
Synthesis, Antitumor Activities, and Apoptosis-Inducing Activities of Schiff’s Bases Incorporating Imidazolidine-2,4-dione Scaffold: Molecular Docking Studies and Enzymatic Inhibition Activities

 , ,
, ,  ,
,  and
and
Abstract

1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. In Vitro Cytotoxicity Assay and SAR Study
2.3. In Vitro Cytotoxicity Against Normal Human Cells (WI-38)
2.4. Kinase Inhibition Activity
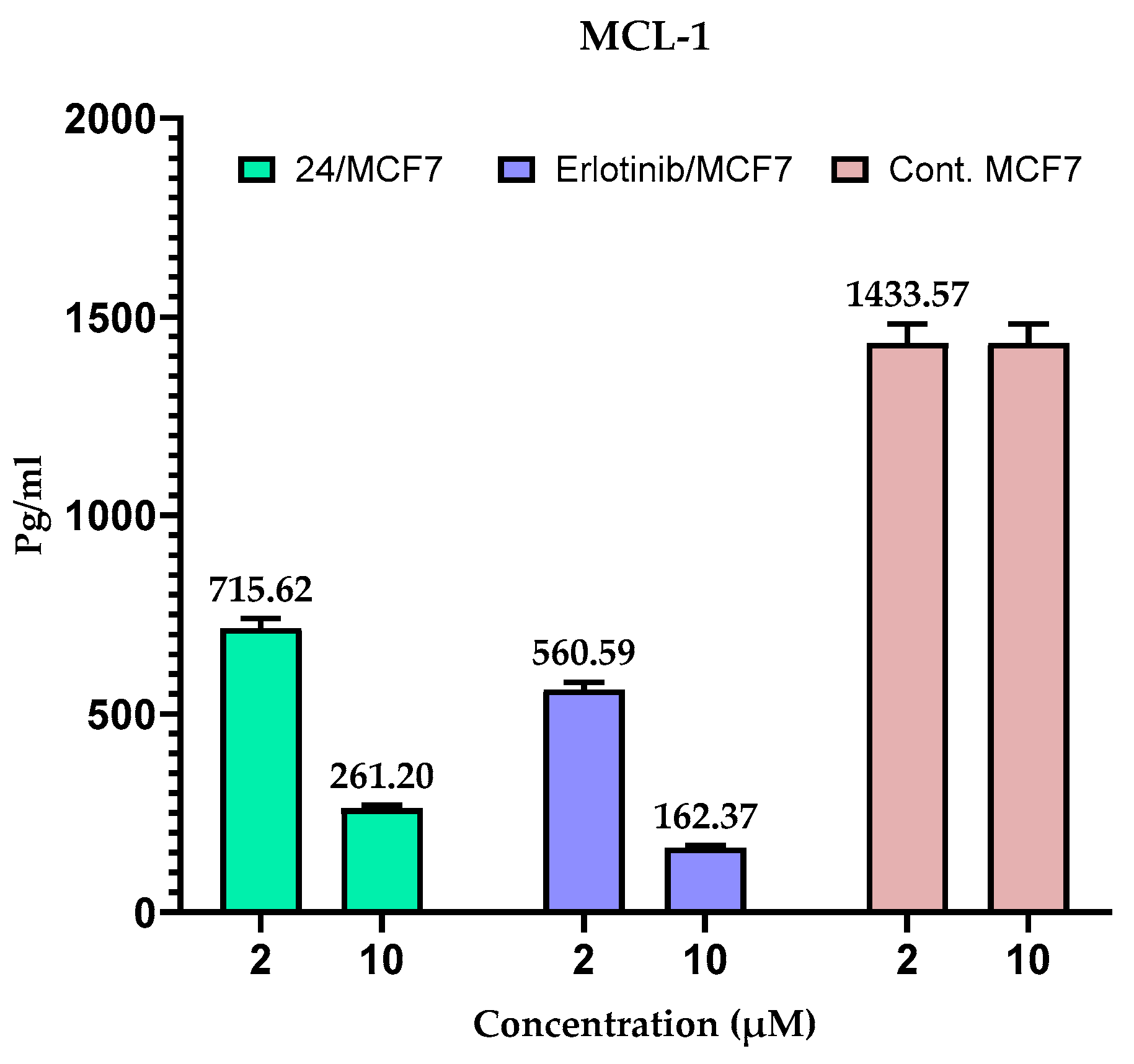
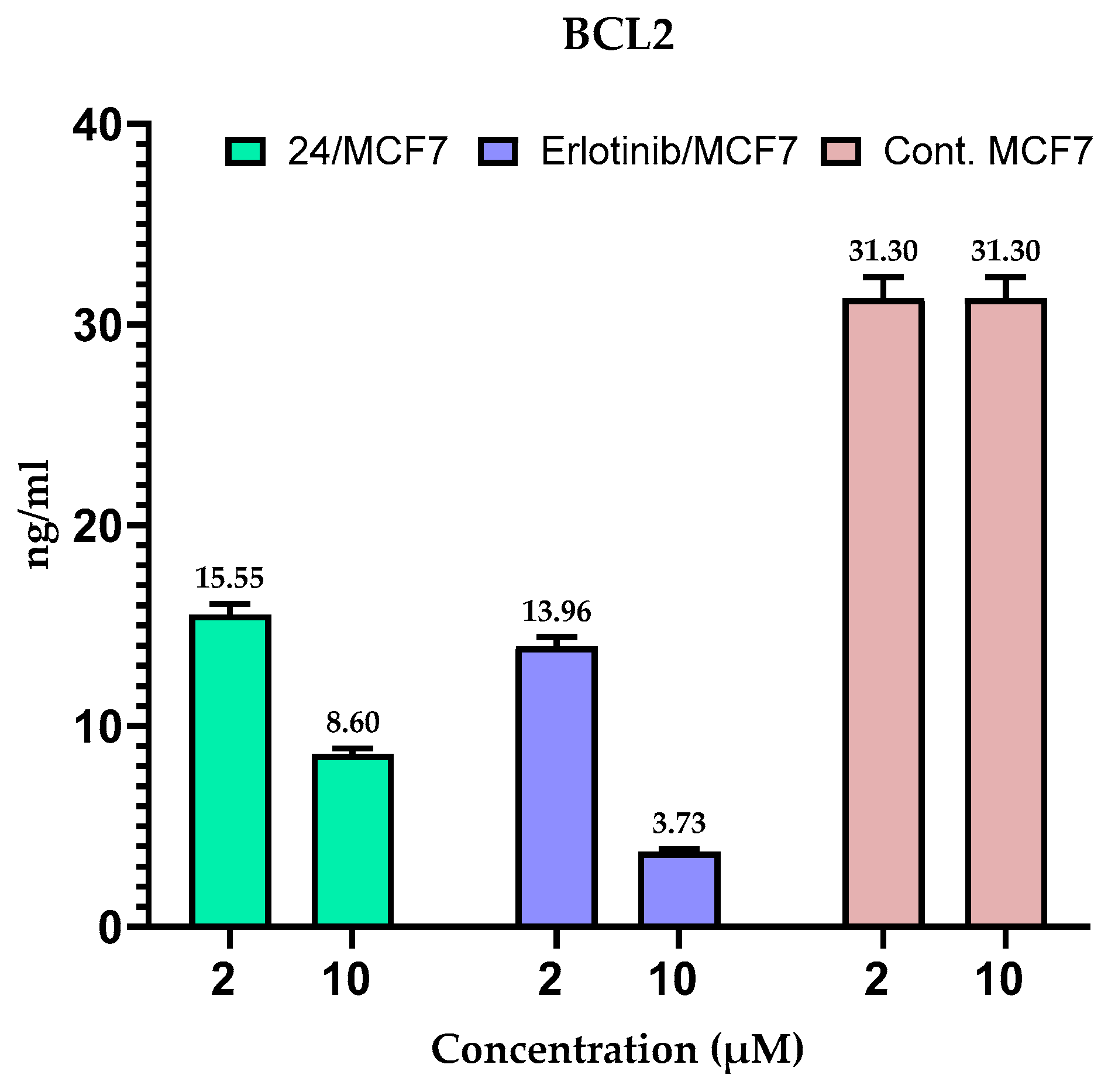
2.5. Measuring the Expression of Anti-Apoptotic Proteins (MCL-1 and BCL-2) in MCF-7 Cells
2.6. Cell Cycle Arrest Analysis
2.7. Drug Likeness
2.8. ADMET Properties
2.9. Molecular Docking Studies
2.9.1. Validation of Docking Approach
2.9.2. Molecular Docking
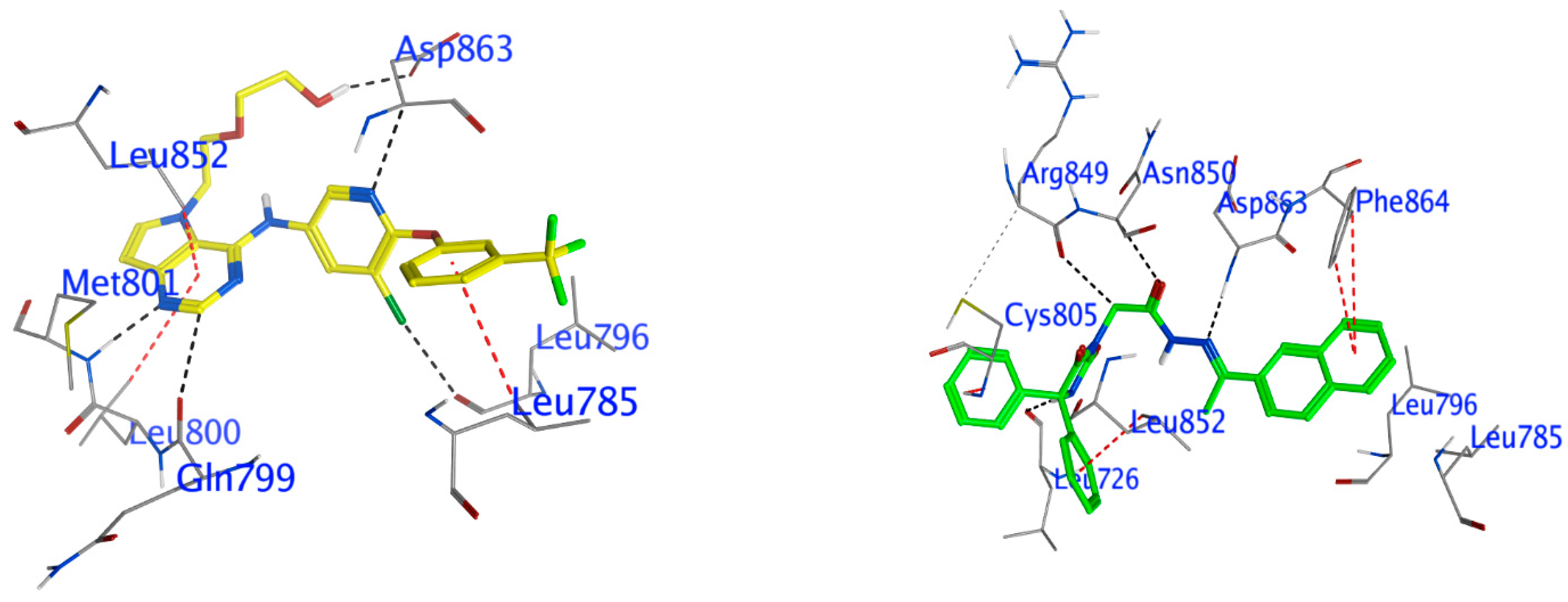
Docking of Compound 24 into HER2
Docking Other Derivatives into HER2
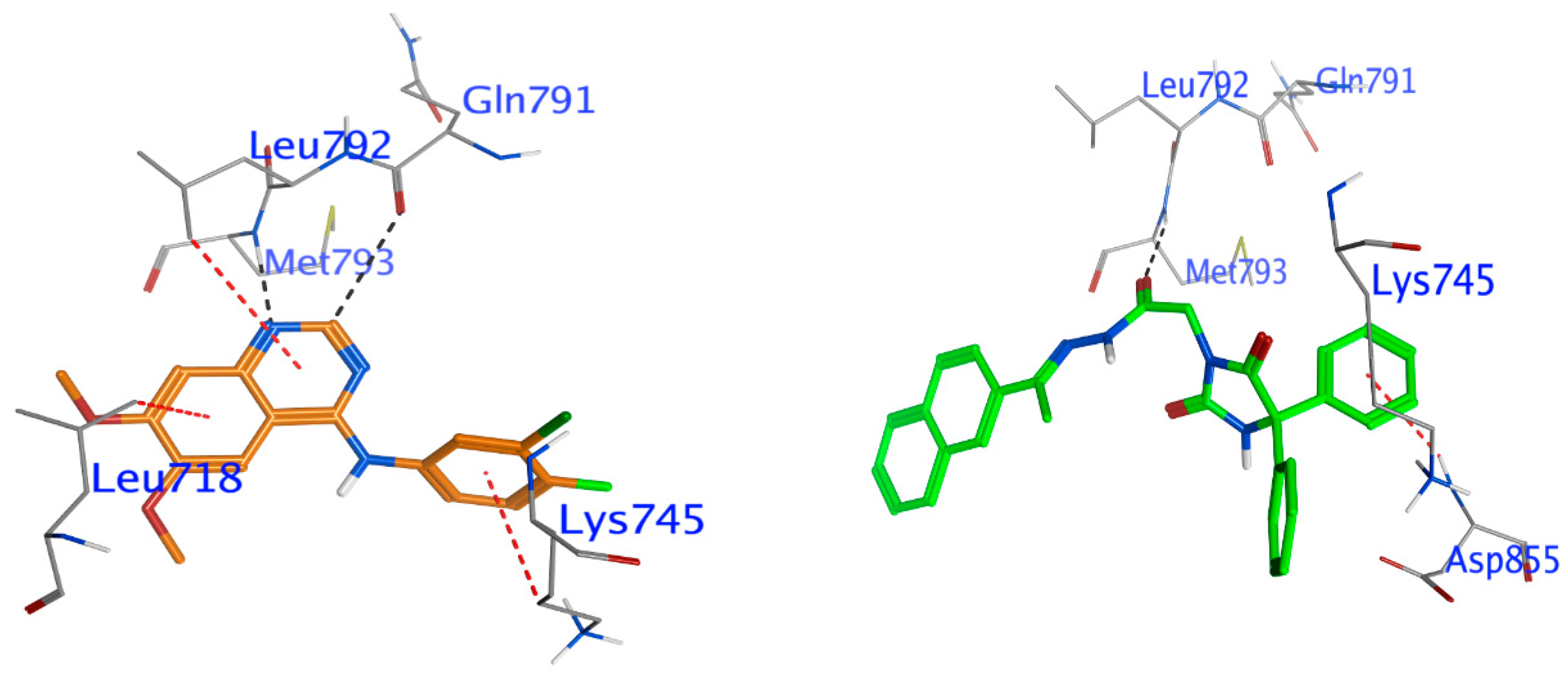
Docking of Compound 24 into EGFR
Docking Other Derivatives into EGFR
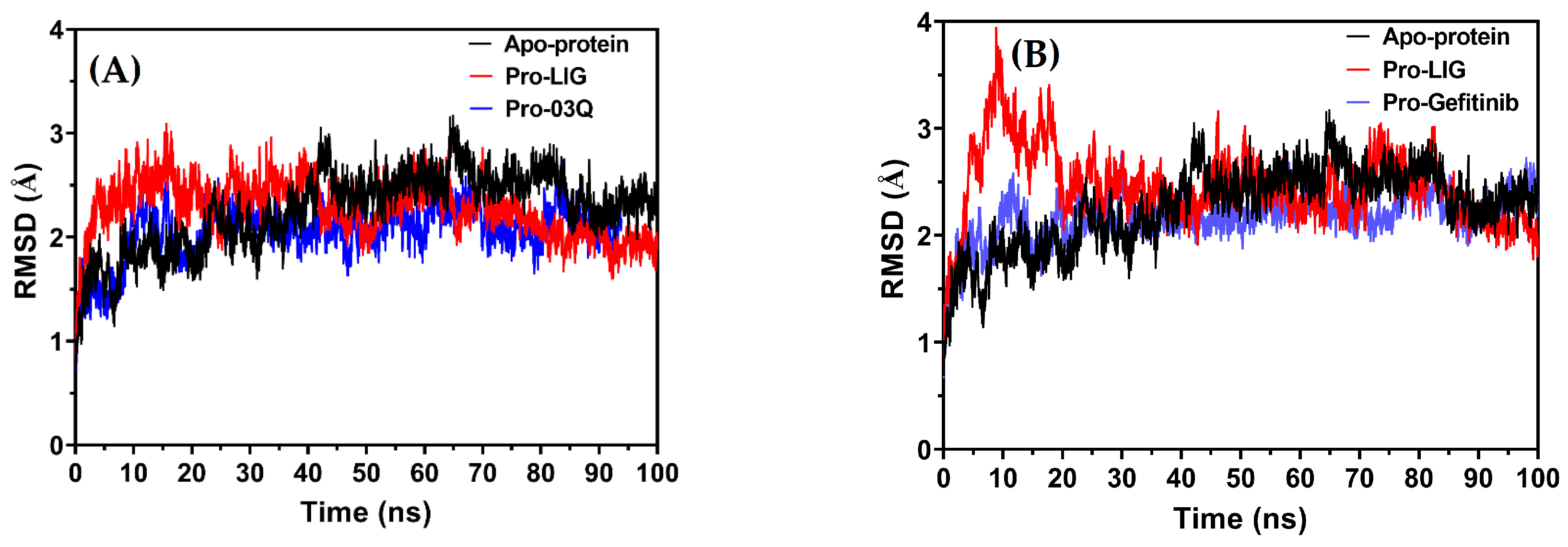
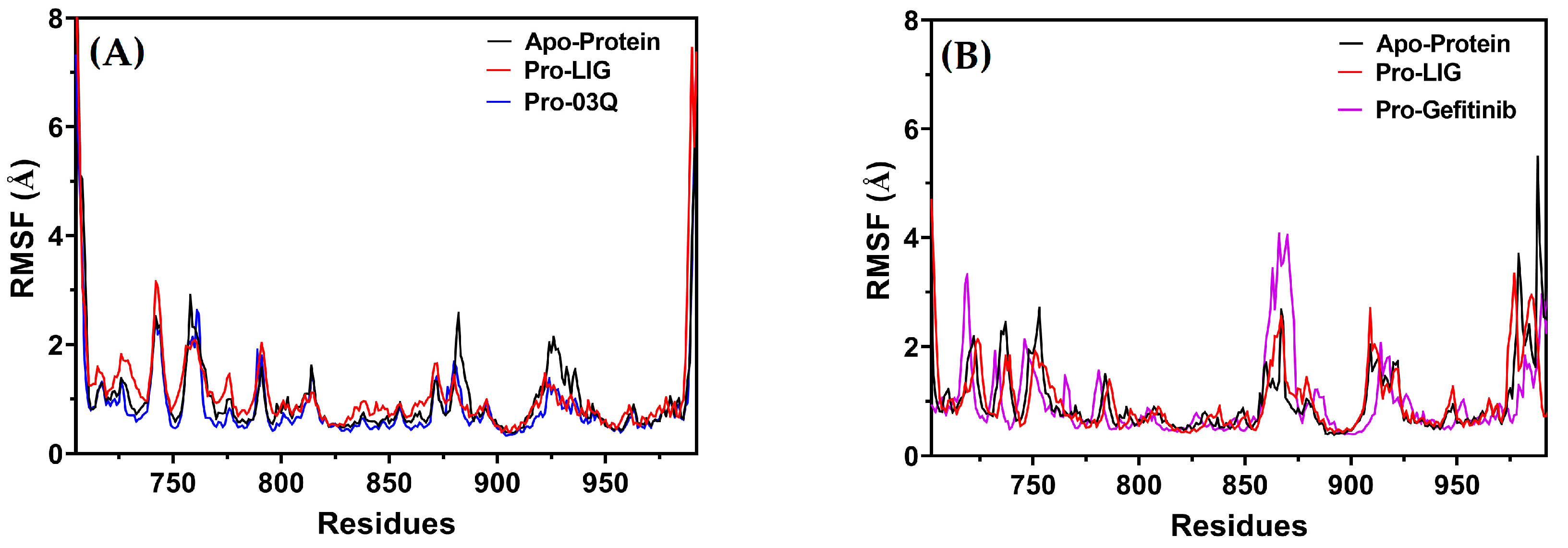
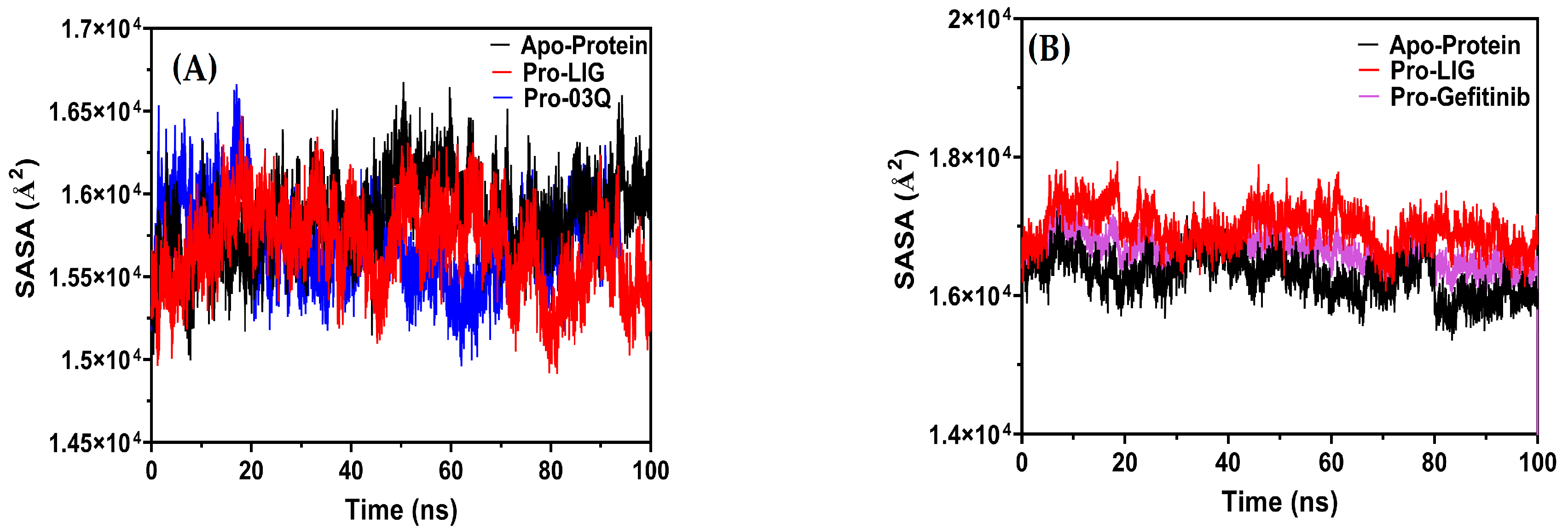
2.9.3. Molecular Dynamics (MD) Simulation
Root Mean Square Deviation (RMSD)
Root Mean Square Fluctuation (RMSF)
Compactness
3. Materials and Methods
3.1. General
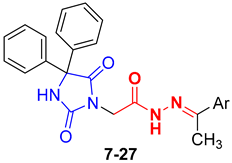
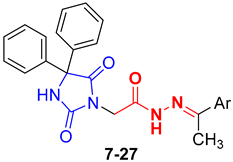
3.1.1. General Procedure for the Synthesis of Compounds 7–27
2-(2,5-Dioxo-4,4-diphenylimidazolidin-1-yl)-N′-(1-phenylethylidene)acetohydrazide (7)
2-(2,5-Dioxo-4,4-diphenylimidazolidin-1-yl)-N′-(1-(o-tolyl)ethylidene)acetohydrazide (8)
2-(2,5-Dioxo-4,4-diphenylimidazolidin-1-yl)-N′-(1-(p-tolyl)ethylidene)acetohydrazide (9)
N′-(1-(4-Chlorophenyl)ethylidene)-2-(2,5-dioxo-4,4-diphenylimidazolidin-1-yl)acetohydrazide (10)
2-(2,5-Dioxo-4,4-diphenylimidazolidin-1-yl)-N′-(1-(4-fluorophenyl)ethylidene)acetohydrazide (11)
2-(2,5-Dioxo-4,4-diphenylimidazolidin-1-yl)-N′-(1-(4-(trifluoromethyl)phenyl)ethylidene)acetohydrazide (12)
2-(2,5-Dioxo-4,4-diphenylimidazolidin-1-yl)-N′-(1-(4-(trifluoromethoxy)phenyl)ethylidene)acetohydrazide (13)
2-(2,5-Dioxo-4,4-diphenylimidazolidin-1-yl)-N′-(1-(2-methoxyphenyl)ethylidene)acetohydrazide (14)
2-(2,5-Dioxo-4,4-diphenylimidazolidin-1-yl)-N′-(1-(3-methoxyphenyl)ethylidene)acetohydrazide (15)
2-(2,5-Dioxo-4,4-diphenylimidazolidin-1-yl)-N′-(1-(4-methoxyphenyl)ethylidene)acetohydrazide (16)
N′-(1-(3,4-Dimethoxyphenyl)ethylidene)-2-(2,5-dioxo-4,4-diphenylimidazolidin-1-yl)acetohydrazide (17)
2-(2,5-Dioxo-4,4-diphenylimidazolidin-1-yl)-N′-(1-(3,4,5-trimethoxyphenyl)ethylidene)acetohydrazide (18)
2-(2,5-Dioxo-4,4-diphenylimidazolidin-1-yl)-N′-(1-(4-nitrophenyl)ethylidene)acetohydrazide (19)
2-(2,5-Dioxo-4,4-diphenylimidazolidin-1-yl)-N′-(1-(pyridin-2-yl)ethylidene)acetohydrazide (20)
2-(2,5-Dioxo-4,4-diphenylimidazolidin-1-yl)-N′-(1-(pyridin-3-yl)ethylidene)acetohydrazide (21)
2-(2,5-Dioxo-4,4-diphenylimidazolidin-1-yl)-N′-(1-(pyridin-4-yl)ethylidene)acetohydrazide (22)
2-(2,5-Dioxo-4,4-diphenylimidazolidin-1-yl)-N′-(1-(naphthalen-1-yl)ethylidene)acetohydrazide (23)
2-(2,5-Dioxo-4,4-diphenylimidazolidin-1-yl)-N′-(1-(naphthalen-2-yl)ethylidene)acetohydrazide (24)
2-(2,5-Dioxo-4,4-diphenylimidazolidin-1-yl)-N′-(1-(furan-2-yl)ethylidene)acetohydrazide (25)
