

Cardiac Fibrosis: Mechanistic Discoveries Linked to SGLT2 Inhibitors

{kind=link}
{kind=link}
Abstract
1. Introduction
Literature Search Strategy
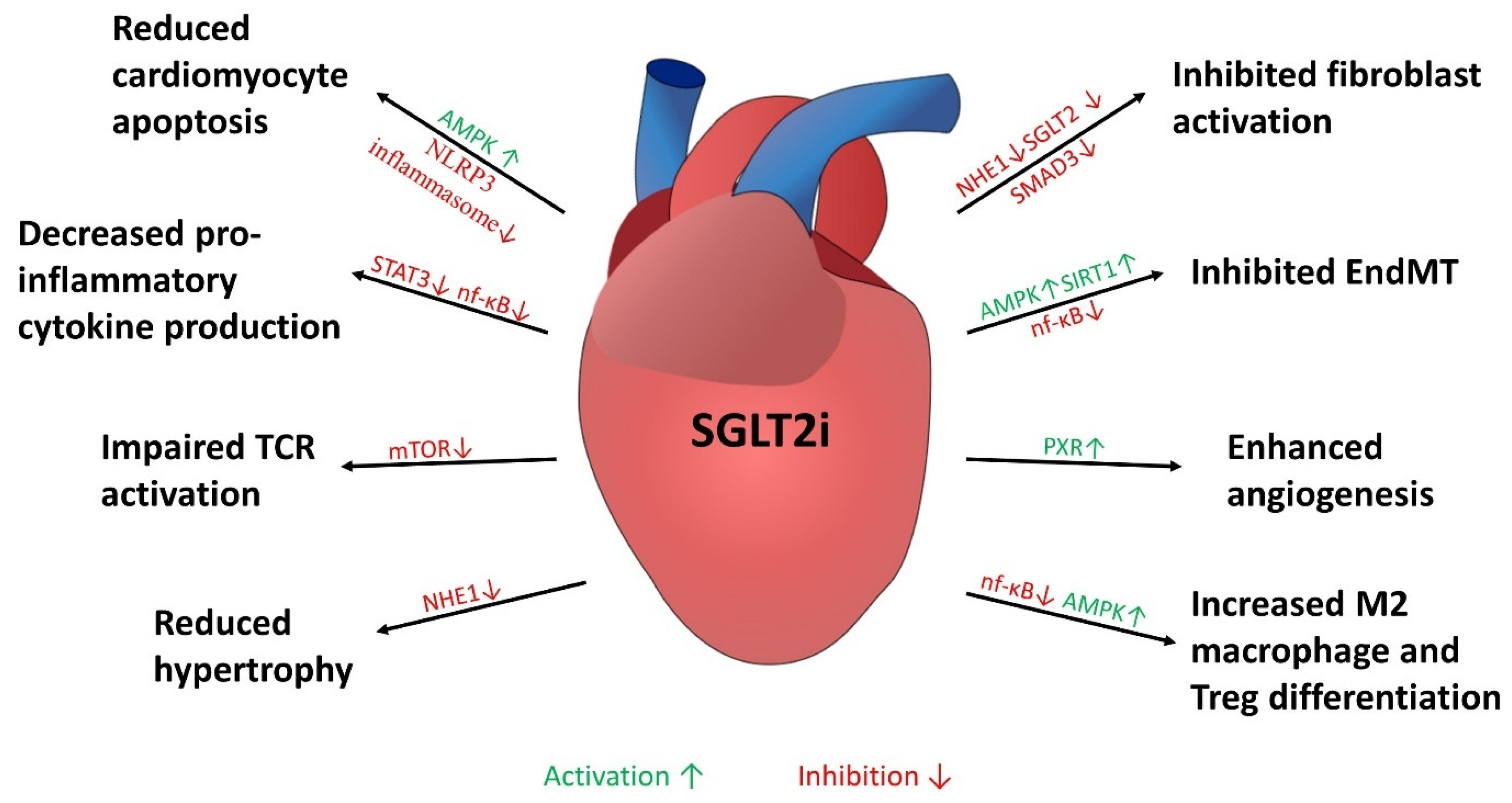
2. Animal Studies
3. Clinical Trials
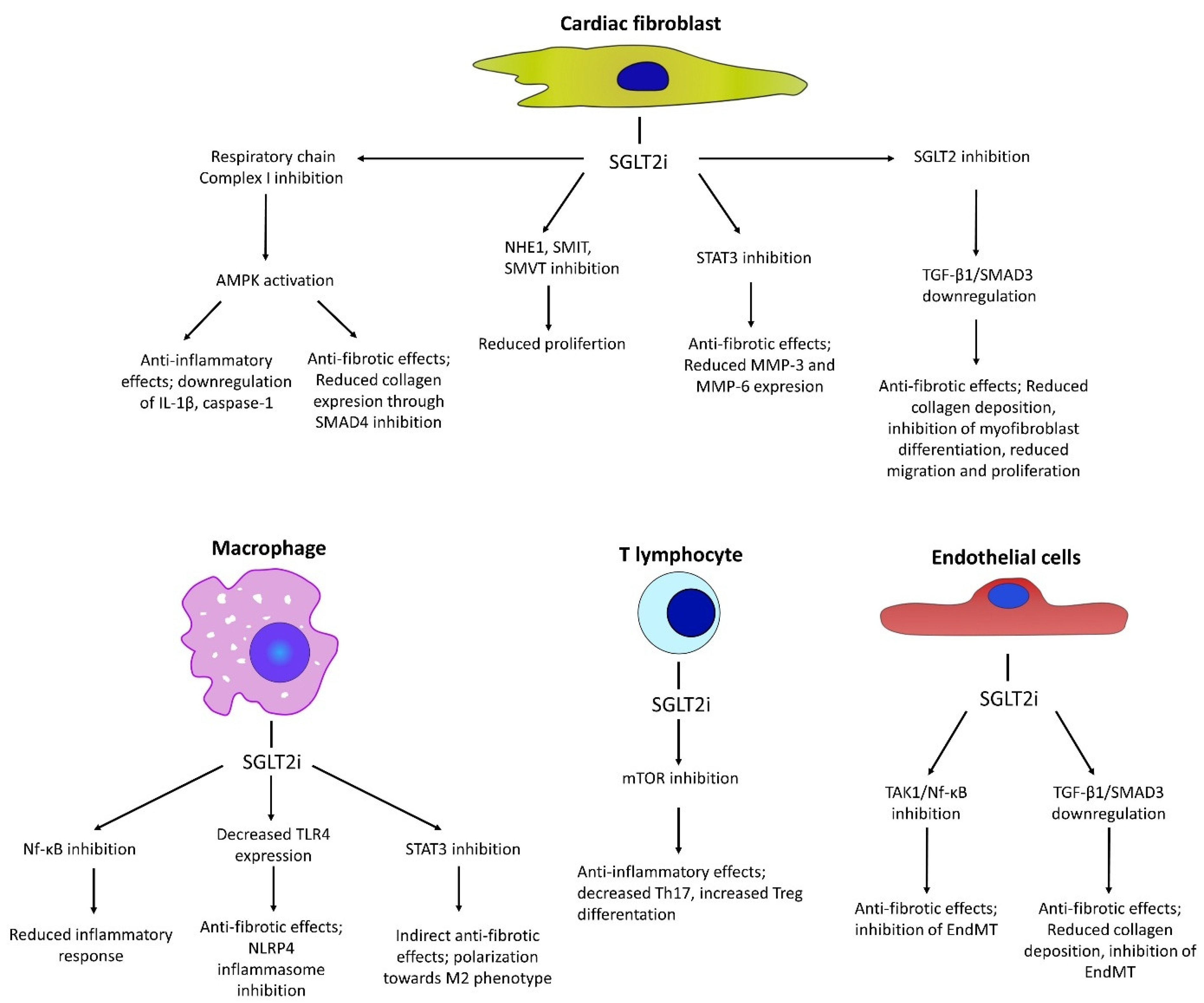
4. Cellular Mechanisms of Anti-Fibrotic Effects Mediated by SGLT2i
4.1. Anti-Fibrotic Effects in Fibroblasts
4.2. Anti-Fibrotic Effects Through Effect on Endothelial Cells
4.3. Modulation of Inflammatory Responses
4.4. Modulation of MicroRNA Profile
5. Indirect Cardioprotective Effects Mediated by SGLT2is
5.1. Promotion of Renal Glucose Excretion
5.2. Reduction in Cardiac Workload and Blood Pressure
5.3. Metabolic Modulation and Ketogenesis
5.4. Electrolyte Balance and Renal Protection
5.5. Reduction in Oxidative Stress
6. Conclusions
Funding
Conflicts of Interest
References
- Jasleen, B.; Vishal, G.K.; Sameera, M.; Fahad, M.; Brendan, O.; Deion, S.; Pemminati, S. Sodium-Glucose Cotransporter 2 (SGLT2) Inhibitors: Benefits Versus Risk. Cureus 2023, 15, e33939. [Google Scholar] [CrossRef]
- Fatima, A.; Rasool, S.; Devi, S.; Talha, M.; Waqar, F.; Nasir, M.; Khan, M.R.; Ibne, S.M.; Jaffari, A.; Haider, A.; et al. Exploring the Cardiovascular Benefits of Sodium-Glucose Cotransporter-2 (SGLT2) Inhibitors: Expanding Horizons Beyond Diabetes Management. Cureus 2023, 15, e46243. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Cardiac fibrosis. Cardiovasc. Res. 2021, 117, 1450–1488. [Google Scholar]
- Travers, J.G.; Kamal, F.A.; Robbins, J.; Yutzey, K.E.; Blaxall, B.C. Cardiac Fibrosis. Circ. Res. 2016, 118, 1021–1040. [Google Scholar] [CrossRef]
- Thomas, T.P.; Grisanti, L.A. The Dynamic Interplay Between Cardiac Inflammation and Fibrosis. Front. Physiol. 2020, 11, 529075. [Google Scholar] [CrossRef] [PubMed]
- Ciampi, C.M.; Sultana, A.; Ossola, P.; Farina, A.; Fragasso, G.; Spoladore, R. Current experimental and early investigational agents for cardiac fibrosis: Where are we at? Expert. Opin. Investig. Drugs 2024, 33, 389–404. [Google Scholar] [CrossRef] [PubMed]
