
Bridging the Gap Between hiPSC-CMs Cardiotoxicity Assessment and Clinical LVEF Decline Risk: A Case Study of 21 Tyrosine Kinase Inhibitors


Abstract
1. Introduction
2. Results
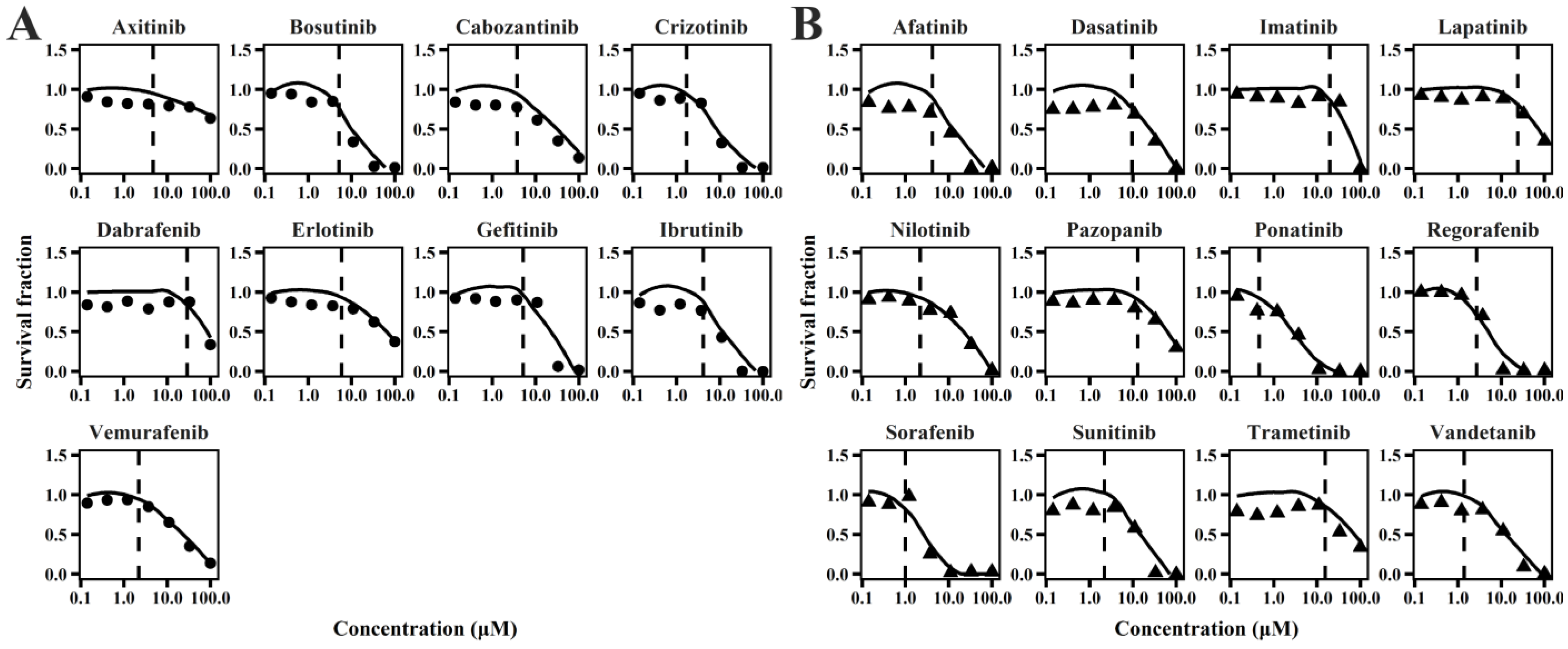
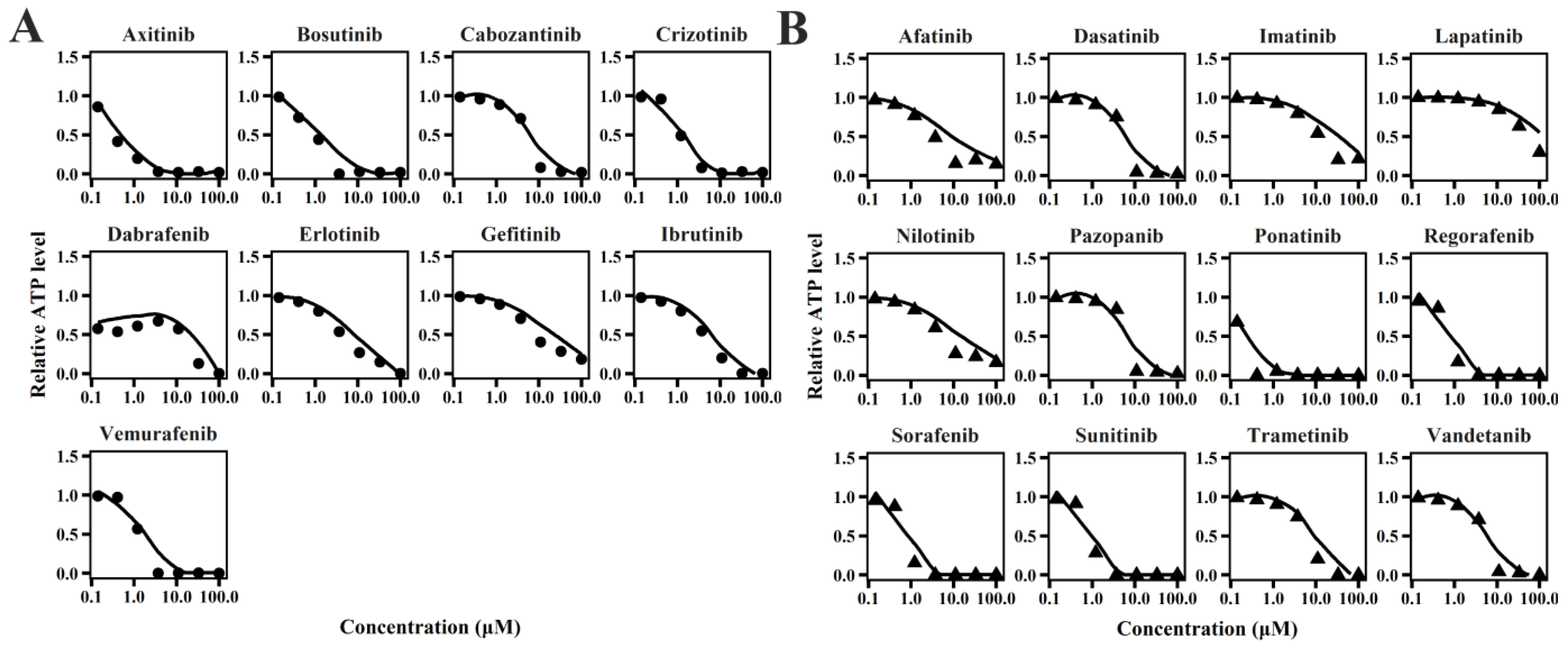
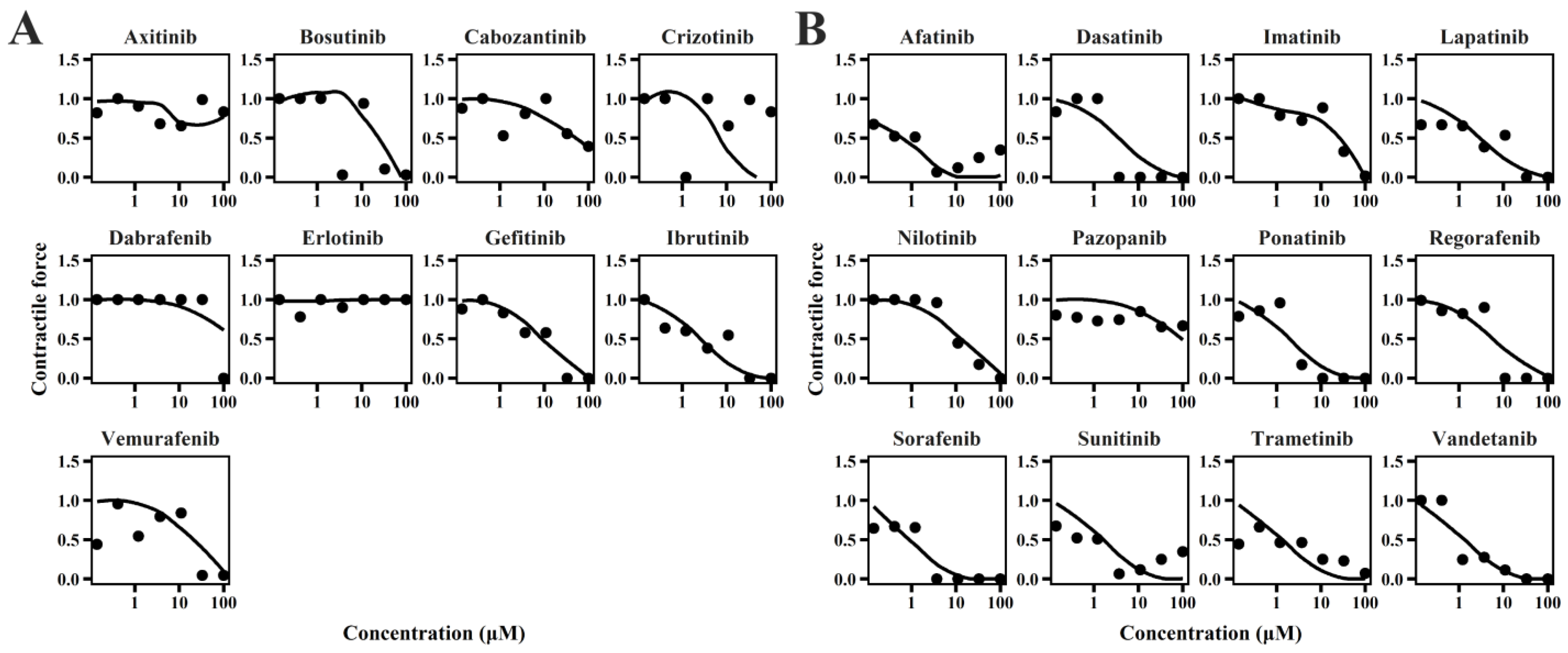
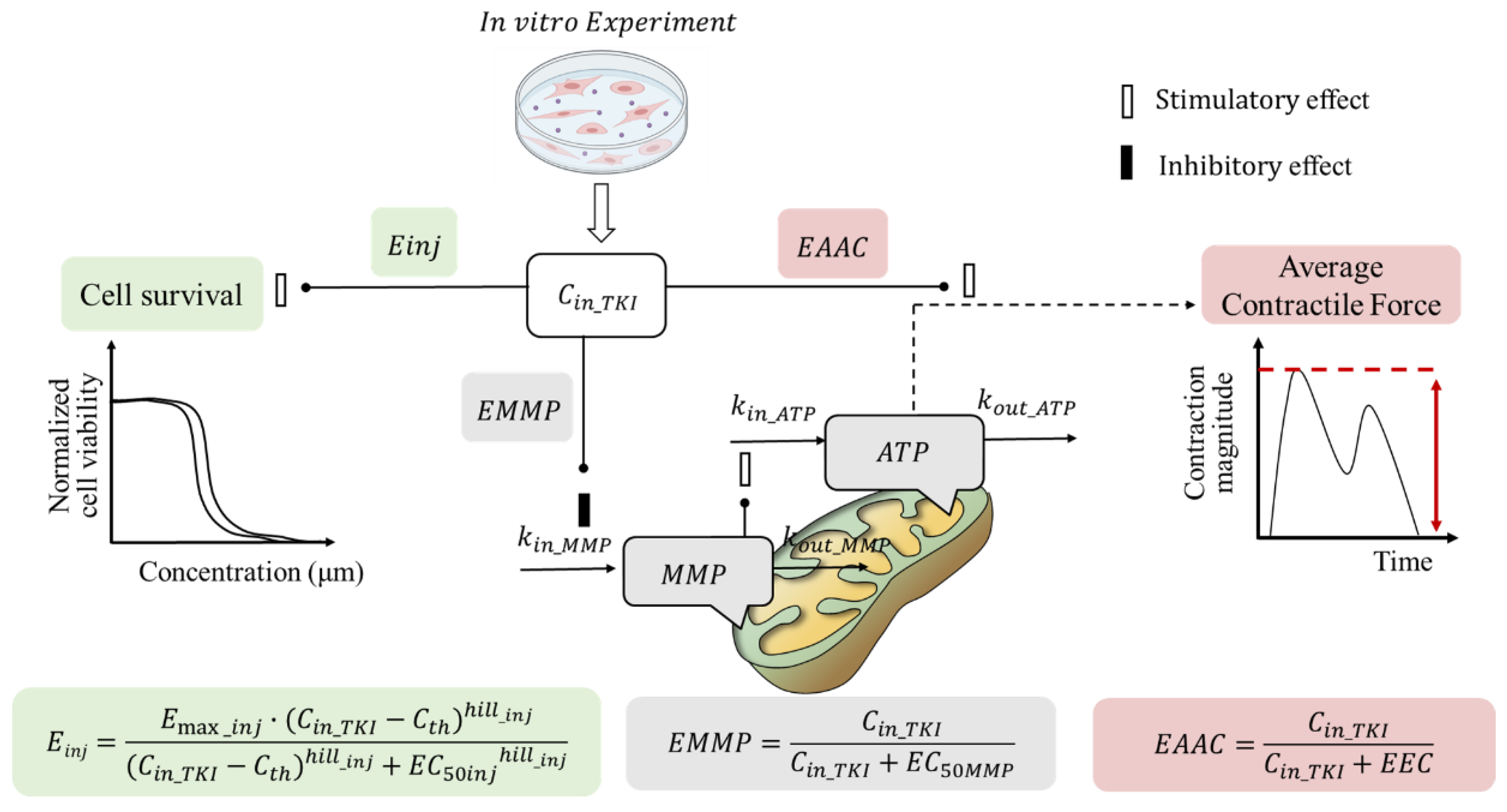
2.1. TD Model of TKIs
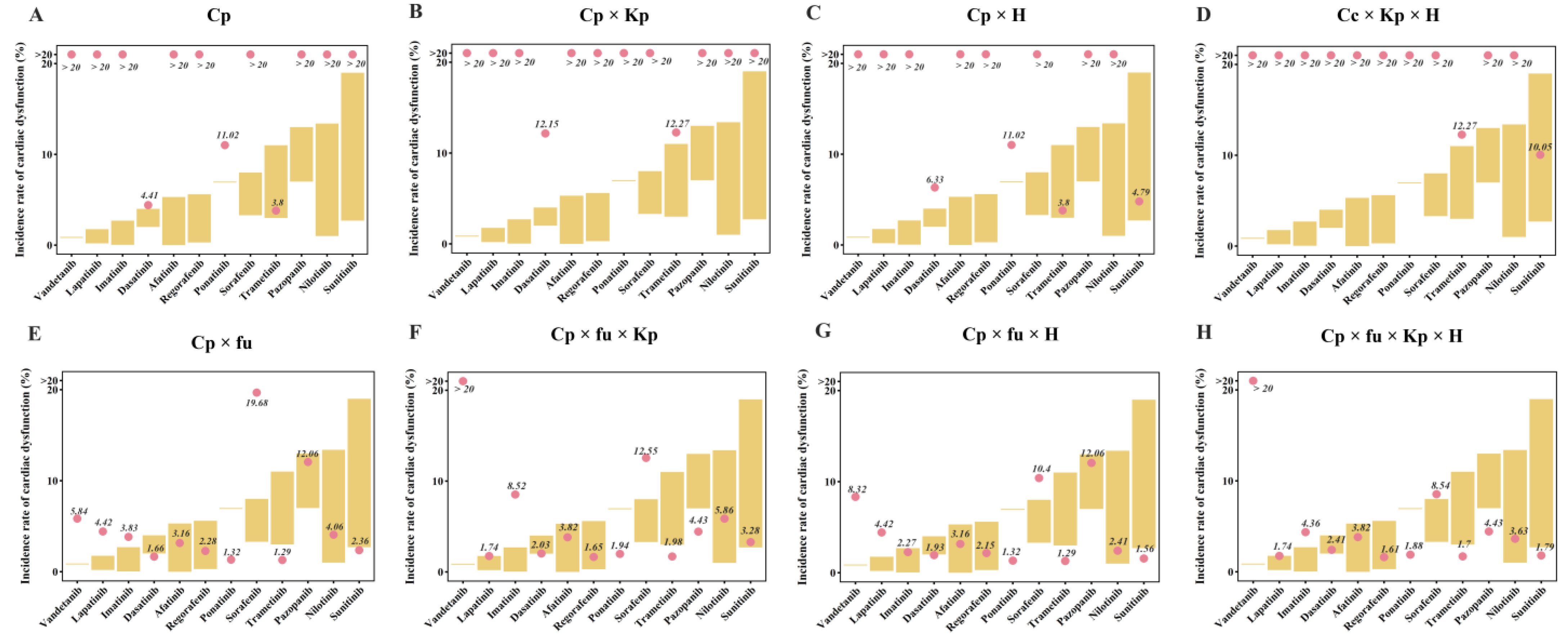
2.2. Quantitative Evaluation of TKI-Induced Cardiac Dysfunction
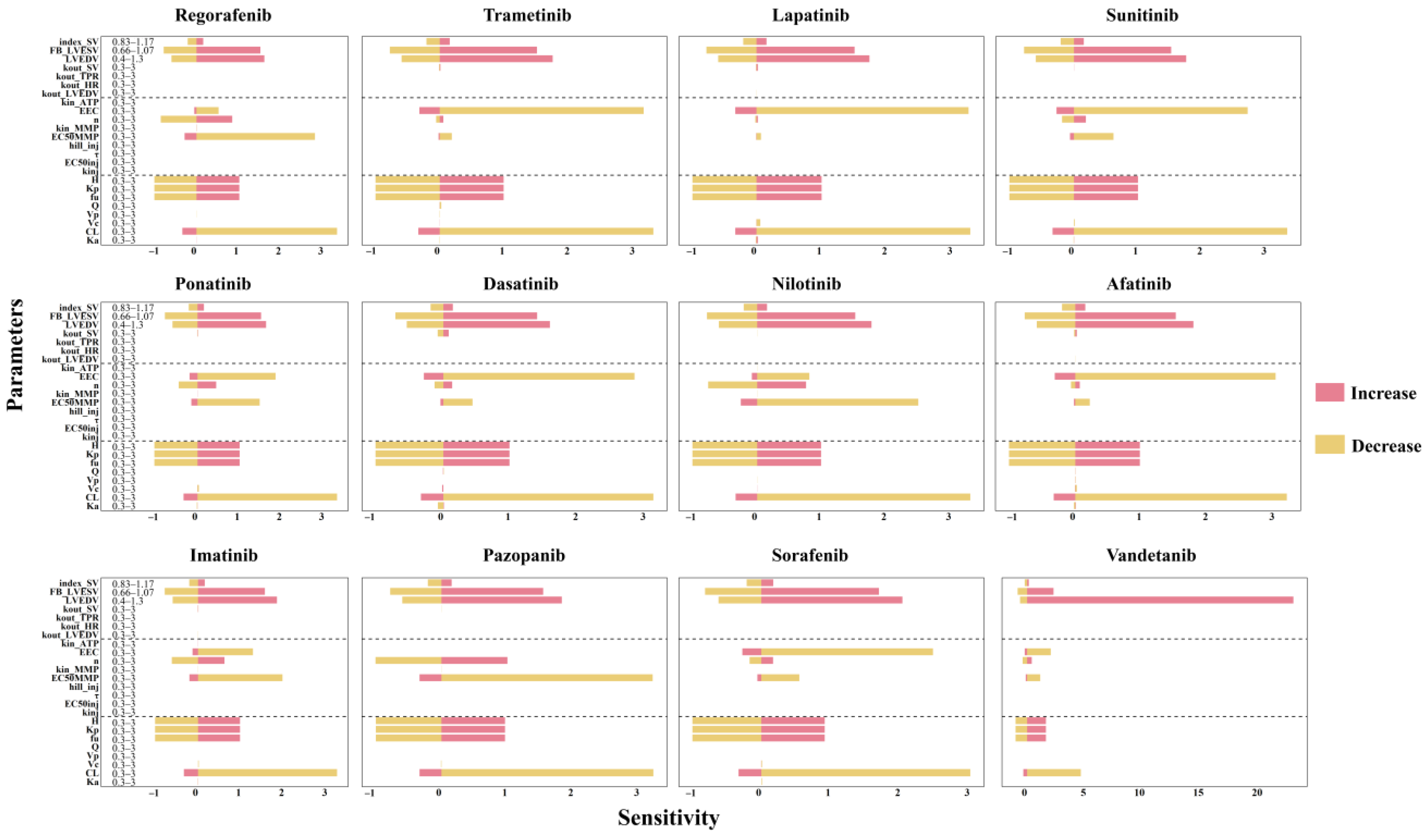
2.3. Results of Sensitivity Analysis
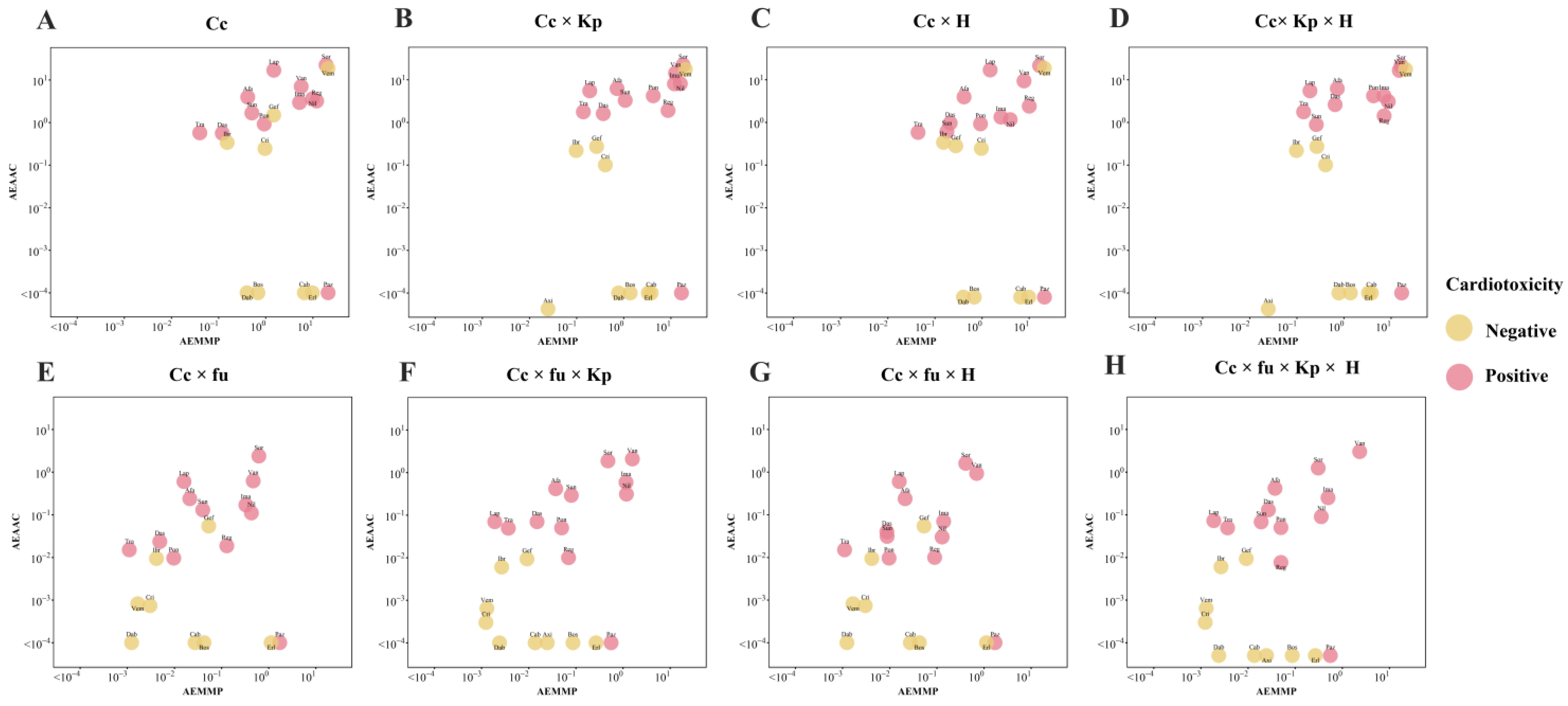
2.4. Qualitative Assessment of TKI-Induced Cardiac Dysfunction
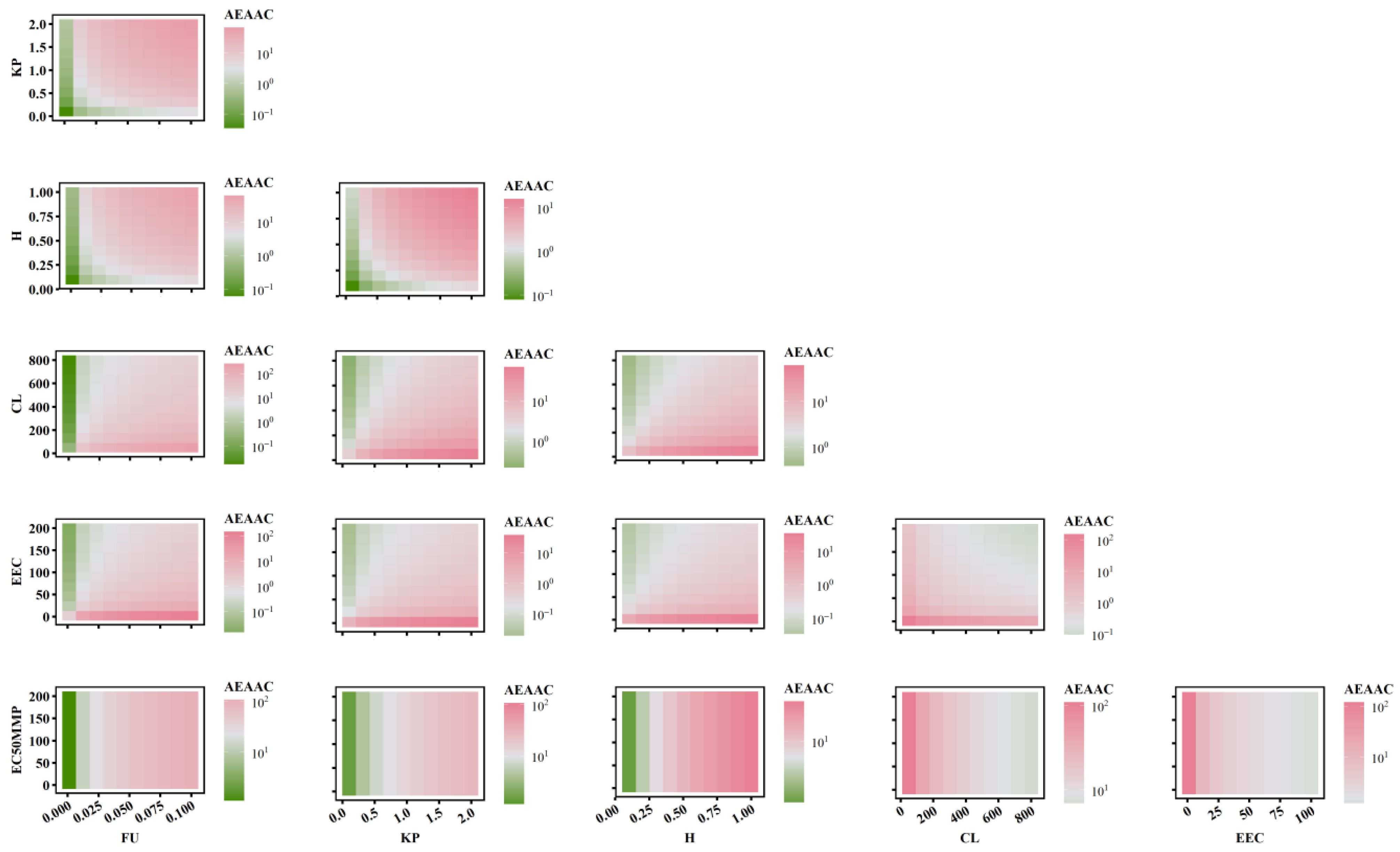
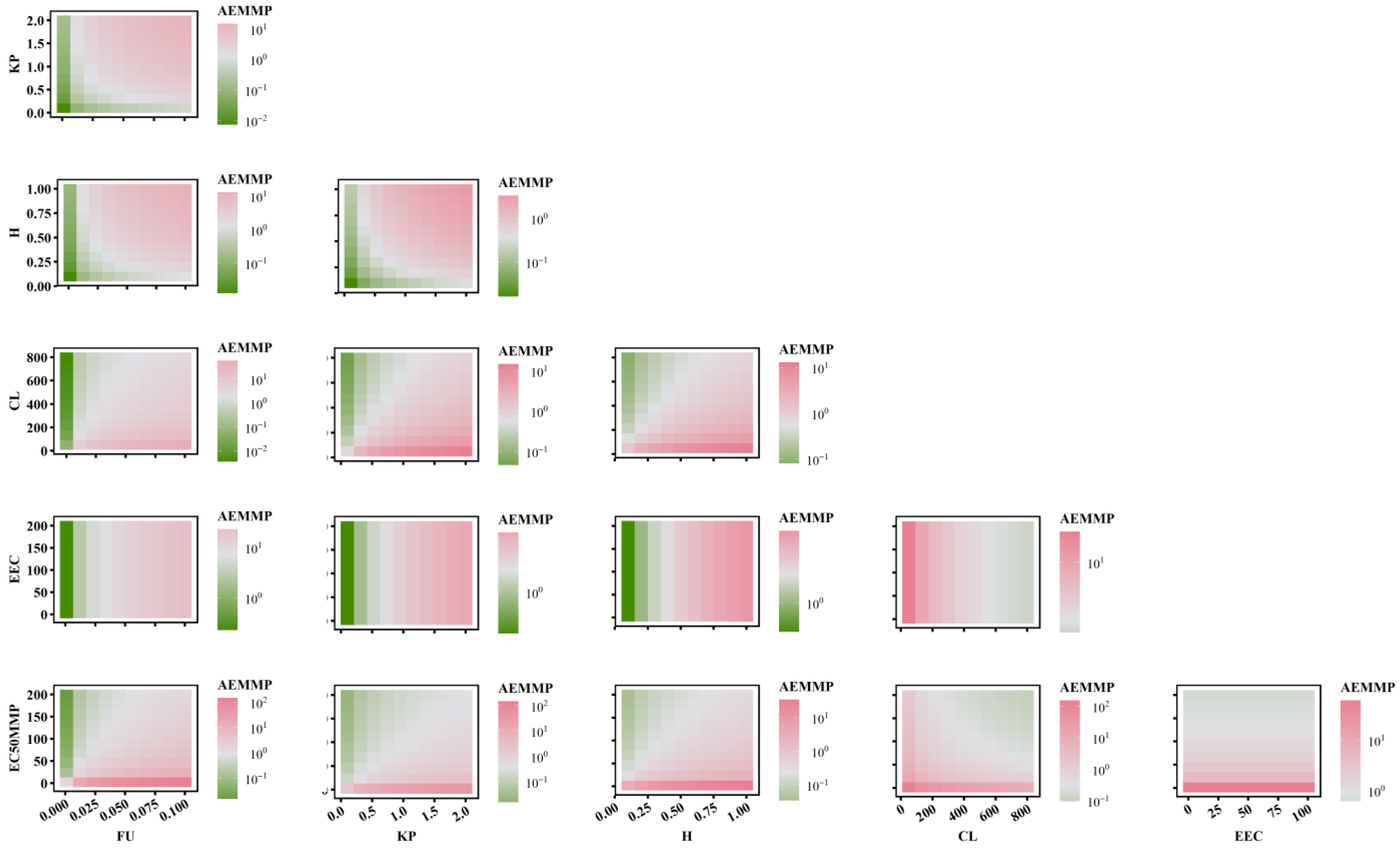
2.5. The Combined Effects of PK-TD Parameters on AEMMP and AEAAC
3. Discussion
4. Materials and Methods
4.1. Data Collection
4.1.1. PK Models of TKIs
4.1.2. In Vitro Data on TKI Cardiotoxicity with hiPSC-CMs
4.1.3. Clinical Incidence of TKI-Induced Cardiac Dysfunction
4.2. In Vitro TD Model
4.2.1. Sub-Model for Cardiomyocyte Viability
4.2.2. Sub-Model for Cardiomyocyte ATP and MMP
4.2.3. Sub-Model for Contractility of Cardiomyocytes
4.2.4. Estimation of TD Model Parameters
4.3. Quantitative Systems Pharmacology (QSP) Model
4.4. IVIVE of TKI-Induced Cardiotoxicity
4.4.1. Quantitative Prediction of the Incidence of Cardiac Dysfunction
4.4.2. Qualitative Identification of Cardiotoxic Drugs
4.5. Factors Affecting Effective Concentration in IVIVE
4.5.1. Plasma Protein Binding
4.5.2. Tissue–Plasma Partition
4.5.3. Drug Heterogeneous Distribution
4.6. Sensitivity Analysis
4.7. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Santoni, M.; Guerra, F.; Conti, A.; Lucarelli, A.; Rinaldi, S.; Belvederesi, L.; Capucci, A.; Berardi, R. Incidence and risk of cardiotoxicity in cancer patients treated with targeted therapies. Cancer Treat. Rev. 2017, 59, 123–131. [Google Scholar]
