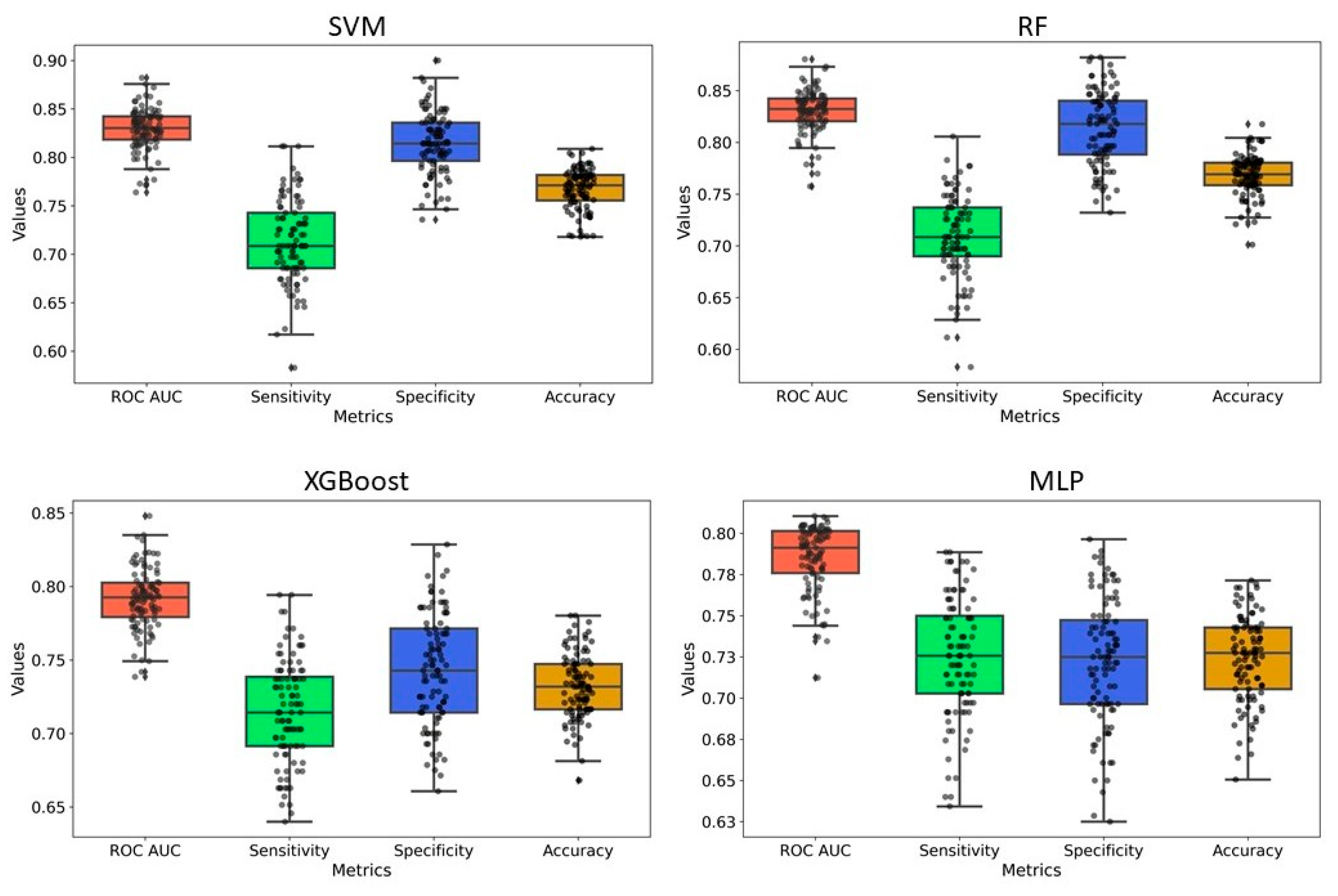
2.2. Model Development and Evaluation
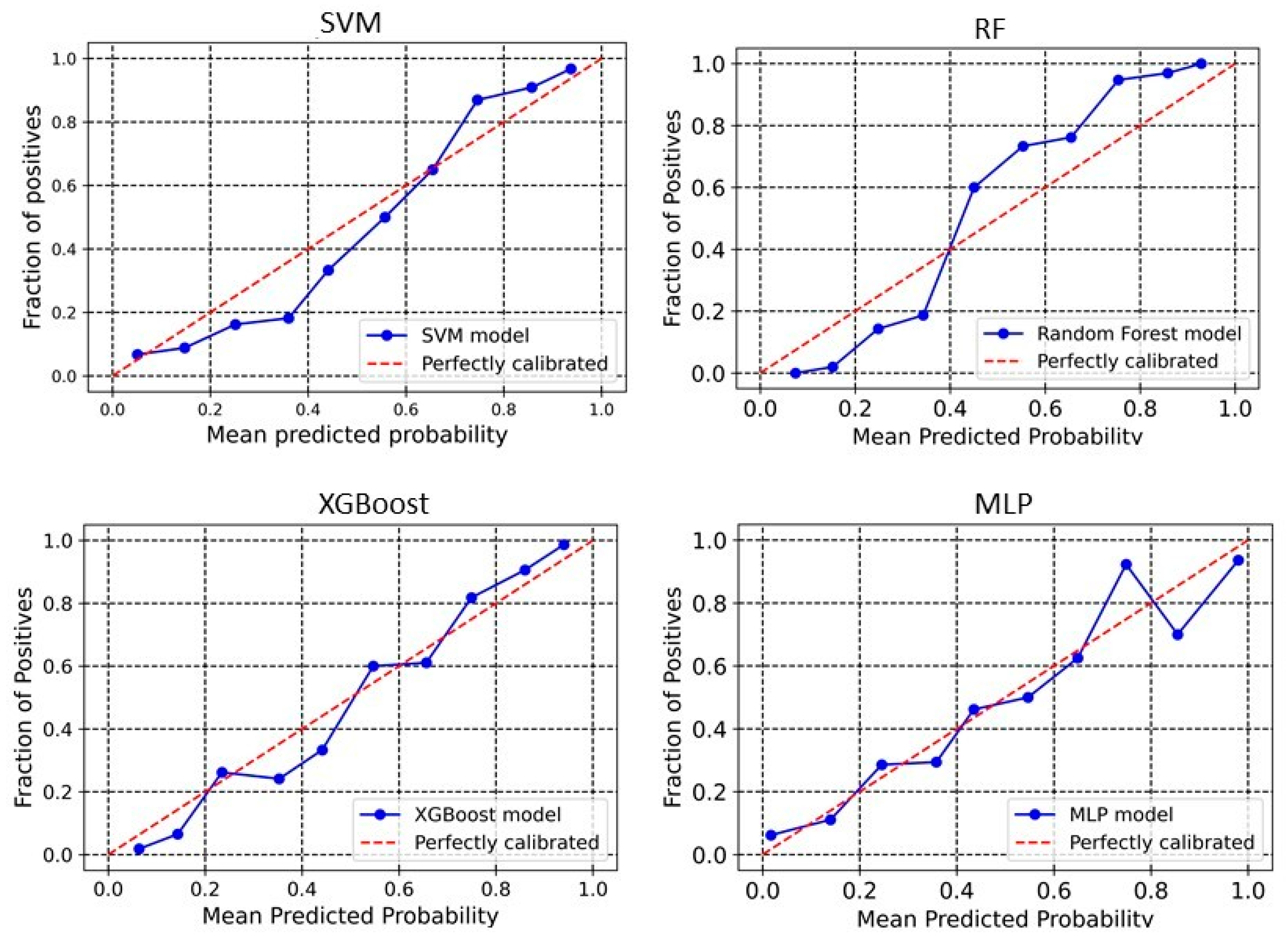
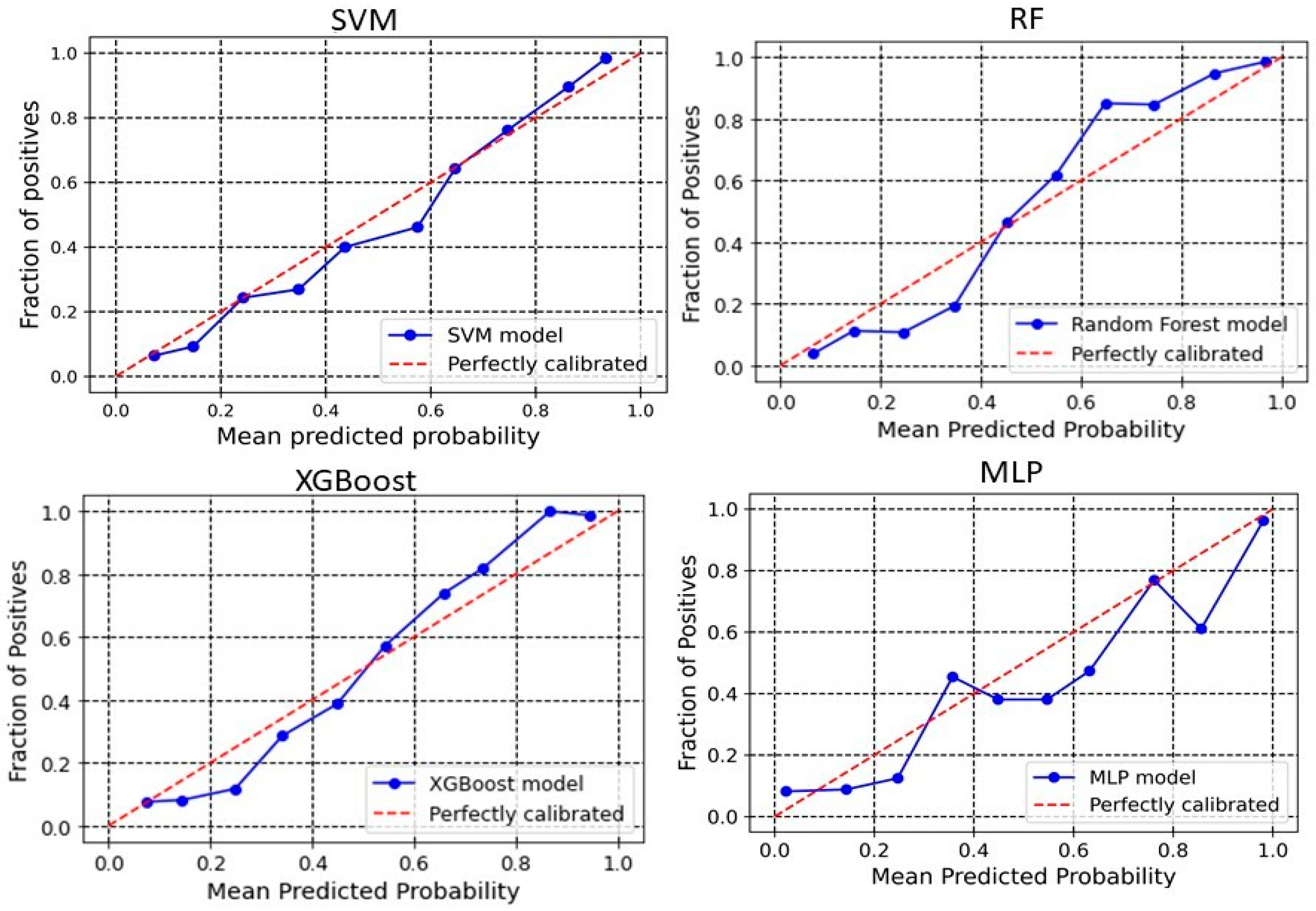
2.3. Calibration Plot
2.4. Y-Randomization
2.5. Consensus Model
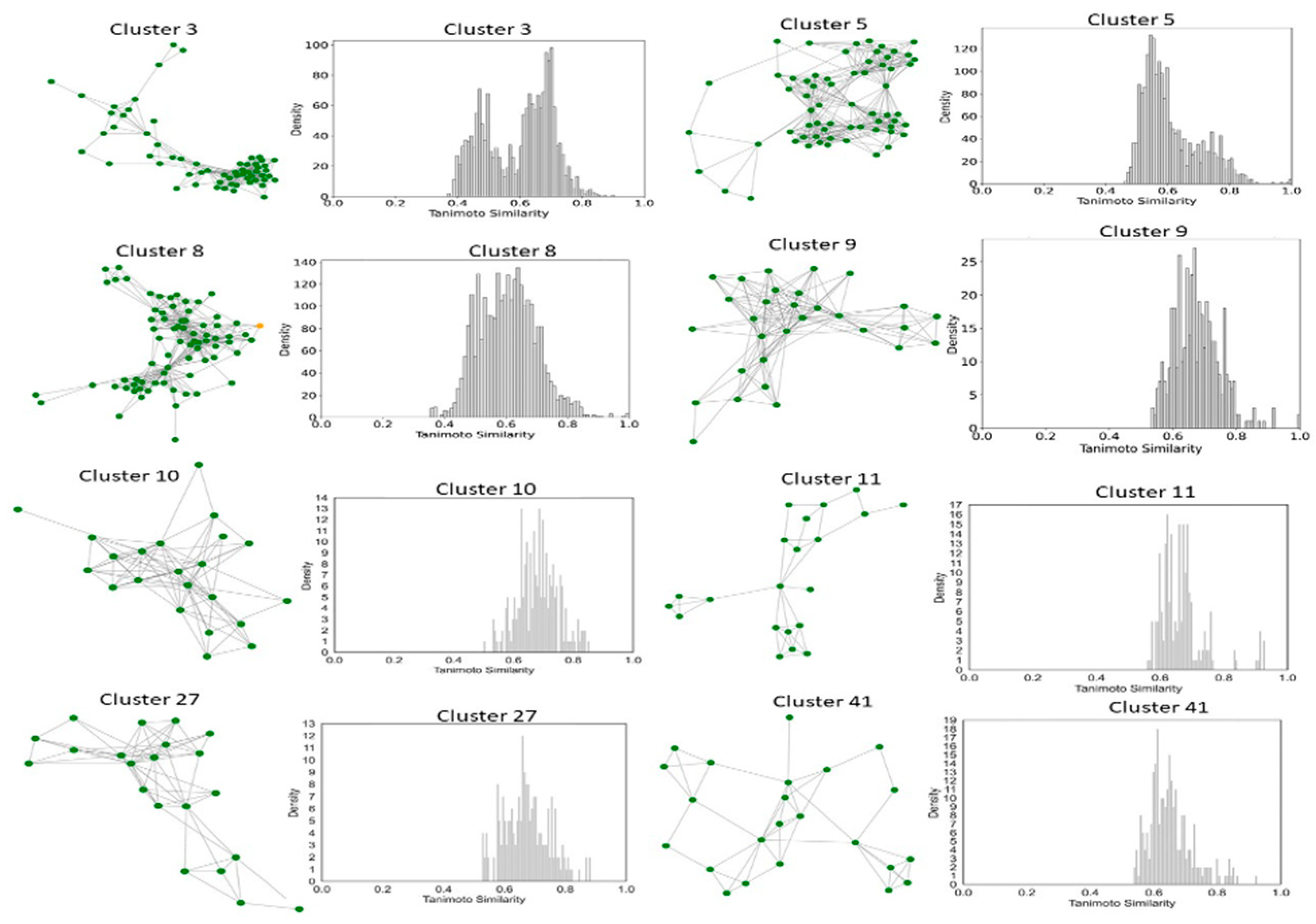
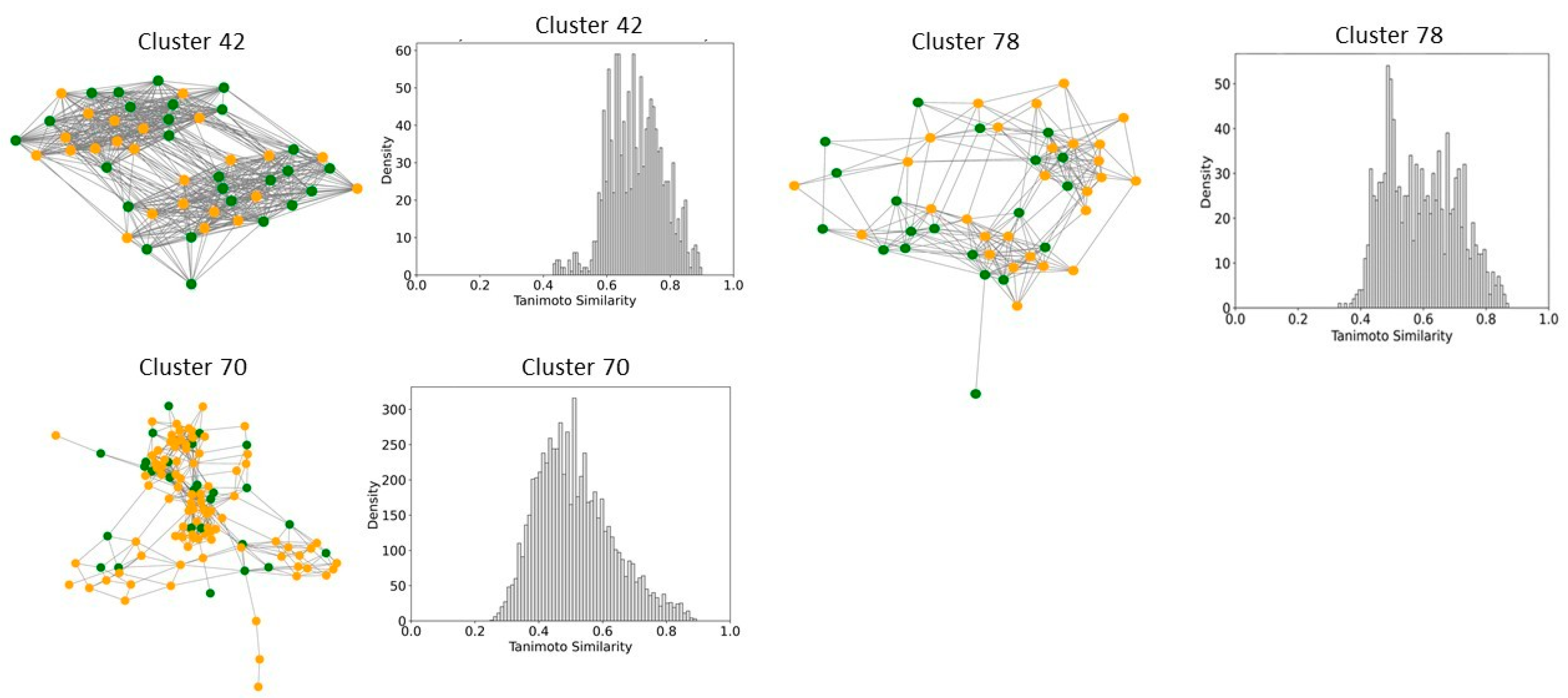
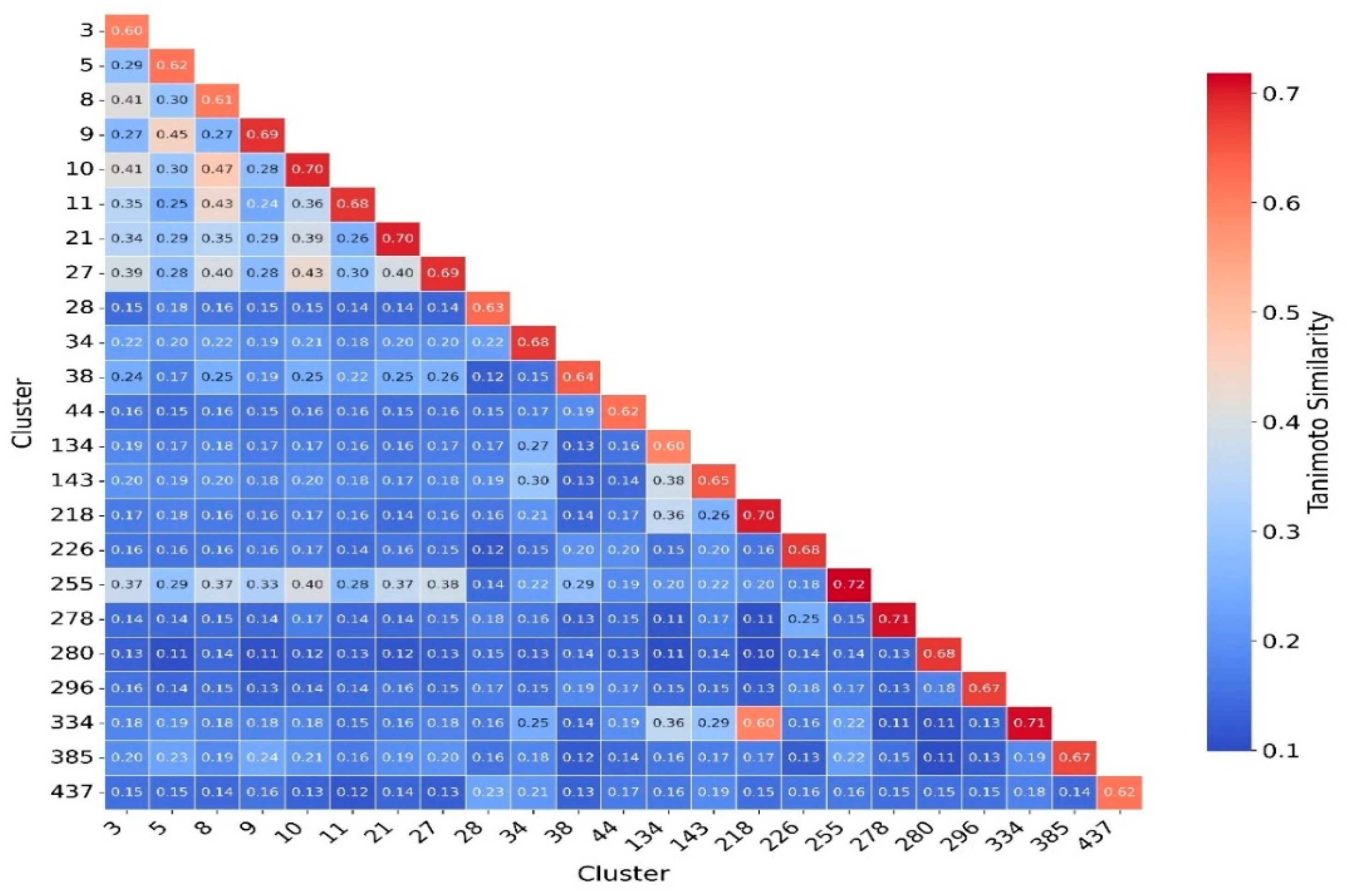
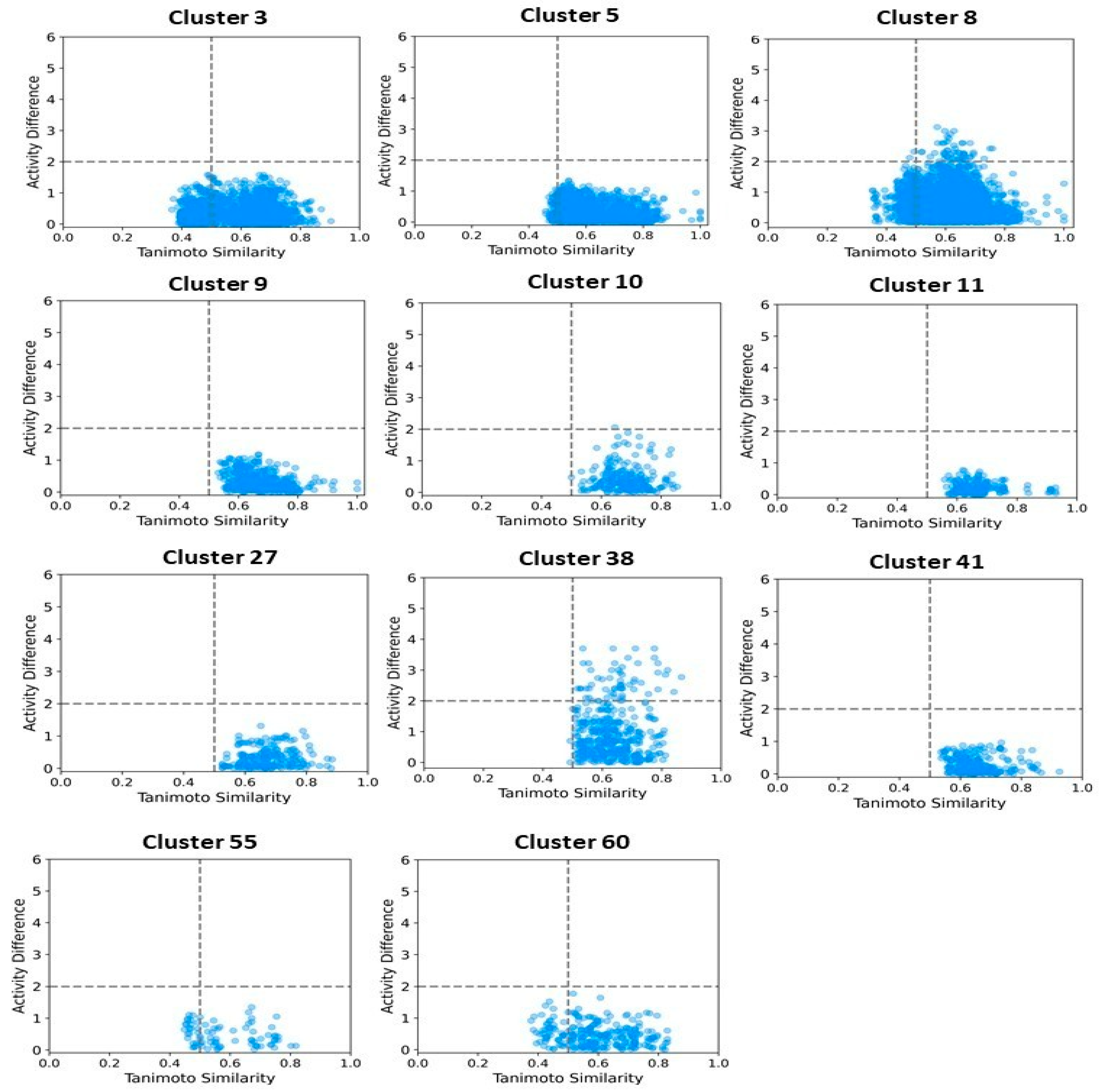
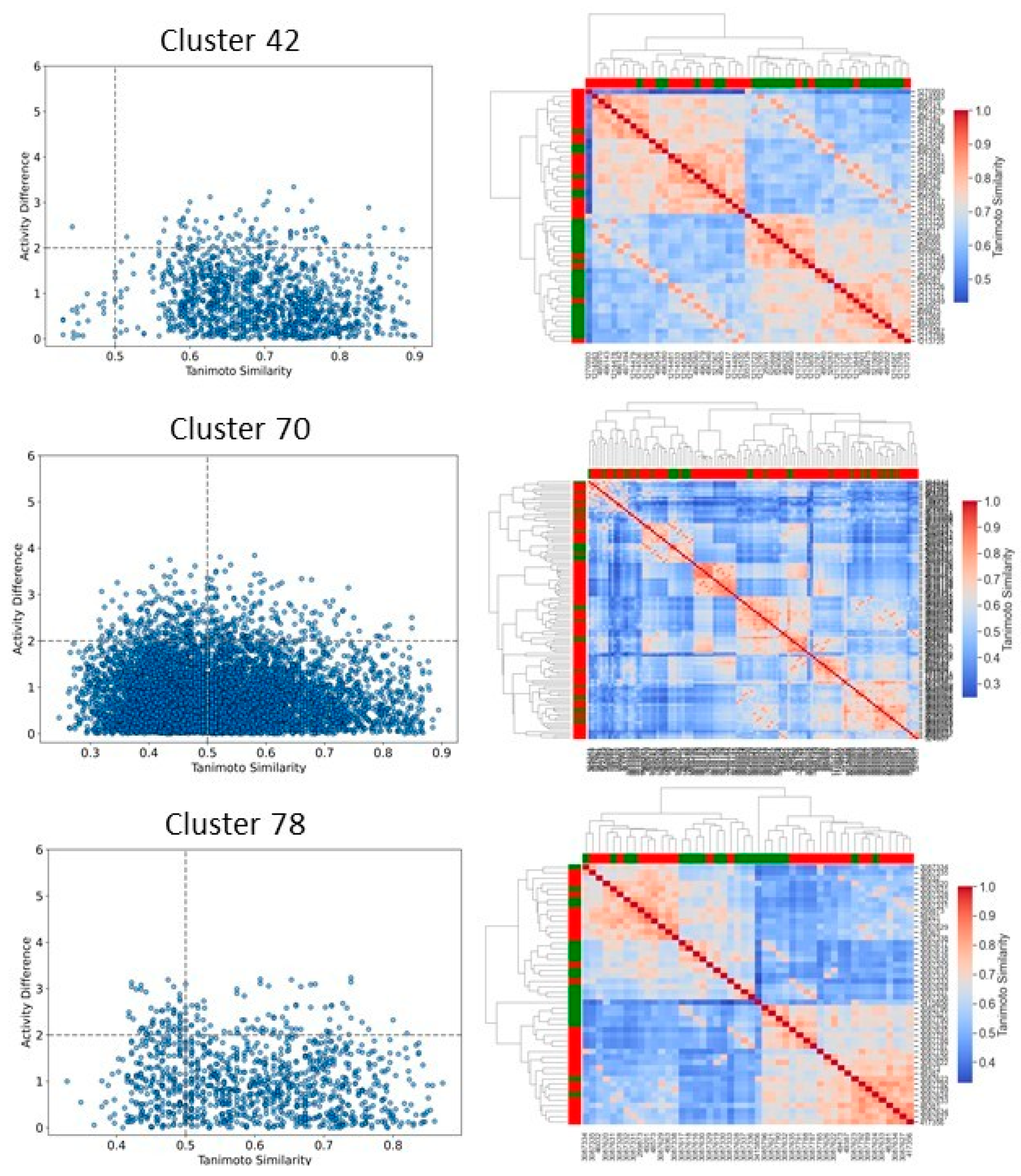
2.6. Clustering, Scaffold Analysis, and Activity Landscape Assessment
3. Discussion
4. Materials and Methods
4.1. Data Preparation
4.2. Molecular Descriptors and Selections
4.3. Model Building
4.4. Robustness of Models
4.5. Model Evaluation
4.6. Consensus Modeling
4.7. Clustering Analyses
4.8. Scaffold and Structural Activity Variations
4.9. Compound Similarity
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gayle, H.D.; Hill, G.L. Global impact of human immunodeficiency virus and AIDS. Clin. Microbiol. Rev. 2001, 14, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Deeks, S.G.; Overbaugh, J.; Phillips, A.; Buchbinder, S. HIV infection. Nat. Rev. Dis. Primers 2015, 1, 15035. [Google Scholar] [CrossRef]
- Gunthard, H.F.; Saag, M.S.; Benson, C.A.; del Rio, C.; Eron, J.J.; Gallant, J.E.; Hoy, J.F.; Mugavero, M.J.; Sax, P.E.; Thompson, M.A.; et al. Antiretroviral Drugs for Treatment and Prevention of HIV Infection in Adults: 2016 Recommendations of the International Antiviral Society-USA Panel. JAMA 2016, 316, 191–210. [Google Scholar] [CrossRef] [PubMed]
- Menendez-Arias, L.; Delgado, R. Update and latest advances in antiretroviral therapy. Trends Pharmacol. Sci. 2022, 43, 16–29. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.L.; Kutluay, S.B. Going beyond Integration: The Emerging Role of HIV-1 Integrase in Virion Morphogenesis. Viruses 2020, 12, 1005. [Google Scholar] [CrossRef]