- Santos-Gallego, C.G.; Requena-Ibanez, J.A.; San Antonio, R.; Garcia-Ropero, A.; Ishikawa, K.; Watanabe, S.; Picatoste, B.; Vargas-Delgado, A.P.; Flores-Umanzor, E.J.; Sanz, J.; et al. Empagliflozin Ameliorates Diastolic Dysfunction and Left Ventricular Fibrosis/Stiffness in Nondiabetic Heart Failure: A Multimodality Study. JACC Cardiovasc. Imaging 2021, 14, 393–407. [Google Scholar] [CrossRef] [PubMed]
- Requena-Ibáñez, J.A.; Santos-Gallego, C.G.; Rodriguez-Cordero, A.; Vargas-Delgado, A.P.; Mancini, D.; Sartori, S.; Atallah-Lajam, F.; Giannarelli, C.; Macaluso, F.; Lala, A.; et al. Mechanistic Insights of Empagliflozin in Nondiabetic Patients with HFrEF: From the EMPA-TROPISM Study. JACC Heart Fail. 2021, 9, 578–589. [Google Scholar] [CrossRef]
- Mason, T.; Coelho-Filho, O.R.; Verma, S.; Chowdhury, B.; Zuo, F.; Quan, A.; Thorpe, K.E.; Bonneau, C.; Teoh, H.; Gilbert, R.E.; et al. Empagliflozin Reduces Myocardial Extracellular Volume in Patients with Type 2 Diabetes and Coronary Artery Disease. JACC Cardiovasc. Imaging 2021, 14, 1164–1173. [Google Scholar] [CrossRef] [PubMed]
- Schauer, A.; Adams, V.; Kämmerer, S.; Langner, E.; Augstein, A.; Barthel, P.; Männel, A.; Fabig, G.; Nascimento Alves, P.K.; Günscht, M.; et al. Empagliflozin Improves Diastolic Function in HFpEF by Restabilizing the Mitochondrial Respiratory Chain. Circ. Heart Fail. 2024, 17, e011107. [Google Scholar] [CrossRef]
- Anker, S.D.; Butler, J.; Filippatos, G.; Ferreira, J.P.; Bocchi, E.; Böhm, M.; Brunner–La Rocca, H.-P.; Choi, D.-J.; Chopra, V.; Chuquiure-Valenzuela, E.; et al. Empagliflozin in Heart Failure with a Preserved Ejection Fraction. N. Engl. J. Med. 2021, 385, 1451–1461. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Pocock, S.J.; Carson, P.; Januzzi, J.; Verma, S.; Tsutsui, H.; Brueckmann, M.; et al. Cardiovascular and Renal Outcomes with Empagliflozin in Heart Failure. N. Engl. J. Med. 2020, 383, 1413–1424. [Google Scholar] [CrossRef]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Bělohlávek, J.; et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef] [PubMed]
- Wiviott, S.D.; Raz, I.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Silverman, M.G.; Zelniker, T.A.; Kuder, J.F.; Murphy, S.A.; et al. Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2019, 380, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; de Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthews, D.R. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 644–657. [Google Scholar] [CrossRef] [PubMed]