- Kenigsberg, B.; Wellstein, A.; Barac, A. Left Ventricular Dysfunction in Cancer Treatment: Is it Relevant? JACC Heart Fail. 2018, 6, 87–95. [Google Scholar] [PubMed]
- Banke, A.; Fosbøl, E.L.; Ewertz, M.; Videbæk, L.; Dahl, J.S.; Poulsen, M.K.; Cold, S.; Jensen, M.-B.; Gislason, G.H.; Schou, M.; et al. Long-Term Risk of Heart Failure in Breast Cancer Patients After Adjuvant Chemotherapy With or Without Trastuzumab. JACC Heart Fail. 2019, 7, 217–224. [Google Scholar] [PubMed]
- Kemp, C.D.; Conte, J.V. The pathophysiology of heart failure. Cardiovasc. Pathol. 2012, 21, 365–371. [Google Scholar]
- Sequeira, V.; van der Velden, J. Historical perspective on heart function: The Frank-Starling Law. Biophys. Rev. 2015, 7, 421–447. [Google Scholar]
- Mamoshina, P.; Rodriguez, B.; Bueno-Orovio, A. Toward a broader view of mechanisms of drug cardiotoxicity. Cell Rep. Med. 2021, 2, 100216. [Google Scholar] [CrossRef] [PubMed]
- Doherty, K.R.; Talbert, D.R.; Trusk, P.B.; Moran, D.M.; Shell, S.A.; Bacus, S. Structural and functional screening in human induced-pluripotent stem cell-derived cardiomyocytes accurately identifies cardiotoxicity of multiple drug types. Toxicol. Appl. Pharmacol. 2015, 285, 51–60. [Google Scholar] [CrossRef]
- Chen, Z.I.; Ai, D.I. Cardiotoxicity associated with targeted cancer therapies. Mol. Clin. Oncol. 2016, 4, 675–681. [Google Scholar]
- Shyam Sunder, S.A.-O.; Sharma, U.C.; Pokharel, S. Adverse effects of tyrosine kinase inhibitors in cancer therapy: Pathophysiology, mechanisms and clinical management. Signal Transduct. Target. Ther. 2023, 8, 262. [Google Scholar]
- Singh, A.P.; Glennon, M.S.; Umbarkar, P.; Gupte, M.; Galindo, C.L.; Zhang, Q.; Force, Q.; Becker, J.R.; Lal, H. Ponatinib-induced cardiotoxicity: Delineating the signalling mechanisms and potential rescue strategies. Cardiovasc. Res. 2019, 115, 966–997. [Google Scholar] [CrossRef]