- Zheng, Y.; Yao, X. Posttranslational modifications of HIV-1 integrase by various cellular proteins during viral replication. Viruses 2013, 5, 1787–1801. [Google Scholar] [CrossRef]
- Delelis, O.; Carayon, K.; Saib, A.; Deprez, E.; Mouscadet, J.F. Integrase and integration: Biochemical activities of HIV-1 integrase. Retrovirology 2008, 5, 114. [Google Scholar] [CrossRef]
- Di Santo, R. Inhibiting the HIV integration process: Past, present, and the future. J. Med. Chem. 2014, 57, 539–566. [Google Scholar] [CrossRef]
- Sayyed, S.K.; Quraishi, M.; Prabakaran, D.S.; Chandrasekaran, B.; Ramesh, T.; Rajasekharan, S.K.; Raorane, C.J.; Sonawane, T.; Ravichandran, V. Exploring Zinc C295 as a Dual HIV-1 Integrase Inhibitor: From Strand Transfer to 3′-Processing Suppression. Pharmaceuticals 2024, 18, 30. [Google Scholar] [CrossRef]
- Arts, E.J.; Hazuda, D.J. HIV-1 antiretroviral drug therapy. Cold Spring Harb. Perspect. Med. 2012, 2, a007161. [Google Scholar] [CrossRef]
- Himmel, D.M.; Arnold, E. Non-Nucleoside Reverse Transcriptase Inhibitors Join Forces with Integrase Inhibitors to Combat HIV. Pharmaceuticals 2020, 13, 122. [Google Scholar] [CrossRef]
- Wang, Y.; Gu, S.X.; He, Q.; Fan, R. Advances in the development of HIV integrase strand transfer inhibitors. Eur. J. Med. Chem. 2021, 225, 113787. [Google Scholar] [CrossRef] [PubMed]
- Scarsi, K.K.; Havens, J.P.; Podany, A.T.; Avedissian, S.N.; Fletcher, C.V. HIV-1 Integrase Inhibitors: A Comparative Review of Efficacy and Safety. Drugs 2020, 80, 1649–1676. [Google Scholar] [CrossRef]