- Steiner, S. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. Z. Gefassmedizin 2016, 13, 17–18. [Google Scholar]
- Tang, J.; Ye, L.; Yan, Q.; Zhang, X.; Wang, L. Effects of Sodium-Glucose Cotransporter 2 Inhibitors on Water and Sodium Metabolism. Front. Pharmacol. 2022, 13, 800490. [Google Scholar] [CrossRef]
- Wallenius, K.; Kroon, T.; Hagstedt, T.; Löfgren, L.; Sörhede-Winzell, M.; Boucher, J.; Lindén, D.; Oakes, N.D. The SGLT2 inhibitor dapagliflozin promotes systemic FFA mobilization, enhances hepatic β-oxidation, and induces ketosis. J. Lipid Res. 2022, 63, 63–66. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Zhang, M.; Suo, M.; Liu, D.; Wang, X.; Liu, M.; Pan, J.; Jin, T.; An, F. Dapagliflozin alleviates cardiac fibrosis through suppressing EndMT and fibroblast activation via AMPKα/TGF-β/Smad signalling in type 2 diabetic rats. J. Cell Mol. Med. 2021, 25, 7642–7659. [Google Scholar] [CrossRef]
- Hodrea, J.; Saeed, A.; Molnar, A.; Fintha, A.; Barczi, A.; Wagner, L.J.; Szabo, A.J.; Fekete, A.; Balogh, D.B. SGLT2 inhibitor dapagliflozin prevents atherosclerotic and cardiac complications in experimental type 1 diabetes. PLoS ONE 2022, 17, e0263285. [Google Scholar] [CrossRef]
- Ye, Y.; Bajaj, M.; Yang, H.C.; Perez-Polo, J.R.; Birnbaum, Y. SGLT-2 Inhibition with Dapagliflozin Reduces the Activation of the Nlrp3/ASC Inflammasome and Attenuates the Development of Diabetic Cardiomyopathy in Mice with Type 2 Diabetes. Further Augmentation of the Effects with Saxagliptin, a DPP4 Inhibitor. Cardiovasc. Drugs Ther. 2017, 31, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhang, J.; Xue, M.; Li, X.; Han, F.; Liu, X.; Xu, L.; Lu, Y.; Cheng, Y.; Li, T.; et al. SGLT2 inhibition with empagliflozin attenuates myocardial oxidative stress and fibrosis in diabetic mice heart. Cardiovasc. Diabetol. 2019, 18, 15. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.G.; Kim, D.; Lee, J.J.; Lee, H.J.; Moon, R.K.; Lee, Y.J.; Lee, S.J.; Lee, O.H.; Kim, C.; Oh, J.; et al. Dapagliflozin attenuates diabetes-induced diastolic dysfunction and cardiac fibrosis by regulating SGK1 signaling. BMC Med. 2022, 20, 309. [Google Scholar] [CrossRef]
