
Oxytocin Protects PC12 Cells Against β-Amyloid-Induced Cell Injury

 , , ,
, , ,  , ,
, ,  , and
, and 
Abstract
1. Introduction
2. Results
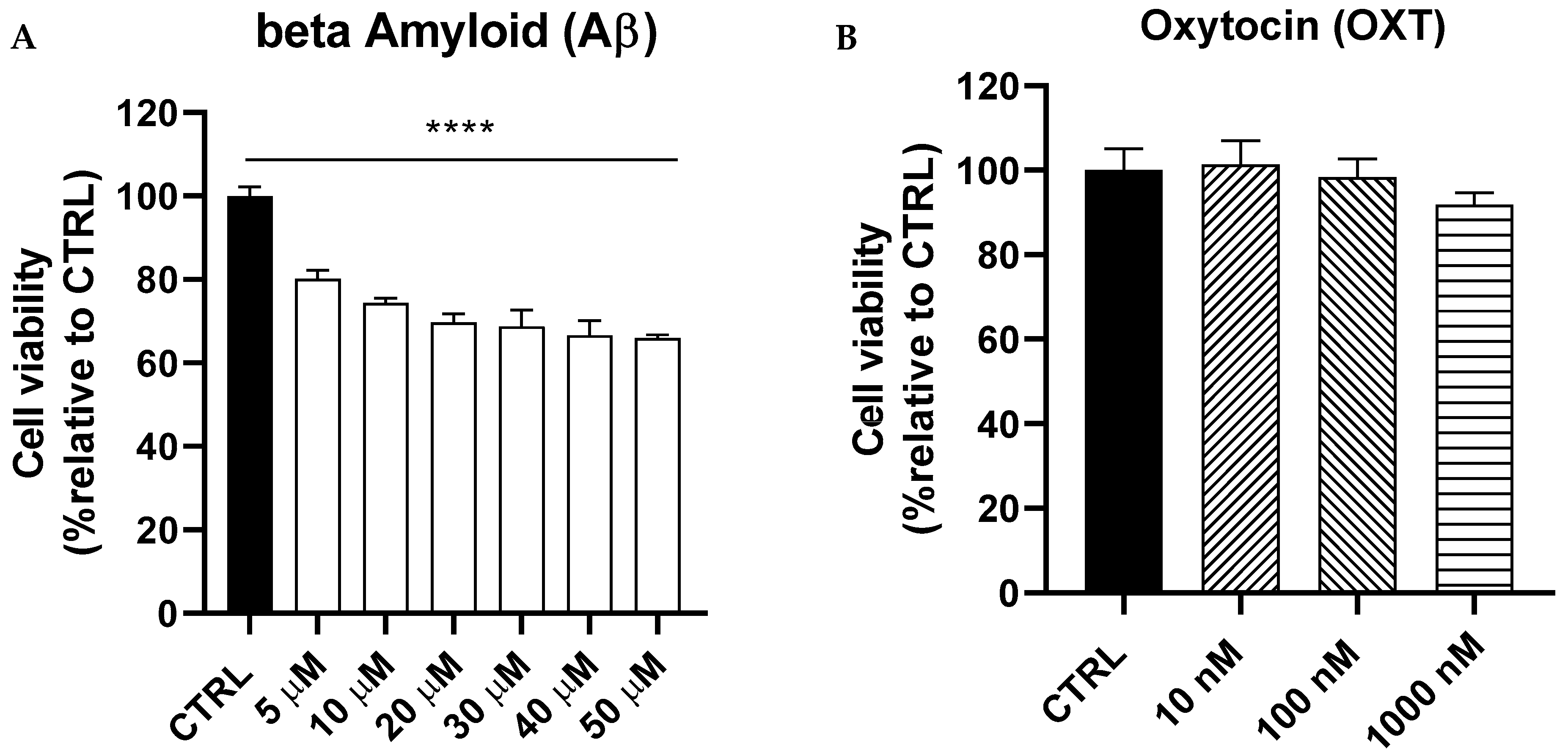
2.1. Dose–Response Curves of Amyloid Beta (Aβ) and Oxytocin (OXT) on PC12 Cells
2.2. Investigating the Protective Effect of OXT Against Aβ-Induced Cytotoxicity
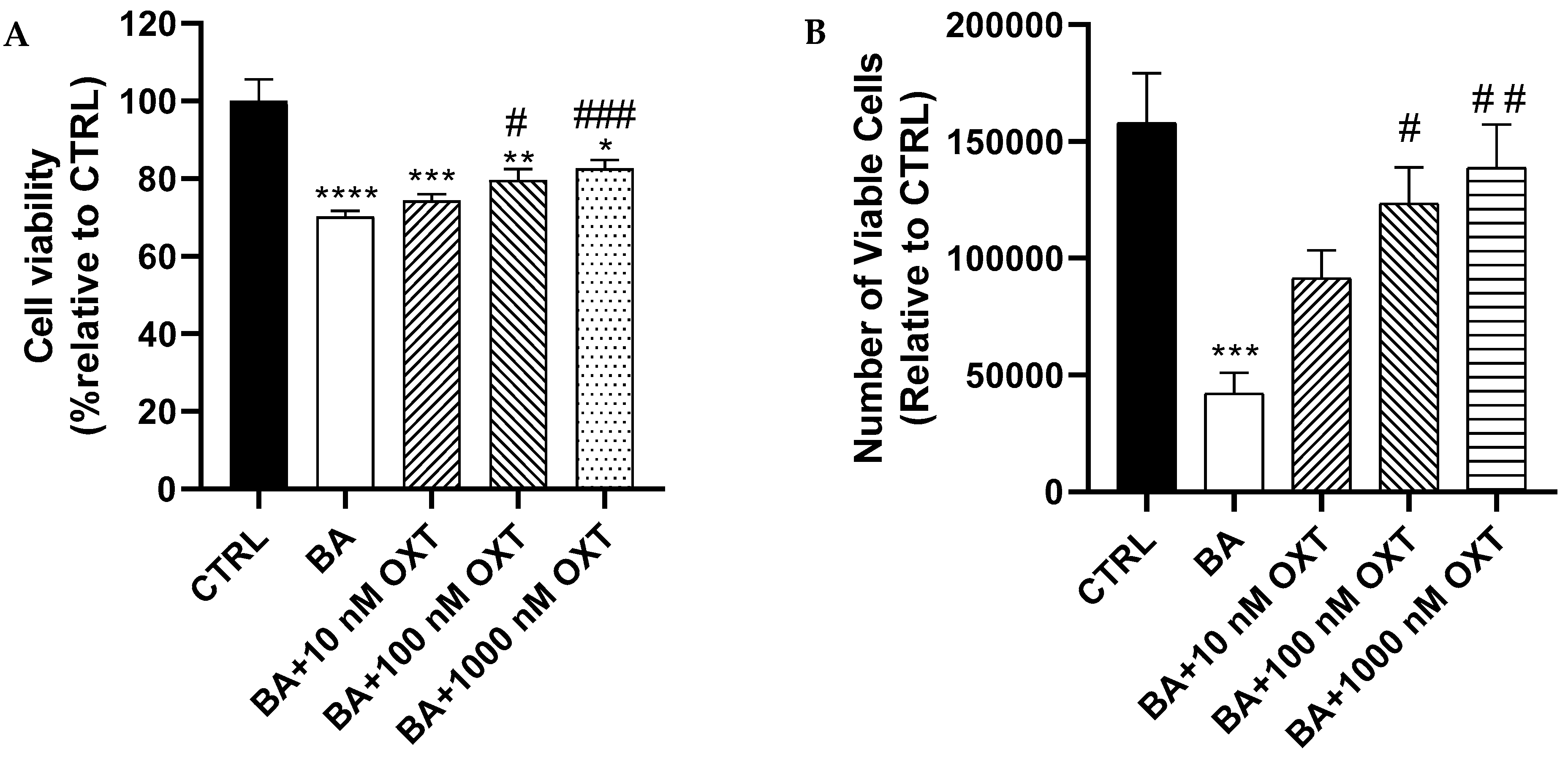
2.2.1. OXT Protects PC12 Cells Against Aβ-Induced Cytotoxicity
2.2.2. OXT Protects PC12 Cells Against Aβ-Induced Cytotoxicity (Trypan Blue Staining Assay)
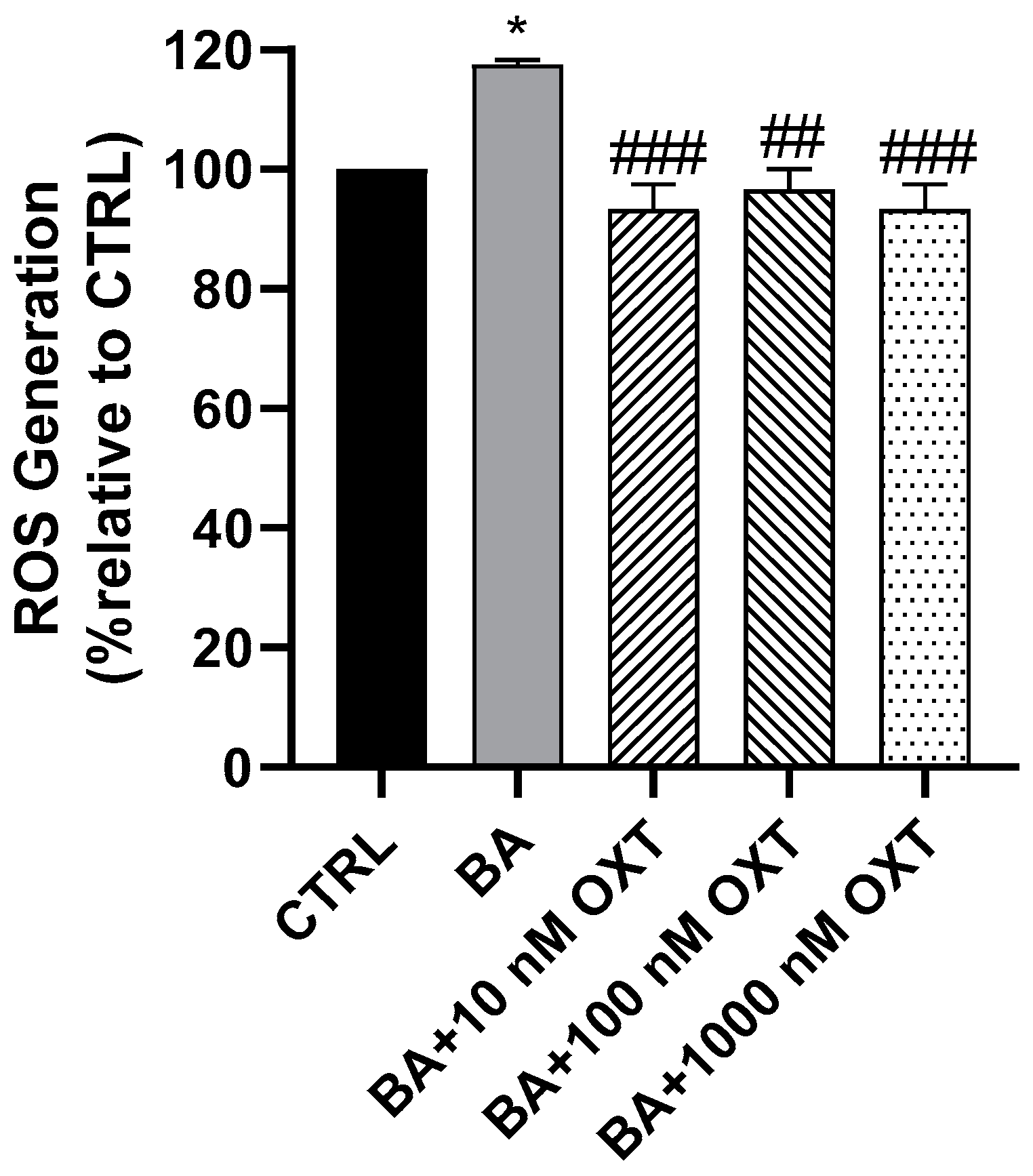
2.2.3. OXT Protects PC12 Cells Against Aβ-Induced Oxidative Stress
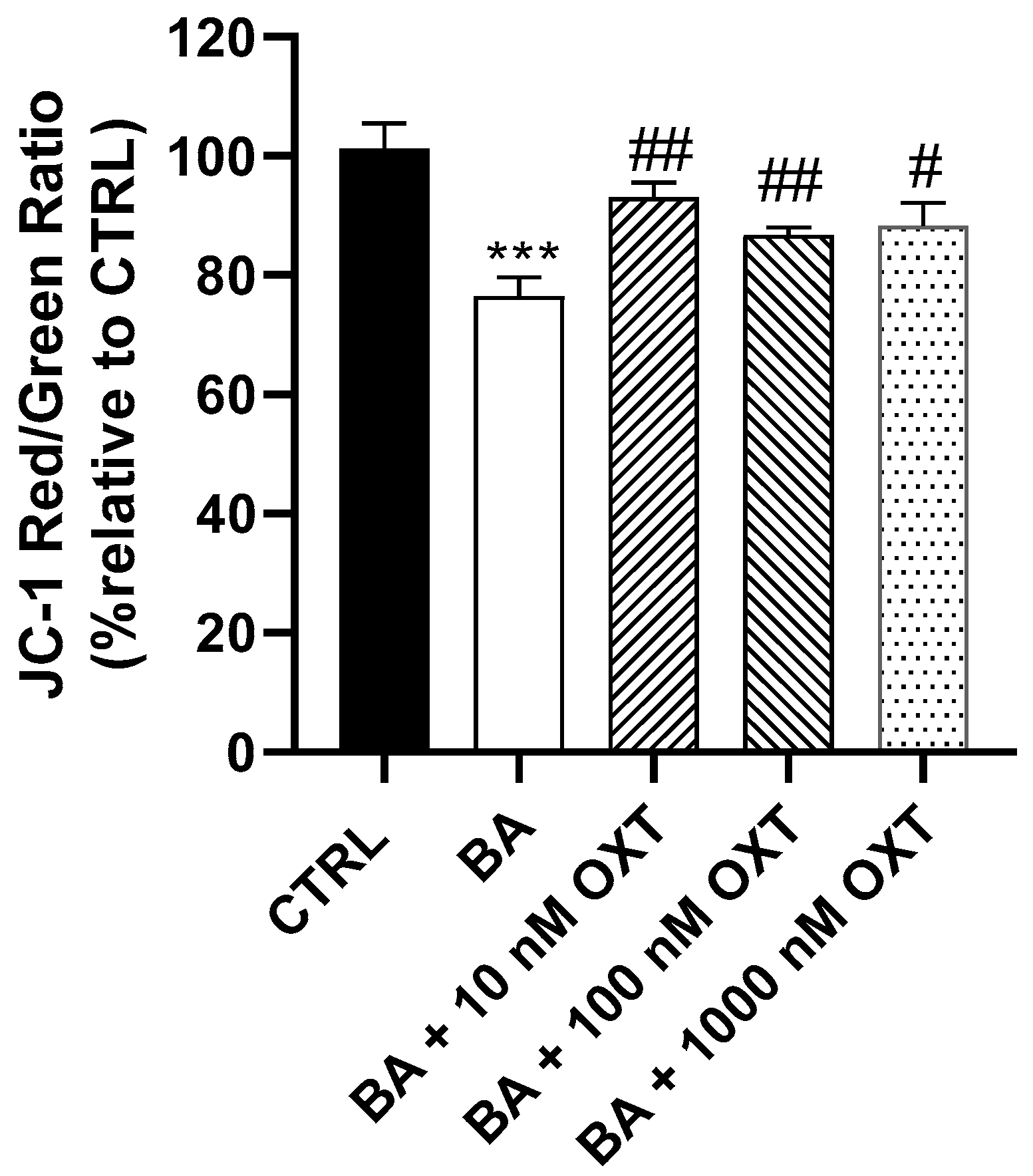
2.2.4. OXT Protects PC12 Cells Against Aβ-Induced Mitochondrial Dysfunction
2.3. Investigating the Protective Effect of OXT Against Aβ-Induced Cytotoxicity Through Modulation of Mitochondrial Apoptosis and MAPK Pathways
2.3.1. Effects of OXT on ERK1/2 Phosphorylation
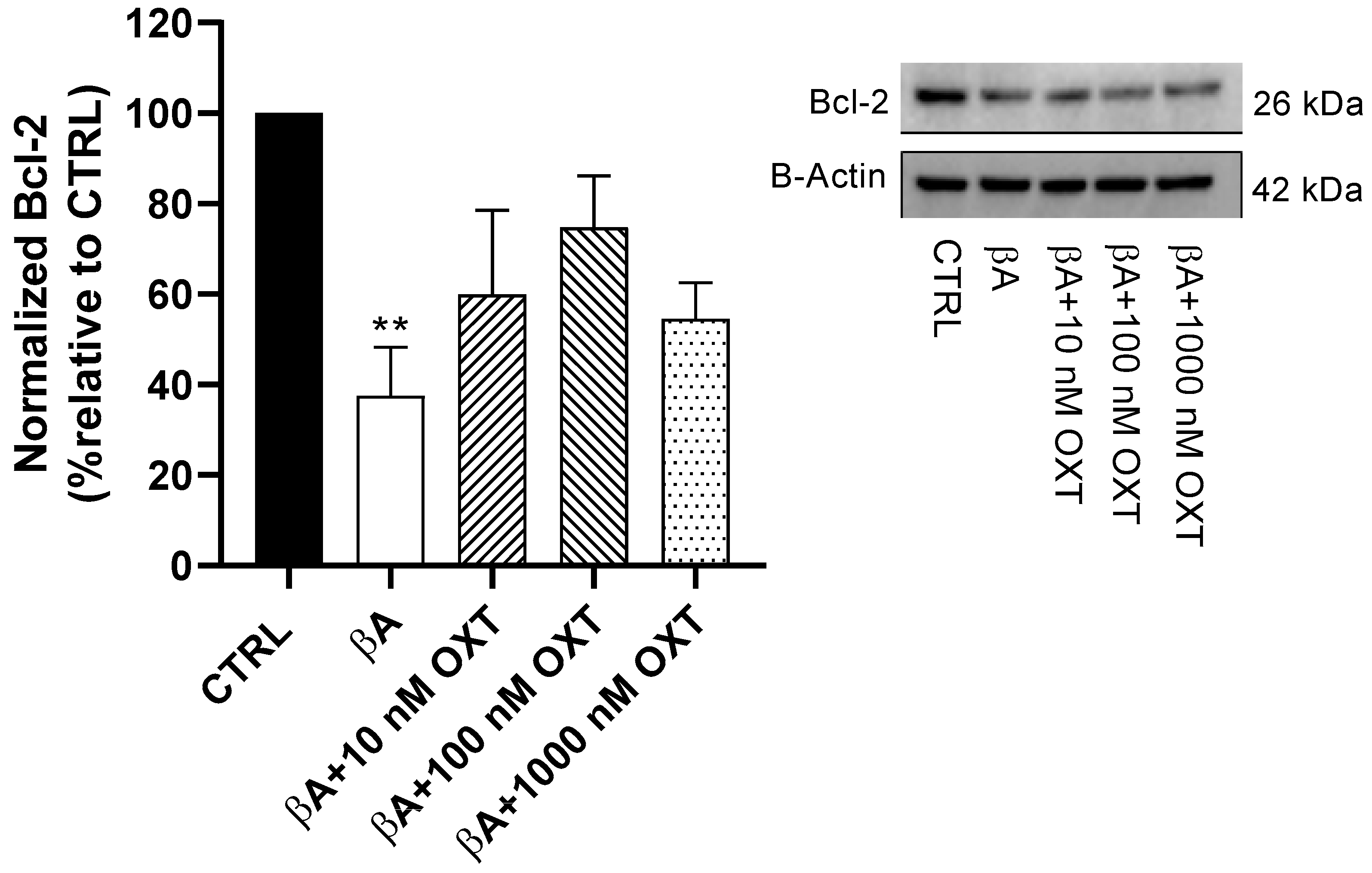
2.3.2. Effect of OXT on Bcl-2 Expression
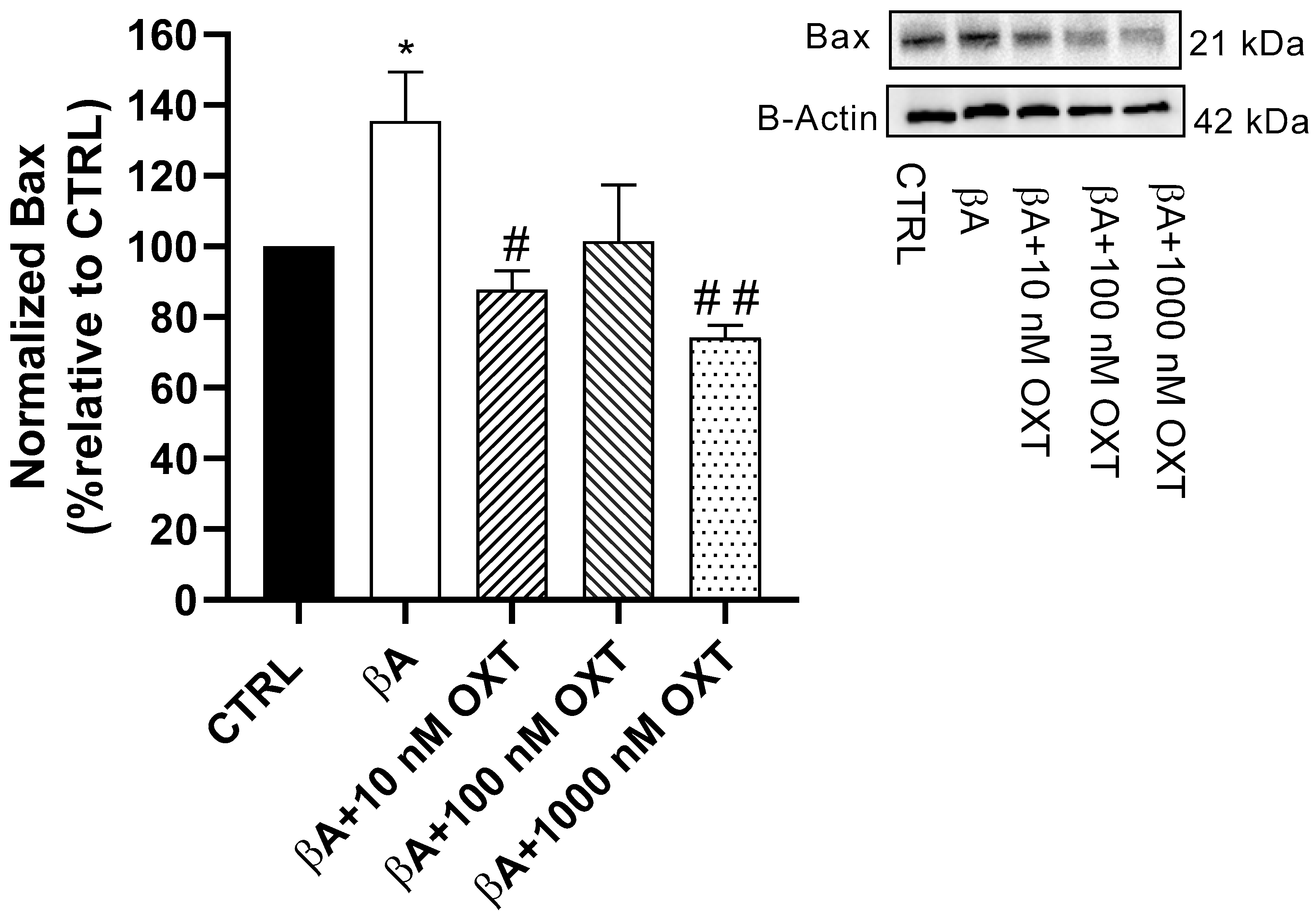
2.3.3. Effect of OXT on BAX Expression
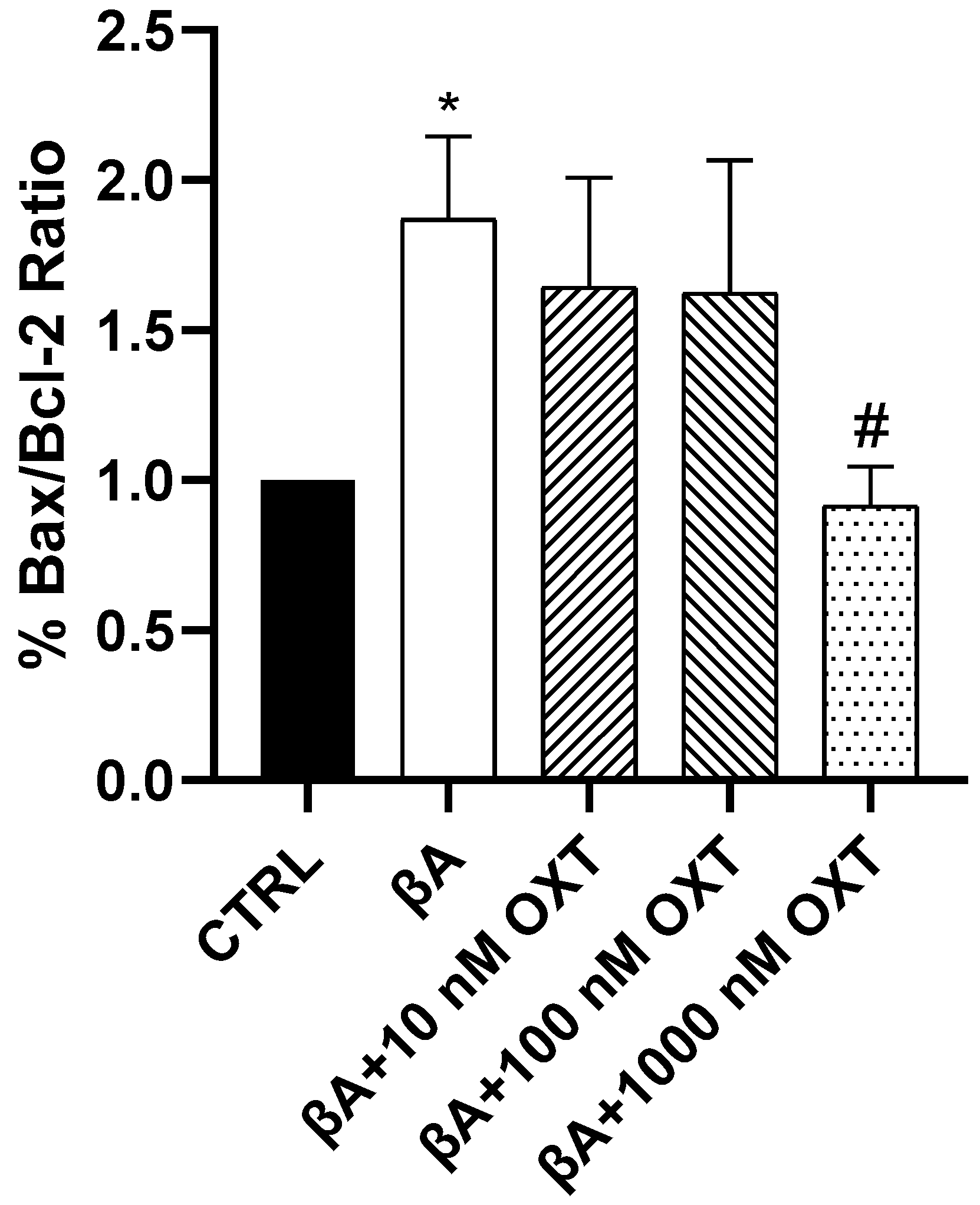
2.3.4. Effect of OXT on BAX/Bcl-2 Ratio
2.3.5. Effect of OXT on Caspase-3 Expression
3. Discussion
3.1. Investigating the Protective Effect of OXT Against Aβ-Amyloid-Induced Cytotoxicity on PC-12 Cells: Effect on Cell Proliferation and Viability
OXT Protects PC12 Cells Against Aβ-Induced Cytotoxicity
3.2. Investigating the Protective Effect of OXT on PC-12 Cells Against Aβ-Amyloid-Induced ROS Generation
OXT Protects PC12 Cells Against Aβ-Induced ROS
3.3. Investigating the Protective Effect of OXT on PC-12 Cells Against Aβ-Amyloid-Induced Mitochondrial Dysfunction
OXT Protects PC12 Cells Against Aβ-Induced Mitochondrial Membrane Disruption
3.4. Investigating Whether the Protective Effect of OXT on PC-12 Cells Against Aβ-Induced Cytotoxicity Involves Modulation of the MAPK and Mitochondrial Apoptosis Pathways
3.4.1. OXT Promotes Cell Proliferation of PC12 Cells by Activation of the ERK1/2 MAPK Signaling Pathway
