2.2.5. Drug Carrier–Excipients Compatibility Testing
The drug and excipient compatibility studies were carried out by checking the physical description and using the Fourier-transform infrared spectroscopy analytical technique. The interaction studies were carried out to ascertain any incompatibility of the drug with the excipients used in the preparation of multi-drug-containing tablets of FR and RT solid dispersions.
An FT-IR spectrophotometer was used for infrared investigations of samples. About 4–5 mg of specimen was mixed with dry potassium bromide (KBr), and the specimen was examined at transmission mode over the wave number range of 4000–400 cm−1.
Fourier-transform infrared (FTIR) spectroscopy was performed to evaluate the potential chemical interactions between the active pharmaceutical ingredients (APIs), FR and RT, and the excipients used in the fixed-dose combination (FDC) formulations. The FTIR spectra of the pure drugs, individual excipients, and final formulations (F1 and F2) were compared to assess any potential changes in functional group vibrations, which could indicate interactions, degradation, or incompatibilities. The characteristic peaks of Favipiravir, including strong absorption bands corresponding to the C=O stretching vibration of the carboxyl and amide groups (~1700 cm
−1 and 1660 cm
−1, respectively), were observed in both the physical mixture and the final formulation, confirming the stability of the drug within the matrix (
Figure 7). Similarly, Roflumilast exhibited its distinctive absorption peaks, including the C=O stretching vibration at ~1730 cm
−1, C-N stretching at ∼1200 cm
−1, and aromatic ring vibrations at ~1600 cm
−1, all of which were retained in the formulations, indicating no significant interactions affecting the drug’s molecular structure (
Figure 8).
Klucel HXF and Klucel EXF, being cellulose derivatives, exhibited characteristic –OH stretching vibrations around ~3400 cm−1, which were also present in the final formulation, indicating no disruption in the polymeric integrity. Compritol 888 ATO, a lipid-based release-retarding agent, showed characteristic ester functional group absorptions at ~1735 cm−1, confirming its structural stability within the formulation. The presence of microcrystalline cellulose and calcium carbonate in the formulations was evident from their respective absorption bands at ~2900 cm−1 (C-H stretching) and ~1400 cm−1 (carbonate ion stretching).
