

Biomarker-Driven Pharmacokinetics and Efficacy of Polymyxin B in Critically Ill Patients with XDR-GN Pneumonia

 ,
, 
Abstract
1. Introduction
2. Results
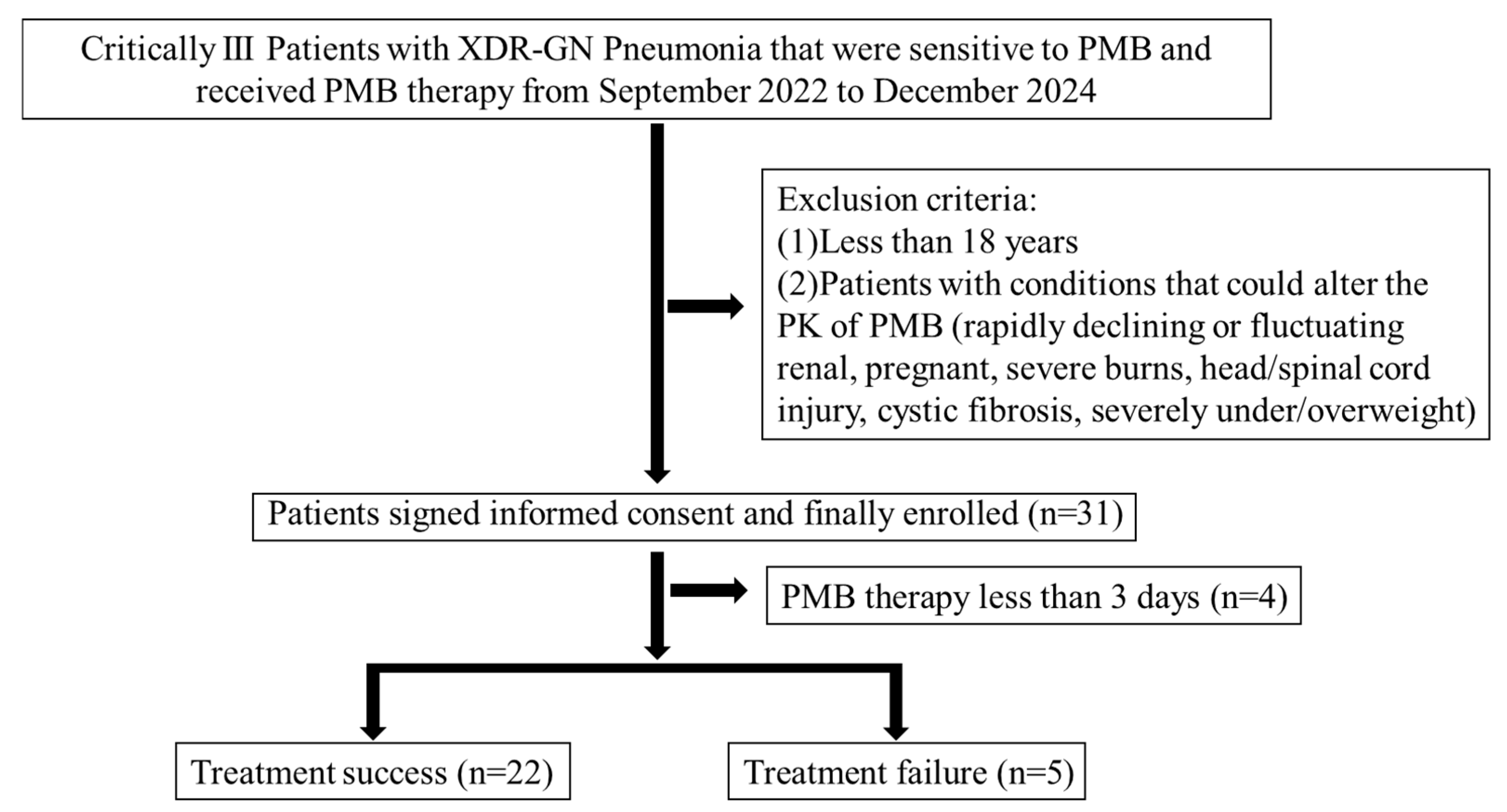
2.1. Clinical Characteristics of the Patients
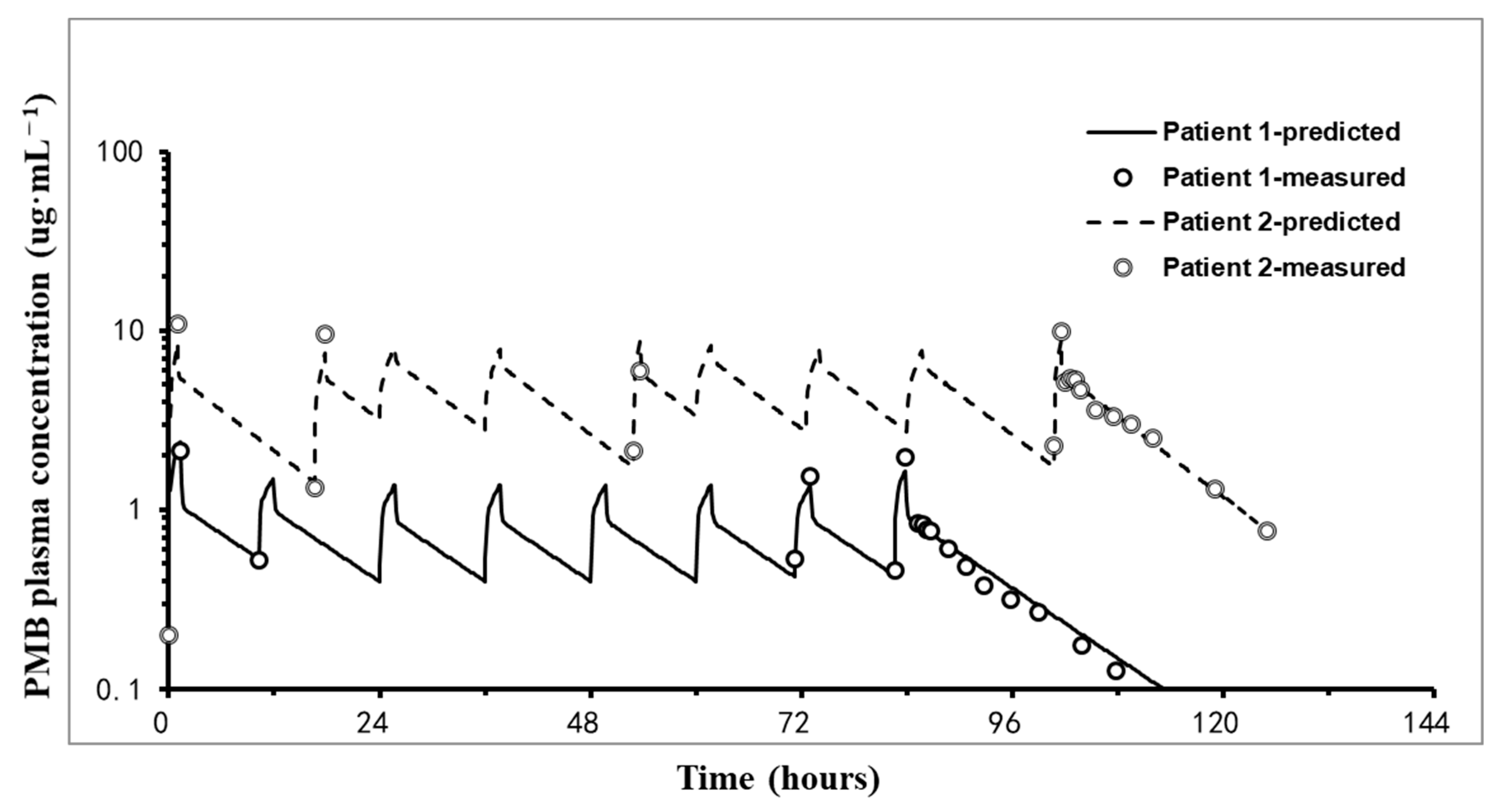
2.2. PK Results
2.3. Pulmonary Concentrations
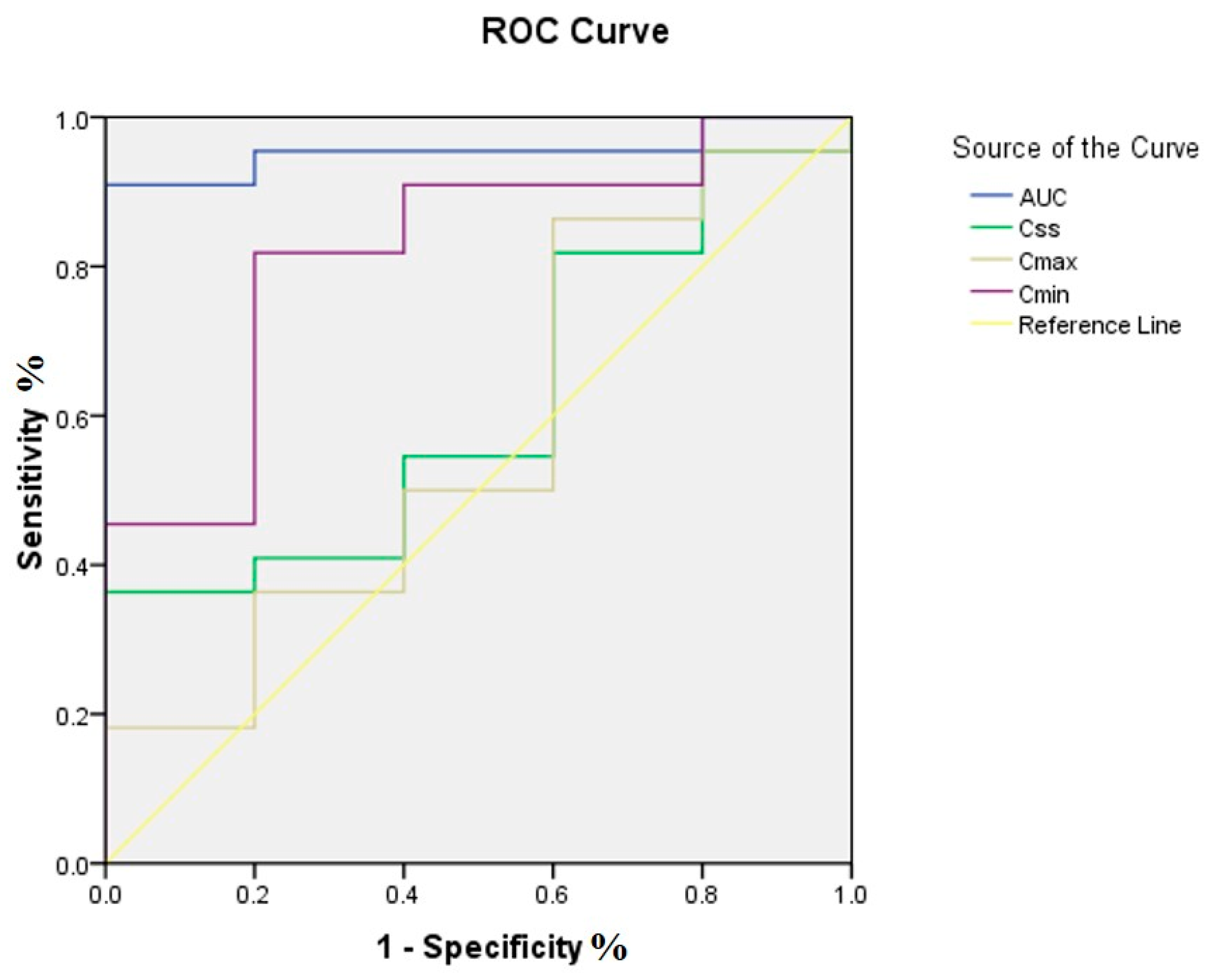
2.4. PK/PD Targets Attainment and Clinical Efficacy
2.5. Correlations Between Patients’ Candidate Biomarkers and PK Parameters
3. Discussion
3.1. PK–PD Characteristics
3.2. Biomarkers Predicting Tissue Pharmacokinetics
3.3. PMB Penetration into the Lung
3.4. PMB Safety
3.5. Limitations
4. Materials and Methods
4.1. Patients and Ethics
4.2. Data Collection
4.3. PMB Administration and Sample Collection
4.4. Quantification of PMB Concentrations in Plasma and BAL Fluid
4.5. Quantification of PMB Concentrations in ELF
4.6. Binding of PMB in Plasma
4.7. PK Analysis
4.8. PD Study
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
| References | Populations | Infection Types | Regimen | Analytical Model | PK Parameters |
|---|---|---|---|---|---|
| Miglis et al., 2018 [45] | American | Acutely ill patients with suspected or documented Gram-negative infection | Loading dose = 2.79 (0.34–3.45) mg/kg/day Maintenance dose = 2.42 (0.34–3.45) mg/kg/day | Two-compartment model (PMB1 and PMB2) | V1 = 33.77 ± 15.21 L; V2 = 78.20 ± 37.46 L; CL = 2.63 ± 1.41 L/h |
| Kubin et al., 2018 [46] | American | Acutely ill patients with suspected or documented Gram-negative infection | Loading doses = 2.5 to 3 mg/kg Maintenance dose = 2.8 (1.5–3) mg/kg/day (infusion for at least 1 h) | One-compartment model | V1 = 34.4 L; CL = 2.37 L/h |
| Manchandani et al., 2018 [47] | Asian (from Thailand and Singapore), American | Patients with suspected or documented Gram-negative infection | 2.1 ± 0.4 mg/kg/day and every 12 h to 24 h (infusion for over 1–3 h) | One-compartment model (PMB1, B2, B3, and isoleucine-B1) | CL = 2.5 ± 1.1 L/h; V = 34.3 ± 16.4 L; t1/2 = 10.1 h; AUC in patients with nephrotoxicity = 52.3 ± 14.8 mg·h/L; AUC in patients without nephrotoxicity = 45.1 ± 17.3 mg.h/L |
| Sandri et al., 2013 [48] | Brazilian, Australian | Critically ill patients with suspected or documented Gram-negative infection | 0.45–3.38 mg/kg/day every 12 h or every 24 h (infusion for 1–4 h) | Nonlinear mixed-effects model S-ADAPT platform (PMB1 and PMB2) | AUCss,0–24h = 66.9 ± 21.6 mg.hour/L; Css,avg = 2.79 ± 0.90 mg/L; CL = 0.0276 L/h/kg; V1 = 0.0939 L/kg; V2 = 0.330 L/kg |
| Wang et al., 2021 [49] | Chinese | MDR Gram-negative bacterial infected patients with and without renal insufficiency | Loading dose = 100–150 mg Maintaining dose 2.0 ± 0.5 mg/kg/day (infusion for least 1 h) | Two-compartmental model (PMB1 and PMB2) | Css,min = 0.27 to 8.42 μg/mL; AUCss,0–24h = 80.14 ± 52.70 mg·h/L; Css,avg = 3.33 ± 2.20 μg/mL; Normal renal function: AUCss, 0–24h = 68.63 ± 43.43 mg·h/L; Renal insufficiency: AUCss,0–24h = 93.04 ± 59.52 mg·h/L |
| Wang et al., 2020 [50] | Chinese | MDR Gram-negative bacterial infected patients | Maintaining dose = 100 to 200 mg/day (infusion for 1–2 h) | One- and two-compartment models (PMB1 and PMB2) | V1 = 6.218 L; V2 = 11.922 L; CL = 1.786 L/h; AUCss,0–12 h = 43.64 ± 27.68 mg·h/L |
| Yu et al., 2020 [51] | Chinese | Suspected or documented Gram-negative bacterial infected patients with various renal functions | 100–200 mg/day | One- and two-compartment models | CL = 1.59 ± 0.051 L/h; Vd = 20.5 ± 0.116 L |
| Thamlikitkul et al., 2016 [52] | Thai, American | Suspected or documented Gram-negative bacterial infected patients with and without renal insufficiency | 1.5–2.5 mg/kg/day | One- and two-compartment models (PMB1, B2, B3, and isoleucine-B1) | Normal renal function: AUC = 63.5 ± 16.6 mg·h/L; CL = 2.5 ± 0.4 L/h Renal insufficiency: AUC = 56.0 ± 17.5 mg·h/L; CL = 2.0 ± 0.6 L/h |
| Surovoy et al., 2022 [53] | Russian | Critically ill patients with secondary bacterial infections caused by COVID-19-associated pneumonia | Loading dose = 200–300 mg Maintaining dose = 2.5–3 mg/kg/day | Noncompartmentalism model ELISA | ECMO: AUCss,0–12 h = 43.38 mg·h/L; AUCss,0–24h = 96.8 mg·h/L; CSS,avg = 4.03 mg/L; Cmax = 5.96 mg/L; Cmin = 2.4 mg/L; Tmax = 5 ± 24 min; Vd = 19.73 L Non-ECMO: AUCss,0–12 h = 34.7 mg·h/L; CSS,avg = 2.9 mg/L; Cmax = 5.08 mg/L; Cmin = 2.15 mg/L; Tmax = 5 ± 9; CL = 1.76 L/h; Vd = 30.4 L |
| Luo et al., 2022 [54] | Chinese | Patients with severe CRO infections | Loading dose = 1.73 (1.25–2.73) mg/kg/day; Maintain dose = 2.22 (1.52–3.00) mg/kg/day | Two-compartment model (PMB1 and PMB2) | V1 = 11.7 ± 6.1 L, V2 = 17.9 ± 17.9 L, Non-CRRT: CL = 1.5 ± 4.9 L/h, CRRT: CL = 1.95 ± 9.9 L/h |
| Yang et al., 2022 [55] | Chinese | Patients with CRO infections | 1.5–3.0 mg/kg/day | Two-compartment model (PMB1 and PMB2) | AUCss,0–24h = 58.5 (IQR: 40.6, 77.2) mg·h/L C0 h = 1.24 (IQR: 0.74, 1.93) mg/L |
| Yu et al., 2022 [56] | Chinese | Patients with CRKP bloodstream infections | Loading dose = 2.5 mg/kg (infusion at 3 h) Maintain dose = 1.25 mg/kg every 12 h (infusion for 2 h) | Non and two-compartment models (PMB1, B2, and isoleucine-B1) | Following the loading dose: Cmin = 1.62 ± 0.41 mg/L; Cmax = 5.53 ± 1.80 mg/L; Css,avg = 3.35 ± 1.06 mg/L; AUCss,0–24h = 79.6 ± 25.0 mg·h/L; CL = 1.6 L/h; V1 = 38.6 L; V2 = 7.13 L |
| Pi et al., 2023 [57] | Chinese | Critically ill patients with severe infection, sepsis, or septic shock | Loading dose = 100–200 mg Maintenance dose = 50–100 mg every 12 h | Two-compartment population PK model (PMB1 and PMB2) | AUCss,0–12 h = 17.73 (IQR, 4.10–76.01) mg·h/L, t1/2 = 5.29 (IQR: 0.72, 21.54) h, Css,avg = 1.48 (IQR: 0.34, 6.33) mg/L, Cmax = 4.59 (IQR: 1.61, 14.53) mg/L, CL = 1.67 L/h, V1 = 7.857 L, V2 = 12.668 L |
