
Fructose-1,6-Bisphosphate Reduces Chronic Constriction Injury Neuropathic Pain in Mice by Targeting Dorsal Root Ganglia Nociceptive Neuron Activation

 , , , , ,
, , , , ,  ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
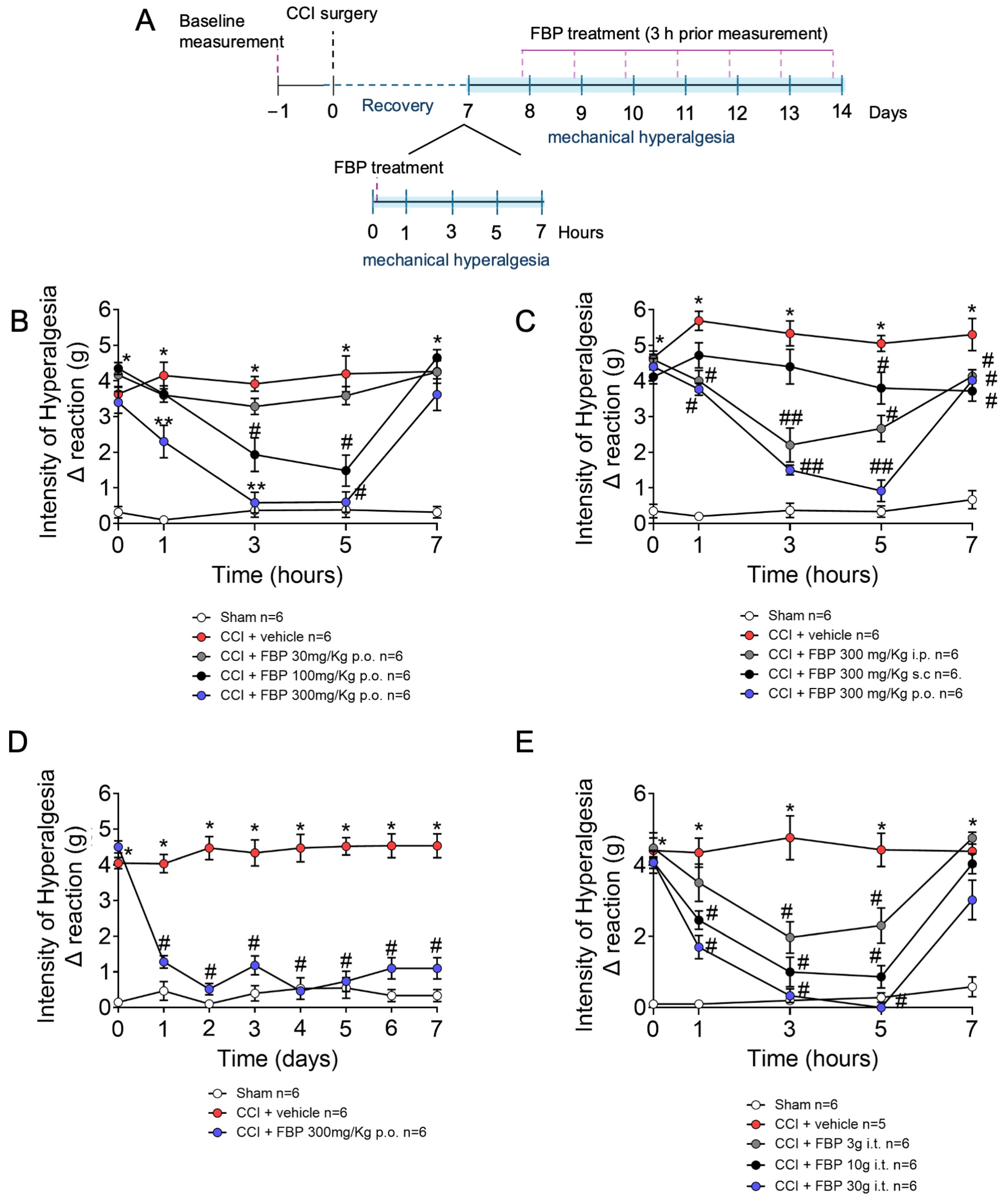
2.1. FBP Reduces Mechanical Hyperalgesia Induced by CCI in a Dose-Dependent Manner
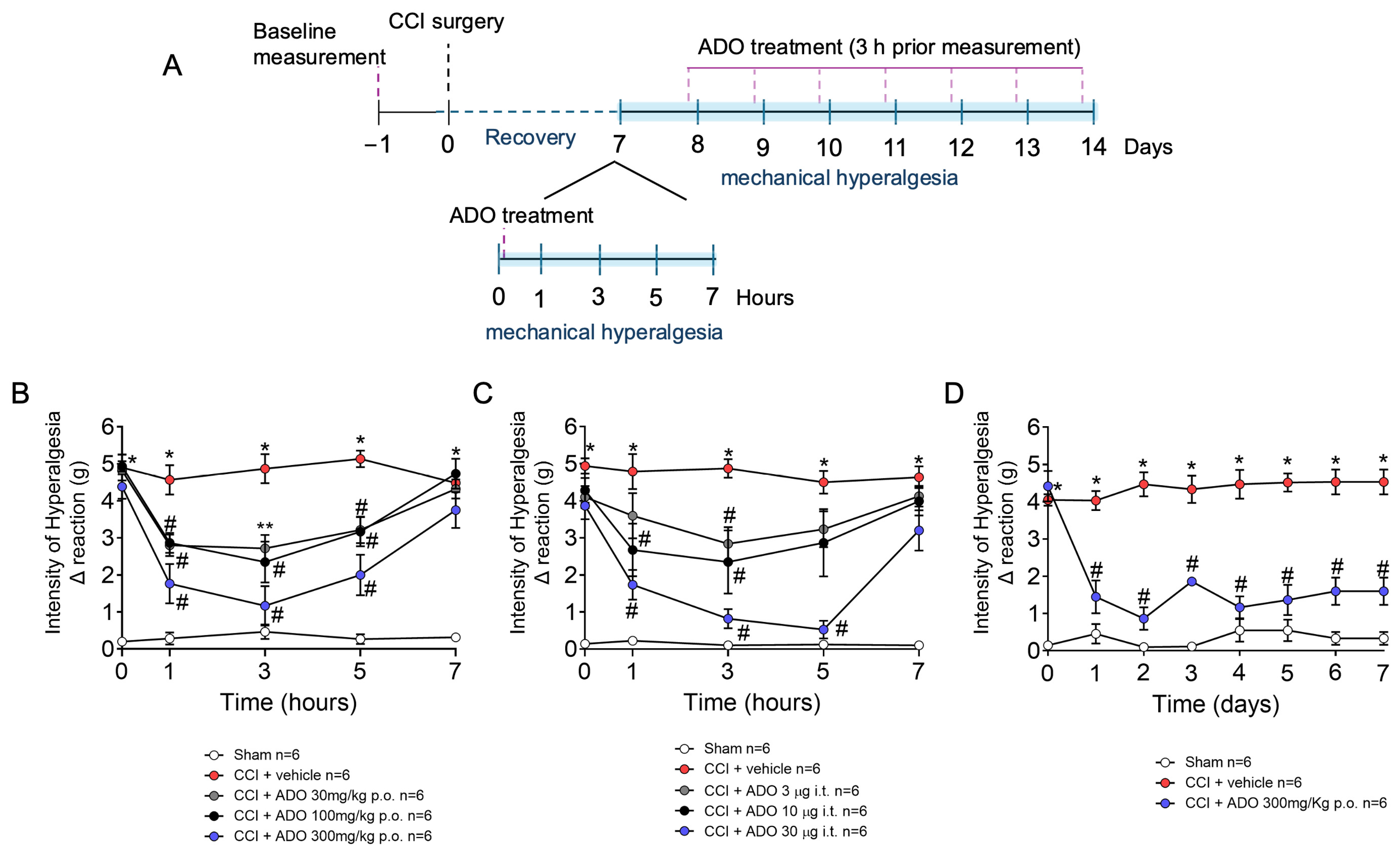
2.2. ADO Reduces Mechanical Hyperalgesia Induced by CCI in a Dose-Dependent Manner
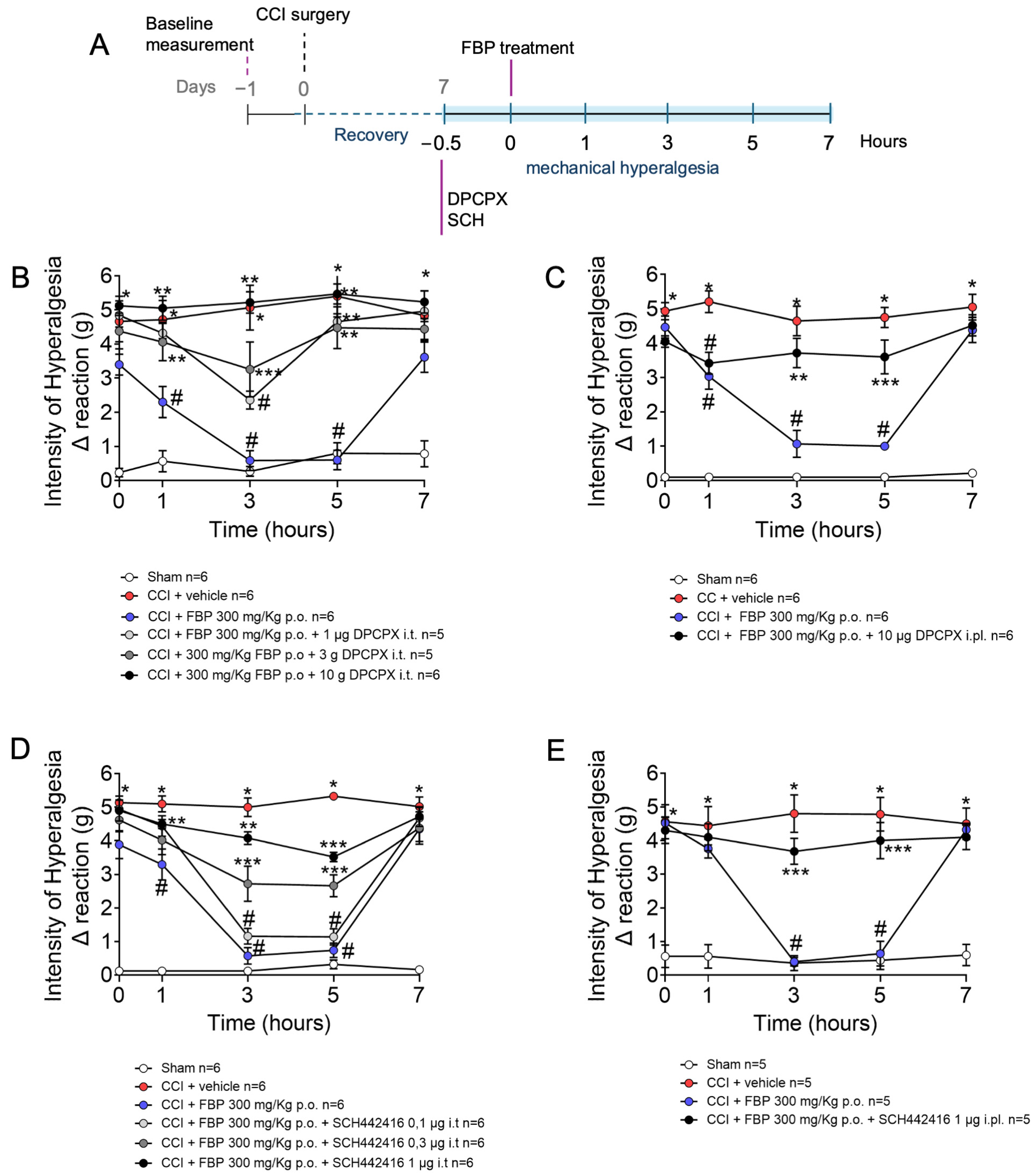
2.3. Analgesic Effect of FBP on CCI-Induced Mechanical Hyperalgesia Is Dependent on Adenosine A1 and A2A Receptors
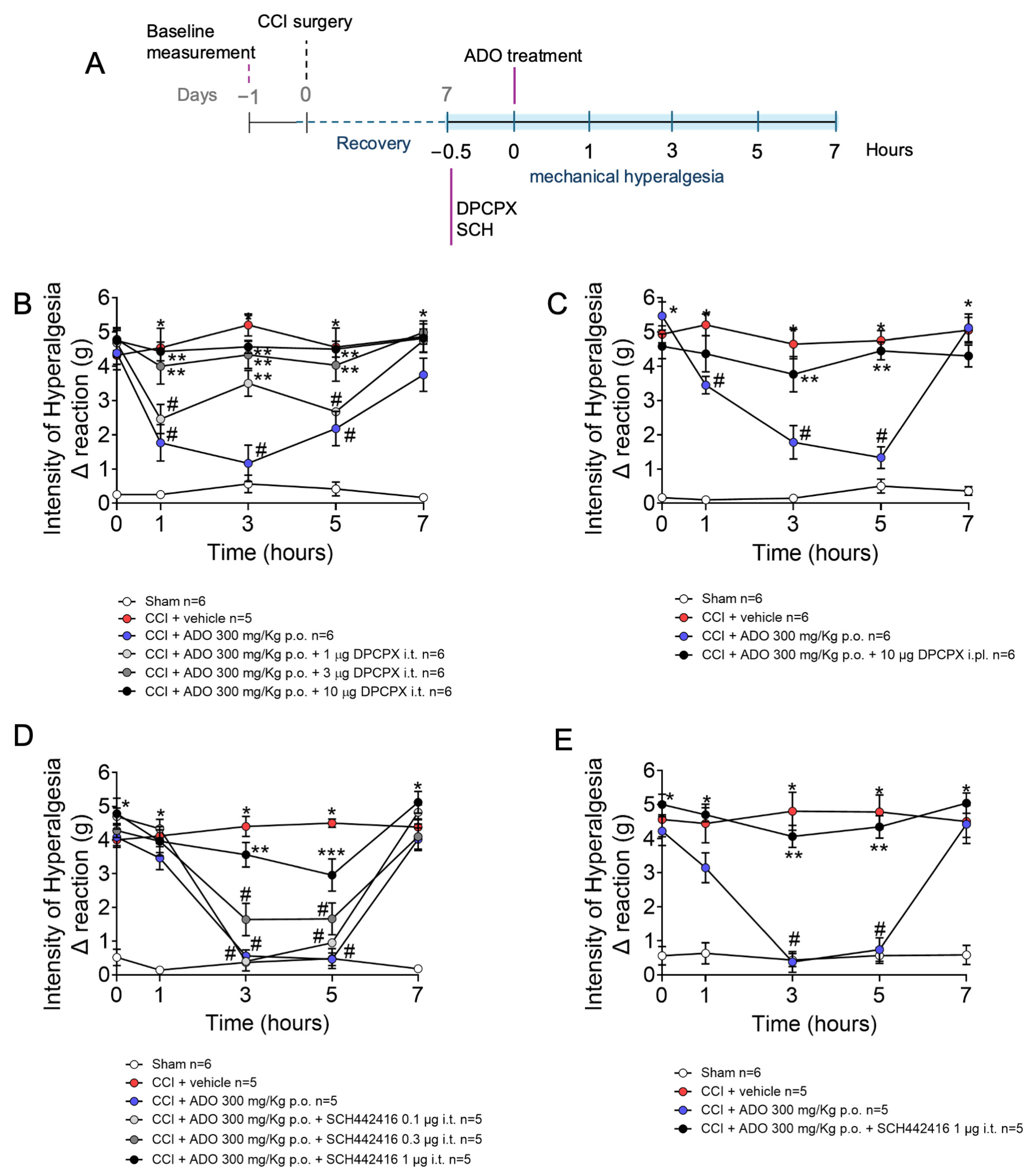
2.4. Analgesic Effect of ADO on CCI-Induced Mechanical Hyperalgesia Is Dependent on Adenosine A1 and A2A Receptors
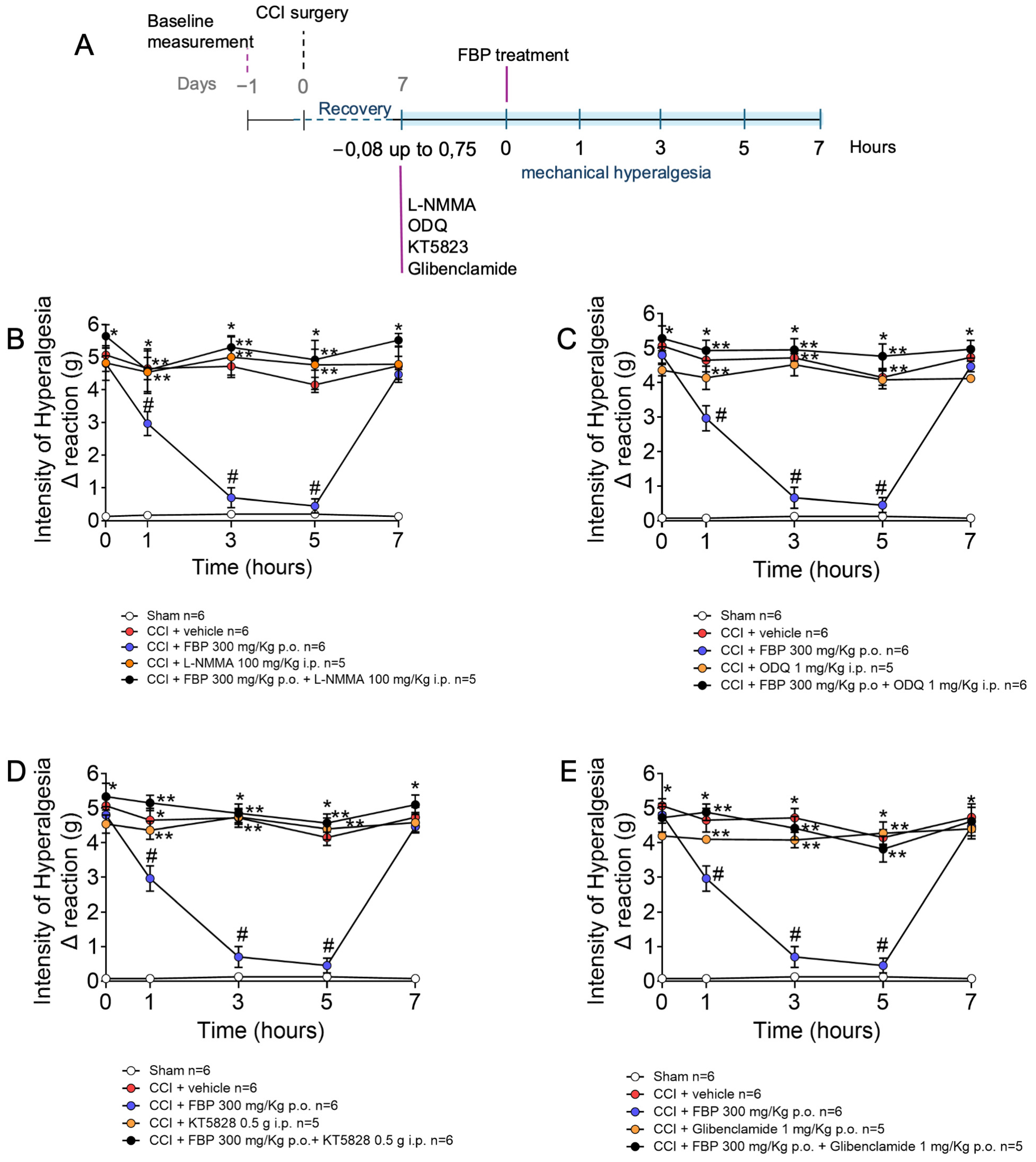
2.5. Analgesic Effect of FBP on CCI-Induced Mechanical Hyperalgesia Depends on the Activation of NO/cGMP/PKG/K+ATP Signaling Pathway
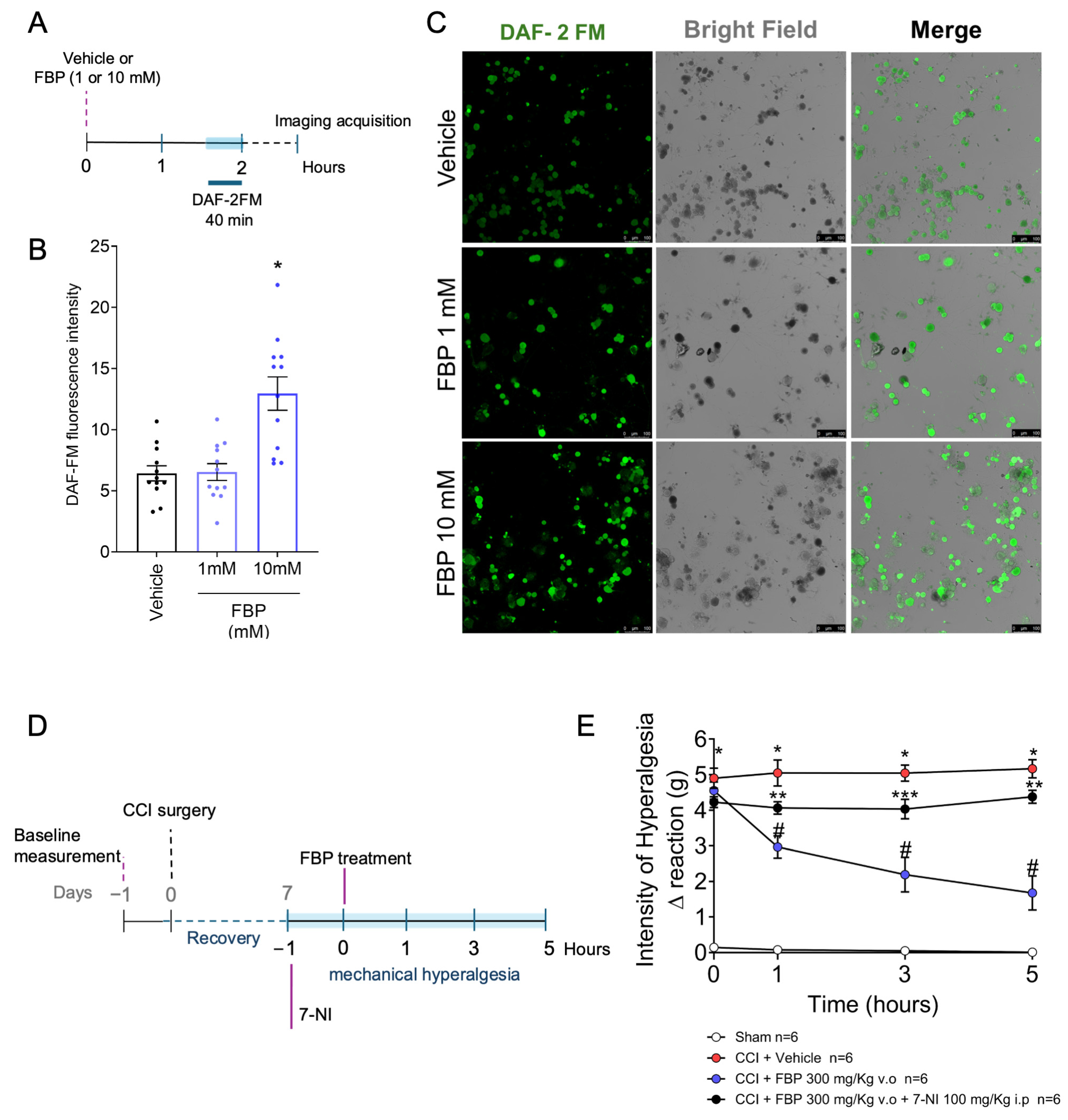
2.6. FBP Induces NO Production in DRG Neurons In Vitro
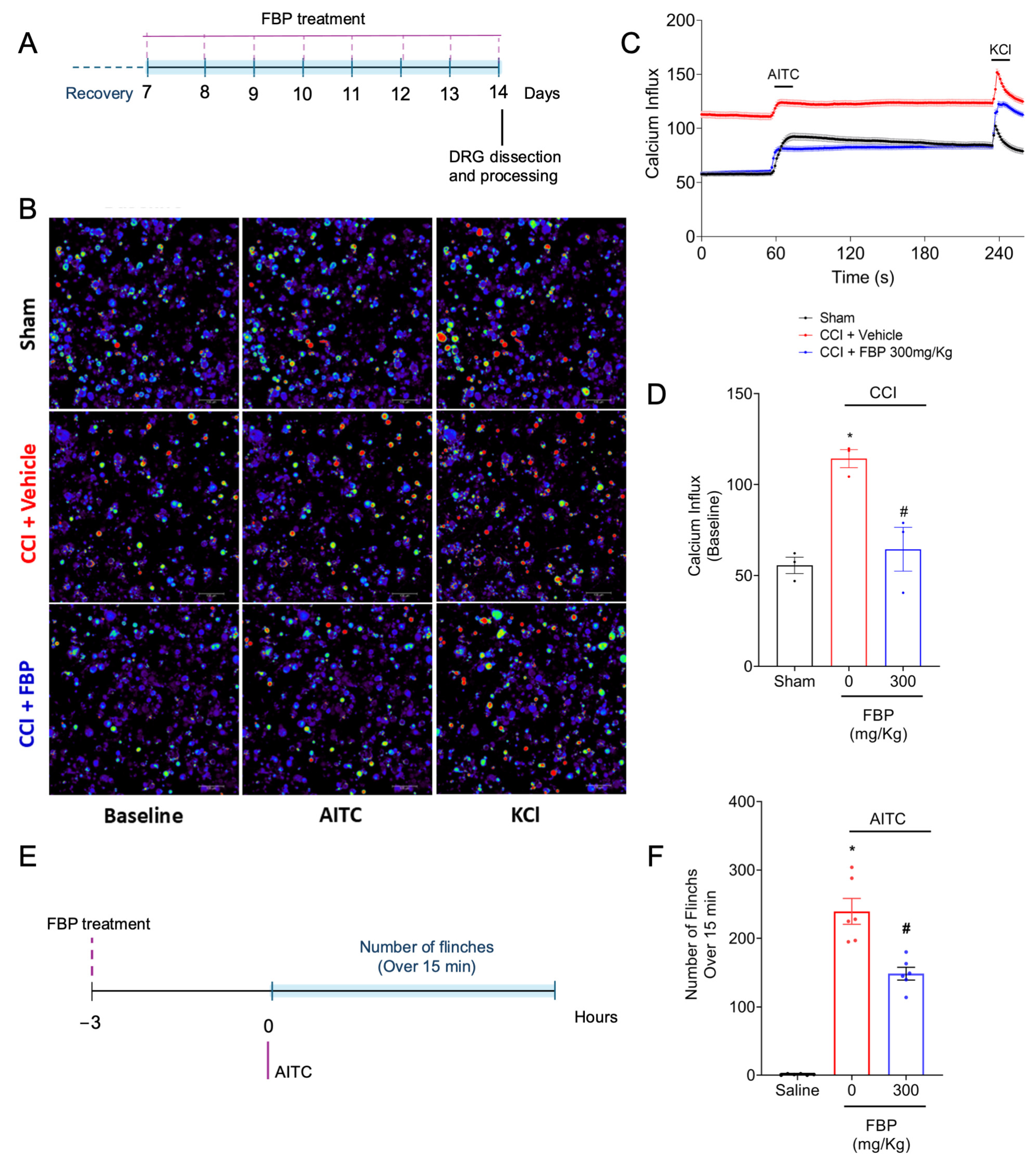
2.7. FBP Reduces the CCI Activation of TRPA1+ DRG Neurons Population and Reduces the Overt Pain-like Behavior Induced by TRPA1 Agonist in Mice
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Drugs
4.3. Experimental Procedures
4.4. Chronic Constriction Injury (CCI)
4.5. Mechanical Hyperalgesia Evaluation
4.6. Overt Pain-like Behaviors
4.7. Intracellular Nitric Oxide
4.8. Calcium Influx Imaging
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Kwan, K.Y.; Allchorne, A.J.; Vollrath, M.A.; Christensen, A.P.; Zhang, D.S.; Woolf, C.J.; Corey, D.P. TRPA1 Contributes to Cold, Mechanical, and Chemical Nociception but Is Not Essential for Hair-Cell Transduction. Neuron 2006, 50, 277–289. [Google Scholar] [CrossRef]
- Karashima, Y.; Damann, N.; Prenen, J.; Talavera, K.; Segal, A.; Voets, T.; Nilius, B. Bimodal Action of Menthol on the Transient Receptor Potential Channel TRPA1. J. Neurosci. 2007, 27, 9874–9884. [Google Scholar] [CrossRef] [PubMed]
- McNamara, C.R.; Mandel-Brehm, J.; Bautista, D.M.; Siemens, J.; Deranian, K.L.; Zhao, M.; Hayward, N.J.; Chong, J.A.; Julius, D.; Moran, M.M.; et al. TRPA1 Mediates Formalin-Induced Pain. Proc. Natl. Acad. Sci. USA. 2007, 104, 13525–13530. [Google Scholar] [CrossRef]
- Bautista, D.M.; Jordt, S.E.; Nikai, T.; Tsuruda, P.R.; Read, A.J.; Poblete, J.; Yamoah, E.N.; Basbaum, A.I.; Julius, D. TRPA1 Mediates the Inflammatory Actions of Environmental Irritants and Proalgesic Agents. Cell 2006, 124, 1269–1282. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.P.; Vase, L.; Hooten, W.M. Chronic Pain: An Update on Burden, Best Practices, and New Advances. Lancet 2021, 397, 2082–2097. [Google Scholar] [CrossRef] [PubMed]
- Shraim, M.A.; Massé-Alarie, H.; Hall, L.M.; Hodges, P.W. Systematic Review and Synthesis of Mechanism-Based Classification Systems for Pain Experienced in the Musculoskeletal System. Clin. J. Pain 2020, 36, 793–812. [Google Scholar] [CrossRef]
- Souza Monteiro de Araujo, D.; Nassini, R.; Geppetti, P.; De Logu, F. TRPA1 as a Therapeutic Target for Nociceptive Pain. Expert Opin. Ther. Targets 2020, 24, 997–1008. [Google Scholar] [CrossRef]