3.4.2. OXT Protects PC12 Cells Against Apoptosis Mediated by Aβ Through Modulation of Mitochondrial Apoptosis Pathway
4. Materials and Methods
4.1. Instrumentation
4.2. Reagents
4.3. Aβ25–35 Preparation
4.4. OXT Preparation
4.5. Experimental Groups
4.6. Cell Culture
4.7. Cell Proliferation and Viability Experiments
4.7.1. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium Bromide (MTT) Assay
4.7.2. Trypan Blue Staining Assay
4.8. Measurement of ROS Level
4.9. Measurement of Mitochondrial Membrane Potential
4.10. Western Blotting
4.11. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gitler, A.D.; Dhillon, P.; Shorter, J. Neurodegenerative disease: Models, mechanisms, and a new hope. Dis. Model. Mech. 2017, 10, 499. [Google Scholar] [CrossRef] [PubMed]
- Schachter, A.S.; Davis, K.L. Alzheimer’s disease. Curr. Treat. Options Neurol. 2000, 2, 51–60. [Google Scholar] [CrossRef]
- Massoud, F.; Gauthier, S. Update on the pharmacological treatment of Alzheimer’s disease. Curr. Neuropharmacol. 2010, 8, 69–80. [Google Scholar] [CrossRef]
- Norton, S.; Matthews, F.E.; Barnes, D.E.; Yaffe, K.; Brayne, C. Potential for primary prevention of Alzheimer’s disease: An analysis of population-based data. Lancet Neurol. 2014, 13, 788–794. [Google Scholar] [CrossRef]
- Kumar, A.; Singh, A.; Ekavali. A review on Alzheimer’s disease pathophysiology and its management: An update. Pharmacol. Rep. 2015, 67, 195–203. [Google Scholar] [CrossRef]
- Masters, C.L.; Simms, G.; Weinman, N.A.; Multhaup, G.; McDonald, B.L.; Beyreuther, K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. USA 1985, 82, 4245–4249. [Google Scholar] [CrossRef] [PubMed]
- Clementi, M.E.; Marini, S.; Coletta, M.; Orsini, F.; Giardina, B.; Misiti, F. Aβ(31–35) and Aβ(25–35) fragments of amyloid beta-protein induce cellular death through apoptotic signals: Role of the redox state of methionine-35. FEBS Lett. 2005, 579, 2913–2918. [Google Scholar] [CrossRef] [PubMed]
- Kubo, T.; Nishimura, S.; Kumagae, Y.; Kaneko, I. In vivo conversion of racemized β-amyloid ([D-Ser26]Aβ1–40) to truncated and toxic fragments ([D-Ser26]Aβ25–35/40) and fragment presence in the brains of Alzheimer’s patients. J. Neurosci. Res. 2002, 70, 474–483. [Google Scholar] [CrossRef]