2-(2,5-Dioxo-4,4-diphenylimidazolidin-1-yl)-N′-(1-(thiophen-2-yl)ethylidene)acetohydrazide (26)
N′-(1-(5-Chlorothiophen-2-yl)ethylidene)-2-(2,5-dioxo-4,4-diphenylimidazolidin-1-yl)acetohydrazide (27)
3.2. Biological Activity
3.2.1. In Vitro Cytotoxicity Assay
3.2.2. In Vitro Kinase Inhibition Assay
3.2.3. Apoptosis Assay
3.2.4. Cell Cycle Analysis
3.3. Molecular Docking Methodology of the Selected Compound
3.3.1. Protein Selection and Preparation
3.3.2. Compound and Reference Inhibitor Preparation
3.3.3. Docking Process
3.3.4. MD Simulation Studies
3.4. ADMET Prediction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yahya, E.B.; Alqadhi, A.M. Recent trends in cancer therapy: A review on the current state of gene delivery. Life Sci. 2021, 269, 119087. [Google Scholar] [PubMed]
- Senwar, K.R.; Reddy, T.S.; Thummuri, D.; Sharma, P.; Naidu, V.; Srinivasulu, G.; Shankaraiah, N. Design, synthesis and apoptosis inducing effect of novel (Z)-3-(3′-methoxy-4′-(2-amino-2-oxoethoxy)-benzylidene) indolin-2-ones as potential antitumour agents. Eur. Med. Chem. 2016, 118, 34–46. [Google Scholar]
- Iwaloye, O.; Ottu, P.O.; Olawale, F.; Babalola, O.O.; Elekofehinti, O.O.; Kikiowo, B.; Adegboyega, A.E.; Ogbonna, H.N.; Adeboboye, C.F.; Folorunso, I.M.; et al. Computer-aided drug design in anti-cancer drug discovery: What have we learnt and what is the way forward? Inform. Med. Unlocked 2023, 41, 101332. [Google Scholar] [CrossRef]
- Li, G.; Li, T.; Fu, W.; Hu, S. Chapter 16—EGFR- and VEGF(R)-targeted small molecules show synergistic activity in colorectal cancer models refractory to combinations of monoclonal antibodies. In Novel Sensitizing Agents for Therapeutic Anti-EGFR Antibodies; Hu, S., Ed.; Academic Press: Cambridge, MA, USA, 2023; pp. 119–123. [Google Scholar]
- Cha, M.Y.; Lee, K.; Kim, M.; Song, J.Y.; Lee, K.H.; Park, J.; Chae, Y.J.; Kim, Y.H.; Suh, K.H.; Lee, G.S. Antitumor activity of HM781-36B, a highly effective pan-HER inhibitor in erlotinib-resistant NSCLC and other EGFR-dependent cancer models. Int. J. Cancer 2012, 130, 2445–2454. [Google Scholar]
- Dallavalle, S.; Dobričić, V.; Lazzarato, L.; Gazzano, E.; Machuqueiro, M.; Pajeva, I.; Tsakovska, I.; Zidar, N.; Fruttero, R. Improvement of conventional anti-cancer drugs as new tools against multidrug resistant tumors. Drug Resist. Updat. 2020, 50, 100682. [Google Scholar] [PubMed]
- Ferrara, N. Role of vascular endothelial growth factor in physiologic and pathologic angiogenesis: Therapeutic implications. Semin. Oncol. 2002, 29, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Lichtenberger, B.M.; Tan, P.K.; Niederleithner, H.; Ferrara, N.; Petzelbauer, P.; Sibilia, M. Autocrine VEGF signaling synergizes with EGFR in tumor cells to promote epithelial cancer development. Cell 2010, 140, 268–279. [Google Scholar]
- Li, W.; Zhang, K.; Wang, W.; Liu, Y.; Huang, J.; Zheng, M.; Li, L.; Zhang, X.; Xu, M.; Chen, G. Combined inhibition of HER2 and VEGFR synergistically improves therapeutic efficacy via PI3K-AKT pathway in advanced ovarian cancer. J. Exp. Clin. Cancer Res. 2024, 43, 56. [Google Scholar] [PubMed]
- Harris, A.L.; Generali, D.G. Chapter 15—Inhibitors of tumor angiogenesis. In Cancer Drug Design and Discovery; Neidle, S., Ed.; Academic Press: New York, NJ, USA, 2008; pp. 351–381. [Google Scholar]