| Chen et al., 2021 [58] | Chinese | Patients with severe pneumonia | 50 mg every 12 h (infusion for 1 h) | Noncompartment model (PMB1, B2, B3, and isoleucine-B1) | AUCss,0–24h = 72.7 ± 28.9 mg·h/L; Cmax = 5.5 ± 1.9 μg/mL; t1/2 = 8.69 ± 2.49 h |
| Tang et al., 2023 [59] | Chinese | Critically ill patients with nosocomial pneumonia caused by CRO | Loading dose = 100–200 mg Maintenance dose = 40–100 mg every 12 h (infusion for at least 1 h) Aerosol delivery: 25 mg/50 mg twice daily | Population PK model with the method of first-order conditional estimation-extended least square method (PMB1 and PMB2) | CL =1.56 ± 4.5 L/h; V1 = 12.5 ±5.67 L; V2 = 29.9 ± 17.3 L; Cmin,ss = 1.9 (IQR: 1.4, 2.7) mg/L; Cmax,ss = 5.9 (IQR: 4.6, 7.1) mg/L; AUCss,0–24h = 67.2 (IQR: 54.8, 84.2) mg·h/L |
| Zhang et al., 2023 [60] | Chinese | Critically ill patients with nosocomial pneumonia caused by CRO | Loading dose = 100–200 mg Maintenance dose = 40–100 mg every 12 h (infusion for at least 1 h) | Population PK model with the method of first-order conditional estimation-extended least square method (PMB1 and PMB2) | CL = 1.56 L/h; V1 = 12.5 L; V2 = 29.9 L; Cmin,ss = 1.9 (IQR: 1.4, 2.7) mg/L; Cmax,ss = 5.9 (IQR: 4.6, 7.1) mg/L; AUCss,0–24h = 67.2 (IQR: 54.8, 84.2) mg·h/L |
References
- Falagas, M.E.; Kasiakou, S.K.; Saravolatz, L.D. Colistin: The pharmacokinetics/pharmacodynamics of systemically administered revival of polymyxins for the management of multidrug-resistant gram-negative bacterial infections. Clin. Infect. Dis. 2005, 40, 1333–1341. [Google Scholar] [CrossRef] [PubMed]
- Storm, D.R.; Rosenthal, K.S.; Swanson, P.E. Polymyxin and Related Peptide Antibiotics. Annu. Rev. Biochem. 1977, 46, 723–763. [Google Scholar] [CrossRef]
- Koch-Weser, J.; Sidel, V.W.; Federman, E.B.; Kanarek, P.; Finer, D.C.; Eaton, A.E. Adverse effects of sodium colistimethate. Manifestations and specific reaction rates during 317 courses of therapy. Ann. Intern. Med. 1970, 72, 857–868. [Google Scholar] [CrossRef]
- Tallgren, L.G.; Liewendahl, K.; Kuhlbaeck, B. The Therapeutic Success and Nephrotoxicity of Colistin in Acute and Chronic Nephropathies with Impaired Renal Function. Acta Medica Scand. 1965, 177, 717–728. [Google Scholar] [CrossRef]
- Baines, R.D.; Rifkind, D., Jr. Intravenous Administration of Sodium Colistimethate. JAMA 1964, 190, 278–281. [Google Scholar] [CrossRef]
- Fekety, F.R.; Norman, P.S., Jr.; Cluff, L.E. The treatment of gram-negative bacillary infections with colistin. The toxicity and efficacy of large doses in forty-eight patients. Ann. Intern. Med. 1962, 57, 214–229. [Google Scholar] [CrossRef] [PubMed]
- Olesen, S.; Madsen, P.O. Intravenous administration of sodium colistimethate in urinary tract infections. Curr. Ther. Res. Clin. Exp. 1967, 9, 283–287. [Google Scholar] [PubMed]
- Beringer, P. The clinical use of colistin in patients with cystic fibrosis. Curr. Opin. Pulm. Med. 2001, 7, 434–440. [Google Scholar] [CrossRef]
- Gales, A.C.; Jones, R.N.; Sader, H.S. Global assessment of the antimicrobial activity of polymyxin B against 54731 clinical isolates of gram-negative bacilli: Report from the SENTRY antimicrobial surveillance programme (2001–2004). Clin. Microbiol. Infect. 2006, 12, 315–321. [Google Scholar] [CrossRef]
- Bratu, S.; Landman, D.; Haag, R.; Recco, R.; Eramo, A.; Alam, M.; Quale, J. Rapid spread of carbapenem resistant Klebsiella pneumoniae in New York City: A new threat to our antibiotic armamentarium. Arch. Intern. Med. 2005, 165, 1430–1435. [Google Scholar] [CrossRef]
- Prioritization of Pathogens to Guide Discovery, Research and Development of New Antibiotics for Drug-Resistant Bacterial Infections, Including Tuberculosis. Available online: https://www.who.int/publications/i/item/WHO-EMP-IAU-2017.12 (accessed on 3 April 2025).
- Li, J.; Nation, R.L. Old polymyxins are back, is resistance close? Clin. Infect. Dis. 2006, 43, 663–664. [Google Scholar] [CrossRef] [PubMed]
- Bedford Laboratories. Polymyxin B for Injection [Package Insert]; Bedford Laboratories: Bedford, OH, USA, 2007. [Google Scholar]
- Tsuji, B.T.; Pogue, J.M.; Zavascki, A.P.; Paul, M.; Daikos, G.L.; Forrest, A.; Giacobbe, D.R.; Viscoli, C.; Giamarellou, H.; Karaiskos, I.; et al. International consensus guidelines for the optimal use of the polymyxins: Endorsed by the American college of clinical pharmacy (ACCP), European society of clinical microbiology and infectious diseases (ESCMID), Infectious diseases society of America (IDSA), International society for anti-infective pharmacology (ISAP), Society of critical care medicine (SCCM), and Society of infectious diseases pharmacists (SIDP). Pharmacotherapy 2019, 39, 10–39. [Google Scholar]
- Bergen, P.J.; Li, J.; Rayner, C.R.; Nation, R.L. Colistin methanesulfonate is an inactive prodrug of colistin against Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2006, 50, 1953–1958. [Google Scholar] [CrossRef] [PubMed]
- Zavascki, A.P.; Goldani, L.Z.; Li, J.; Nation, R.L. Polymyxin B for the treatment of multidrug-resistant pathogens: A critical review. J. Antimicrob. Chemother. 2007, 60, 1206–1215. [Google Scholar] [CrossRef] [PubMed]
- Kwa, A.; Kasiakou, S.K.; Tam, V.H.; Falagas, M.E. Polymyxin B: Similarities to and differences from colistin (polymyxin E). Expert Rev. Anti-Infect. Ther. 2007, 5, 811–821. [Google Scholar] [CrossRef]
- Nation, R.L.; Velkov, T.; Li, J. Colistin and polymyxin B: Peas in a pod, or chalk and cheese? Clin. Infect. Dis. 2014, 59, 88–94. [Google Scholar] [CrossRef]