Importantly, no significant peak shifts, disappearance, or new peak formations were observed in the FTIR spectra of the FDC formulations compared to the individual components, suggesting the absence of strong chemical interactions or degradation products. This confirms that both F1 and F2 formulations maintained the structural integrity of the APIs and excipients, ensuring the stability and compatibility of the developed fixed-dose combination. The retention of all characteristic peaks in the final formulations suggests that the formulation process did not induce any unwanted physicochemical changes, reinforcing the suitability of the selected excipients in preserving the desired pharmaceutical attributes of the drug product. The FTIR results, therefore, support the feasibility of the developed FDC formulations, ensuring their potential efficacy, stability, and reliability for therapeutic application in COVID-19 treatment.
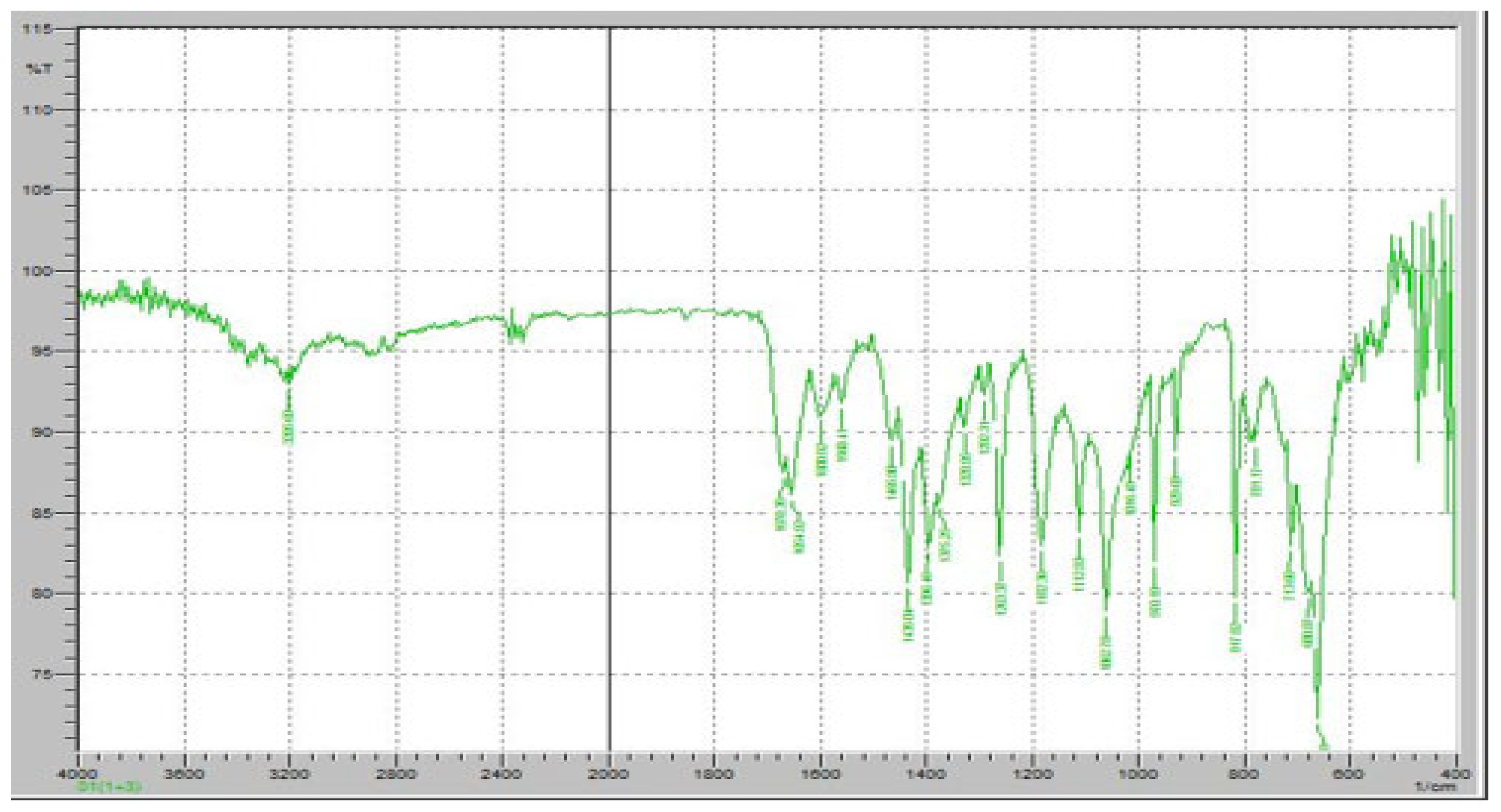
Figure 7 demonstrates the FTIR results of pure FR. The FTIR spectrum of FR exhibits distinct absorption bands corresponding to its characteristic functional groups, confirming its chemical identity and structural stability. A strong and sharp absorption peak was observed at approximately 1700 cm
−1, corresponding to the C=O stretching vibration of the carboxyl group, which is a key functional moiety responsible for the drug’s antiviral activity. Additionally, an absorption band at 1660 cm
−1 was detected, indicating the presence of amide C=O stretching, which is crucial for the molecular conformation of FR. The presence of an intense peak around 1250 cm
−1 corresponds to C-F stretching vibrations, confirming the fluorine substitution in the pyridine ring of Favipiravir. Furthermore, the broad absorption band in the 3100–3500 cm
−1 range is attributed to O-H and N-H stretching vibrations, indicative of intermolecular hydrogen bonding in the solid-state structure of FR. Peaks at 1400–1500 cm
−1 correspond to the C=C and C=N stretching within the pyrazine core of the molecule, further confirming its aromatic structure. The presence of peaks around 2900 cm
−1 represents C-H stretching, which is commonly associated with aliphatic and aromatic hydrogen vibrations.