- da Silva, M.D.V.; Martelossi-Cebinelli, G.; Yaekashi, K.M.; Carvalho, T.T.; Borghi, S.M.; Casagrande, R.; Verri, W.A. A Narrative Review of the Dorsal Root Ganglia and Spinal Cord Mechanisms of Action of Neuromodulation Therapies in Neuropathic Pain. Brain Sci. 2024, 14, 589. [Google Scholar] [CrossRef]
- Bouhassira, D.; Lantéri-Minet, M.; Attal, N.; Laurent, B.; Touboul, C. Prevalence of Chronic Pain with Neuropathic Characteristics in the General Population. Pain 2008, 136, 380–387. [Google Scholar] [CrossRef]
- Van Hecke, O.; Austin, S.K.; Khan, R.A.; Smith, B.H.; Torrance, N. Neuropathic Pain in the General Population: A Systematic Review of Epidemiological Studies. Pain 2014, 155, 654–662. [Google Scholar] [CrossRef]
- Bouhassira, D. Neuropathic Pain: Definition, Assessment and Epidemiology. Rev. Neurol. 2019, 175, 16–25. [Google Scholar] [CrossRef]
- Bouchenaki, H.; Bégou, M.; Magy, L.; Hajj, R.; Demiot, C. Pharmacological Management of Neuropathic Pain. Therapie 2019, 74, 633–643. [Google Scholar] [CrossRef]
- Ciucă Anghel, D.M.; Nițescu, G.V.; Tiron, A.T.; Guțu, C.M.; Baconi, D.L. Understanding the Mechanisms of Action and Effects of Drugs of Abuse. Molecules 2023, 28, 4969. [Google Scholar] [CrossRef]
- Nafziger, A.N.; Barkin, R.L. Opioid Therapy in Acute and Chronic Pain. J. Clin. Pharmacol. 2018, 58, 1111–1122. [Google Scholar] [CrossRef]
- Bannister, K.; Sachau, J.; Baron, R.; Dickenson, A.H. Neuropathic Pain: Mechanism-Based Therapeutics. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 257–274. [Google Scholar] [CrossRef]
- Markov, A.K.; Neely, W.A.; Didlake, R.H.; Terry, J.; Causey, A.; Lehan, P.H. Metabolic Responses to Fructose-1,6-Diphosphate in Healthy Subjects. Metabolism 2000, 49, 698–703. [Google Scholar] [CrossRef]
- Kirtley, M.E.; McKay, M. Fructose-1,6-Bisphosphate, a Regulator of Metabolism. Mol. Cell. Biochem. 1977, 18, 141–149. [Google Scholar] [CrossRef]
- Alva, N.; Alva, R.; Carbonell, T. Fructose 1,6-Bisphosphate: A Summary of Its Cytoprotective Mechanism. Curr. Med. Chem. 2016, 23, 4396–4417. [Google Scholar] [CrossRef]
- Alves Filho, J.C.F.; Santos, R.C.V.; Castaman, T.A.; De Oliveira, J.R. Anti-Inflammatory Effects of Fructose-1,6-Bisphosphate on Carrageenan-Induced Pleurisy in Rat. Pharmacol. Res. 2004, 49, 245–248. [Google Scholar] [CrossRef]
- Ahn, S.M.; Yoon, H.Y.; Lee, B.G.; Park, K.C.; Chung, J.H.; Moon, C.H.; Lee, S.H. Fructose-1,6-Diphosphate Attenuates Prostaglandin E2 Production and Cyclo-Oxygenase-2 Expression in UVB-Irradiated HaCaT Keratinocytes. Br. J. Pharmacol. 2002, 137, 497–503. [Google Scholar] [CrossRef]
- Valério, D.A.; Ferreira, F.I.; Cunha, T.M.; Alves-Filho, J.C.; Lima, F.O.; De Oliveira, J.R.; Ferreira, S.H.; Cunha, F.Q.; Queiroz, R.H.; Verri, W.A. Fructose-1,6-Bisphosphate Reduces Inflammatory Pain-like Behaviour in Mice: Role of Adenosine Acting on A 1 Receptors. Br. J. Pharmacol. 2009, 158, 558–568. [Google Scholar] [CrossRef]
- Fredholm, B.B.; IJzerman, A.P.; Jacobson, K.A.; Linden, J.; Müller, C.E. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and Classification of Adenosine Receptors—An Update. Pharmacol. Rev. 2011, 63, 1–34. [Google Scholar] [CrossRef]