- Nation, R.L.; Rigatto, M.H.P.; Falci, D.R.; Zavascki, A.P. Polymyxin Acute Kidney Injury: Dosing and Other Strategies to Reduce Toxicity. Antibiotics 2019, 8, 24. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Jie, F.; Ding, Y.; Qu, H.; Chen, D.; Huang, J. Real-world data on the effects of colistin sulfate and polymyxin B sulfate in the treatment of pneumonia induced by CR-GNB. J. Infect. Dev. Ctries. 2024, 18, 1050–1057. [Google Scholar] [CrossRef]
- Oliveira, M.S.; Prado, G.V.; Costa, S.F.; Grinbaum, R.S.; Levin, A.S. Polymyxin B and colistimethate are comparable as to efficacy and renal toxicity. Diagn. Microbiol. Infect. Dis. 2009, 65, 431–434. [Google Scholar] [CrossRef]
- Akajagbor, D.S.; Wilson, S.L.; Shere-Wolfe, K.D.; Dakum, P.; Charurat, M.E.; Gilliam, B.L. Higher incidence of acute kidney injury with intravenous colistimethate sodium compared with polymyxin B in critically ill patients at a tertiary care medical center. Clin. Infect. Dis. 2013, 57, 1300–1303. [Google Scholar] [CrossRef]
- Phe, K.; Lee, Y.; McDaneld, P.M.; Prasad, N.; Yin, T.; Figueroa, D.A.; Musick, W.L.; Cottreau, J.M.; Hu, M.; Tam, V.H. In vitro assessment and multicenter cohort study of comparative nephrotoxicity rates associated with colistimethate versus polymyxin B therapy. Antimicrob. Agents Chemother. 2014, 5, 2740–2746. [Google Scholar] [CrossRef] [PubMed]
- Tuon, F.; Rigatto, M.H.; Lopes, C.; Kamei, L.K.; Rocha, J.L.; Zavascki, A.P. Risk factors for acute kidney injury in patients treated with polymyxin B or colistin methanesulfonate sodium. Int. J. Antimicrob. Agents 2014, 43, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Rigatto, M.H.; Oliveira, M.S.; Perdigao-Neto, L.V.; Levin, A.S.; Carrilho, C.M.; Tanita, M.T.; Tuon, F.F.; Cardoso, D.E.; Lopes, N.T.; Falci, D.R.; et al. Multicenter prospective cohort study of renal failure in patients treated with colistin versus polymyxin B. Antimicrob. Agents Chemother. 2016, 60, 2443–2449. [Google Scholar] [CrossRef]
- Vardakas, K.Z.; Falagas, M.E. Colistin versus polymyxin B for the treatment of patients with multidrug-resistant gram-negative infections: A systematic review and meta-analysis. Int. J. Antimicrob. Agents 2017, 49, 233–238. [Google Scholar] [CrossRef]
- Crass, R.L.; Rutter, W.C.; Burgess, D.R.; Martin, C.A.; Burgess, D.S. Nephrotoxicity in patients with or without cystic fibrosis treated with polymyxin B compared to colistin. Antimicrob. Agents Chemother. 2017, 61, e02329-16. [Google Scholar] [CrossRef] [PubMed]
- Cheah, S.E.; Wang, J.; Nguyen, V.T.; Turnidge, J.D.; Li, J.; Nation, R.L. New pharmacokinetic/pharmacodynamic studies of systemically administered colistin against Pseudomonas aeruginosa and Acinetobacter baumannii in mouse thigh and lung infection models: Smaller response in lung infection. J. Antimicrob. Chemother. 2015, 12, 3291–3297. [Google Scholar]
- Landersdorfer, C.B.; Wang, J.; Wirth, V.; Chen, K.; Kaye, K.S.; Tsuji, B.; Li, J.; Nation, R.L. Pharmacokinetics/pharmacodynamics of systemically administered polymyxin B against Klebsiella pneumoniae in mouse thigh and lung infection models. J. Antimicrob. Chemother. 2017, 73, 462–468. [Google Scholar] [CrossRef]
- Ly, N.S.; Bulman, Z.P.; Bulitta, J.B.; Baron, C.; Rao, G.G.; Holden, P.N.; Li, J.; Sutton, M.D.; Tsuji, B.T. Optimization of polymyxin B in combination with doripenem to combat mutator Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2016, 60, 2870–2880. [Google Scholar] [CrossRef]
- Smith, N.M.; Bulman, Z.P.; Sieron, A.O.; Bulitta, J.B.; Holden, P.N.; Nation, R.L.; Li, J.; Wright, G.D.; Tsuji, B.T. Pharmacodynamics of dose-escalated ‘front-loading’ polymyxin B regimens against polymyxin-resistant mcr-1-harbouring Escherichia coli. J. Antimicrob. Chemother. 2017, 72, 2297–2303. [Google Scholar] [CrossRef]
- Bulman, Z.P.; Chen, L.; Walsh, T.J.; Satlin, M.J.; Qian, Y.; Bulitta, J.B.; Peloquin, C.A.; Holden, P.N.; Nation, R.L.; Li, J.; et al. Polymyxin combinations combat Escherichia coli harboring mcr-1 and blaNDM-5: Preparation for a postantibiotic era. mBio 2017, 8, e00540-17. [Google Scholar] [CrossRef]
- Zhao, M.; Bulman, Z.P.; Lenhard, J.R.; Satlin, M.J.; Kreiseirth, B.N.; Walsh, T.J.; Marrocco, A.; Bergen, P.J.; Nation, R.L.; Zhang, J.; et al. Pharmacodynamics of colistin and fosfomycin: A ‘treasure trove’ combination combats KPC-producing Klebsiella pneumoniae. J. Antimicrob. Chemother. 2017, 72, 1985–1990. [Google Scholar] [CrossRef] [PubMed]
- Hanafin, P.O.; Kwa, A.; Zavaschi, A.P.; Sandri, A.M.; Scheetz, M.H.; Kubin, C.J.; Shah, J.; Cherng, B.P.Z.; Yin, M.T.; Wang, J.; et al. A population pharmacokinetic model of polymyxin B based on prospective clinical data to inform dosing in hospitalized patients. Clin. Microbiol. Infect. 2023, 29, 1174–1181. [Google Scholar] [CrossRef] [PubMed]
- Kwa, A.L.; Lim, T.P.; Low, J.G.; Hou, J.G.; Kurup, A.; Prince, R.A.; Tam, V.H. Pharmacokinetics of polymyxin B1 in patients with multidrug-resistant gram-negative bacterial infections. Diagn. Microbiol. Infect. Dis. 2008, 60, 163–167. [Google Scholar] [CrossRef]
- Kwa, A.L.; Abdelraouf, K.; Low, J.G.; Tam, V.H. Pharmacokinetics of polymyxin B in a patient with renal insufficiency: A case report. Clin. Infect. Dis. 2011, 52, 1280–1281. [Google Scholar] [CrossRef] [PubMed]
- Surovoy, Y.A.; Burkin, M.A.; Galvidis, I.A.; Sobolev, M.A.; Rende, O.C.; Tsarenko, S.V. Comparative polymyxin B pharmacokinetics in critically ill patients with renal insufficiency and in continuous veno-venous hemodialysis. Eur. J. Clin. Pharmacol. 2023, 79, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Zhang, Z.; Qi, X.; Zhong, Y.; Sun, T.; Chen, L.; Zhu, J.; Lv, X.; Ma, P. Efficiency of polymyxin B treatment against nosocomial infection: A systematic review and meta-analysis. Front. Med. (Lausanne) 2024, 11, 1400757. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.C.; Guo, J.H.; Xie, J.; Xu, M.; Hao, X.; Ma, K.; Rao, Y.F. Population pharmacokinetics of polymyxin B: A systematic review. Ann. Transl. Med. 2022, 10, 231. [Google Scholar] [CrossRef]