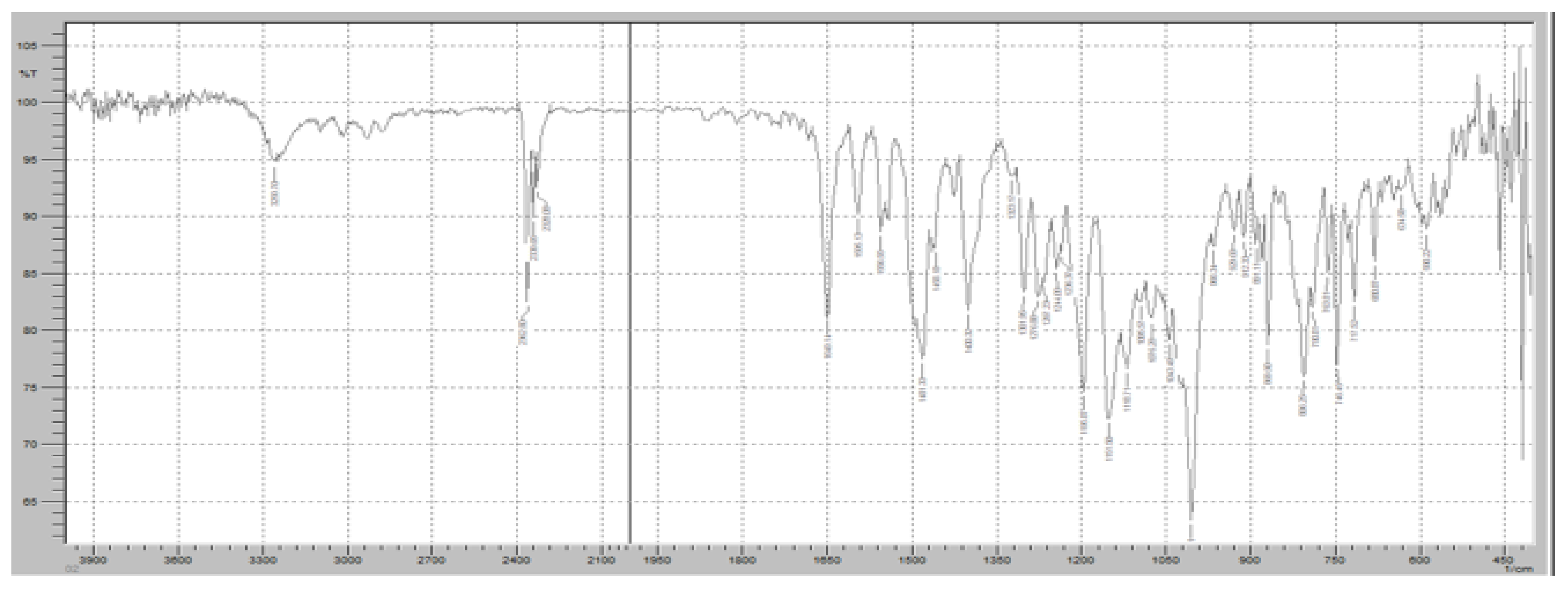
Figure 8 demonstrates the FTIR spectrum of pure RT. The FTIR spectrum exhibits several distinct peaks corresponding to the molecular vibrations of the functional groups present in RT, ensuring its chemical stability in the solid state. A strong absorption band was observed at approximately 1730 cm
−1, corresponding to the C=O stretching vibration of the cyclic carboxylester group, which is a key functional group responsible for the drug’s pharmacological activity as a phosphodiesterase-4 (PDE4) inhibitor. Additionally, a significant peak at 1665 cm
−1 was detected, representing C=O stretching of the amide group, indicating the presence of a lactam moiety in the molecular structure of RT. The characteristic C-N stretching vibrations of the pyridine ring were observed in the range of 1200–1300 cm
−1, confirming the presence of the nitrogen heterocyclic system within the RT molecule. Another prominent absorption band was identified at 1600 cm
−1, corresponding to aromatic C=C stretching, which is associated with the benzamide core structure of RT. Furthermore, a broad absorption band in the 3200–3400 cm
−1 range was observed, attributed to N-H stretching vibrations, confirming the presence of amide functionalities that contribute to RT’s molecular interactions. Peaks in the 2800–3000 cm
−1 range were also noted, corresponding to C-H stretching vibrations of both aliphatic and aromatic groups, further verifying the integrity of the hydrocarbon framework of RT.
Figure 9 demonstrates the FTIR spectrum of the FR and Klucel HXF mixture to assess potential interactions between the active pharmaceutical ingredient (API) and the sustained-release polymer. The FTIR scan exhibits characteristic peaks of both FR and Klucel HXF, providing insights into their compatibility and any possible molecular interactions. The characteristic C=O stretching peak of FR was observed at approximately 1700 cm
−1, confirming the preservation of its carboxyl functional group. The amide C=O stretching band at 1660 cm
−1 remained unchanged, indicating no significant interaction affecting the amide structure. The C-F stretching vibration around 1250 cm
−1 was also detected, confirming the stability of the fluorine-substituted pyridine ring. Broad O-H and N-H stretching vibrations in the 3100–3500 cm
−1 range were present, consistent with the hydrogen bonding properties of FR. The broad O-H stretching vibration of Klucel HXF, attributed to hydrogen bonding in hydroxypropyl cellulose, was observed in the 3200–3600 cm
−1 range. A peak around 1100 cm
−1 corresponding to C-O-C stretching vibrations of the ether groups in Klucel HXF was present, confirming the intact polymer structure. No major shifts, disappearance, or formation of new peaks were detected, suggesting that FR and Klucel HXF did not undergo significant chemical interactions or degradation. The presence of all characteristic peaks of both components indicates physical rather than chemical interaction, implying that Klucel HXF acts as a carrier without altering the molecular integrity of FR.