- Balasubramanyan, S.; Sharma, S.S. Protective Effect of Adenosine in Diabetic Neuropathic Pain Is Mediated through Adenosine A1-Receptors. Indian J. Physiol. Pharmacol. 2008, 52, 233–242. [Google Scholar]
- Lavand’homme, P.M.; Eisenach, J.C. Exogenous and Endogenous Adenosine Enhance the Spinal Antiallodynic Effects of Morphine in a Rat Model of Neuropathic Pain. Pain 1999, 80, 31–36. [Google Scholar] [CrossRef]
- Eisenach, J.C.; Curry, R.; Hood, D.D. Dose Response of Intrathecal Adenosine in Experimental Pain and Allodynia. Anesthesiology 2002, 97, 938–942. [Google Scholar] [CrossRef]
- Eisenach, J.C.; Rauck, R.L.; Curry, R. Intrathecal, but Not Intravenous Adenosine Reduces Allodynia in Patients with Neuropathic Pain. Pain 2003, 105, 65–70. [Google Scholar] [CrossRef]
- Lynch, M.E.; Clark, A.J.; Sawynok, J. Intravenous Adenosine Alleviates Neuropathic Pain: A Double Blind Placebo Controlled Crossover Trial Using an Enriched Enrolment Design. Pain 2003, 103, 111–117. [Google Scholar] [CrossRef]
- Belfrage, M.; Segerdahl, M.; Arnér, S.; Sollevi, A. The Safety and Efficacy of Intrathecal Adenosine in Patients with Chronic Neuropathic Pain. Anesth. Analg. 1999, 89, 136–142. [Google Scholar] [CrossRef]
- Haskó, G.; Pacher, P.; Deitch, E.A.; Vizi, E.S. Shaping of Monocyte and Macrophage Function by Adenosine Receptors. Pharmacol. Ther. 2007, 113, 264–275. [Google Scholar] [CrossRef]
- Stockwell, J.; Jakova, E.; Cayabyab, F.S. Adenosine A1 and A2A Receptors in the Brain: Current Research and Their Role in Neurodegeneration. Molecules 2017, 22, 676. [Google Scholar] [CrossRef]
- Lima, F.O.; Souza, G.R.; Verri, W.A.; Parada, C.A.; Ferreira, S.H.; Cunha, F.Q.; Cunha, T.M. Direct Blockade of Inflammatory Hypernociception by Peripheral A1 Adenosine Receptors: Involvement of the NO/CGMP/PKG/KATP Signaling Pathway. Pain 2010, 151, 506–515. [Google Scholar] [CrossRef]
- Nascimento, F.P.; Macedo-Júnior, S.J.; Pamplona, F.A.; Luiz-Cerutti, M.; Córdova, M.M.; Constantino, L.; Tasca, C.I.; Dutra, R.C.; Calixto, J.B.; Reid, A.; et al. Adenosine A1 Receptor-Dependent Antinociception Induced by Inosine in Mice: Pharmacological, Genetic and Biochemical Aspects. Mol. Neurobiol. 2015, 51, 1368–1378. [Google Scholar] [CrossRef]
- Yamamoto, S.; Nakanishi, O.; Matsui, T.; Shinohara, N.; Kinoshita, H.; Lambert, C.; Ishikawa, T. Intrathecal Adenosine A1 Receptor Agonist Attenuates Hyperalgesia without Inhibiting Spinal Glutamate Release in the Rat. Cell. Mol. Neurobiol. 2003, 23, 175–185. [Google Scholar] [CrossRef]
- Cui, J.G.; Sollevi, A.; Linderoth, B.; Meyerson, B.A. Adenosine Receptor Activation Suppresses Tactile Hypersensitivity and Potentiates Spinal Cord Stimulation in Mononeuropathic Rats. Neurosci. Lett. 1997, 223, 173–176. [Google Scholar] [CrossRef]
- Zhang, M.; Dai, Q.; Liang, D.; Li, D.; Chen, S.; Chen, S.; Han, K.; Huang, L.; Wang, J. Involvement of Adenosine A1 Receptor in Electroacupuncture-Mediated Inhibition of Astrocyte Activation during Neuropathic Pain. Arq. Neuropsiquiatr. 2018, 76, 736–742. [Google Scholar] [CrossRef]