- de Korte, T.; Katili, P.A.; Mohd Yusof, N.A.N.; van Meer, B.J.; Saleem, U.; Burton, F.L.; Smith, G.L.; Clements, P.; Mummery, C.L.; Eschenhagen, T.; et al. Unlocking Personalized Biomedicine and Drug Discovery with Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes: Fit for Purpose or Forever Elusive? Annu. Rev. Pharmacol. Toxicol. 2020, 60, 529–551. [Google Scholar] [CrossRef]
- Sharma, A.A.-O.; Burridge, P.W.; McKeithan, W.L.; Serrano, R.A.-O.; Shukla, P.; Sayed, N.; Churko, J.M.; Kitani, T.; Wu, H.; Holmström, A.; et al. High-throughput screening of tyrosine kinase inhibitor cardiotoxicity with human induced pluripotent stem cells. Sci. Transl. Med. 2017, 9, eaaf2584. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.A.-O.; McKeithan, W.A.-O.; Serrano, R.; Kitani, T.; Burridge, P.W.; del Álamo, J.C.; Mercola, M.; Wu, J.C. Use of human induced pluripotent stem cell-derived cardiomyocytes to assess drug cardiotoxicity. Nat. Protoc. 2018, 13, 3018–3041. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.A.; Fau, D.L.; Kerns, E.H. The effect of plasma protein binding on in vivo efficacy: Misconceptions in drug discovery. Nat. Rev. Drug Discov. 2010, 9, 929–939. [Google Scholar] [CrossRef]
- Di, L. An update on the importance of plasma protein binding in drug discovery and development. Expert. Opin. Drug Discov. 2021, 16, 1453–1465. [Google Scholar] [CrossRef] [PubMed]
- Gaohua, L.; Miao, X.; Dou, L. Crosstalk of physiological pH and chemical pKa under the umbrella of physiologically based pharmacokinetic modeling of drug absorption, distribution, metabolism, excretion, and toxicity. Expert Opin. Drug Metab. Toxicol. 2021, 17, 1103–1124. [Google Scholar] [CrossRef]
- Sirenko, O.; Hancock, M.K.; Crittenden, C.; Hammer, M.; Keating, S.; Carlson, C.B.; Chandy, G. Phenotypic Assays for Characterizing Compound Effects on Induced Pluripotent Stem Cell-Derived Cardiac Spheroids. Assay Drug Dev. Technol. 2017, 15, 280–296. [Google Scholar] [CrossRef]