- He, R.R.; Yue, G.L.; Dong, M.L.; Wang, J.Q.; Cheng, C. Sepsis Biomarkers: Advancements and Clinical Applications—A Narrative Review. Int. J. Mol. Sci. 2024, 25, 9010. [Google Scholar] [CrossRef]
- Póvoa, P.; Coelho, L.; Dal-Pizzol, F.; Ferrrer, R.; Huttner, A.; Morris, A.C.; Nobre, V.; Ramirez, P.; Rouze, A.; Salluh, J.; et al. How to use biomarkers of infection or sepsis at the bedside: Guide to clinicians. Intensive Care Med. 2023, 49, 142–153. [Google Scholar] [CrossRef]
- Tosca, E.M.; Carlo, A.D.; Ronchi, D.; Magni, P. Model-Informed Reinforcement Learning for Enabling Precision Dosing Via Adaptive Dosing. Clin. Pharmacol. Ther. 2024, 116, 619–636. [Google Scholar] [CrossRef]
- Sun, J.; Su, J.; Xie, Y.; Yin, M.T.; Huang, Y.; Xu, L.; Zhou, Q.; Zhu, B. Plasma IL-6/IL-10 ratio and IL-8, LDH, and HBDH level predict the severity and the risk of death in AIDS patients with pneumocystis pneumonia. J. Immunol. Res. 2016, 2016, 1583951. [Google Scholar] [CrossRef] [PubMed]
- Kaneda, H.; Waddell, T.K.; De Perrot, M.; Bai, X.H.; Gutierrez, C.; Arenovich, T.; Chaparro, C.; Liu, M.; Keshavjee, S. Pre-implantation multiple cytokine mRNA expression analysis of donor lung grafts predicts survival after lung transplantation in humans. Am. J. Transplant. 2006, 6, 544–551. [Google Scholar] [CrossRef] [PubMed]
- Miglis, C.; Rhodes, N.J.; Avedissian, S.N.; Kubin, C.J.; Yin, M.T.; Nelson, B.C.; Pai, M.P.; Scheetz, M.H. Population pharmacokinetics of polymyxin B in acutely ill adult patients. Antimicrob. Agents Chemother. 2018, 62, e01475-17. [Google Scholar] [CrossRef]
- Kubin, C.J.; Nelson, B.C.; Miglis, C.; Scheetz, M.H.; Rhodes, N.J.; Avedissian, S.N.; Cremers, S.; Yin, M.T. Population pharmacokinetics of intravenous polymyxin B from clinical samples. Antimicrob. Agents Chemother. 2018, 62, e01493-17. [Google Scholar] [CrossRef]
- Manchandani, P.; Thamlikitkul, V.; Dubrovskaya, Y.; Babic, J.T.; Lye, D.C.; Lee, L.S.; Tam, V.H. Population pharmacokinetics of polymyxin B. Clin. Pharmacol. Ther. 2018, 104, 534–538. [Google Scholar] [CrossRef] [PubMed]
- Sandri, A.M.; Landersdorfer, C.B.; Jacob, J.; Boniatti, M.M.; Dalarosa, M.G.; Falci, D.R.; Behle, T.F.; Bordinhao, R.C.; Wang, J.; Forrest, A.; et al. Population pharmacokinetics of intravenous polymyxin B in critically ill patients: Implications for selection of dosage regimens. Clin. Infect. Dis. 2013, 57, 524–531. [Google Scholar] [CrossRef]
- Wang, P.; Zhang, Q.; Zhu, Z.; Pei, H.; Feng, M.; Sun, T.; Yang, J.; Zhang, X. Comparing the population pharmacokinetics of and acute kidney injury due to polymyxin B in Chinese patients with or without renal insufficiency. Antimicrob. Agents Chemother. 2021, 65, e01900-20. [Google Scholar] [CrossRef]
- Wang, P.; Zhang, Q.; Zhu, Z.; Feng, M.; Sun, T.; Yang, J.; Zhang, X. Population pharmacokinetics and limited sampling strategy for therapeutic drug monitoring of polymyxin B in Chinese patients with multidrug-resistant gram-negative bacterial infections. Front. Pharmacol. 2020, 5, 829. [Google Scholar] [CrossRef]
- Yu, X.B.; Jiao, Z.; Zhang, C.H.; Dai, Y.; Zhou, Z.Y.; Han, L.; Wen, X.; Sheng, C.C.; Lin, G.Y.; Pan, J.Y. Population pharmacokinetic and optimization of polymyxin B dosing in adult patients with various renal functions. Br. J. Clin. Pharmacol. 2020, 87, 1869–1877. [Google Scholar] [CrossRef]
- Thamlikitkul, V.; Dubrovskaya, Y.; Manchandani, P.; Ngamprasertchai, T.; Boonyasiri, A.; Babic, J.T.; Tam, V.H. Dosing and pharmacokinetics of polymyxin B in patients with renal insufficiency. Antimicrob. Agents Chemother. 2016, 61, e01337. [Google Scholar]
- Surovoy, Y.A.; Burkin, M.A.; Galvidis, I.A.; Bochkov, P.O.; Oganesyan, A.V.; Tsarenko, S.V. Comparative polymyxin B pharmacokinetics in patients receiving extracorporeal membrane oxygenation. J. Antimicrob. Chemother. 2022, 77, 1379–1384. [Google Scholar] [CrossRef]
- Luo, X.; Zhang, Y.; Liang, P.; Zhu, H.; Li, M.; Ding, X.; Zhang, J. Population pharmacokinetics of polymyxin B and dosage strategy in critically ill patients with/without continuous renal replacement therapy. Eur. J. Pharm. Sci. 2022, 175, 106214. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liu, S.; Lu, J.; Sun, T.; Wang, P.; Zhang, X. An area under the concentration–time curve threshold as a predictor of efficacy and nephrotoxicity for individualizing polymyxin B dosing in patients with carbapenem-resistant gram-negative bacteria. Crit. Care. 2022, 26, 320. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Liu, X.; Du, X.; Chen, H.; Zhao, F.; Zhou, Z.; Wang, Y.; Zheng, Y.; Bergen, P.J.; Li, X.; et al. Pharmacokinetics/pharmacodynamics of polymyxin B in patients with bloodstream infection caused by carbapenem-resistant Klebsiella pneumoniae. Front. Pharmacol. 2022, 13, 975066. [Google Scholar] [CrossRef]
- Pi, M.Y.; Cai, C.J.; Zuo, L.Y.; Zheng, J.T.; Zhang, M.L.; Lin, X.B.; Chen, X.; Zhong, G.P.; Xia, Y.Z. Population pharmacokinetics and limited sampling strategies of polymyxin B in critically ill patients. J. Antimicrob. Chemother. 2023, 78, 792–801. [Google Scholar] [CrossRef]
- Chen, W.; Liu, H.; Wang, Q.; Wang, X.; Kong, X.; Wang, X.; Zhang, X.; Zhan, Q.; Li, P. Estimation of the area under concentration-time curve of polymyxin B based on limited sampling concentrations in Chinese patients with severe pneumonia. Eur. J. Clin. Pharmacol. 2021, 77, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.; Li, Y.; Xu, P.; Zhong, Y.; Yang, M.; Ma, W.; Xiang, D.; Zhang, B.; Zhou, Y. Optimization of polymyxin B regimens for the treatment of carbapenem-resistant organism nosocomial pneumonia: A real-world prospective study. Crit. Care 2023, 27, 164. [Google Scholar] [CrossRef]