- Yang, Z.; Li, T.; Xian, J.; Chen, J.; Huang, Y.; Zhang, Q.; Lin, X.; Lu, H.; Lin, Y. SGLT2 inhibitor dapagliflozin attenuates cardiac fibrosis and inflammation by reverting the HIF-2α signaling pathway in arrhythmogenic cardiomyopathy. FASEB J. 2022, 36, e22410. [Google Scholar] [CrossRef]
- Chung, C.C.; Lin, Y.K.; Chen, Y.C.; Kao, Y.H.; Yeh, Y.H.; Trang, N.N.; Chen, Y.J. Empagliflozin suppressed cardiac fibrogenesis through sodium-hydrogen exchanger inhibition and modulation of the calcium homeostasis. Cardiovasc. Diabetol. 2023, 22, 27. [Google Scholar] [CrossRef]
- Koizumi, T.; Watanabe, M.; Yokota, T.; Tsuda, M.; Handa, H.; Koya, J.; Nishino, K.; Tatsuta, D.; Natsui, H.; Kadosaka, T.; et al. Empagliflozin suppresses mitochondrial reactive oxygen species generation and mitigates the inducibility of atrial fibrillation in diabetic rats. Front. Cardiovasc. Med. 2023, 10, 1005408. [Google Scholar] [CrossRef] [PubMed]
- Travers, J.G.; Tharp, C.A.; Rubino, M.; McKinsey, T.A. Therapeutic targets for cardiac fibrosis: From old school to next-gen. J. Clin. Investig. 2022, 132, e148554. [Google Scholar] [CrossRef]
- Savarese, G.; Lund, L.H. Global Public Health Burden of Heart Failure. Card. Fail. Rev. 2017, 3, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Ambrosy, A.P.; Fonarow, G.C.; Butler, J.; Chioncel, O.; Greene, S.J.; Vaduganathan, M.; Nodari, S.; Lam, C.S.P.; Sato, N.; Shah, A.N.; et al. The global health and economic burden of hospitalizations for heart failure: Lessons learned from hospitalized heart failure registries. J. Am. Coll. Cardiol. 2014, 63, 1123–1133. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Zhao, C.; Fang, S. SGLT2 promotes cardiac fibrosis following myocardial infarction and is regulated by miR-141. Exp. Ther. Med. 2021, 22, 715. [Google Scholar] [CrossRef]
- Marfella, R.; Scisciola, L.; D’Onofrio, N.; Maiello, C.; Trotta, M.C.; Sardu, C.; Panarese, I.; Ferraraccio, F.; Capuano, A.; Barbieri, M.; et al. Sodium-glucose cotransporter-2 (SGLT2) expression in diabetic and non-diabetic failing human cardiomyocytes. Pharmacol. Res. 2022, 184, 106448. [Google Scholar] [CrossRef]