- Hu, L.; Fan, M.; Shi, S.; Song, X.; Wang, F.; He, H.; Qi, B. Dual target inhibitors based on EGFR: Promising anticancer agents for the treatment of cancers (2017-). Eur. J. Med. Chem. 2022, 227, 113963. [Google Scholar]
- Sharma, P.S.; Sharma, R.; Tyagi, T. Receptor tryosine kinase inhibitors as potent weapons in war against cancers. Curr. Pharm. Des. 2009, 15, 758–776. [Google Scholar]
- Shaban, N.; Kamashev, D.; Emelianova, A.; Buzdin, A. Targeted inhibitors of EGFR: Structure, biology, biomarkers, and clinical applications. Cells 2023, 13, 47. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.K.; Thakur, G.S.; Jain, S.K. Recent Development in Hydantoins, Thiohydantoins, and Selenohydantoins as Anticancer Agents: Structure-activity Relationship and Design Strategies. Mini Rev. Med. Chem. 2025, 25, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Kim, S.; Shin, D. Recent applications of hydantoin and thiohydantoin in medicinal chemistry. Eur. J. Med. Chem. 2019, 164, 517–545. [Google Scholar] [CrossRef]
- Hmuda, S.; Trišović, N.; Rogan, J.; Poleti, D.; Vitnik, Ž.; Vitnik, V.; Valentić, N.; Božić, B.; Ušćumlić, G. New derivatives of hydantoin as potential antiproliferative agents: Biological and structural characterization in combination with quantum chemical calculations. Monatshefte Für Chem. Chem. Mon. 2014, 145, 821–833. [Google Scholar] [CrossRef]
- Azizmohammadi, M.; Khoobi, M.; Ramazani, A.; Emami, S.; Zarrin, A.; Firuzi, O.; Miri, R.; Shafiee, A. 2H-chromene derivatives bearing thiazolidine-2, 4-dione, rhodanine or hydantoin moieties as potential anticancer agents. Eur. J. Med. Chem. 2013, 59, 15–22. [Google Scholar] [CrossRef]
- Zuliani, V.; Carmi, C.; Rivara, M.; Fantini, M.; Lodola, A.; Vacondio, F.; Bordi, F.; Plazzi, P.V.; Cavazzoni, A.; Galetti, M. 5-Benzylidene-hydantoins: Synthesis and antiproliferative activity on A549 lung cancer cell line. Eur. J. Med. Chem. 2009, 44, 3471–3479. [Google Scholar] [CrossRef]
- Meyers, M.J.; Anderson, E.J.; McNitt, S.A.; Krenning, T.M.; Singh, M.; Xu, J.; Zeng, W.; Qin, L.; Xu, W.; Zhao, S. Evaluation of spiropiperidine hydantoins as a novel class of antimalarial agents. Bioorganic Med. Chem. 2015, 23, 5144–5150. [Google Scholar] [CrossRef]
- Nishinami, S.; Ikeda, K.; Nagao, T.; Koyama, A.H.; Arakawa, T.; Shiraki, K. Aromatic interaction of hydantoin compounds leads to virucidal activities. Biophys. Chem. 2021, 275, 106621. [Google Scholar]
- Zhang, M.; Liang, Y.; Li, H.; Liu, M.; Wang, Y. Design, synthesis, and biological evaluation of hydantoin bridged analogues of combretastatin A-4 as potential anticancer agents. Bioorganic Med. Chem. 2017, 25, 6623–6634. [Google Scholar] [CrossRef]
- Żesławska, E.; Kincses, A.; Spengler, G.; Nitek, W.; Wyrzuc, K.; Kieć-Kononowicz, K.; Handzlik, J. The 5-aromatic hydantoin-3-acetate derivatives as inhibitors of the tumour multidrug resistance efflux pump P-glycoprotein (ABCB1): Synthesis, crystallographic and biological studies. Bioorganic Med. Chem. 2016, 24, 2815–2822. [Google Scholar] [CrossRef]