- Trivedi, J.; Mahajan, D.; Jaffe, R.J.; Acharya, A.; Mitra, D.; Byrareddy, S.N. Recent Advances in the Development of Integrase Inhibitors for HIV Treatment. Curr. HIV/AIDS Rep. 2020, 17, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Zhao, A.V.; Crutchley, R.D.; Guduru, R.C.; Ton, K.; Lam, T.; Min, A.C. A clinical review of HIV integrase strand transfer inhibitors (INSTIs) for the prevention and treatment of HIV-1 infection. Retrovirology 2022, 19, 22. [Google Scholar] [CrossRef] [PubMed]
- Boomgarden, A.C.; Upadhyay, C. Progress and Challenges in HIV-1 Vaccine Research: A Comprehensive Overview. Vaccines 2025, 13, 148. [Google Scholar] [CrossRef]
- Carracedo-Reboredo, P.; Linares-Blanco, J.; Rodriguez-Fernandez, N.; Cedron, F.; Novoa, F.J.; Carballal, A.; Maojo, V.; Pazos, A.; Fernandez-Lozano, C. A review on machine learning approaches and trends in drug discovery. Comput. Struct. Biotechnol. J. 2021, 19, 4538–4558. [Google Scholar] [CrossRef]
- Dara, S.; Dhamercherla, S.; Jadav, S.S.; Babu, C.M.; Ahsan, M.J. Machine Learning in Drug Discovery: A Review. Artif. Intell. Rev. 2022, 55, 1947–1999. [Google Scholar] [CrossRef]
- Hashemi, S.; Vosough, P.; Taghizadeh, S.; Savardashtaki, A. Therapeutic peptide development revolutionized: Harnessing the power of artificial intelligence for drug discovery. Heliyon 2024, 10, e40265. [Google Scholar] [CrossRef]
- Soares, T.A.; Nunes-Alves, A.; Mazzolari, A.; Ruggiu, F.; Wei, G.W.; Merz, K. The (Re)-Evolution of Quantitative Structure-Activity Relationship (QSAR) Studies Propelled by the Surge of Machine Learning Methods. J. Chem. Inf. Model. 2022, 62, 5317–5320. [Google Scholar] [CrossRef]
- Das, R.N.; Roy, K. QSPR with extended topochemical atom (ETA) indices. 4. Modeling aqueous solubility of drug like molecules and agrochemicals following OECD guidelines. Struct. Chem. 2012, 24, 303–331. [Google Scholar] [CrossRef]