- Heneka, M.T. Neuroinflammation in Alzheimer’s Disease. Lancet Neurol. 2008, 14, 564–573. [Google Scholar] [CrossRef]
- Solito, E.; Sastre, M. Microglia function in Alzheimer’s disease. Front. Pharmacol. 2012, 3, 14. [Google Scholar] [CrossRef]
- Kaminsky, Y.G.; Kosenko, E.A. Effects of amyloid-beta peptides on hydrogen peroxide-metabolizing enzymes in rat brain in vivo. Free Radic. Res. 2008, 42, 564–573. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Youle, R.J. The Role of Mitochondria in Apoptosis. Annu. Rev. Genet. 2004, 43, 3647–3652. [Google Scholar] [CrossRef] [PubMed]
- Shariff, K.; Ghosal, S.; Matouschek, A. The force exerted by the membrane potential during protein import into the mitochondrial matrix. Biophys. J. 2004, 86, 3647–3652. [Google Scholar] [CrossRef]
- Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Orr, A.L.; Brand, M.D. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 2013, 1, 304–312. [Google Scholar] [CrossRef]
- Behl, C.; Davis, J.B.; Lesley, R.; Schubert, D. Hydrogen peroxide mediates amyloid beta protein toxicity. Cell 1994, 77, 817–827. [Google Scholar] [CrossRef]
- Hensley, K.; Carney, J.M.; Mattson, M.P.; Aksenova, M.; Harris, M.; Wu, J.F.; Floyd, R.A.; Butterfield, D.A. A model for beta-amyloid aggregation and neurotoxicity based on free radical generation by the peptide: Relevance to Alzheimer disease. Proc. Natl. Acad. Sci. USA 1994, 91, 3270. [Google Scholar] [CrossRef] [PubMed]
- Shearman, M.S.; Ragan, C.I.; Iversen, L.L. Inhibition of PC12 cell redox activity is a specific, early indicator of the mechanism of beta-amyloid-mediated cell death. Proc. Natl. Acad. Sci. USA 1994, 91, 1470. [Google Scholar] [CrossRef]
- Imai, Y.; Soda, M.; Inoue, H.; Hattori, N.; Mizuno, Y.; Takahashi, R. An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of Parkin. Cell 2001, 105, 891–902. [Google Scholar] [CrossRef]