- Alkahtani, H.M.; Alanazi, M.M.; Aleanizy, F.S.; Alqahtani, F.Y.; Alhoshani, A.; Alanazi, F.E.; Almehizia, A.A.; Abdalla, A.N.; Alanazi, M.G.; El-Azab, A.S.; et al. Synthesis, anticancer, apoptosis-inducing activities and EGFR and VEGFR2 assay mechanistic studies of 5,5-diphenylimidazolidine-2,4-dione derivatives: Molecular docking studies. Saudi Pharm. J. 2019, 27, 682–693. [Google Scholar] [CrossRef] [PubMed]
- Alanazi, A.M.; El-Azab, A.S.; Al-Swaidan, I.A.; Maarouf, A.R.; El-Bendary, E.R.; Abu El-Enin, M.A.; Abdel-Aziz, A.A. Synthesis, single-crystal, in vitro antitumor evaluation and molecular docking of 3-substitued 5, 5-diphenylimidazolidine-2, 4-dione derivatives. Med. Chem. Res. 2013, 22, 6129–6142. [Google Scholar] [CrossRef]
- Aqeel, A.W.; Al-Sha’er, M.A.; Ayoub, R.; Jarrar, Q.; Alelaimat, M.A. Novel hydantoin derivatives: Synthesis and biological activity evaluation. Results Chem. 2023, 6, 101118. [Google Scholar] [CrossRef]
- da Silva, C.M.; Silva, M.M.; Reis, F.S.; Ruiz, A.L.T.G.; de Carvalho, J.E.; Santos, J.C.C.; Figueiredo, I.M.; Alves, R.B.; Modolo, L.V.; de Fátima, Â. Studies on free radical scavenging, cancer cell antiproliferation, and calf thymus DNA interaction of Schiff bases. J. Photochem. Photobiol. B Biol. 2017, 172, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.; Kuman, M.M.; Palvai, S.; Sengupta, P.; Basu, S. Impairing powerhouse in colon cancer cells by hydrazide–hydrazone-based small molecule. ACS Omega 2018, 3, 1470–1481. [Google Scholar] [CrossRef]
- Han, M.İ.; Atalay, P.; Tunç, C.Ü.; Ünal, G.; Dayan, S.; Aydın, Ö.; Küçükgüzel, Ş.G. Design and synthesis of novel (S)-Naproxen hydrazide-hydrazones as potent VEGFR-2 inhibitors and their evaluation in vitro/in vivo breast cancer models. Bioorganic Med. Chem. 2021, 37, 116097. [Google Scholar] [CrossRef]
- Handzlik, J.; Bajda, M.; Zygmunt, M.; Maciąg, D.; Dybała, M.; Bednarski, M.; Filipek, B.; Malawska, B.; Kieć-Kononowicz, K. Antiarrhythmic properties of phenylpiperazine derivatives of phenytoin with α1-adrenoceptor affinities. Bioorganic Med. Chem. 2012, 20, 2290–2303. [Google Scholar] [CrossRef]
- Pfeffer, C.M.; Singh, A.T. Apoptosis: A target for anticancer therapy. Int. J. Mol. Sci. 2018, 19, 448. [Google Scholar] [CrossRef]
- Coloff, J.L.; Macintyre, A.N.; Nichols, A.G.; Liu, T.; Gallo, C.A.; Plas, D.R.; Rathmell, J.C. Akt-dependent glucose metabolism promotes Mcl-1 synthesis to maintain cell survival and resistance to Bcl-2 inhibition. Cancer Res. 2011, 71, 5204–5213. [Google Scholar] [CrossRef]
- Han, Z.; Liang, J.; Li, Y.; He, J. Drugs and clinical approaches targeting the antiapoptotic protein: A Review. BioMed Res. Int. 2019, 2019, 1212369. [Google Scholar] [CrossRef]
- Peng, Z.; Gillissen, B.; Richter, A.; Sinnberg, T.; Schlaak, M.S.; Eberle, J. Enhanced Apoptosis and Loss of Cell Viability in Melanoma Cells by Combined Inhibition of ERK and Mcl-1 Is Related to Loss of Mitochondrial Membrane Potential, Caspase Activation and Upregulation of Proapoptotic Bcl-2 Proteins. Int. J. Mol. Sci. 2023, 24, 4961. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Gurkan-Cavusoglu, E. A comprehensive review of computational cell cycle models in guiding cancer treatment strategies. NPJ Syst. Biol. Appl. 2024, 10, 71. [Google Scholar]