- Zhang, B.; Li, X.; Chen, Y.; Chen, B.; Cheng, Y.; Lin, H.; Que, W.; Liu, M.; Zhou, L.; Zhang, H.; et al. Determination of polymyxin B in human plasma and epithelial lining fluid using LC-MS/MS and its clinical application in therapeutic drug monitoring. J. Pharm. Biomed. Anal. 2023, 227, 115291. [Google Scholar] [CrossRef]
- Zavascki, A.P.; Goldani, L.Z.; Cao, G.; Superti, S.V.; Lutz, L.; Barth, A.L.; Ramos, F.; Boniatti, M.M.; Nation, R.L.; Li, J. Pharmacokinetics of intravenous polymyxin B in critically ill patients. Clin. Infect. Dis. 2008, 47, 1298–1304. [Google Scholar] [CrossRef]
- Inoue, S.; Howgate, E.M.; Rowland-Yeo, K.; Shimada, T.; Yamazaki, H.; Tucker, G.T.; Rostami-Hodjegan, A. Prediction of in vivo drug clearance from in vitro data. II: Potential inter-ethnic differences. Xenobiotica 2006, 36, 499–513. [Google Scholar] [CrossRef]
- Bart, G.; Lenz, S.; Straka, R.J.; Brundage, R.C. Ethnic and genetic factors in methadone pharmacokinetics: A population pharmacokinetic study. Drug Alcohol Depend. 2014, 145, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Ngaimisi, E.; Habtewold, A.; Minzi, O.; Makonnen, E.; Mugusi, S.; Amogne, W.; Yimer, G.; Riedel, K.D.; Janabi, M.; Aderaye, G.; et al. Importance of ethnicity, CYP2B6 and ABCB1 genotype for efavirenz pharmacokinetics and treatment outcomes: A parallel-group prospective cohort study in two sub-Saharan Africa populations. PLoS ONE 2013, 8, e67946. [Google Scholar] [CrossRef] [PubMed]
- Andrews, L.M.; De Winter, B.C.; Van Gelder, T.; Hesselink, D.A. Consideration of the ethnic prevalence of genotypes in the clinical use of tacrolimus. Pharmacogenomics 2016, 17, 1737–1740. [Google Scholar] [CrossRef]
- Ye, C.; Jin, H.; Zhang, R.; Sun, Y.; Wang, Z.; Sun, W.; Sun, W.; Peng, Q.; Liu, R.; Huang, Y. Variability of warfarin dose response associated with CYP2C9 and VKORC1 gene polymorphisms in Chinese patients. J. Int. Med. Res. 2014, 42, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Ahsan, C.H.; Renwick, A.G.; Macklin, B.; Challenor, V.F.; Waller, D.G.; George, C.F. Ethnic differences in the pharmacokinetics of oral nifedipine. Br. J. Clin. Pharmacol. 1991, 31, 399–403. [Google Scholar] [CrossRef]
- Guo, T.; Mao, G.F.; Xia, D.Y.; Su, X.Y.; Zhao, L.S. Pharmacokinetics of midazolam tablet in different Chinese ethnic groups. J. Clin. Pharm. Ther. 2011, 36, 406–411. [Google Scholar] [CrossRef]
- Tornatore, K.M.; Meaney, C.J.; Wilding, G.E.; Chang, S.S.; Gundroo, A.; Cooper, L.M.; Gray, V.; Shin, K.; Fetterly, G.J.; Prey, J.; et al. Influence of sex and race on mycophenolic acid pharmacokinetics in stable African American and Caucasian renal transplant recipients. Clin. Pharmacokinet. 2015, 54, 423–434. [Google Scholar] [CrossRef]
- Staiano, A.E.; Broyles, S.T.; Gupta, A.K.; Katzmarzyk, P.T. Ethnic and sex differences in visceral, subcutaneous, and total body fat in children and adolescents. Obesity 2013, 21, 1251–1255. [Google Scholar] [CrossRef]
- Liu, A.; Byrne, N.M.; Kagawa, M.; Ma, G.; Kijboonchoo, K.; Nasreddine, L.; Poh, B.K.; Ismil, M.N.; Hills, A.P. Ethnic differences in body fat distribution among Asian pre-pubertal children: A cross-sectional multicenter study. BMC Public Health 2011, 11, 500. [Google Scholar] [CrossRef]
- Deurenberg, P.; Tagliabue, A.; Wang, J.; Wolde-Gebriel, Z. Multi-frequency bioelectrical impedance for the prediction of body water compartments: Validation in different ethnic groups. Asia Pac. J. Clin. Nutr. 1996, 5, 217–221. [Google Scholar]
- Johnson, J.A.; Livingston, T.N. Differences between blacks and whites in plasma protein binding of drugs. Eur. J. Clin. Pharmacol. 1997, 51, 485–488. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.M.; Wang, J.; Pierson, R.N.; Wang, Z.M.; Heymsfield, S.B.; Sardinha, L.B.; Heshka, S. Extracellular water: Greater expansion with age in African Americans. J. Appl. Physiol. 2005, 99, 261–267. [Google Scholar] [CrossRef]
- Kitada, M. Genetic polymorphism of cytochrome P450 enzymes in Asian populations: Focus on CYP2D6. Int. J. Clin. Pharmacol. Res. 2003, 23, 313–315. [Google Scholar]
- Daniel, L.H. Polymorphisms of cytochrome P450 are potential candidates that could potentially help clinicians on the treatment of cardiovascular diseases among Asian populations. Indian Heart J. 2017, 69, 655–656. [Google Scholar] [CrossRef] [PubMed]
- Cropp, C.D.; Yee, S.W.; Giacomini, K.M. Genetic variation in drug transporters in ethnic populations. Clin. Pharmacol. Ther. 2008, 84, 412–416. [Google Scholar] [CrossRef]
- Tanaka, Y.; Manabe, A.; Fukushima, H.; Suzuki, R.; Nakadate, H.; Kondoh, K.; Nakamura, K.; Koh, K.; Fukushima, T.; Tsuchida, M.; et al. Multidrug resistance protein 4 (MRP4) polymorphisms impact the 6-mercaptopurine dose tolerance during maintenance therapy in Japanese childhood acute lymphoblastic leukemia. Pharmacogenom. J. 2015, 15, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Abla, N.; Chinn, L.W.; Nakamura, T.; Liu, L.; Huang, C.C.; Johns, S.J.; Kawamoto, M.; Stryke, D.; Taylor, T.R.; Ferrin, T.E.; et al. The human multidrug resistance protein 4 (MRP4, ABCC4): Functional analysis of a highly polymorphic gene. J. Pharmacol. Exp. Ther. 2008, 325, 859–868. [Google Scholar] [CrossRef]
- Sasaki, T.; Takane, H.; Ogawa, K.; Isagawa, S.; Hirota, T.; Higuchi, S.; Horii, T.; Otsubo, K.; Ieiri, I. Population pharmacokinetic and pharmacodynamic analysis of linezolid and a hematologic side effect, thrombocytopenia, in Japanese patients. Antimicrob. Agents Chemother. 2011, 55, 1867–1873. [Google Scholar] [CrossRef]