- Curros-Criado, M.M.; Herrero, J.F. The Antinociceptive Effects of the Systemic Adenosine A1 Receptor Agonist CPA in the Absence and in the Presence of Spinal Cord Sensitization. Pharmacol. Biochem. Behav. 2005, 82, 721–726. [Google Scholar] [CrossRef]
- Gong, Q.J.; Li, Y.Y.; Xin, W.J.; Wei, X.H.; Cui, Y.; Wang, J.; Liu, Y.; Liu, C.C.; Li, Y.Y.; Liu, X.G. Differential Effects of Adenosine A1 Receptor on Pain-Related Behavior in Normal and Nerve-Injured Rats. Brain Res. 2010, 1361, 23–30. [Google Scholar] [CrossRef]
- Boison, D.; Chen, J.F.; Fredholm, B.B. Adenosine Signaling and Function in Glial Cells. Cell Death Differ. 2010, 17, 1071–1082. [Google Scholar] [CrossRef]
- Haskó, G.; Cronstein, B. Regulation of Inflammation by Adenosine. Front. Immunol. 2013, 4, 00085. [Google Scholar] [CrossRef]
- Gessi, S.; Varani, K.; Merighi, S.; Ongini, E.; Borea, P.A. A(2A) Adenosine Receptors in Human Peripheral Blood Cells. Br. J. Pharmacol. 2000, 129, 2–11. [Google Scholar] [CrossRef]
- Loram, L.C.; Taylor, F.R.; Strand, K.A.; Harrison, J.A.; RzasaLynn, R.; Sholar, P.; Rieger, J.; Maier, S.F.; Watkins, L.R. Intrathecal Injection of Adenosine 2A Receptor Agonists Reversed Neuropathic Allodynia through Protein Kinase (PK)A/PKC Signaling. Brain. Behav. Immun. 2013, 33, 112–122. [Google Scholar] [CrossRef]
- Bantel, C.; Tobin, J.R.; Li, X.; Childers, S.R.; Chen, S.R.; Eisenach, J.C. Intrathecal Adenosine Following Spinal Nerve Ligation in Rat: Short Residence Time in Cerebrospinal Fluid and No Change in A(1) Receptor Binding. Anesthesiology 2002, 96, 103–108. [Google Scholar] [CrossRef]
- Picolo, G.; Cury, Y. Peripheral Neuronal Nitric Oxide Synthase Activity Mediates the Antinociceptive Effect of Crotalus Durissus Terrificus Snake Venom, a δ- and κ-Opioid Receptor Agonist. Life Sci. 2004, 75, 559–573. [Google Scholar] [CrossRef]
- Sachs, D.; Cunha, F.Q.; Ferreira, S.H. Peripheral Analgesic Blockade of Hypernociception: Activation of Arginine/NO/CGMP/Protein Kinase G/ATP-Sensitive K+ Channel Pathway. Proc. Natl. Acad. Sci. USA. 2004, 101, 3680–3685. [Google Scholar] [CrossRef]
- Bertozzi, M.M.; Rossaneis, A.C.; Fattori, V.; Longhi-Balbinot, D.T.; Freitas, A.; Cunha, F.Q.; Alves-Filho, J.C.; Cunha, T.M.; Casagrande, R.; Verri, W.A. Diosmin Reduces Chronic Constriction Injury-Induced Neuropathic Pain in Mice. Chem. Biol. Interact. 2017, 273, 180–189. [Google Scholar] [CrossRef]
- Schulte, G.; Robertson, B.; Fredholm, B.B.; Delander, G.E.; Shortland, P.; Molander, C. Distribution of Antinociceptive Adenosine A1 Receptors in the Spinal Cord Dorsal Horn, and Relationship to Primary Afferents and Neuronal Subpopulations. Neuroscience 2003, 121, 907–916. [Google Scholar] [CrossRef]
- Funez, M.I.; Ferrari, L.F.; Duarte, D.B.; Sachs, D.; Cunha, F.Q.; Lorenzetti, B.B.; Parada, C.A.; Ferreira, S.H. Teleantagonism: A Pharmacodynamic Property of the Primary Nociceptive Neuron. Proc. Natl. Acad. Sci. USA. 2008, 105, 19038–19043. [Google Scholar] [CrossRef]
- Haddad, M.; Cherchi, F.; Alsalem, M.; Al-saraireh, Y.M.; Madae’en, S. Adenosine Receptors as Potential Therapeutic Analgesic Targets. Int. J. Mol. Sci. 2023, 24, 13160. [Google Scholar] [CrossRef]