- Sukhachev, V.S.; Dmitriev, A.V.; Ivanov, S.M.; Savosina, P.I.; Druzhilovskiy, D.S.; Filimonov, D.A.; Poroikov, V.V. Assessment of the Efficiency of Selecting Promising Compounds During Virtual Screening Based on Various Estimations of Drug-Likeness. Pharm. Chem. J. 2025, 58, 1388–1396. [Google Scholar]
- Kar, S.; Leszczynski, J. Recent advances of computational modeling for predicting drug metabolism: A perspective. Curr. Drug Metab. 2017, 18, 1106–1122. [Google Scholar] [PubMed]
- Patel, H.M.; Noolvi, M.N.; Sharma, P.; Jaiswal, V.; Bansal, S.; Lohan, S.; Kumar, S.S.; Abbot, V.; Dhiman, S.; Bhardwaj, V. Quantitative structure–activity relationship (QSAR) studies as strategic approach in drug discovery. Med. Chem. Res. 2014, 23, 4991–5007. [Google Scholar]
- Yunta, M.J.R. Using molecular modelling to study interactions between molecules with biological activity. In Bioinformatics; IntechOpen: Rijeka, Croatia, 2012; p. 92. [Google Scholar]
- Guerrab, W.; Akachar, J.; Jemli, M.E.; Abudunia, A.; Ouaabou, R.; Alaoui, K.; Ibrahimi, A.; Ramli, Y. Synthesis, molecular docking, ADMET evaluation and in vitro cytotoxic activity evaluation on RD and L20B cell lines of 3-substituted 5, 5-diphenylimidazolidine-2, 4-dione derivatives. J. Biomol. Struct. Dyn. 2023, 41, 4592–4600. [Google Scholar]
- Al-Suwaidan, I.A.; Alanazi, A.M.; Alaa, A.; Mohamed, M.A.; El-Azab, A.S. Design, synthesis and biological evaluation of 2-mercapto-3-phenethylquinazoline bearing anilide fragments as potential antitumor agents: Molecular docking study. Bioorganic Med. Chem. Lett. 2013, 23, 3935–3941. [Google Scholar]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar]
- Alkahtani, H.M.; Abdalla, A.N.; Obaidullah, A.J.; Alanazi, M.M.; Almehizia, A.A.; Alanazi, M.G.; Ahmed, A.Y.; Alwassil, O.I.; Darwish, H.W.; Alaa, A. Synthesis, cytotoxic evaluation, and molecular docking studies of novel quinazoline derivatives with benzenesulfonamide and anilide tails: Dual inhibitors of EGFR/HER2. Bioorganic Chem. 2020, 95, 103461. [Google Scholar]
- Abdel-Aziz, A.A.; El-Azab, A.S.; AlSaif, N.A.; Obaidullah, A.J.; Al-Obaid, A.M.; Al-Suwaidan, I.A. Synthesis, potential antitumor activity, cell cycle analysis, and multitarget mechanisms of novel hydrazones incorporating a 4-methylsulfonylbenzene scaffold: A molecular docking study. J. Enzyme Inhib. Med. Chem. 2021, 36, 1520–1538. [Google Scholar]
- Filipe, H.A.; Loura, L.M. Molecular dynamics simulations: Advances and applications. Molecules 2022, 27, 2105. [Google Scholar] [CrossRef] [PubMed]
- Arnittali, M.; Rissanou, A.N.; Harmandaris, V. Structure of biomolecules through molecular dynamics simulations. Procedia Comp. Sci. 2019, 156, 69–78. [Google Scholar]
- Weiner, S.J.; Kollman, P.A.; Case, D.A.; Singh, U.C.; Ghio, C.; Alagona, G.; Profeta, S.; Weiner, P. A new force field for molecular mechanical simulation of nucleic acids and proteins. J. Am. Chem. Soc. 1984, 106, 765–784. [Google Scholar]
- Sneha, P.; Doss, C.G.P. Molecular dynamics: New frontier in personalized medicine. Adv. Protein Chem. Struct. Biol. 2016, 102, 181–224. [Google Scholar]
- Ausaf Ali, S.; Imtaiyaz Hassan, M.; Islam, A.; Ahmad, F. A review of methods available to estimate solvent-accessible surface areas of soluble proteins in the folded and unfolded states. Curr. Protein Pept. Sci. 2014, 15, 456–476. [Google Scholar]