- Roy, K.; Kabir, H. QSPR with extended topochemical atom (ETA) indices: Modeling of critical micelle concentration of non-ionic surfactants. Chem. Eng. Sci. 2012, 73, 86–98. [Google Scholar] [CrossRef]
- Roy, K.; Ambure, P.; Kar, S. How Precise Are Our Quantitative Structure-Activity Relationship Derived Predictions for New Query Chemicals? ACS Omega 2018, 3, 11392–11406. [Google Scholar] [CrossRef]
- Klingspohn, W.; Mathea, M.; Ter Laak, A.; Heinrich, N.; Baumann, K. Efficiency of different measures for defining the applicability domain of classification models. J. Cheminform. 2017, 9, 44. [Google Scholar] [CrossRef] [PubMed]
- Rucker, C.; Rucker, G.; Meringer, M. y-Randomization and its variants in QSPR/QSAR. J. Chem. Inf. Model. 2007, 47, 2345–2357. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, X. QSPR model for Caco-2 cell permeability prediction using a combination of HQPSO and dual-RBF neural network. RSC Adv. 2020, 10, 42938–42952. [Google Scholar] [CrossRef]
- Lemay, A.; Hoebel, K.; Bridge, C.P.; Befano, B.; De Sanjose, S.; Egemen, D.; Rodriguez, A.C.; Schiffman, M.; Campbell, J.P.; Kalpathy-Cramer, J. Improving the repeatability of deep learning models with Monte Carlo dropout. NPJ Digit. Med. 2022, 5, 174. [Google Scholar] [CrossRef]
- Pate, A.; Sperrin, M.; Riley, R.D.; Peek, N.; Van Staa, T.; Sergeant, J.C.; Mamas, M.A.; Lip, G.Y.H.; O’Flaherty, M.; Barrowman, M.; et al. Calibration plots for multistate risk predictions models. Stat. Med. 2024, 43, 2830–2852. [Google Scholar] [CrossRef]
- Roth, J.P.; Bajorath, J. Relationship between prediction accuracy and uncertainty in compound potency prediction using deep neural networks and control models. Sci. Rep. 2024, 14, 6536. [Google Scholar] [CrossRef]