- Eid, S.R.; Crown, E.D.; Moore, E.L.; Liang, H.A.; Choong, K.C.; Dima, S.; Henze, D.A.; Kane, S.A.; Urban, M.O. HC-030031, a TRPA1 Selective Antagonist, Attenuates Inflammatory- and Neuropathy-Induced Mechanical Hypersensitivity. Mol. Pain 2008, 4, 1744–8069. [Google Scholar] [CrossRef]
- Pinheiro, F.D.V.; Villarinho, J.G.; Silva, C.R.; Oliveira, S.M.; Pinheiro, K.D.V.; Petri, D.; Rossato, M.F.; Guerra, G.P.; Trevisan, G.; Antonello Rubin, M.; et al. The Involvement of the TRPA1 Receptor in a Mouse Model of Sympathetically Maintained Neuropathic Pain. Eur. J. Pharmacol. 2015, 747, 105–113. [Google Scholar] [CrossRef]
- Veras, F.P.; Peres, R.S.; Saraiva, A.L.L.; Pinto, L.G.; Louzada-Junior, P.; Cunha, T.M.; Paschoal, J.A.R.; Cunha, F.Q.; Alves-Filho, J.C. Fructose 1,6-Bisphosphate, a High-Energy Intermediate of Glycolysis, Attenuates Experimental Arthritis by Activating Anti-Inflammatory Adenosinergic Pathway. Sci. Rep. 2015, 5, 15171. [Google Scholar] [CrossRef]
- Planas, M.E.; Sánchez, S.; González, P.; de Oliveira, J.R.; Bartrons, R. Protective Effect of Fructose 1,6-Bisphosphate against Carrageenan-Induced Inflammation. Eur. J. Pharmacol. 1993, 237, 251–255. [Google Scholar] [CrossRef]
- Sola, A.; Panés, J.; Xaus, C.; Hotter, G. Fructose-1,6-Biphosphate and Nucleoside Pool Modifications Prevent Neutrophil Accumulation in the Reperfused Intestine. J. Leukoc. Biol. 2003, 73, 74–81. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Sitkovsky, M.V.; Robson, S.C. Purinergic Signaling during Inflammation. N. Engl. J. Med. 2012, 367, 2322–2333. [Google Scholar] [CrossRef]
- Cury, Y.; Picolo, G.; Gutierrez, V.P.; Ferreira, S.H. Pain and Analgesia: The Dual Effect of Nitric Oxide in the Nociceptive System. Nitric Oxide—Biol. Chem. 2011, 25, 243–254. [Google Scholar] [CrossRef]
- Zaninelli, T.H.; Mizokami, S.S.; Bertozzi, M.M.; Saraiva-Santos, T.; Pinho-Ribeiro, F.A.; de Oliveira, G.I.; Streck, R.; Araújo, E.J.A.; Arakawa, N.S.; Borghi, S.M.; et al. Kaurenoic Acid Reduces Ongoing Chronic Constriction Injury-Induced Neuropathic Pain: Nitric Oxide Silencing of Dorsal Root Ganglia Neurons. Pharmaceuticals 2023, 16, 343. [Google Scholar] [CrossRef]
- Mori, A.; Cross, B.; Uchida, S.; Kerrick Walker, J.; Ristuccia, R. How Are Adenosine and Adenosine A2A Receptors Involved in the Pathophysiology of Amyotrophic Lateral Sclerosis? Biomedicines 2021, 9, 1027. [Google Scholar] [CrossRef]
- Bastia, E.; Varani, K.; Monopoli, A.; Bertorelli, R. Effects of A1 and A2A Adenosine Receptor Ligands in Mouse Acute Models of Pain. Neurosci. Lett. 2002, 328, 241–244. [Google Scholar] [CrossRef]
- Montes, G.C.; Hammes, N.; Da Rocha, M.D.; Montagnoli, T.L.; Fraga, C.A.M.; Barreiro, E.J.; Sudo, R.T.; Zapata-Sudo, G. Treatment with Adenosine Receptor Agonist Ameliorates Pain Induced by Acute and Chronic Inflammation. J. Pharmacol. Exp. Ther. 2016, 358, 315–323. [Google Scholar] [CrossRef]
- Regaya, I.; Pham, T.; Andreotti, N.; Sauze, N.; Carrega, L.; Martin-Eauclaire, M.F.; Jouirou, B.; Peragut, J.C.; Vacher, H.; Rochat, H.; et al. Small Conductance Calcium-Activated K+ Channels, SkCa, but Not Voltage-Gated K+ (Kv) Channels, Are Implicated in the Antinociception Induced by CGS21680, a A2A Adenosine Receptor Agonist. Life Sci. 2004, 76, 367–377. [Google Scholar] [CrossRef]