- Bu, L.A.-O.; Hu, M.; Lu, W.; Wang, Z.; Lu, G.A.-O. Printing Semiconductor-Insulator Polymer Bilayers for High-Performance Coplanar Field-Effect Transistors. Adv. Mater. 2018, 30, 1704695. [Google Scholar] [CrossRef]
- Sang, L.; Zhou, Z.; Luo, S.; Zhang, Y.; Qian, H.; Zhou, Y.; He, H.; Hao, K. An In Silico Platform to Predict Cardiotoxicity Risk of Anti-tumor Drug Combination with hiPSC-CMs Based In Vitro Study. Pharm. Res. 2023, 41, 247–262. [Google Scholar] [CrossRef]
- Serrano, C.; Cortés, J.; De Mattos-Arruda, L.; Bellet, M.; Gómez, P.; Saura, C.; Pérez, J.; Vidal, M.; Muñoz-Couselo, E.; Carreras, M.J.; et al. Trastuzumab-related cardiotoxicity in the elderly: A role for cardiovascular risk factors. Ann. Oncol. 2012, 23, 897–902. [Google Scholar] [CrossRef]
- Colatsky, T.; Fermini, B.; Gintant, G.; Pierson, J.B.; Sager, P.; Sekino, Y.; Strauss, D.G.; Stockbridge, N. The Comprehensive in Vitro Proarrhythmia Assay (CiPA) initiative—Update on progress. J. Pharmacol. Toxicol. Methods 2016, 81, 15–20. [Google Scholar] [PubMed]
- Ridder, B.J.; Leishman, D.J.; Bridgland-Taylor, M.; Samieegohar, M.; Han, X.; Wu, W.W.; Randolph, A.; Tran, P.; Sheng, J.; Danker, T.; et al. A systematic strategy for estimating hERG block potency and its implications in a new cardiac safety paradigm. Toxicol. Appl. Pharmacol. 2020, 394, 114961. [Google Scholar]
- Sang, L.; Yuan, Y.; Zhou, Y.; Zhou, Z.; Jiang, M.; Liu, X.; Hao, K.; He, H. A quantitative systems pharmacology approach to predict the safe-equivalent dose of doxorubicin in patients with cardiovascular comorbidity. CPT Pharmacomet. Syst. Pharmacol. 2021, 10, 1512–1524. [Google Scholar]
- Goßmann, M.; Linder, P.; Thomas, U.; Juhasz, K.; Lemme, M.; George, M.; Fertig, N.; Dragicevic, E.; Stoelzle-Feix, S. Integration of mechanical conditioning into a high throughput contractility assay for cardiac safety assessment. J. Pharmacol. Toxicol. Methods 2020, 105, 106892. [Google Scholar]