- Nishitoh, H.; Matsuzawa, A.; Tobiume, K.; Saegusa, K.; Takeda, K.; Inoue, K.; Hori, S.; Kakizuka, A.; Ichijo, H. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002, 16, 1345–1355. [Google Scholar] [CrossRef]
- Liao, L.; Su, H.; Tan, F.; Qiu, J.; Zhu, P.; Chen, Y.; Lin, S.; Wu, H.; Mao, P.; Hu, J.; et al. Salidroside protects PC-12 cells against amyloid β-induced apoptosis by activation of the ERK1/2 and AKT signaling pathways. Int. J. Mol. Med. 2019, 43, 1769–1777. [Google Scholar] [CrossRef]
- Paradis, E.; Douillard, H.; Koutroumanis, M.; Goodyer, C.; LeBlanc, A. Amyloid β Peptide of Alzheimer’s Disease Downregulates Bcl-2 and Upregulates Bax Expression in Human Neurons. J. Neurosci. 1996, 16, 7533. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.-E.; Huang, C.-C.; Hwang, J.-P.; Yang, A.C.; Tu, P.-C.; Yeh, H.-L.; Hong, C.-J.; Liou, Y.-J.; Chen, J.-F.; Lin, C.-P.; et al. Effect of Bcl-2 rs956572 SNP on regional gray matter volumes and cognitive function in elderly males without dementia. Age 2013, 35, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Indrigo, M.; Morella, I.; Orellana, D.; d’Isa, R.; Papale, A.; Parra, R.; Gurgone, A.; Lecca, D.; Cavaccini, A.; Tigaret, C.M. Nuclear ERK1/2 signaling potentiation enhances neuroprotection and cognition via Importinα1/KPNA2. EMBO Mol. Med. 2023, 15, e15984. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.-P.; Wang, L.; Han, L.; Wang, S.C. Nonsocial functions of hypothalamic oxytocin. ISRN Neurosci. 2013, 2013, 179272. [Google Scholar] [CrossRef]
- Buemann, B.; Uvnäs-Moberg, K. Oxytocin may have a therapeutical potential against cardiovascular disease: Possible pharmaceutical and behavioral approaches. Med. Hypotheses 2020, 138, 109597. [Google Scholar] [CrossRef]
- Jameson, H.; Bateman, R.; Byrne, P.; Dyavanapalli, J.; Wang, X.; Jain, V.; Mendelowitz, D. Oxytocin neuron activation prevents hypertension that occurs with chronic intermittent hypoxia/hypercapnia in rats. Am. J. Physiol. Hear. Circ. Physiol. 2016, 310, H1549–H1557. [Google Scholar] [CrossRef]
- Wang, P.; Wang, S.C.; Yang, H.; Lv, C.; Jia, S.; Liu, X.; Wang, X.; Meng, D.; Qin, D.; Zhu, H.; et al. Therapeutic Potential of Oxytocin in Atherosclerotic Cardiovascular Disease: Mechanisms and Signaling Pathways. Front. Neurosci. 2019, 13, 454. [Google Scholar] [CrossRef]