- Loram, L.C.; Harrison, J.A.; Sloane, E.M.; Hutchinson, M.R.; Sholar, P.; Taylor, F.R.; Berkelhammer, D.; Coats, B.D.; Poole, S.; Milligan, E.D.; et al. Enduring Reversal of Neuropathic Pain by a Single Intrathecal Injection of Adenosine 2A Receptor Agonists: A Novel Therapy for Neuropathic Pain. J. Neurosci. 2009, 29, 14015. [Google Scholar] [CrossRef]
- Nascimento, F.P.; Figueredo, S.M.; Marcon, R.; Martins, D.F.; Macedo, S.J.; Lima, D.A.N.; Almeida, R.C.; Ostroski, R.M.; Rodrigues, A.L.S.; Santos, A.R.S. Inosine Reduces Pain-Related Behavior in Mice: Involvement of Adenosine A1 and A2A Receptor Subtypes and Protein Kinase C Pathways. J. Pharmacol. Exp. Ther. 2010, 334, 590–598. [Google Scholar] [CrossRef]
- Ledent, C.; Vaugeoist, J.M.; Schiffmann, S.N.; Pedrazzini, T.; El Yacoubi, M.; Vanderhaeghen, J.J.; Costentin, J.; Heath, J.K.; Vassart, G.; Parmentier, M. Aggressiveness, Hypoalgesia and High Blood Pressure in Mice Lacking the Adenosine A2a Receptor. Nature 1997, 388, 674–678. [Google Scholar] [CrossRef]
- Doak, G.J.; Sawynok, J. Complex Role of Peripheral Adenosine in the Genesis of the Response to Subcutaneous Formalin in the Rat. Eur. J. Pharmacol. 1995, 281, 311–318. [Google Scholar] [CrossRef]
- Cunha, T.M.; Roman-Campos, D.; Lotufo, C.M.; Duarte, H.L.; Souza, G.R.; Verri, W.A.; Funez, M.I.; Dias, Q.M.; Schivo, I.R.; Domingues, A.C.; et al. Morphine Peripheral Analgesia Depends on Activation of the PI3Kγ/AKT/NNOS/NO/KATP Signaling Pathway. Proc. Natl. Acad. Sci. USA. 2010, 107, 4442–4447. [Google Scholar] [CrossRef]
- Tonussi, C.R.; Ferreira, S.H. Mechanism of Diclofenac Analgesia: Direct Blockade of Inflammatory Sensitization. Eur. J. Pharmacol. 1994, 251, 173–179. [Google Scholar] [CrossRef]
- Cecílio, N.T.; Souza, G.R.; Alves-Filho, J.C.; Cunha, F.Q.; Cunha, T.M. The PI3Kγ/AKT Signaling Pathway Mediates Peripheral Antinociceptive Action of Dipyrone. Fundam. Clin. Pharmacol. 2021, 35, 364–370. [Google Scholar] [CrossRef]
- Longhi-Balbinot, D.T.; Rossaneis, A.C.; Pinho-Ribeiro, F.A.; Bertozzi, M.M.; Cunha, F.Q.; Alves-Filho, J.C.; Cunha, T.M.; Peron, J.P.S.; Miranda, K.M.; Casagrande, R.; et al. The Nitroxyl Donor, Angeli’s Salt, Reduces Chronic Constriction Injury-Induced Neuropathic Pain. Chem. Biol. Interact. 2016, 256, 1–8. [Google Scholar] [CrossRef]
- Ventura-Martínez, R.; Déciga-Campos, M.; Díaz-Reval, M.I.; González-Trujano, M.E.; López-Muñoz, F.J. Peripheral Involvement of the Nitric Oxide–CGMP Pathway in the Indomethacin-Induced Antinociception in Rat. Eur. J. Pharmacol. 2004, 503, 43–48. [Google Scholar] [CrossRef]
- Menéndez, L.; Juárez, L.; García, V.; Hidalgo, A.; Baamonde, A. Involvement of Nitric Oxide in the Inhibition of Bone Cancer-Induced Hyperalgesia through the Activation of Peripheral Opioid Receptors in Mice. Neuropharmacology 2007, 53, 71–80. [Google Scholar] [CrossRef]
- De Carvalho Veloso, C.; Rodrigues, V.G.; Ferreira, R.C.M.; Duarte, L.P.; Klein, A.; Duarte, I.D.; Romero, T.R.L.; De Castro Perez, A. Tingenone, a Pentacyclic Triterpene, Induces Peripheral Antinociception Due to NO/CGMP and ATP-Sensitive K(+) Channels Pathway Activation in Mice. Eur. J. Pharmacol. 2015, 755, 1–5. [Google Scholar] [CrossRef]