- Crass, R.L.; Cojutti, P.G.; Pai, M.P.; Pea, F. Reappraisal of linezolid dosing in renal impairment to improve safety. Antimicrob. Agents Chemother. 2019, 63, e00605-19. [Google Scholar] [CrossRef]
- Ide, T.; Takesue, Y.; Ikawa, K.; Morikawa, N.; Ueda, T.; Takahashi, Y.; Nakajima, K.; Takeda, K.; Nishi, S. Population pharmacokinetics/pharmacodynamics of linezolid in sepsis patients with and without continuous renal replacement therapy. Int. J. Antimicrob. Agents 2018, 51, 745–751. [Google Scholar] [CrossRef]
- Taubert, M.; Zoller, M.; Maier, B.; Frechen, S.; Scharf, C.; Holdt, L.M.; Frey, L.; Vogeser, M.; Fuhr, U.; Zander, J. Predictors of Inadequate Linezolid Concentrations after Standard Dosing in Critically Ill Patients. Antimicrob. Agents Chemother. 2016, 60, 5254–5261. [Google Scholar] [CrossRef] [PubMed]
- Manchandani, P.; Zhou, J.; Ledesma, K.R.; Truong, L.D.; Chow, D.S.; Eriksen, J.L.; Tam, V.H. Characterization of Polymyxin B Biodistribution and Disposition in an Animal Model. Antimicrob. Agents Chemother. 2016, 60, 1029–1034. [Google Scholar] [CrossRef] [PubMed]
- Chousterman, B.G.; Swirski, F.K.; Weber, G.F. Cytokine storm and sepsis disease pathogenesis. Semin. Immunopathol. 2017, 39, 517–528. [Google Scholar] [CrossRef]
- Ríos-Toro, J.J.; Márquez-Coello, M.; García-Álvarez, J.M.; Martin-Aspas, A.; Rivera-Fernandez, R.; de Benito, A.S.; Giron-Gonzalez, J.A. Soluble membrane receptors, interleukin 6, procalcitonin and C reactive protein as prognostic markers in patients with severe sepsis and septic shock. PLoS ONE 2017, 12, e0175254. [Google Scholar] [CrossRef]
- Gibot, S.; Béné, M.C.; Noel, R.; Massin, F.; Guy, J.; Cravoisy, A.; Barraud, D.; Bittencourt, M.D.C.; Quenot, J.P.; Bollaert, P.E.; et al. Combination biomarkers to diagnose sepsis in the critically ill patient. Am. J. Respir. Crit. Care Med. 2012, 186, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Dolin, H.H.; Papadimos, T.J.; Stepkowski, S.; Chen, X.; Pan, Z.K. A novel combination of biomarkers to herald the onset of sepsis prior to the manifestation of symptoms. Shock 2018, 49, 364–370. [Google Scholar] [CrossRef]
- Blot, S.I.; Pea, F.; Lipman, J. The effect of pathophysiology on pharmacokinetics in the critically ill patient—Concepts appraised by the example of antimicrobial agents. Adv. Drug Deliv. Rev. 2014, 77, 3–11. [Google Scholar] [CrossRef]
- Fabre, V.; Karaba, S.; Amoah, J.; Robinson, M.; Jones, G.; Dzintars, K.; Katz, M.; Landrum, B.M.; Qasba, S.; Gupta, P.; et al. The role of procalcitonin in antibiotic decision-making in COVID-19 infection. Infect. Control Hosp. Epidemiol. 2021, 43, 570–575. [Google Scholar] [CrossRef]
- Thorsted, A.; Nielsen, E.I.; Friberg, L.E. Pharmacodynamics of immune response biomarkers of interest for evaluation of treatment effects in bacterial infections. Int. J. Antimicrob. Agents 2020, 56, 106059. [Google Scholar] [CrossRef]
- Li, T.; Zinger, J.; D T Sassen, S.; Juffermans, N.P.; C P Koch, B.; Endeman, H. The relation between inflammatory biomarkers and drug pharmacokinetics in the critically ill patients: A scoping review. Crit. Care 2024, 28, 376. [Google Scholar] [CrossRef]
- Boisson, M.; Jacobs, M.; Gregoire, N.; Gobin, P.; Marchand, S.; Couet, W.; Mimoz, O. Comparison of intrapulmonary and systemic pharmacokinetics of colistin methanesulfonate (CMS) and colistin after aerosol delivery and intravenous administration of CMS in critically ill patients. Antimicrob. Agents Chemother. 2014, 58, 7331–7339. [Google Scholar] [CrossRef] [PubMed]
- Athanassa, Z.E.; Markantonis, S.L.; Fousteri, M.Z.; Myrianthefs, P.M.; Boutzouka, E.G.; Tsakris, A.; Baltopoulos, G.J. Pharmacokinetics of inhaled colistimethate sodium (CMS) in mechanically ventilated critically ill patients. Intensive Care Med. 2012, 38, 1779–1786. [Google Scholar] [CrossRef] [PubMed]
- Honeybourne, D.; Tobin, C.; Jevons, G.; Andrews, J.; Wise, R. Intrapulmonary penetration of linezolid. J. Antimicrob. Chemother. 2003, 51, 1431–1434. [Google Scholar] [CrossRef] [PubMed]
- Honeybourne, D. Antibiotic penetration in the respiratory tract and implications for the selection of antimicrobial therapy. Curr. Opin. Pulm. Med. 1997, 3, 170–174. [Google Scholar] [CrossRef]
- Nix, D.E. Intrapulmonary concentrations of antimicrobial agents. Infect. Dis. Clin. N. Am. 1998, 12, 631–646. [Google Scholar] [CrossRef]
- Baldwin, D.R.; Honeybourne, D.; Wise, R. Pulmonary disposition of antimicrobial agents: In vivo observations and clinical relevance. Antimicrob. Agents Chemother. 1992, 36, 1176–1180. [Google Scholar] [CrossRef]
- Dhanani, J.; Roberts, J.A.; Chew, M.; Lipman, J.; Boots, R.J.; Paterson, D.L.; Fraser, J.F. Antimicrobial chemotherapy and lung microdialysis: A review. Int. J. Antimicrob. Agents 2010, 36, 491–500. [Google Scholar] [CrossRef]
- Zeitlinger, M.; Müller, M.; Joukhadar, C. Lung microdialysis—A powerful tool for the determination of exogenous and endogenous compounds in the lower respiratory tract (mini-review). AAPS J. 2005, 7, E600–E608. [Google Scholar] [CrossRef]
- Kiem, S.; Schentag, J.J. Interpretation of antibiotic concentration ratios measured in epithelial lining fluid. Antimicrob. Agents Chemother. 2008, 52, 24–36. [Google Scholar] [CrossRef]
- Baldwin, D.R.; Honeybourne, D.; Wise, R. Pulmonary disposition of antimicrobial agents: Methodological considerations. Antimicrob. Agents Chemother. 1992, 36, 1171–1175. [Google Scholar] [CrossRef]