- Rokach, L. Ensemble-based classifiers. Artif. Intell. Rev. 2010, 33, 1–39. [Google Scholar] [CrossRef]
- Fuadah, Y.N.; Pramudito, M.A.; Firdaus, L.; Vanheusden, F.J.; Lim, K.M. QSAR Classification Modeling Using Machine Learning with a Consensus-Based Approach for Multivariate Chemical Hazard End Points. ACS Omega 2024, 9, 50796–50808. [Google Scholar] [CrossRef] [PubMed]
- Valsecchi, C.; Grisoni, F.; Consonni, V.; Ballabio, D. Consensus versus Individual QSARs in Classification: Comparison on a Large-Scale Case Study. J. Chem. Inf. Model. 2020, 60, 1215–1223. [Google Scholar] [CrossRef]
- Hu, Y.; Bajorath, J. Target family-directed exploration of scaffolds with different SAR profiles. J. Chem. Inf. Model. 2011, 51, 3138–3148. [Google Scholar] [CrossRef]
- Hu, Y.; Stumpfe, D.; Bajorath, J. Computational Exploration of Molecular Scaffolds in Medicinal Chemistry. J. Med. Chem. 2016, 59, 4062–4076. [Google Scholar] [CrossRef]
- Velkoborsky, J.; Hoksza, D. Scaffold analysis of PubChem database as background for hierarchical scaffold-based visualization. J. Cheminform. 2016, 8, 74. [Google Scholar] [CrossRef] [PubMed]
- Saldivar-Gonzalez, F.I.; Huerta-Garcia, C.S.; Medina-Franco, J.L. Chemoinformatics-based enumeration of chemical libraries: A tutorial. J. Cheminform. 2020, 12, 64. [Google Scholar] [CrossRef] [PubMed]
- Jorda, R.; Krajcovicova, S.; Kralova, P.; Soural, M.; Krystof, V. Scaffold hopping of the SYK inhibitor entospletinib leads to broader targeting of the BCR signalosome. Eur. J. Med. Chem. 2020, 204, 112636. [Google Scholar] [CrossRef]
- Bemis, G.W.; Murcko, M.A. The properties of known drugs. 1. Molecular frameworks. J. Med. Chem. 1996, 39, 2887–2893. [Google Scholar] [CrossRef]