- Nassini, R.; Materazzi, S.; Benemei, S.; Geppetti, P. The TRPA1 Channel in Inflammatory and Neuropathic Pain and Migraine. Rev. Physiol. Biochem. Pharmacol. 2014, 167, 1–43. [Google Scholar] [CrossRef]
- Boiko, N.; Medrano, G.; Montano, E.; Jiang, N.; Williams, C.R.; Madungwe, N.B.; Bopassa, J.C.; Kim, C.C.; Parrish, J.Z.; Hargreaves, K.M.; et al. TrpA1 Activation in Peripheral Sensory Neurons Underlies the Ionic Basis of Pain Hypersensitivity in Response to Vinca Alkaloids. PLoS ONE 2017, 12, e0186888. [Google Scholar] [CrossRef]
- Sorge, R.E.; Mapplebeck, J.C.S.; Rosen, S.; Beggs, S.; Taves, S.; Alexander, J.K.; Martin, L.J.; Austin, J.S.; Sotocinal, S.G.; Chen, D.; et al. Different Immune Cells Mediate Mechanical Pain Hypersensitivity in Male and Female Mice. Nat. Neurosci. 2015, 18, 1081–1083. [Google Scholar] [CrossRef]
- Fan, C.Y.; McAllister, B.B.; Stokes-Heck, S.; Harding, E.K.; Pereira de Vasconcelos, A.; Mah, L.K.; Lima, L.V.; van den Hoogen, N.J.; Rosen, S.F.; Ham, B.; et al. Divergent Sex-Specific Pannexin-1 Mechanisms in Microglia and T Cells Underlie Neuropathic Pain. Neuron 2025, 113, 896–911.e9. [Google Scholar] [CrossRef]
- Guazelli, C.F.S.; Fattori, V.; Colombo, B.B.; Ludwig, I.S.; Vicente, L.G.; Martinez, R.M.; Georgetti, S.R.; Urbano, A.; Casagrande, R.; Baracat, M.M.; et al. Development of Trans-Chalcone Loaded Pectin/Casein Biodegradable Microcapsules: Efficacy Improvement in the Management of Experimental Colitis. Int. J. Pharm. 2023, 642, 123206. [Google Scholar] [CrossRef]
- Zimmermann, M. Ethical Guidelines for Investigations of Experimental Pain in Conscious Animals. Pain 1983, 16, 109–110. [Google Scholar] [CrossRef]
- Cunha, T.M.; Verri, W.A.; Vivancos, G.G.; Moreira, I.F.; Reis, S.; Parada, C.A.; Cunha, F.Q.; Ferreira, S.H. Mouse Paw Pressure-Meter Test Brazilian. J. Med. Biol. Res. 2004, 37, 401–407. [Google Scholar]
- da Rocha Lapa, F.; da Silva, M.D.; de Almeida Cabrini, D.; Santos, A.R.S. Anti-Inflammatory Effects of Purine Nucleosides, Adenosine and Inosine, in a Mouse Model of Pleurisy: Evidence for the Role of Adenosine A2 Receptors. Purinergic Signal. 2012, 8, 693–704. [Google Scholar] [CrossRef]
- Khandwala, H.; Zhang, Z.; Loomis, C.W. Inhibition of Strychnine-Allodynia Is Mediated by Spinal Adenosine A1- but Not A2-Receptors in the Rat. Brain Res. 1998, 808, 106–109. [Google Scholar] [CrossRef]
- Sawynok, J.; Reid, A.R.; Liu, J. Spinal and Peripheral Adenosine A1 Receptors Contribute to Antinociception by Tramadol in the Formalin Test in Mice. Eur. J. Pharmacol. 2013, 714, 373–378. [Google Scholar] [CrossRef]
- Staurengo-Ferrari, L.; Zarpelon, A.C.; Longhi-Balbinot, D.T.; Marchesi, M.; Cunha, T.M.; Alves-Filho, J.C.; Cunha, F.Q.; Ferreira, S.H.; Casagrande, R.; Miranda, K.M.; et al. Nitroxyl Inhibits Overt Pain-like Behavior in Mice: Role of CGMP/PKG/ATP-Sensitive Potassium Channel Signaling Pathway. Pharmacol. Rep. 2014, 66, 691. [Google Scholar] [CrossRef]
- Manchope, M.F.; Calixto-Campos, C.; Coelho-Silva, L.; Zarpelon, A.C.; Pinho-Ribeiro, F.A.; Georgetti, S.R.; Baracat, M.M.; Casagrande, R.; Verri, W.A. Naringenin Inhibits Superoxide Anion-Induced Inflammatory Pain: Role of Oxidative Stress, Cytokines, Nrf-2 and the NO−cGMP−PKG−KATPChannel Signaling Pathway. PLoS ONE 2016, 11, e0153015. [Google Scholar] [CrossRef]