- Wang, H.; Sheehan, R.P.; Palmer, A.C.; Everley, R.A.; Boswell, S.A.; Ron-Harel, N.; Ringel, A.E.; Holton, K.M.; Jacobson, C.A.; Erickson, A.R.; et al. Adaptation of Human iPSC-Derived Cardiomyocytes to Tyrosine Kinase Inhibitors Reduces Acute Cardiotoxicity via Metabolic Reprogramming. Cell Syst. 2019, 8, 412–426. [Google Scholar]
- Li, X.; Lin, Y.; Lin, S.; Huang, J.; Ruan, Z. Advancements in understanding cardiotoxicity of EGFR- TKIs in non-small cell lung cancer treatment and beyond. Front. Pharmacol. 2024, 15, 1404692. [Google Scholar]
- Chaar, M.; Kamta, J.; Ait-Oudhia, S. Mechanisms, monitoring, and management of tyrosine kinase inhibitors-associated cardiovascular toxicities. Oncol. Targets Ther. 2018, 11, 6227–6237. [Google Scholar]
- Paraskevaidis, I.; Kourek, C.; Farmakis, D.; Tsougos, E. Heart Failure: A Deficiency of Energy-A Path Yet to Discover and Walk. Biomedicines 2024, 12, 2589. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Karwi, Q.G.; Tian, R.; Wende, A.R.; Abel, E.D. Cardiac Energy Metabolism in Heart Failure. Circ. Res. 2021, 128, 1487–1513. [Google Scholar] [CrossRef]
- Crump, K.S.; Chen, C.; Louis, T.A. The future use of in vitro data in risk assessment to set human exposure standards: Challenging problems and familiar solutions. Environ. Health Perspect. 2010, 118, 1350–1354. [Google Scholar]
- Wetmore, B.A.; Wambaugh, J.F.; Ferguson, S.S.; Li, L.; Clewell, H.J., III; Judson, R.S.; Judson, R.S.; Freeman, K.; Bao, W.; Sochaski, M.A.; et al. Relative impact of incorporating pharmacokinetics on predicting in vivo hazard and mode of action from high-throughput in vitro toxicity assays. Toxicol. Sci. 2013, 132, 327–346. [Google Scholar] [CrossRef] [PubMed]
- Wetmore, B.A. Quantitative in vitro-to-in vivo extrapolation in a high-throughput environment. Toxicology 2015, 332, 94–101. [Google Scholar] [PubMed]