- Lamer, C.; De Beco, V.; Soler, P.; Calvat, S.; Fagon, J.Y.; Dombret, M.C.; Farinotti, R.; Chastre, J.; Gibert, C. Analysis of vancomycin entry into pulmonary lining fluid by bronchoalveolar lavage in critically ill patients. Antimicrob. Agents Chemother. 1993, 37, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Brillault, J.; De Castro, W.V.; Harnois, T.; Kitzis, A.; Olivier, J.C.; Couet, W. P-glycoprotein-mediated transport of moxifloxacin in a Calu-3 lung epithelial cell model. Antimicrob. Agents Chemother. 2009, 53, 1457–1462. [Google Scholar] [CrossRef] [PubMed]
- Deguchi, Y.; Sun, J.; Tauchi, Y.; Sakai, S.; Morimoto, K. Distribution characteristics of grepafloxacin, a fluoroquinolone antibiotic, in lung epithelial lining fluid and alveolar macrophage. Drug Metab. Pharmacokinet. 2003, 18, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Deguchi, Y.; Tauchi, Y.; He, Z.; Cheng, G.; Morimoto, K. Distribution characteristics of orally administered olamufloxacin, a newly synthesized fluoroquinolone antibacterial, in lung epithelial lining fluid and alveolar macrophage in rats. Eur. J. Pharm. Biopharm. 2006, 64, 238–245. [Google Scholar] [CrossRef]
- Aoki, M.; Iguchi, M.; Hayashi, H.; Shibasaki, S.; Kurosawa, T.; Hayashi, M. Active uptake of ulifloxacin in plasma to lung that controls its concentrations in epithelial lining fluid. Biol. Pharm. Bull. 2009, 32, 1095–1100. [Google Scholar] [CrossRef]
- Patel, K.B.; Xuan, D.; Tessier, P.R.; Russomanno, J.H.; Quinitiliani, R.; Nightingale, C.H. Comparison of bronchopulmonary pharmacokinetics of clarithromycin and azithromycin. Antimicrob. Agents Chemother. 1996, 40, 2375–2379. [Google Scholar] [CrossRef]
- Conte, J.E.; Golden, J.; Duncan, S.; McKenna, E.; Lin, E.; Zurlinden, E. Single-dose intrapulmonary pharmacokinetics of azithromycin, clarithromycin, ciprofloxacin and cefuroxime in volunteer subjects. Antimicrob Agents Chemother. 1996, 40, 1617–1622. [Google Scholar] [CrossRef]
- Wildfeuer, A.; Laufen, H.; Zimmermann, T. Uptake of azithromycin by various cells and its intracellular activity under in vitro conditions. Antimicrob. Agents Chemother. 1996, 40, 75–79. [Google Scholar] [CrossRef]
- Tomazic, J.M.; Maticic, M.; Kotnik, V.; Simcic, S.; Wraber, B.; Zakotnik, B. Ex vivio effect of azithromycin in human leukocyte bactericidal functions. Antimicrob. Agents Chemother. 1995, 39, 1906. [Google Scholar] [CrossRef]
- Girard, A.E.; Cimochowski, C.R.; Faiella, J.A. Correlation of increased azithromycin concentrations with phagocyte infiltration into sites of localized infection. J. Antimicrob. Chemother. 1996, 37 (Suppl. C), 9–19. [Google Scholar] [CrossRef]
- Tulkens, P.M. Intracellular pharmacokinetics and localization of antibiotics as predictors of their efficacy against intraphagocytic infections. Scand. J. Infect. Dis. Suppl. 1990, 74, 209–217. [Google Scholar]
- Lenhard, J.R.; Thamlikitkul, V.; Silveira, F.P.; Garonzik, S.M.; To, X.; Forrest, A.; Shin, B.S.; Kaye, K.S.; Bulitta, J.B.; Nation, R.L.; et al. Polymyxin-resistant, carbapenem-resistant Acinetobacter baumannii is eradicated by a triple combination of agents that lack individual activity. J. Antimicrob. Chemother. 2017, 72, 1415–1420. [Google Scholar] [CrossRef] [PubMed]
- Landersdorfer, C.B.; Ly, N.S.; Xu, H.; Tsuji, B.T.; Bulitta, J.B. Quantifying subpopulation synergy for antibiotic combinations via mechanism-based modeling and a sequential dosing design. Antimicrob. Agents Chemother. 2013, 57, 2343–2351. [Google Scholar] [CrossRef]
- Lenhard, J.R.; Smith, N.M.; Bulman, Z.P.; Tao, X.; Thamlikitkul, V.; Shin, B.S.; Nation, R.L.; Li, J.; Bulitta, J.B.; Tsuji, B.T. High dose ampicillin/sulbactam combinations combat polymyxin-resistant Acinetobacter baumannii in a hollow-fiber infection model. Antimicrob. Agents Chemother. 2017, 61, e01268-16. [Google Scholar] [CrossRef] [PubMed]
- Qiao, L.Y.; Zuo, W.; Yang, Y.; Liu, X.; Wang, Q.; Yu, J.; Wu, J.; Xu, T.; Jiang, J.; Zhang, B.; et al. Clinical Outcomes and Safety of Intravenous Polymyxin B-based Treatment in Critically Ill Patients with Carbapenem-resistant Acinetobacter baumannii Nosocomial Pneumonia. Int. J. Antimicrob. Agents 2023, 62, 106880. [Google Scholar] [CrossRef] [PubMed]
- The World Medical Association (WMA). Available online: https://www.wma.net/policies-post/wma-declaration-of-helsinki/ (accessed on 27 January 2025).
- Kwon, S.H.; Ahn, H.L.; Han, O.Y.; La, H.O. Efficacy and safety profile comparison of colistin and tigecycline on the extensively drug resistant Acinetobacter baumannii. Biol. Pharm. Bull. 2014, 37, 340–346. [Google Scholar] [CrossRef]
- Conte, J.J.; Golden, J.A.; Kelly, M.G.; Zurlinden, E. Steady-state serum and intrapulmonary pharmacokinetics and pharmacodynamics of tigecycline. Int. J. Antimicrob. Agents 2005, 25, 523–529. [Google Scholar] [CrossRef]
- Conte, J.J.; Golden, J.A.; Kipps, J.; Zurlinden, E. Steady-state plasma and intrapulmonary pharmacokinetics and pharmacodynamics of cethromycin. Antimicrob. Agents Chemother. 2004, 48, 3508–3515. [Google Scholar] [CrossRef]
- Rennard, S.I.; Basset, G.; Lecossier, D.; O’Donnell, K.M.; Pinkston, P.; Martin, P.G.; Crystal, R.G. Estimation of volume of epithelial lining fluid recovered by lavage using urea as marker of dilution. J. Appl. Physiol. 1986, 60, 532–538. [Google Scholar] [CrossRef]
- Sandri, A.M.; Landersdorfer, C.B.; Jacob, J.; Boniatti, M.M.; Dalarosa, M.G.; Falci, D.R.; Behle, T.F.; Saitovitch, D.; Wang, J.; Forrest, A.; et al. Pharmacokinetics of polymyxin B in patients on continuous venovenous haemodialysis. J. Antimicrob. Chemother. 2012, 68, 674–677. [Google Scholar] [CrossRef]
- IBM Support. Available online: https://www.ibm.com/support/pages/how-cite-ibm-spss-statistics-or-earlier-versions-spss (accessed on 27 January 2025).