The FTIR spectrum of the Roflumilast (RT) and Klucel EXF mixture was analyzed to assess potential molecular interactions and confirm the compatibility of the immediate-release polymer with the active pharmaceutical ingredient (
Figure 10). The characteristic peaks of Roflumilast were well retained in the spectrum, with the strong C=O stretching vibration of the cyclic carboxylester group observed at 1730 cm
−1, confirming the integrity of the ester functionality. Additionally, the amide C=O stretching peak at 1665 cm
−1 remained unchanged, indicating the structural stability of the amide bond, while the aromatic C=C stretching vibrations at 1600 cm
−1 validated the presence of the benzamide core. The C-N stretching vibrations of the pyridine ring were also evident in the 1200–1300 cm
−1 range, further confirming the stability of the drug. Similarly, Klucel EXF exhibited its characteristic broad O-H stretching vibration in the 3200–3600 cm
−1 range, attributed to hydrogen bonding in hydroxypropyl cellulose, along with a distinct peak around 1100 cm
−1 representing C-O-C stretching vibrations of the ether groups, ensuring the intact polymer structure. The presence of all major peaks from both RT and Klucel EXF suggests no significant chemical interactions, as no major shifts, disappearance, or formation of new peaks were observed. This indicates that the interaction between RT and Klucel EXF is primarily physical rather than chemical, confirming that Klucel EXF acts as a suitable immediate-release carrier without altering the structural integrity of Roflumilast. The FTIR analysis, therefore, supports the stability and compatibility of this formulation component, ensuring effective drug release while maintaining the physicochemical properties of RT.
The FTIR spectrum of the FR and Compritol ATO 888 mixture was analyzed to evaluate potential interactions and confirm the compatibility of the lipid-based release-retarding agent with the active pharmaceutical ingredient (
Figure 11). The characteristic peaks of Favipiravir remained intact, with the strong C=O stretching vibration of the carboxyl functional group observed at 1715 cm
−1, confirming the preservation of its keto-amide structure. The amide N-H bending peak at 1625 cm
−1 and the aromatic C=C stretching vibrations around 1550 cm
−1 further validated the structural stability of FR. The broad O-H stretching vibration in the range of 3100–3500 cm
−1 was also evident, indicating the presence of hydrogen bonding. Compritol ATO 888, a lipid-based excipient, exhibited its characteristic C-H stretching vibrations in the range of 2800–2950 cm
−1, corresponding to the long-chain fatty acids in glyceryl behenate, and a prominent ester C=O stretching peak at 1735 cm
−1. No significant peak shifts, disappearance, or formation of new peaks were observed in the FTIR spectrum of the mixture, suggesting the absence of strong chemical interactions or degradation. The findings indicate that Compritol ATO 888 and Favipiravir remain compatible, with only physical interactions contributing to the formulation. This confirms that Compritol ATO 888 can effectively function as a release-retarding agent in the formulation without compromising the chemical stability of Favipiravir, ensuring controlled drug release while maintaining the physicochemical properties of the active ingredient.
The FTIR spectrum of the FR and Crospovidone (crosslinked polyvinylpyrrolidone) mixture was analyzed to assess potential molecular interactions and confirm the compatibility of the super disintegrant with the active pharmaceutical ingredient (
Figure 12). The characteristic peaks of Favipiravir remained unchanged, with a strong C=O stretching vibration observed at 1715 cm
−1, confirming the stability of its keto-amide functional group. The amide N-H bending peak at 1625 cm
−1 and the aromatic C=C stretching vibrations around 1550 cm
−1 further validated the structural integrity of FR. Additionally, a broad O-H stretching band in the 3100–3500 cm
−1 range was present, suggesting the potential for hydrogen bonding. Crospovidone exhibited its characteristic C=O stretching peak at 1650 cm
−1, attributed to the pyrrolidone ring, along with C-N stretching vibrations in the 1200–1300 cm
−1 range. Importantly, no significant peak shifts, disappearance, or formation of new peaks were observed in the FTIR spectrum of the FR-Crospovidone mixture, indicating the absence of strong chemical interactions. The results suggest that the interaction between FR and Crospovidone is primarily physical rather than chemical, ensuring the stability of the active ingredient. This confirms that Crospovidone can be safely incorporated as a superdisintegrant in the formulation without affecting the physicochemical integrity of Favipiravir, thereby facilitating rapid disintegration and improved drug dissolution in the final dosage form.