- Dix, D.J.; Houck, K.A.; Martin, M.T.; Richard, A.M.; Setzer, R.W.; Kavlock, R.J. The ToxCast program for prioritizing toxicity testing of environmental chemicals. Toxicol. Sci. 2007, 95, 5–12. [Google Scholar]
- Knudsen, T.; Martin, M.; Chandler, K.; Kleinstreuer, N.; Judson, R.; Sipes, N. Predictive models and computational toxicology. Methods Mol. Biol. 2013, 947, 343–374. [Google Scholar]
- Blaauboer, B.J. The long and winding road of progress in the use of in vitro data for risk assessment purposes: From “carnation test” to integrated testing strategies. Toxicology 2015, 332, 4–7. [Google Scholar]
- Wilk-Zasadna, I.; Bernasconi, C.; Pelkonen, O.; Coecke, S. Biotransformation in vitro: An essential consideration in the quantitative in vitro-to-in vivo extrapolation (QIVIVE) of toxicity data. Toxicology 2015, 332, 8–19. [Google Scholar] [PubMed]
- Henneberger, L.A.-O.; Huchthausen, J.A.-O.; Wojtysiak, N.; Escher, B.A.-O.X. Quantitative In Vitro-to-In Vivo Extrapolation: Nominal versus Freely Dissolved Concentration. Chem. Res. Toxicol. 2021, 34, 1175–1182. [Google Scholar]
- Groothuis, F.A.; Heringa, M.B.; Nicol, B.; Hermens, J.L.; Blaauboer, B.J.; Kramer, N.I. Dose metric considerations in in vitro assays to improve quantitative in vitro-in vivo dose extrapolations. Toxicology 2015, 332, 30–40. [Google Scholar] [CrossRef]
- Ando, H.; Yoshinaga, T.; Yamamoto, W.; Asakura, K.; Uda, T.; Taniguchi, T.; Ojima, A.; Shinkyo, R.; Kikuchi, K.; Osada, T.; et al. A new paradigm for drug-induced torsadogenic risk assessment using human iPS cell-derived cardiomyocytes. J. Pharmacol. Toxicol. Methods 2017, 84, 111–127. [Google Scholar] [CrossRef]
- FDA. NEXAVAR (Sorafenib) Tablets, for Oral Use; The U.S. Food and Drug Administration: Silver Spring, MD, USA, 2020. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/021923s024lblrpl.pdf (accessed on 6 June 2020).