- Bianco, M.; Marinho, D.; Hoelz, L.V.B.; Bastos, M.M.; Boechat, N. Pyrroles as Privileged Scaffolds in the Search for New Potential HIV Inhibitors. Pharmaceuticals 2021, 14, 893. [Google Scholar] [CrossRef]
- Sun, L.; Gao, P.; Zhan, P.; Liu, X. Pyrazolo[1,5-a]pyrimidine-based macrocycles as novel HIV-1 inhibitors: A patent evaluation of WO2015123182. Expert. Opin. Ther. Pat. 2016, 26, 979–986. [Google Scholar] [CrossRef]
- Chen, L.; Ao, Z.; Jayappa, K.D.; Kobinger, G.; Liu, S.; Wu, G.; Wainberg, M.A.; Yao, X. Characterization of antiviral activity of benzamide derivative AH0109 against HIV-1 infection. Antimicrob. Agents Chemother. 2013, 57, 3547–3554. [Google Scholar] [CrossRef]
- Castillo Millán, J.; Portilla, J. Recent Advances in the Synthesis of New Pyrazole Derivatives; Italian Society of Chemistry: Rome, Italy, 2019; Volume 22. [Google Scholar]
- Arias-Gomez, A.; Godoy, A.; Portilla, J. Functional Pyrazolo[1,5-a]pyrimidines: Current Approaches in Synthetic Transformations and Uses As an Antitumor Scaffold. Molecules 2021, 26, 2708. [Google Scholar] [CrossRef]
- Kuznietsova, H.; Dziubenko, N.; Byelinska, I.; Hurmach, V.; Bychko, A.; Lynchak, O.; Milokhov, D.; Khilya, O.; Rybalchenko, V. Pyrrole derivatives as potential anti-cancer therapeutics: Synthesis, mechanisms of action, safety. J. Drug Target. 2020, 28, 547–563. [Google Scholar] [CrossRef] [PubMed]
- Abd El-Hameed, R.H.; Sayed, A.I.; Mahmoud Ali, S.; Mosa, M.A.; Khoder, Z.M.; Fatahala, S.S. Synthesis of novel pyrroles and fused pyrroles as antifungal and antibacterial agents. J. Enzyme Inhib. Med. Chem. 2021, 36, 2183–2198. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.S.; Zhang, L.; Schmidt, R.; Cai, Z.W.; Wei, D.; Williams, D.K.; Lombardo, L.J.; Trainor, G.L.; Xie, D.; Zhang, Y.; et al. Discovery of pyrrolopyridine-pyridone based inhibitors of Met kinase: Synthesis, X-ray crystallographic analysis, and biological activities. J. Med. Chem. 2008, 51, 5330–5341. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.J.; Lee, H.L.; Kim, S.J.; Jeong, J.H.; Ji, S.H.; Kim, H.B.; Kang, M.; Chung, H.W.; Park, C.S.; Choo, H.; et al. Identification of novel pyrrolopyrimidine and pyrrolopyridine derivatives as potent ENPP1 inhibitors. J. Enzyme Inhib. Med. Chem. 2022, 37, 2434–2451. [Google Scholar] [CrossRef]
- Maehigashi, T.; Ahn, S.; Kim, U.I.; Lindenberger, J.; Oo, A.; Koneru, P.C.; Mahboubi, B.; Engelman, A.N.; Kvaratskhelia, M.; Kim, K.; et al. A highly potent and safe pyrrolopyridine-based allosteric HIV-1 integrase inhibitor targeting host LEDGF/p75-integrase interaction site. PLoS Pathog. 2021, 17, e1009671. [Google Scholar] [CrossRef]
- Wang, S.; Fang, K.; Dong, G.; Chen, S.; Liu, N.; Miao, Z.; Yao, J.; Li, J.; Zhang, W.; Sheng, C. Scaffold Diversity Inspired by the Natural Product Evodiamine: Discovery of Highly Potent and Multitargeting Antitumor Agents. J. Med. Chem. 2015, 58, 6678–6696. [Google Scholar] [CrossRef]
- Zdrazil, B.; Felix, E.; Hunter, F.; Manners, E.J.; Blackshaw, J.; Corbett, S.; de Veij, M.; Ioannidis, H.; Lopez, D.M.; Mosquera, J.F.; et al. The ChEMBL Database in 2023: A drug discovery platform spanning multiple bioactivity data types and time periods. Nucleic Acids Res. 2024, 52, D1180–D1192. [Google Scholar] [CrossRef]
- Yap, C.W. PaDEL-descriptor: An open source software to calculate molecular descriptors and fingerprints. J. Comput. Chem. 2011, 32, 1466–1474. [Google Scholar] [CrossRef]
- Landrum, G. RDKit: Open-Source Cheminformatics. 22 May 2006. Available online: http://www.rdkit.org (accessed on 31 August 2015).