- Alaa, A.; El-Azab, A.S.; Abou-Zeid, L.A.; ElTahir, K.E.H.; Abdel-Aziz, N.I.; Ayyad, R.R.; Al-Obaid, A.M. Synthesis, anti-inflammatory, analgesic and COX-1/2 inhibition activities of anilides based on 5, 5-diphenylimidazolidine-2, 4-dione scaffold: Molecular docking studies. Eur. J. Med. Chem. 2016, 115, 121–131. [Google Scholar]
- Denizot, F.; Lang, R. Rapid colorimetric assay for cell growth and survival: Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J. Immunol. Methods 1986, 89, 271–277. [Google Scholar] [CrossRef]
- Supino, R. MTT assays. In In Vitro Toxicity Testing Protocols; Springer Nature: Berlin/Heidelberg, Germany, 1995; pp. 137–149. [Google Scholar]
- Nakamura, J.L. The epidermal growth factor receptor in malignant gliomas: Pathogenesis and therapeutic implications. Expert Opin. Ther. Targets 2007, 11, 463–472. [Google Scholar]
- Tai, W.; Mahato, R.; Cheng, K. The role of HER2 in cancer therapy and targeted drug delivery. J. Control. Release 2010, 146, 264–275. [Google Scholar]
- Takahashi, H.; Chen, M.C.; Pham, H.; Matsuo, Y.; Ishiguro, H.; Reber, H.A.; Takeyama, H.; Hines, O.J.; Eibl, G. Simultaneous knock-down of Bcl-xL and Mcl-1 induces apoptosis through Bax activation in pancreatic cancer cells. Biochim. Biophys. Acta 2013, 1833, 2980–2987. [Google Scholar]
- Nicoletti, I.; Migliorati, G.; Pagliacci, M.C.; Grignani, F.; Riccardi, C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J. Immunol. Methods 1991, 139, 271–279. [Google Scholar] [CrossRef] [PubMed]
- McTigue, M.; Murray, B.W.; Chen, J.H.; Deng, Y.; Solowiej, J.; Kania, R.S. Molecular conformations, interactions, and properties associated with drug efficiency and clinical performance among VEGFR TK inhibitors. Proc. Natl. Acad. Sci. USA 2012, 109, 18281–18289. [Google Scholar] [PubMed]
- Sabry, M.A.; Ghaly, M.A.; Maarouf, A.R.; El-Subbagh, H.I. New thiazole-based derivatives as EGFR/HER2 and DHFR inhibitors: Synthesis, molecular modeling simulations and anticancer activity. Eur. J. Med. Chem. 2022, 241, 114661. [Google Scholar]
- Hamdi, A.; Elhusseiny, W.M.; Othman, D.I.; Haikal, A.; Bakheit, A.H.; El-Azab, A.S.; Al-Agamy, M.H.; Alaa, A. Synthesis, antitumor, and apoptosis-inducing activities of novel 5-arylidenethiazolidine-2, 4-dione derivatives: Histone deacetylases inhibitory activity and molecular docking study. Eur. J. Med. Chem. 2022, 244, 114827. [Google Scholar]
- Bhat, M.A.; Naglah, A.M.; Bakheit, A.H.; Al-Omar, M.A.; Ansari, S.A.; Alkahtani, H.M.; Aleanizy, F.S.; Eltayb, E.K.; Alqahtani, F.Y. Novel indole derivatives of dihydropyrimidinone: Synthesis, characterization, molecular docking and antimicrobial activity. J. Mol. Struct. 2023, 1291, 136091. [Google Scholar]
- Brooks, B.R.; Brooks, C.L., III; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [PubMed]
- Bakheit, A.H.; Saquib, Q.; Ahmed, S.; Ansari, S.M.; Al-Salem, A.M.; Al-Khedhairy, A.A. Covalent inhibitors from saudi medicinal plants target RNA-dependent RNA polymerase (RdRp) of SARS-CoV-2. Viruses 2023, 15, 2175. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; He, X.; Vanommeslaeghe, K.; MacKerell, A.D., Jr. Extension of the CHARMM general force field to sulfonyl-containing compounds and its utility in biomolecular simulations. J. Comput. Chem. 2012, 33, 2451–2468. [Google Scholar]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Bakheit, A.H.; Alkahtani, H.M. Integrated structural, functional, and ADMET analysis of 2-methoxy-4, 6-diphenylnicotinonitrile: The convergence of X-ray diffraction, molecular docking, dynamic simulations, and advanced computational insights. Molecules 2023, 28, 6859. [Google Scholar] [CrossRef] [PubMed]