- FDA. STIVARGA (Regorafenib) Oral Tablets; The U.S. Food and Drug Administration: Silver Spring, MD, USA, 2020. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/203085s013lbl.pdf (accessed on 2 February 2020).
- Lin, J.H. Tissue distribution and pharmacodynamics: A complicated relationship. Curr. Drug Metab. 2006, 7, 39–65. [Google Scholar] [CrossRef]
- Wang, J.; Zhu, K.; Zhang, M.; Zhou, Q.; Ji, W.; Yao, Z.; Li, D. Pharmacokinetics, tissue distribution, and subacute toxicity of oral carrageenan in mice. Int. J. Biol. Macromol. 2024, 226 Pt 1, 130725. [Google Scholar] [CrossRef]
- Ozbey, A.A.-O.; Combarel, D.; Poinsignon, V.; Lovera, C.; Saada, E.; Mir, O.A.-O.; Paci, A. Population Pharmacokinetic Analysis of Pazopanib in Patients and Determination of Target AUC. Pharmaceuticals 2021, 14, 927. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, J.T.; Haap, M.; Kopp, H.G.; Lipp, H.P. Tyrosine kinase inhibitors—A review on pharmacology, metabolism and side effects. Curr. Drug Metab. 2009, 10, 470–481. [Google Scholar] [CrossRef] [PubMed]
- Utsey, K.; Gastonguay, M.S.; Russell, S.; Freling, R.; Riggs, M.M.; Elmokadem, A. Quantification of the Impact of Partition Coefficient Prediction Methods on Physiologically Based Pharmacokinetic Model Output Using a Standardized Tissue Composition. Drug Metab. Dispos. 2020, 48, 903–916. [Google Scholar] [CrossRef] [PubMed]
- Poulin, P. Drug Distribution to Human Tissues: Prediction and Examination of the Basic Assumption in In Vivo Pharmacokinetics-Pharmacodynamics (PK/PD) Research. J. Pharm. Sci. 2015, 104, 2110–2118. [Google Scholar] [CrossRef]
- Ahmed, H.A.-O.; Böhmdorfer, M.; Eberl, S.; Jäger, W.; Zeitlinger, M.A.-O. Interspecies variability in protein binding of antibiotics basis for translational PK/PD studies-a case study using cefazolin. Antimicrob. Agents Chemother. 2024, 20, e0164723. [Google Scholar] [CrossRef]
- Correia, C.A.-O.; Koshkin, A.; Duarte, P.; Hu, D.; Carido, M.; Sebastião, M.A.-O.; Gomes-Alves, P.; Elliott, D.A.; Domian, I.J.; Teixeira, A.P.; et al. 3D aggregate culture improves metabolic maturation of human pluripotent stem cell derived cardiomyocytes. Biotechnol. Bioeng. 2018, 115, 630–644. [Google Scholar] [CrossRef]
- Archer, C.R.; Sargeant, R.; Basak, J.; Pilling, J.; Barnes, J.R.; Pointon, A. Characterization and Validation of a Human 3D Cardiac Microtissue for the Assessment of Changes in Cardiac Pathology. Sci. Rep. 2018, 8, 10160. [Google Scholar] [CrossRef]
- Le, X.; Nilsson, M.; Goldman, J.; Reck, M.; Nakagawa, K.; Kato, T.; Ares, L.P.; Frimodt-Moller, B.; Wolff, K.; Visseren-Grul, C.; et al. Dual EGFR-VEGF Pathway Inhibition: A Promising Strategy for Patients With EGFR-Mutant NSCLC. J. Thorac. Oncol. 2021, 16, 205–215. [Google Scholar] [CrossRef]
- Cortes, J.E.; Kantarjian, H.; Shah, N.P.; Bixby, D.; Mauro, M.J.; Flinn, I.; O’Hare, T.; Hu, S.; Narasimhan, N.I.; Rivera, V.M.; et al. Ponatinib in refractory Philadelphia chromosome-positive leukemias. N. Engl. J. Med. 2012, 367, 2075–2088. [Google Scholar] [CrossRef]
- Garg, S.; de Lemos, J.A.; Matulevicius, S.A.; Ayers, C.; Pandey, A.; Neeland, I.J.; Berry, J.D.; McColl, R.; Maroules, C.; Peshock, R.M.; et al. Association of Concentric Left Ventricular Hypertrophy with Subsequent Change in Left Ventricular End-Diastolic Volume: The Dallas Heart Study. Circ. Heart Fail. 2017, 10, e003959. [Google Scholar]
- Zhao, P.; Zhang, L.; Grillo, J.A.; Liu, Q.; Bullock, J.M.; Moon, Y.J.; Song, P.; Brar, S.S.; Madabushi, R.; Wu, T.C.; et al. Applications of physiologically based pharmacokinetic (PBPK) modeling and simulation during regulatory review. Clin. Pharmacol. Ther. 2011, 89, 259–267. [Google Scholar]
- Landersdorfer, C.B.; Kinzig, M.; Höhl, R.; Kempf, P.; Nation, R.L.; Sörgel, F. Physiologically Based Population Pharmacokinetic Modeling Approach for Ciprofloxacin in Bone of Patients Undergoing Orthopedic Surgery. ACS Pharmacol. Transl. Sci. 2020, 3, 444–454. [Google Scholar] [CrossRef] [PubMed]
- Jones, H.M.; Parrott, N.; Jorga, K.; Lavé, T. A novel strategy for physiologically based predictions of human pharmacokinetics. Clin. Pharmacokinet. 2006, 45, 511–542. [Google Scholar]
- Godinas, L.; Guignabert, C.; Seferian, A.; Perros, F.; Bergot, E.; Sibille, Y.; Humbert, M.; Montani, D. Tyrosine kinase inhibitors in pulmonary arterial hypertension: A double-edge sword? Semin. Respir. Crit. Care Med. 2013, 34, 714–724. [Google Scholar] [PubMed]
- Jin, Y.; Xu, Z.; Yan, H.; He, Q.; Yang, X.; Luo, P. A Comprehensive Review of Clinical Cardiotoxicity Incidence of FDA-Approved Small-Molecule Kinase Inhibitors. Front. Pharmacol. 2020, 11, 891. [Google Scholar]
- Lyon, A.R.; López-Fernández, T.; Couch, L.S.; Asteggiano, R.; Aznar, M.C.; Bergler-Klein, J.; Boriani, G.; Cardinale, D.; Cordoba, R.; Cosyns, B.; et al. 2022 ESC Guidelines on cardio-oncology developed in collaboration with the European Hematology Association (EHA), the European Society for Therapeutic Radiology and Oncology (ESTRO) and the International Cardio-Oncology Society (IC-OS). Eur. Heart J. 2022, 43, 4229–4361. [Google Scholar]
- Hua, Q.; Fan, L.; Wang, Z.W.; Li, J. 2023 Guideline for the management of hypertension in the elderly population in China. J. Geriatr. Cardiol. 2024, 21, 589–630. [Google Scholar]
- Drafts, B.C.; Twomley, K.M.; D’Agostino, R.; Lawrence, J.; Avis, N.; Ellis, L.R.; Thohan, V.; Jordan, J.; Melin, S.A.; Frank, M.; et al. Low to moderate dose anthracycline-based chemotherapy is associated with early noninvasive imaging evidence of subclinical cardiovascular disease. JACC Cardiovasc. Imaging 2013, 6, 877–885. [Google Scholar]
- Kosaraju, A.; Goyal, A.; Grigorova, Y.; Makaryus, A.N. Left Ventricular Ejection Fraction; StatPearls: Treasure Island, FL, USA, 2024. [Google Scholar]
- Nabeta, T.; Inomata, T.; Ishii, S.; Yazaki, M.; Fujita, T.; Iida, Y.; Ikeda, Y.; Maekawa, E.; Naruke, T.; Koitabashi, T.; et al. Dilated cardiomyopathy with re-worsening left ventricular ejection fraction. Heart Vessel. 2019, 34, 95–103. [Google Scholar]
- Waagstein, F.; Caidahl, K.; Wallentin, I.; Bergh, C.H.; Hjalmarson, A. Long-term beta-blockade in dilated cardiomyopathy. Effects of short- and long-term metoprolol treatment followed by withdrawal and readministration of metoprolol. Circulation 1989, 80, 551–563. [Google Scholar] [PubMed]
- WHO. Global Report on Hypertension: The Race Against a Silent Killer; World Health Organization: Geneva, Switzerland, 2023; Available online: https://www.who.int/publications/i/item/9789240081062 (accessed on 19 September 2023).