- Alanazi, M.M.; Havranek, T.; Bakos, J.; Cubeddu, L.X.; Castejon, A.M. Cell proliferation and anti-oxidant effects of oxytocin and oxytocin receptors: Role of extracellular signal-regulating kinase in astrocyte-like cells. Endocr. Regul. 2020, 54, 172–182. [Google Scholar] [CrossRef]
- Sánchez-Vidaña, D.I.; Chan, N.-M.J.; Chan, A.H.; Hui, K.K.; Lee, S.; Chan, H.-Y.; Law, Y.S.; Sze, M.Y.; Tsui, W.-C.S.; Fung, T.K.; et al. Repeated treatment with oxytocin promotes hippocampal cell proliferation, dendritic maturation and affects socio-emotional behavior. Neuroscience 2016, 333, 65–77. [Google Scholar] [CrossRef]
- Hsieh, F.F.; Korsunsky, I.; Shih, A.J.; Moss, M.A.; Chatterjee, P.K.; Deshpande, J.; Xue, X.; Madankumar, S.; Kumar, G.; Rochelson, B.; et al. Maternal oxytocin administration modulates gene expression in the brains of perinatal mice. J. Perinat. Med. 2022, 50, 207–218. [Google Scholar] [CrossRef]
- Sola, C.; Zanelli, V.; Molinari, M.A.; Casadio, C.; Ricci, F.; Carpentiero, O.; Tondelli, M.; Lui, F.; Nichelli, P.F.; Benuzzi, F.; et al. Understanding basic and social emotions in Alzheimer’s disease and frontotemporal dementia. Front. Psychol. 2025, 16, 1535722. [Google Scholar] [CrossRef] [PubMed]
- Diamantopoulos, M. Behavioral and Psychological Changes in Alzheimer’s and Other Neurodegenerative Disorders. In Handbook of Computational Neurodegeneration; Springer International Publishing: Cham, Switzerland, 2022; pp. 1–30. [Google Scholar]
- Ghazy, A.A.; Soliman, O.A.; Elbahnasi, A.I.; Alawy, A.Y.; Mansour, A.M.; Gowayed, M.A. Role of Oxytocin in Different Neuropsychiatric, Neurodegenerative, and Neurodevelopmental Disorders. Rev. Physiol. Biochem. Pharmacol. 2023, 186, 95–134. [Google Scholar] [PubMed]
- Düşünceli, F.; Işeri, S.Ö.; Ercan, F.; Gedik, N.; Yeǧen, C.; Yeǧen, B.Ç. Oxytocin alleviates hepatic ischemia-reperfusion injury in rats. Peptides 2008, 29, 1216–1222. [Google Scholar] [CrossRef] [PubMed]