The FTIR spectrum of the Roflumilast (RT) and Crospovidone (crosslinked polyvinylpyrrolidone) mixture was analyzed to evaluate possible molecular interactions and ensure the compatibility of the superdisintegrant with the active pharmaceutical ingredient (
Figure 13). The characteristic peaks of Roflumilast remained intact, with the strong C=O stretching vibration of the cyclic carboxylester group observed at 1730 cm
−1, confirming the structural stability of the ester functionality. Additionally, the amide C=O stretching peak at 1665 cm
−1 remained unaltered, while the aromatic C=C stretching vibrations at 1600 cm
−1 validated the integrity of the benzamide core. The C-N stretching vibrations of the pyridine ring were also retained in the 1200–1300 cm
−1 range, further confirming the absence of degradation or chemical interaction. Crospovidone exhibited its characteristic C=O stretching peak at 1650 cm
−1, attributed to the pyrrolidone ring, along with C-N stretching vibrations in the fingerprint region. Importantly, the FTIR spectrum of the RT-Crospovidone mixture did not show any significant shifts, disappearance, or formation of new peaks, indicating that no strong chemical interactions or incompatibilities occurred. This suggests that the interaction between RT and Crospovidone is primarily physical rather than chemical, ensuring the stability of Roflumilast in the formulation. These findings confirm that Crospovidone can be safely incorporated as a superdisintegrant in the immediate-release layer without affecting the physicochemical integrity of Roflumilast, thereby facilitating rapid tablet disintegration and enhancing drug dissolution performance. Amorphous solid dispersion of excipients–drug and excipients to the drug was prepared by employing the solvent evaporation method. FR and RT solutions were prepared by dissolving 20 g of the sample, constituting 60%
w/
w drugs and 40%
w/
w excipients in 100 mL of methanol. The solvent was then evaporated using a rotary evaporator at 45 Celsius. The samples were then treated to remove any residual solvent.
To compare the stability of the amorphous state of RT and FR in each granule type, 10 g of each sample was kept in containers at 75% RH, with an ambient temperature of 25 °C, for the specified periods (2 weeks). The samples were then measured by FTIR upon removal of the lid at each time point.
The FTIR spectra of FR and RT after the preparation of amorphous solid dispersions (ASDs) using the solvent evaporation method were analyzed to assess any potential changes in their molecular interactions and structural integrity. The primary concern in ASD formation is whether hydrogen bonding, molecular dispersion, or interactions with excipients lead to significant peak shifts, disappearance, or the formation of new peaks (
Figure 14). For FR, the characteristic carbonyl (C=O) stretching vibration at 1715 cm
−1, associated with the keto-amide functional group, remained largely unchanged, indicating that no significant chemical transformation occurred (
Figure 14). However, a slight broadening of the O-H stretching band (3100–3500 cm
−1) was observed, which suggests the possible formation of hydrogen bonds with the excipients in the amorphous state. The amide N-H bending peak (1625 cm
−1) and aromatic C=C stretching vibrations (1550 cm
−1) were retained, confirming the molecular stability of FR.
For RT, the FTIR spectrum showed that the strong ester C=O stretching peak at 1730 cm
−1 remained intact, confirming that the cyclic carboxylester functionality was not degraded (
Figure 15). The amide C=O stretching peak at 1665 cm
−1 and aromatic C=C peaks around 1600 cm
−1 were also preserved. However, similar to FR, a broadening of the O-H stretching band in the 3100–3500 cm
−1 range was observed, indicating potential hydrogen bonding interactions between RT and the excipients (
Figure 16).
After two weeks of stability testing at 75% RH and 25 °C, the FTIR spectra of both FR and RT showed no significant new peak formations, no disappearance of characteristic peaks, and no major peak shifts beyond minor broadening in the hydrogen bonding region. This confirms that the amorphous solid dispersions maintained the structural integrity of both drugs, with only physical interactions occurring. The results indicate that the solvent evaporation method successfully transformed FR and RT into an amorphous state while maintaining their chemical stability and compatibility with the excipients.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}