- Meanwell, N.A. Improving drug candidates by design: A focus on physicochemical properties as a means of improving compound disposition and safety. Chem. Res. Toxicol. 2011, 24, 1420–1456. [Google Scholar] [CrossRef] [PubMed]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A. ADMETlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| Compound | Ar | IC50 (μM) a | ||
| HCT-116 | HePG-2 | MCF-7 | ||
| 7 | Ph | 47.46 ± 2.8 | 55.81 ± 3.3 | 38.30 ± 2.2 |
| 8 | 2-CH3-Ph | 37.61 ± 2.3 | 25.43 ± 1.8 | 34.06 ± 2.0 |
| 9 | 4-CH3-Ph | 71.23 ± 3.8 | 29.73 ± 2.1 | 69.01 ± 3.9 |
| 10 | 4-Cl-Ph | 17.90 ± 1.4 | 10.69 ± 0.9 | 11.18 ± 0.9 |
| 11 | 4-F-Ph | 28.33 ± 2.1 | 21.06 ± 1.8 | 14.25 ± 1.2 |
| 12 | 4-CF3-Ph | 52.48 ± 3.0 | 47.71 ± 2.7 | 45.83 ± 2.4 |
| 13 | 4-OCF3-Ph | 20.11 ± 1.7 | 13.94 ± 1.2 | 9.58 ± 0.8 |
| 14 | 2-OCH3-Ph | 74.03 ± 3.9 | 63.27 ± 3.8 | 59.14 ± 3.4 |
| 15 | 3-OCH3-Ph | 53.85 ± 3.1 | 45.12 ± 2.5 | 55.33 ± 3.2 |
| 16 | 4-OCH3-Ph | 36.52 ± 2.4 | 27.28 ± 2.0 | 35.82 ± 2.1 |
| 17 | 3,4-Di-OCH3-Ph | 26.75 ± 2.0 | 19.63 ± 1.7 | 15.71 ± 1.3 |
| 18 | 3,4,5-Tri-OCH3-Ph | 39.63 ± 2.4 | 37.24 ± 2.3 | 26.02 ± 1.6 |
| 19 | 4-NO2-Ph | 31.27 ± 2.1 | 19.90 ± 1.8 | 20.42 ± 1.5 |
| 20 | 2-Pyridyl | 46.72 ± 2.7 | 49.10 ± 2.8 | 32.39 ± 1.9 |
| 21 | 3-Pyridyl | 19.25 ± 1.5 | 11.50 ± 1.0 | 8.61 ± 0.6 |
| 22 | 4-Pyridyl | 25.96 ± 1.9 | 17.18 ± 1.6 | 13.11 ± 1.1 |
| 23 | 1-Naphthyl | 34.39 ± 2.3 | 31.82 ± 2.1 | 28.13 ± 1.7 |
| 24 | 2-Naphthyl | 12.83 ± 0.9 | 9.07 ± 0.8 | 4.92 ± 0.3 |
| 25 | 2-Furyl | 49.94 ± 2.9 | 34.56 ± 2.1 | 47.28 ± 2.6 |
| 26 | 2-Thiophenyl | 87.50 ± 4.6 | 32.26 ± 2.2 | 76.83 ± 4.2 |
| 27 | 5-Cl-2-thiophenyl | 33.18 ± 2.2 | 16.84 ± 1.6 | 19.72 ± 1.5 |
| Doxorubicin | - | 5.23 ± 0.3 | 4.50 ± 0.2 | 4.17 ± 0.2 |
| Erlotinib | - | 8.53 ± 0.5 | 7.90 ± 0.6 | 5.18 ± 0.4 |
| Comp. | WI-38 | MCF-7 | *SI | HCT-116 | SI | HePG-2 | SI |
|---|---|---|---|---|---|---|---|
| 10 | 54.21 | 11.18 | 4.84 | 17.9 | 3.02 | 10.69 | 5.07 |
| 13 | 45.39 | 9.58 | 4.73 | 20.11 | 2.25 | 13.94 | 3.25 |
| 21 | 60.79 | 8.61 | 7.06 | 19.25 | 3.15 | 11.5 | 5.28 |
| 24 | 39.52 | 4.92 | 8.03 | 12.83 | 3.08 | 9.07 | 4.35 |
| Doxorubicin | 6.72 | 4.17 | 1.61 | 5.23 | 1.28 | 4.5 | 1.49 |
| Erlotinib | 46.52 | 5.18 | 8.98 | 8.53 | 5.45 | 7.9 | 5.88 |
 | |||
|---|---|---|---|
| Compound | Ar | Kinase Inhibition IC50 (µM) | |
| EGFR | HER2 | ||
| 10 | 4-ChloroPh | 0.67 ± 0.03 | 0.71 ± 0.02 |
| 13 | 4-TrifluoromethoxyPh | 0.61 ± 0.02 | 0.28 ± 0.01 |
| 21 | 3-Pyridyl | 1.61 ± 0.07 | 1.17 ± 0.04 |
| 24 | 2-Naphthyl | 0.07 ± 0.004 | 0.04 ± 0.001 |
| Erlotinib | - | 0.05 ± 0.002 | nt |
| Lapatinib | - | nt | 0.03 ± 0.001 |
| Concentrations | %G0–G1 | %S | %G2/M | Comment |
|---|---|---|---|---|
| 24 at 2 µM | 52.12 | 22.45 | 25.43 | Cell cycle arrest at G2/M |
| 24 at 10 µM | 50.61 | 15.93 | 33.46 | - |
| Erlotinib at 2 µM | 46.12 | 22.83 | 31.05 | Cell cycle arrest at G2/M |