- Senturk, G.E.; Erkanli, K.; Aydin, U.; Yucel, D.; Isiksacan, N.; Ercan, F.; Arbak, S. The protective effect of oxytocin on ischemia/reperfusion injury in rat urinary bladder. Peptides 2013, 40, 82–88. [Google Scholar] [CrossRef]
- Ghasemnezhad, R.; Mohammadghasemi, F.; Faghani, M.; Bahadori, M.H. Oxytocin can decrease germ cells apoptotic index in testis under acute ischemia reperfusion in a rat model. Iran. J. Reprod. Med. 2015, 13, 283–290. [Google Scholar]
- Mustaly-Kalimi, S.; Gallegos, W.; Marr, R.A.; Peterson, D.A.; Sekler, I.; Stutzmann, G.E. Mitochondrial Dysfunction in Alzheimer’s Disease Is Driven By Excess ER-Calcium Release in Patient-Derived Neurons. bioRxiv 2024. bioRxiv:2024.10.11.617867. [Google Scholar]
- Bhatia, S.; Rawal, R.; Sharma, P.; Singh, T.; Singh, M.; Singh, V. Mitochondrial Dysfunction in Alzheimer’s Disease: Opportunities for Drug Development. Curr. Neuropharmacol. 2022, 20, 675. [Google Scholar] [CrossRef]
- Misrani, A.; Tabassum, S.; Yang, L. Mitochondrial Dysfunction and Oxidative Stress in Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 617588. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Wei, S.; Jo, Y.; Zhang, Y.; Park, W.; Gariani, K.; Oh, C.-M.; Kim, H.H.; Ha, K.-T.; Park, K.S.; et al. Mitochondria-associated programmed cell death as a therapeutic target for age-related disease. Exp. Mol. Med. 2023, 55, 1595–1619. [Google Scholar] [CrossRef]
- Buchholz, T.A.; Davis, D.W.; McConkey, D.J.; Symmans, W.F.; Valero, V.; Jhingran, A.; Tucker, S.L.; Pusztai, L.; Cristofanilli, M.; Esteva, F.J.; et al. Chemotherapy-induced apoptosis and Bcl-2 levels correlate with breast cancer response to chemotherapy. Cancer J. 2003, 9, 33–41. [Google Scholar] [CrossRef]
- Das, A.; Banik, N.L.; Patel, S.J.; Ray, S.K. Dexamethasone protected human glioblastoma U87MG cells from temozolomide induced apoptosis by maintaining Bax:Bcl-2 ratio and preventing proteolytic activities. Mol. Cancer 2004, 3, 36. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.-M.; Sun, Y.; Lu, Y.-Y.; Zhang, H.; Chen, Q.-L.; Su, S.-B. Curcumin reduces mitomycin C resistance in breast cancer stem cells by regulating Bcl-2 family-mediated apoptosis. Cancer Cell Int. 2017, 17, 84. [Google Scholar] [CrossRef] [PubMed]