| Demographics | Total (n = 27) | Treatment Success (n = 22) | Treatment Failure (n = 5) | p |
|---|---|---|---|---|
| Age (years) [IQR] a | 68.0 [63.5, 74.5] | 67.5 [56.8, 72.0] | 75.0 [74.0, 75.0] | 0.218 |
| Male sex, n (%) b | 0.618 | |||
| Male | 19 (70.4) | 15 (68.2) | 4 (80.0) | |
| Female | 8 (29.6) | 7 (31.8) | 1 (20.0) | |
| Weight (kg) [IQR] a | 67.5 [60.0, 76.5] | 66.3 [60.0, 78.5] | 68.0 [56.0, 70.0] | 0.430 |
| BMI [IQR] a | 24.2 [22.2, 25.7] | 24.3 [22.6, 25.9] | 22.7 [21.9, 24.0] | 0.274 |
| Baseline CCr (mL/min) [IQR ] a | 72.5 [34.4–125.6] | 72.8 [34.0–125.4] | 77.8 [45.5–136.7] | 0.350 |
| Cardiovascular diseases, n (%) b | 14 (51.9) | 11 (50.0) | 3 (60.0) | 0.574 |
| Diabetes, n (%) b | 6 (22.2) | 4 (18.2) | 2 (40.0) | 0.320 |
| Renal dysfunction, n (%) b | 12 (44.4) | 9 (40.9) | 3 (60.0) | 0.458 |
| Hepatic dysfunction, n (%) b | 5 (18.5) | 4 (18.2) | 1 (20.0) | 0.510 |
| APACHE II score [IQR] a | 22.0 [17.0, 24.0] | 19.5 [16.0, 22.0] | 22.0 [20.0, 24.0] | 0.049 |
| SOFA score [IQR] a | 12.0 [9.0, 14.0] | 11.0 [9.0, 13.5] | 12.5 [9.5, 15.5] | 0.672 |
| Mechanical ventilation, n (%) b | 25 (92.6) | 20 (90.9) | 5 (100.0) | 0.502 |
| CRRT, n (%) b | 9 (33.3) | 8 (36.4) | 1 (20.0) | 0.460 |
| ECMO, n (%) b | 1 (3.7) | 1 (4.6) | 0 (0.0) | 0.643 |
| Septic shock, n (%) b | 13 (48.2) | 10 (45.5) | 3 (60.0) | 0.574 |
| MODS, n (%) b | 6 (22.2) | 4 (18.2) | 2 (40.0) | 0.340 |
| LOS (days) [IQR] a | 25 [16.0, 50.5] | 22.0 [16.3, 43.3] | 25.5 [16.0, 54.0] | 0.155 |
| ICU LOS (days) [IQR] a | 22 [12.0, 30.0] | 21 [9.3, 26.0] | 22 [16.0, 34.0] | 0.113 |
| Antimicrobial therapy duration (days) [IQR] a | 20 [12.0, 27.0] | 21 [9.3, 27.5] | 16 [16.0, 22.0] | 0.363 |
| PMB therapy duration (days) [IQR] a | 9 [5.0, 14.0] | 7 [4.3, 13.0] | 13 [13.0, 15.0] | 0.002 |
| PMB PK/PD indices [IQR] a | ||||
| AUCss,0–24h | 100.8 [76.9, 137.3] | 111.7 [85.5, 146.9] | 64.7 [58.9, 75.1] | 0.002 |
| Css,avg | 5.2 [3.8, 5.9] | 5.0 [3.7, 5.5] | 6.6 [5.9, 7.7] | 0.903 |
| Css,max | 8.7 [6.6, 10.1] | 8.7 [6.1, 9.7] | 11.1 [9.9, 11.4] | 0.647 |
| Css,min | 2.2 [1.6, 3.1] | 2.1 [1.1, 2.9] | 3.00 [2.9, 3.1] | 0.529 |
| Anti-GNB regimens, n (%) b | ||||
| Monotherapy | 4 (14.8) | 4 (18.2) | 0 (0.0) | 1.000 |
| Two-drug combination | ||||
| PMB + Carbapenase | 9 (33.3) | 7 (31.8) | 2 (40.0) | 1.000 |
| PMB + β-lactam or β-lactam/β-lactamase Inhibitors | 3 (11.1) | 3 (13.6) | 0 (0.0) | 1.000 |
| PMB + Tetracycline | 1 (3.7) | 1 (4.6) | 0 (0.0) | 1.000 |
| PMB + Fluoroquinolone | 3 (11.1) | 3 (13.6) | 0 (0.0) | 1.000 |
| Three or more drug combination | 7 (25.9) | 4 (18.2) | 3 (60.0) | 0.101 |
| Isolated GNM, n (%) b | ||||
| Acinetobacter baumannii | 14 (51.9) | 12 (54.6) | 2 (40.0) | 0.648 |
| Pseudomonas aeruginosa | 5 (18.5) | 3 (13.6) | 2 (40.0) | 1.000 |
| Klebsiella spp. | 9 (33.3) | 8 (36.4) | 1 (20.0) | 0.636 |
| E. coli | 2 (7.4) | 2 (9.1) | 0 (0.0) | 1.000 |
| Burkholderia cepacia | 6 (22.2) | 4 (18.2) | 2 (40.0) | 0.303 |
| Stenotrophomonas maltophilia | 3 (11.1) | 2 (9.1) | 1 (20.0) | 0.474 |
| Others | 6 (22.2) | 5 (22.7) | 1 (20.0.) | 1.000 |
| Isolated GPM, n (%) b | 8 (29.6) | 5 (22.7) | 3 (60.0) | 0.136 |
| Multisites, n (%) b | ||||
| Blood | 6 (22.2) | 2 (9.1) | 4 (80.0) | 0.004 |
| Others | 9 (33.3) | 9 (40.9) | 0 (0.0) | 0.136 |
| Inflammatory factors before PMB [IQR] a | ||||
| TNF-α, (pg/mL) | 17.6 [11.5, 22.5] | 16.6 [11.1, 21.4] | 17.8 [17.5, 23.5] | 0.844 |
| IL-6/IL-10 | 7.7 [2.71, 11.66] | 7.91 [2.9, 14.7] | 5.22 [2.2, 10.8] | 0.424 |
| Inflammatory factors after 3 days of PMB [IQR] a | ||||
| TNF-α, (pg/mL) | 20.6 [10.25, 24.80] | 20.2 [10.0, 25.5] | 21.6 [19.5, 22.6] | 0.604 |
| IL-6/IL-10 | 6.6 [2.36, 13.59] | 6.14 [2.0, 13.0] | 8.3 [6.1, 18.3] | 0.740 |
| Nephrotoxicity, n (%) b | 4 (14.81) | 2 (9.1) | 3 (20.0) | 0.461 |
| Parameter | Mean | SD | CV (%) |
|---|---|---|---|
| Css,max (μg·mL−1) | 8.3 | 0.7 | 8.4 |
| t1/2 (h) | 19.6 | 4.6 | 23.5 |
| Vd (L) | 30.4 | 6.4 | 21.1 |
| CL (L·h−1) | 1.6 | 0.4 | 25.0 |
| Cloading dose (μg·mL−1) | 5.5 | 3.0 | 5.5 |
| Css,avg (μg·mL−1) | 5.1 | 1.1 | 21.6 |
| AUCss,0–24h (h·μg·mL−1) | 110.0 | 8.5 | 15.5 |
| PPBR (%) | 85.7 | 5.1 | 6.0 |
| PK/PD Targets Attainment |
Total (n = 27) |
Treatment Success (n = 22) |
Treatment Failure (n = 5) | p-Value |
|---|---|---|---|---|
| AUCss,0–24h < 50 (h·μg·mL−1), n (%) | 1 (3.70) | 0 (0) | 1 (20.00) | 0.061 |
| AUCss,0–24h of 50–100 (h·μg·mL−1), n (%) | 13 (48.15) | 10 (45.45) | 3 (60.00) | |
| AUCss,0–24h > 100 (h·μg·mL−1), n (%) | 13 (48.15) | 12 (54.55) | 1 (60.00) | |
| Css,avg < 2 (μg·mL−1), n (%) | 1 (3.70) | 0 (0) | 1 (20.00) | 0.140 |
| Css,avg of 2–4 (ug·mL−1), n (%) | 9 (33.33) | 7 (31.82) | 2 (40.00) | |
| Css,avg > 4 (μg·mL−1), n (%) | 17 (62.96) | 15 (68.18) | 2 (40.00) |
| Patient Factor | PMB Concentration | PK Parameters | Pulmonary Distribution | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Cpre | C0 | Tmax | t1/2 | Vd | CL | AUCss,0–24h | ELF/Plasma Ratio (%) | ||
| TNF-α | p-value | 0.287 | 0.363 | 0.59 | 0.726 | 0.034 * | 0.248 | 0.952 | 0.014 * |
| Correlation | −0.146 | 0.182 | 0.109 | 0.071 | 0.42 | −0.23 | 0.012 | −0.485 | |
| IL-6/IL-10 | p-value | 0.042 * | 0.114 | 0.881 | 0.034 * | 0.995 | 0.418 | 0.652 | 0.205 |
| Correlation | −0.638 | −0.311 | −0.03 | −0.62 | 0.023 | 0.162 | −0.091 | 0.182 | |
| Albumin level | p-value | 0.773 | 0.217 | 0.636 | 0.223 | 0.86 | 0.283 | 0.24 | 0.53 |
| Correlation | 0.058 | −0.246 | −0.095 | 0.243 | −0.036 | −0.214 | 0.31 | 0.201 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zuo, W.; Wang, Q.; Su, L.; Yu, J.; Fan, H.; Fu, Q.; Long, Y.; Zhang, B. Biomarker-Driven Pharmacokinetics and Efficacy of Polymyxin B in Critically Ill Patients with XDR-GN Pneumonia. Pharmaceuticals 2025, 18, 586. https://doi.org/10.3390/ph18040586
Zuo W, Wang Q, Su L, Yu J, Fan H, Fu Q, Long Y, Zhang B. Biomarker-Driven Pharmacokinetics and Efficacy of Polymyxin B in Critically Ill Patients with XDR-GN Pneumonia. Pharmaceuticals. 2025; 18(4):586. https://doi.org/10.3390/ph18040586
Chicago/Turabian StyleZuo, Wei, Qianlin Wang, Longxiang Su, Jiaxin Yu, Hongwei Fan, Qiang Fu, Yun Long, and Bo Zhang. 2025. "Biomarker-Driven Pharmacokinetics and Efficacy of Polymyxin B in Critically Ill Patients with XDR-GN Pneumonia" Pharmaceuticals 18, no. 4: 586. https://doi.org/10.3390/ph18040586
APA StyleZuo, W., Wang, Q., Su, L., Yu, J., Fan, H., Fu, Q., Long, Y., & Zhang, B. (2025). Biomarker-Driven Pharmacokinetics and Efficacy of Polymyxin B in Critically Ill Patients with XDR-GN Pneumonia. Pharmaceuticals, 18(4), 586. https://doi.org/10.3390/ph18040586