- Codd, M.B.; Sugrue, D.D.; Gersh, B.J.; Melton, L.J., 3rd. Epidemiology of idiopathic dilated and hypertrophic cardiomyopathy. A population-based study in Olmsted County, Minnesota, 1975–1984. Circulation 1989, 80, 564–572. [Google Scholar]
- Herrmann, J.; Lenihan, D.A.-O.; Armenian, S.A.-O.; Barac, A.A.-O.; Blaes, A.A.-O.; Cardinale, D.A.-O.; Carver, J.; Dent, S.; Ky, B.; Lyon, A.R.; et al. Defining cardiovascular toxicities of cancer therapies: An International Cardio-Oncology Society (IC-OS) consensus statement. Eur. Heart J. 2022, 43, 280–299. [Google Scholar] [PubMed]
- O’Brien, M.E.; Wigler, N.; Inbar, M.; Rosso, R.; Grischke, E.; Santoro, A.; Catane, R.; Kieback, D.G.; Tomczak, P.; Ackland, S.P.; et al. Reduced cardiotoxicity and comparable efficacy in a phase III trial of pegylated liposomal doxorubicin HCl (CAELYX/Doxil) versus conventional doxorubicin for first-line treatment of metastatic breast cancer. Ann. Oncol. 2004, 15, 440–449. [Google Scholar] [PubMed]
- Adachi, K.; Utsumi, M.; Sato, T.; Nakano, H.; Shimizu, M.; Yamazaki, H. Modeled Rat Hepatic and Plasma Concentrations of Chemicals after Virtual Administrations Using Two Sets of in Silico Liver-to-Plasma Partition Coefficients. Biol. Pharm. Bull. 2023, 46, 1316–1323. [Google Scholar]
- Chinese Society of Ultrasound in Medicine and Engineering; Chinese Physicians Association; Echocardiography Committee. Clinical Application Guidelines for Echocardiographic Assessment of Cardiac Systolic and Diastolic Function. Chin. J. Ultrasound Med. Biol. 2020, 29, 461–477. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | 10−2) | (μM) | (μM) | (μM) | (μM) | |
|---|---|---|---|---|---|---|
| Afatinib | 15 | 17.62 | 3 | 4.16 | 4.62 | 0.41 |
| Axitinib | 0.32 | 4 | 1.04 | 4.7 | 50.15 | / |
| Bosutinib | 12 | 14.57 | 3 | 5.16 | 9.53 | / |
| Cabozantinib | 2.7 | 64.72 | 0.67 | 3.7 | 10.84 | / |
| Crizotinib | 4.4 | 12.05 | 1.61 | 1.7 | 10.8 | 44.24 |
| Dabrafenib | 1.9 | 55.25 | 1.18 | 28.6 | 45.75 | / |
| Dasatinib | 3.1 | 19.99 | 0.76 | 9.6 | 13.26 | 2.76 |
| Erlotinib | 1.9 | 93.47 | 0.76 | 5.96 | 5.54 | / |
| Gefitinib | 7.4 | 44.59 | 1.91 | 5.16 | 10.64 | 10.47 |
| Ibrutinib | 27 | 26.39 | 3 | 4.16 | 5.63 | 2.50 |
| Imatinib | 18 | 72.74 | 3 | 19.8 | 16.41 | 32.97 |
| Lapatinib | 1.3 | 27.39 | 1.38 | 24.2 | 69.6 | 1.80 |
| Nilotinib | 6.8 | 97.37 | 1.05 | 2.2 | 7.16 | 28.16 |
| Pazopanib | 1.5 | 72.36 | 0.69 | 13 | 21.49 | / |
| Ponatinib | 11 | 11.6 | 1.54 | 0.46 | 2.54 | 2.57 |
| Regorafenib | 4.4 | 5.38 | 1.58 | 2.7 | 3.2 | 16.27 |
| Sorafenib | 4.5 | 4.36 | 3 | 1 | 3.31 | 0.79 |
| Sunitinib | 10 | 22.02 | 3 | 2.2 | 5.01 | 1.42 |
| Trametinib | 1.3 | 24.77 | 2.61 | 15.4 | 12.59 | 0.94 |
| Vandetanib | 7.1 | 79.89 | 1.05 | 1.38 | 10.72 | 0.94 |
| Vemurafenib | 2.6 | 44.65 | 0.87 | 2.2 | 15.46 | 31.32 |
| Healthy | Hypertension | Dilated Cardiomyopathy | |
|---|---|---|---|
| MAP0 | 94.92 | 106 [60] | 91.54 |
| LVEDV0 | 113 [61] | 100.95 | 146 [53] |
| Index_SV0 | 0.6 [62] | 0.6 | 0.55 [63] |
| FB_LVESV | 0.0042 | 0.0045 | 0.0028 |
| CV_MAP | 0.1 | 0.2 | 0.3 |
| CV_LVEDV | 0.1 | 0.2 | 0.2 |
| CV_Index_SV0 | 0.05 | 0.05 | 0.05 |
| CV_FB_LVESV | 0.124 | 0.124 | 0.124 |
| SV0 | 67.8 | 60.57 | 80.3 |
| TPR0 | 0.02 | 0.025 | 0.02 |
| HR0 | 70 | 70 | 57 [64] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wan, Z.; Wang, C.; Luo, S.; Zhu, J.; He, H.; Hao, K. Bridging the Gap Between hiPSC-CMs Cardiotoxicity Assessment and Clinical LVEF Decline Risk: A Case Study of 21 Tyrosine Kinase Inhibitors. Pharmaceuticals 2025, 18, 450. https://doi.org/10.3390/ph18040450
Wan Z, Wang C, Luo S, Zhu J, He H, Hao K. Bridging the Gap Between hiPSC-CMs Cardiotoxicity Assessment and Clinical LVEF Decline Risk: A Case Study of 21 Tyrosine Kinase Inhibitors. Pharmaceuticals. 2025; 18(4):450. https://doi.org/10.3390/ph18040450
Chicago/Turabian StyleWan, Zhijie, Chenyu Wang, Shizheng Luo, Jinwei Zhu, Hua He, and Kun Hao. 2025. "Bridging the Gap Between hiPSC-CMs Cardiotoxicity Assessment and Clinical LVEF Decline Risk: A Case Study of 21 Tyrosine Kinase Inhibitors" Pharmaceuticals 18, no. 4: 450. https://doi.org/10.3390/ph18040450
APA StyleWan, Z., Wang, C., Luo, S., Zhu, J., He, H., & Hao, K. (2025). Bridging the Gap Between hiPSC-CMs Cardiotoxicity Assessment and Clinical LVEF Decline Risk: A Case Study of 21 Tyrosine Kinase Inhibitors. Pharmaceuticals, 18(4), 450. https://doi.org/10.3390/ph18040450