- Momenabadi, S.; Vafaei, A.A.; Khorasani, M.Z.; Vakili, A. Pre-Ischemic Oxytocin Treatment Alleviated Neuronal Injury via Suppressing NF-κB, MMP-9, and Apoptosis Regulator Proteins in A Mice Model of Stroke. Cell J. 2022, 24, 337–345. [Google Scholar]
- Lindenboim, L.; Haviv, R.; Stein, R. Inhibition of drug-induced apoptosis by survival factors in PC12 cells. J. Neurochem. 1995, 64, 1054–1063. [Google Scholar] [CrossRef]
- Callens, M.; Kraskovskaya, N.; Derevtsova, K.; Annaert, W.; Bultynck, G.; Bezprozvanny, I.; Vervliet, T. The role of Bcl-2 proteins in modulating neuronal Ca2+ signaling in health and in Alzheimer’s disease. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2021, 1868, 118997. [Google Scholar] [CrossRef]
- Finger, E.; Berry, S.; Cummings, J.; Coleman, K.; Hsiung, R.; Feldman, H.H.; Boxer, A. Adaptive crossover designs for assessment of symptomatic treatments targeting behaviour in neurodegenerative disease: A phase 2 clinical trial of intranasal oxytocin for frontotemporal dementia (FOXY). Alzheimer’s Res. Ther. 2018, 10, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Sikich, L.; Kolevzon, A.; King, B.H.; McDougle, C.J.; Sanders, K.B.; Kim, S.-J.; Spanos, M.; Chandrasekhar, T.; Trelles, P.; Rockhill, C.M.; et al. Intranasal Oxytocin in Children and Adolescents with Autism Spectrum Disorder. N. Engl. J. Med. 2021, 385, 1462–1473. [Google Scholar] [CrossRef]
- Lestanova, Z.; Puerta, F.; Alanazi, M.; Bacova, Z.; Kiss, A.; Castejon, A.M.; Bakos, J. Downregulation of Oxytocin Receptor Decreases the Length of Projections Stimulated by Retinoic Acid in the U-87MG Cells. Neurochem. Res. 2017, 42, 1006–1014. [Google Scholar] [CrossRef]
- Zatkova, M.; Bacova, Z.; Puerta, F.; Lestanova, Z.; Alanazi, M.; Kiss, A.; Reichova, A.; Castejon, A.M.; Ostatnikova, D.; Bakos, J. Projection length stimulated by oxytocin is modulated by the inhibition of calcium signaling in U-87MG cells. J. Neural Transm. 2018, 125, 1847–1856. [Google Scholar] [CrossRef]