| Erlotinib at 10 µM | 35.89 | 15.72 | 48.39 | - |
| Cont. MCF-7 | 63.19 | 28.99 | 7.82 | - |
| Compound | Ligand | Receptor | Interaction | Distance (Å) | E (kcal/mol) | S (kcal/mol) |
|---|---|---|---|---|---|---|
| The HER2 kinase domain (PDB code: 3PP0) | ||||||
| Comp. 24 | N 26 | O Leu 726 (A) | H-donor | 2.85 | −1.9 | −8.78 |
| C 29 | O Arg849 (A) | H-donor | 3.44 | −0.7 | ||
| N 35 | N Asp863 (A) | H-acceptor | 3.3 | −1.1 | ||
| O 58 | CA Asn850 (A) | H-acceptor | 3.56 | −0.3 | ||
| 6-ring | N Cys805 (A) | pi-H | 3.47 | −0.5 | ||
| 6-ring | CD2 Leu852 (A) | pi-H | 3.76 | −0.3 | ||
| 6-ring | CB Phe864 (A) | pi-H | 4.94 | −0.3 | ||
| 6-ring | CD2 Phe864 (A) | pi-H | 4.27 | −0.5 | ||
| 03Q | O1 1 | OD2 Asp863 (A) | H-donor | 3.03 | −2 | −10.68 |
| C12 23 | O Gln799 (A) | H-donor | 3.27 | −0.4 | ||
| CL32 50 | O Leu796 (A) | H-donor | 3.21 | −0.3 | ||
| N11 22 | N Met801 (A) | H-acceptor | 2.9 | −5.5 | ||
| N18 32 | CA Asp863 (A) | H-acceptor | 3.19 | −1.3 | ||
| 6-ring | CD1 Leu785 (A) | pi-H | 4.3 | −0.4 | ||
| 6-ring | CD1 Leu800 (A) | pi-H | 4.7 | −0.4 | ||
| 6-ring | CD1 Leu852 (A) | pi-H | 3.42 | −0.4 | ||
| 6-ring | CD2 Phe864 (A) | pi-H | 3.83 | −0.3 | ||
| The EGFR kinase domain (PDB code: 2ITY) | ||||||
| Comp. 24 | O 58 | N Met793 (A) | H-acceptor | 3.14 | −2.3 | −6.71 |
| 6-ring | N Asp855 (A) | pi-H | 4.32 | −0.5 | ||
| IRE | C2 20 | O Gln791 (A) | H-donor | 3.42 | −0.5 | −6.53 |
| N3 19 | N Met793 (A) | H-acceptor | 2.67 | −4.7 | ||
| 6-ring | CD1 Leu718 (A) | pi-H | 4.17 | −0.5 | ||
| 6-ring | CD Lys745 (A) | pi-H | 4.31 | −0.5 | ||
| 6-ring | CD1 Leu792 (A) | pi-H | 4.97 | −0.3 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alanazi, F.S.; Alkahtani, H.M.; Abdel-Aziz, A.A.-M.; El-Azab, A.S.; Asiri, H.H.; Bakheit, A.H.; Al-Omary, F.A. Synthesis, Antitumor Activities, and Apoptosis-Inducing Activities of Schiff’s Bases Incorporating Imidazolidine-2,4-dione Scaffold: Molecular Docking Studies and Enzymatic Inhibition Activities. Pharmaceuticals 2025, 18, 496. https://doi.org/10.3390/ph18040496
Alanazi FS, Alkahtani HM, Abdel-Aziz AA-M, El-Azab AS, Asiri HH, Bakheit AH, Al-Omary FA. Synthesis, Antitumor Activities, and Apoptosis-Inducing Activities of Schiff’s Bases Incorporating Imidazolidine-2,4-dione Scaffold: Molecular Docking Studies and Enzymatic Inhibition Activities. Pharmaceuticals. 2025; 18(4):496. https://doi.org/10.3390/ph18040496
Chicago/Turabian StyleAlanazi, Fhdah S., Hamad M. Alkahtani, Alaa A.-M. Abdel-Aziz, Adel S. El-Azab, Hanadi H. Asiri, Ahmed H. Bakheit, and Fatmah A. Al-Omary. 2025. "Synthesis, Antitumor Activities, and Apoptosis-Inducing Activities of Schiff’s Bases Incorporating Imidazolidine-2,4-dione Scaffold: Molecular Docking Studies and Enzymatic Inhibition Activities" Pharmaceuticals 18, no. 4: 496. https://doi.org/10.3390/ph18040496
APA StyleAlanazi, F. S., Alkahtani, H. M., Abdel-Aziz, A. A.-M., El-Azab, A. S., Asiri, H. H., Bakheit, A. H., & Al-Omary, F. A. (2025). Synthesis, Antitumor Activities, and Apoptosis-Inducing Activities of Schiff’s Bases Incorporating Imidazolidine-2,4-dione Scaffold: Molecular Docking Studies and Enzymatic Inhibition Activities. Pharmaceuticals, 18(4), 496. https://doi.org/10.3390/ph18040496