- Fattori, V.; Zaninelli, T.H.; Ferraz, C.R.; Brasil-silva, L.; Borghi, S.M.; Cunha, J.M.; Chichorro, J.G.; Casagrande, R.; Verri, W.A. Neuropharmacology Maresin 2 Is an Analgesic Specialized Pro-Resolution Lipid Mediator in Mice by Inhibiting Neutrophil and Monocyte Recruitment, Nociceptor Neuron TRPV1 and TRPA1 Activation, and CGRP Release. Neuropharmacology 2022, 216, 109189. [Google Scholar] [CrossRef]
- Bennett, G.J.; Xie, Y.K. A Peripheral Mononeuropathy in Rat That Produces Disorders of Pain Sensation like Those Seen in Man. Pain 1988, 33, 87–107. [Google Scholar] [CrossRef]
- Zarpelon, A.C.; Rodrigues, F.C.; Lopes, A.H.; Souza, G.R.; Carvalho, T.T.; Pinto, L.G.; Xu, D.; Ferreira, S.H.; Alves-Filho, J.C.; McInnes, I.B.; et al. Spinal Cord Oligodendrocyte-Derived Alarmin IL-33 Mediates Neuropathic Pain. FASEB J. 2016, 30, 54–65. [Google Scholar] [CrossRef]
- Fattori, V.; Pinho-Ribeiro, F.A.; Staurengo-Ferrari, L.; Borghi, S.M.; Rossaneis, A.C.; Casagrande, R.; Verri, W.A. The Specialised Pro-Resolving Lipid Mediator Maresin 1 Reduces Inflammatory Pain with a Long-Lasting Analgesic Effect. Br. J. Pharmacol. 2019, 176, 1728–1744. [Google Scholar] [CrossRef]
- Erdfelder, E.; FAul, F.; Buchner, A.; Lang, A.G. Statistical Power Analyses Using G*Power 3.1: Tests for Correlation and Regression Analyses. Behav. Res. Methods 2009, 41, 1149–1160. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dionisio, A.M.; Milanez, P.d.A.O.; Zarpelon-Schutz, A.C.; Mizokami, S.S.; Bertozzi, M.M.; Yaekashi, K.M.; Camilios-Neto, D.; Borghi, S.M.; Casagrande, R.; Verri, W.A. Fructose-1,6-Bisphosphate Reduces Chronic Constriction Injury Neuropathic Pain in Mice by Targeting Dorsal Root Ganglia Nociceptive Neuron Activation. Pharmaceuticals 2025, 18, 660. https://doi.org/10.3390/ph18050660
Dionisio AM, Milanez PdAO, Zarpelon-Schutz AC, Mizokami SS, Bertozzi MM, Yaekashi KM, Camilios-Neto D, Borghi SM, Casagrande R, Verri WA. Fructose-1,6-Bisphosphate Reduces Chronic Constriction Injury Neuropathic Pain in Mice by Targeting Dorsal Root Ganglia Nociceptive Neuron Activation. Pharmaceuticals. 2025; 18(5):660. https://doi.org/10.3390/ph18050660
Chicago/Turabian StyleDionisio, Amanda Martins, Paula de Azevedo Oliveira Milanez, Ana Carla Zarpelon-Schutz, Sandra Satie Mizokami, Mariana Marques Bertozzi, Kelly Megumi Yaekashi, Doumit Camilios-Neto, Sergio Marques Borghi, Rubia Casagrande, and Waldiceu A. Verri. 2025. "Fructose-1,6-Bisphosphate Reduces Chronic Constriction Injury Neuropathic Pain in Mice by Targeting Dorsal Root Ganglia Nociceptive Neuron Activation" Pharmaceuticals 18, no. 5: 660. https://doi.org/10.3390/ph18050660
APA StyleDionisio, A. M., Milanez, P. d. A. O., Zarpelon-Schutz, A. C., Mizokami, S. S., Bertozzi, M. M., Yaekashi, K. M., Camilios-Neto, D., Borghi, S. M., Casagrande, R., & Verri, W. A. (2025). Fructose-1,6-Bisphosphate Reduces Chronic Constriction Injury Neuropathic Pain in Mice by Targeting Dorsal Root Ganglia Nociceptive Neuron Activation. Pharmaceuticals, 18(5), 660. https://doi.org/10.3390/ph18050660