- Meng, M.; Zhang, L.; AI, D.; Wu, H.; Peng, W. β-Asarone Ameliorates β-Amyloid–Induced Neurotoxicity in PC12 Cells by Activating P13K/Akt/Nrf2 Signaling Pathway. Front. Pharmacol. 2021, 12, 659955. [Google Scholar] [CrossRef]
- Li, R.C.; Pouranfar, F.; Lee, S.K.; Morris, M.W.; Wang, Y.; Gozal, D. Neuroglobin protects PC12 cells against β-amyloid-induced cell injury. Neurobiol. Aging 2008, 29, 1815–1822. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group 1 | Group 2 | Group 3 | Group 4 | Group 5 |
|---|---|---|---|---|
| Control | 20 μM Aβ for 48 h | 10 nM OXT for 2 h + 20 μM Aβ for 48 h | 100 nM OXT for 2 h + 20 μM Aβ for 48 h | 1000 nM OXT for 2 h + 20 μM Aβ for 48 h |
| RED (Oligomers) | GREEN (Monomers) |
|---|---|
| Excitation/530 nm | Excitation/485 nm |
| Emission/590 nm | Emission/528 nm |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alanazi, M.M.; Albaker, A.B.; Alzaagi, L.A.; Alsabhan, J.F.; Alasmari, F.; Almutairi, M.M.; Alharbi, M.S.; Alasmari, A.F.; Alqahtani, F.; Alsanea, S. Oxytocin Protects PC12 Cells Against β-Amyloid-Induced Cell Injury. Pharmaceuticals 2025, 18, 390. https://doi.org/10.3390/ph18030390
Alanazi MM, Albaker AB, Alzaagi LA, Alsabhan JF, Alasmari F, Almutairi MM, Alharbi MS, Alasmari AF, Alqahtani F, Alsanea S. Oxytocin Protects PC12 Cells Against β-Amyloid-Induced Cell Injury. Pharmaceuticals. 2025; 18(3):390. https://doi.org/10.3390/ph18030390
Chicago/Turabian StyleAlanazi, Mohammed Mufadhe, Awatif B. Albaker, Lamia A. Alzaagi, Jawza F. Alsabhan, Fawaz Alasmari, Mohammed M. Almutairi, Metab S. Alharbi, Abdullah F. Alasmari, Faleh Alqahtani, and Sary Alsanea. 2025. "Oxytocin Protects PC12 Cells Against β-Amyloid-Induced Cell Injury" Pharmaceuticals 18, no. 3: 390. https://doi.org/10.3390/ph18030390
APA StyleAlanazi, M. M., Albaker, A. B., Alzaagi, L. A., Alsabhan, J. F., Alasmari, F., Almutairi, M. M., Alharbi, M. S., Alasmari, A. F., Alqahtani, F., & Alsanea, S. (2025). Oxytocin Protects PC12 Cells Against β-Amyloid-Induced Cell Injury. Pharmaceuticals, 18(3), 390. https://doi.org/10.3390/ph18030390