- Zhang, Y.; Deng, Q.; Liang, W.; Zou, X. An Efficient Feature Selection Strategy Based on Multiple Support Vector Machine Technology with Gene Expression Data. Biomed. Res. Int. 2018, 2018, 7538204. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, Y.; Muhammad, A.I.; Rabiu, A.M. Optimized SVM—Based Network Anomaly Detection with Genetic Algorithm and Recursive Feature Elimination. In Proceedings of the 2023 2nd International Conference on Multidisciplinary Engineering and Applied Science (ICMEAS), Abuja, Nigeria, 1–3 November 2023; pp. 1–5. [Google Scholar]
- Rodriguez-Perez, R.; Bajorath, J. Evolution of Support Vector Machine and Regression Modeling in Chemoinformatics and Drug Discovery. J. Comput. Aided Mol. Des. 2022, 36, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Chen, T.; Guestrin, C. XGBoost. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; pp. 785–794. [Google Scholar]
- Bishop, C.M. Neural Networks for Pattern Recognition; Oxford University Press, Inc.: Oxford, UK, 1995. [Google Scholar]
- Vracko, M.; Bandelj, V.; Barbieri, P.; Benfenati, E.; Chaudhry, Q.; Cronin, M.; Devillers, J.; Gallegos, A.; Gini, G.; Gramatica, P.; et al. Validation of counter propagation neural network models for predictive toxicology according to the OECD principles: A case study. SAR QSAR Environ. Res. 2006, 17, 265–284. [Google Scholar] [CrossRef]
- Nahm, F.S. Receiver operating characteristic curve: Overview and practical use for clinicians. Korean J. Anesthesiol. 2022, 75, 25–36. [Google Scholar] [CrossRef]
- Hong, H.; Rua, D.; Sakkiah, S.; Selvaraj, C.; Ge, W.; Tong, W. Consensus Modeling for Prediction of Estrogenic Activity of Ingredients Commonly Used in Sunscreen Products. Int. J. Environ. Res. Public Health 2016, 13, 958. [Google Scholar] [CrossRef]
- Jimenes-Vargas, K.; Pazos, A.; Munteanu, C.R.; Perez-Castillo, Y.; Tejera, E. Prediction of compound-target interaction using several artificial intelligence algorithms and comparison with a consensus-based strategy. J. Cheminform. 2024, 16, 27. [Google Scholar] [CrossRef]
- Rogers, D.; Hahn, M. Extended-connectivity fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef]
- Scalfani, V.F.; Patel, V.D.; Fernandez, A.M. Visualizing chemical space networks with RDKit and NetworkX. J. Cheminform. 2022, 14, 87. [Google Scholar] [CrossRef]
- Smith, N.R.; Zivich, P.N.; Frerichs, L.M.; Moody, J.; Aiello, A.E. A Guide for Choosing Community Detection Algorithms in Social Network Studies: The Question Alignment Approach. Am. J. Prev. Med. 2020, 59, 597–605. [Google Scholar] [CrossRef]
- Sheridan, R.P.; Karnachi, P.; Tudor, M.; Xu, Y.; Liaw, A.; Shah, F.; Cheng, A.C.; Joshi, E.; Glick, M.; Alvarez, J. Experimental Error, Kurtosis, Activity Cliffs, and Methodology: What Limits the Predictivity of Quantitative Structure-Activity Relationship Models? J. Chem. Inf. Model. 2020, 60, 1969–1982. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Methods | Data Set | Accuracy | Sensitivity | Specificity | Precision | F1 |
|---|---|---|---|---|---|---|
| SVM | Training Set | 0.88 | 0.83 | 0.91 | 0.87 | 0.85 |
| Test Set | 0.87 | 0.81 | 0.90 | 0.84 | 0.83 | |
| RF | Training Set | 0.97 | 0.97 | 0.98 | 0.98 | 0.97 |
| Test Set | 0.89 | 0.80 | 0.95 | 0.92 | 0.85 | |
| XGB | Training Set | 0.98 | 0.97 | 0.98 | 0.98 | 0.98 |
| Test Set | 0.88 | 0.81 | 0.93 | 0.87 | 0.84 | |
| MLP | Training Set | 0.97 | 0.97 | 0.98 | 0.97 | 0.96 |
| Test Set | 0.87 | 0.81 | 0.91 | 0.85 | 0.83 |
| Methods | Data Set | Accuracy | Sensitivity | Specificity | Precision | F1 |
|---|---|---|---|---|---|---|
| SVM | Training Set | 0.87 | 0.78 | 0.92 | 0.88 | 0.83 |
| Test Set | 0.86 | 0.77 | 0.92 | 0.85 | 0.81 | |
| RF | Training Set | 0.90 | 0.84 | 0.94 | 0.91 | 0.87 |
| Test Set | 0.87 | 0.78 | 0.92 | 0.86 | 0.82 | |
| XGB | Training Set | 0.90 | 0.85 | 0.95 | 0.93 | 0.89 |
| Test Set | 0.85 | 0.77 | 0.91 | 0.85 | 0.80 | |
| MLP | Training Set | 0.92 | 1.00 | 0.99 | 1.00 | 1.00 |
| Test Set | 0.84 | 0.66 | 0.93 | 0.86 | 0.74 |
| Cluster | N | Ns | Nss | Ns/N | Nss/N |
|---|---|---|---|---|---|
| 3 | 61 | 36 | 29 | 0.59 | 0.47 |
| 5 | 64 | 20 | 12 | 0.31 | 0.18 |
| 8 | 75 | 4 | 19 | 0.05 | 0.25 |
| 9 | 30 | 8 | 6 | 0.26 | 0.20 |
| 10 | 22 | 12 | 11 | 0.54 | 0.50 |
| 11 | 21 | 14 | 7 | 0.66 | 0.33 |
| 27 | 21 | 12 | 9 | 0.57 | 0.42 |
| 28 | 29 | 19 | 18 | 0.65 | 0.62 |
| 38 | 30 | 19 | 18 | 0.63 | 0.60 |
| 41 | 23 | 14 | 13 | 0.60 | 0.56 |
| 42 | 51 | 1 | 0 | 0.02 | 0.00 |
| 44 | 15 | 1 | 0 | 0.06 | 0.00 |
| 70 | 116 | 10 | 2 | 0.09 | 0.02 |
| 78 | 48 | 1 | 0 | 0.02 | 0.00 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Danishuddin; Haque, M.A.; Madhukar, G.; Jamal, Q.M.S.; Kim, J.-J.; Ahmad, K. Machine Learning-Driven Consensus Modeling for Activity Ranking and Chemical Landscape Analysis of HIV-1 Inhibitors. Pharmaceuticals 2025, 18, 714. https://doi.org/10.3390/ph18050714
Danishuddin, Haque MA, Madhukar G, Jamal QMS, Kim J-J, Ahmad K. Machine Learning-Driven Consensus Modeling for Activity Ranking and Chemical Landscape Analysis of HIV-1 Inhibitors. Pharmaceuticals. 2025; 18(5):714. https://doi.org/10.3390/ph18050714
Chicago/Turabian StyleDanishuddin, Md Azizul Haque, Geet Madhukar, Qazi Mohammad Sajid Jamal, Jong-Joo Kim, and Khurshid Ahmad. 2025. "Machine Learning-Driven Consensus Modeling for Activity Ranking and Chemical Landscape Analysis of HIV-1 Inhibitors" Pharmaceuticals 18, no. 5: 714. https://doi.org/10.3390/ph18050714
APA StyleDanishuddin, Haque, M. A., Madhukar, G., Jamal, Q. M. S., Kim, J.-J., & Ahmad, K. (2025). Machine Learning-Driven Consensus Modeling for Activity Ranking and Chemical Landscape Analysis of HIV-1 Inhibitors. Pharmaceuticals, 18(5), 714. https://doi.org/10.3390/ph18050714