- Mroueh, A.; Fakih, W.; Gong, D.S.; Auger, C.; Pieper, M.P.; Morel, O.; Mazzucotelli, J.P.; Schini-Kerth, V. SGLT2 expression in the left ventricle of cardiac patients is correlated with low-grade inflammation involving the pro-oxidant AT1R/NADPH oxidases/SGLT2 crosstalk: Potential role in heart failure. Eur. Heart J. 2023, 44, ehad655-3150. [Google Scholar] [CrossRef]
- Nakano, D.; Akiba, J.; Tsutsumi, T.; Kawaguchi, M.; Yoshida, T.; Koga, H.; Kawaguchi, T. Hepatic expression of sodium–glucose cotransporter 2 (SGLT2) in patients with chronic liver disease. Med. Mol. Morphol. 2022, 55, 304. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, L.; Yamashita, S.; Nomiyama, T.; Kawanami, T.; Hamaguchi, Y.; Shigeoka, T.; Horikawa, T.; Tanaka, Y.; Yanase, T.; Kawanami, D.; et al. Sodium-glucose cotransporter 2 inhibitor canagliflozin attenuates lung cancer cell proliferation in vitro. Diabetol. Int. 2021, 12, 389. [Google Scholar] [CrossRef]
- Hu, J.; Teng, J.; Hui, S.; Liang, L. SGLT-2 inhibitors as novel treatments of multiple organ fibrosis. Heliyon 2024, 10, e29486. [Google Scholar] [CrossRef]
- Byrne, N.J.; Matsumura, N.; Maayah, Z.H.; Ferdaoussi, M.; Takahara, S.; Darwesh, A.M.; Levasseur, J.L.; Jahng, J.W.S.; Vos, D.; Parajuli, N.; et al. Empagliflozin Blunts Worsening Cardiac Dysfunction Associated with Reduced NLRP3 (Nucleotide-Binding Domain-Like Receptor Protein 3) Inflammasome Activation in Heart Failure. Circ. Heart Fail. 2020, 13, E006277. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Zou, R.; Shi, W.; Zhou, N.; Chen, S.; Zhou, H.; Chen, X.; Wu, Y. SGLT2 inhibitor dapagliflozin reduces endothelial dysfunction and microvascular damage during cardiac ischemia/reperfusion injury through normalizing the XO-SERCA2-CaMKII-coffilin pathways. Theranostics 2022, 12, 5034–5050. [Google Scholar] [CrossRef] [PubMed]
- Lv, C.; Hu, C.; Zhu, C.; Wan, X.; Chen, C.; Ji, X.; Qin, Y.; Lu, L.; Guo, X. Empagliflozin alleviates the development of autoimmune myocarditis via inhibiting NF-κB-dependent cardiomyocyte pyroptosis. Biomed. Pharmacother. 2024, 170, 115963. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.Y.; Hsu, H.C.; Fang, Y.H.; Liu, P.Y.; Liu, Y.W. Empagliflozin attenuates doxorubicin-induced cardiotoxicity by inhibiting the JNK signaling pathway. Biomed. Pharmacother. 2024, 176, 116759. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Wang, L.; Ren, W.Y.; Xu, H.X.; Wu, N.N.; Yu, D.H.; Reiter, R.J.; Zha, W.L.; Guo, Q.D.; Ren, J. SGLT2 inhibitor empagliflozin alleviates cardiac remodeling and contractile anomalies in a FUNDC1-dependent manner in experimental Parkinson’s disease. Acta Pharmacol. Sin. 2023, 45, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Overberg, K.; Ghouse, Z.; Hollmann, M.W.; Weber, N.C.; Coronel, R.; Zuurbier, C.J. Empagliflozin mitigates cardiac hypertrophy through cardiac RSK/NHE-1 inhibition. Biomed. Pharmacother. 2024, 174, 116477. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C.; Shiou, Y.L.; Jhuo, S.J.; Chang, C.Y.; Liu, P.L.; Jhuang, W.J.; Dai, Z.K.; Chen, W.Y.; Chen, Y.F.; Lee, A.S. The sodium-glucose co-transporter 2 inhibitor empagliflozin attenuates cardiac fibrosis and improves ventricular hemodynamics in hypertensive heart failure rats. Cardiovasc. Diabetol. 2019, 18, 45. [Google Scholar] [CrossRef]
- Chen, X.; Yang, Q.; Bai, W.; Yao, W.; Liu, L.; Xing, Y.; Meng, C.; Qi, P.; Dang, Y.; Qi, X. Dapagliflozin Attenuates Myocardial Fibrosis by Inhibiting the TGF-β1/Smad Signaling Pathway in a Normoglycemic Rabbit Model of Chronic Heart Failure. Front. Pharmacol. 2022, 13, 873108. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, Y.; Zhang, Y.; Ye, T.; Wang, K.; Li, S.; Zhang, Y. SIRT1 mediates the inhibitory effect of Dapagliflozin on EndMT by inhibiting the acetylation of endothelium Notch1. Cardiovasc. Diabetol. 2023, 22, 331. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Liu, X.; Ding, B.; Lu, Q.; Ma, J. SGLT2 inhibitor Dapagliflozin alleviates cardiac dysfunction and fibrosis after myocardial infarction by activating PXR and promoting angiogenesis. Biomed. Pharmacother. 2024, 177, 116994. [Google Scholar] [CrossRef]
- Sabe, S.A.; Xu, C.M.; Sabra, M.; Harris, D.D.; Malhotra, A.; Aboulgheit, A.; Stanley, M.; Abid, M.R.; Sellke, F.W. Canagliflozin Improves Myocardial Perfusion, Fibrosis, and Function in a Swine Model of Chronic Myocardial Ischemia. J. Am. Heart Assoc. 2023, 12, 28623. [Google Scholar] [CrossRef]
- Zhang, T.; Wang, X.; Wang, Z.; Zhai, J.; He, L.; Wang, Y.; Zuo, Q.; Ma, S.; Zhang, G.; Guo, Y. Canagliflozin Ameliorates Ventricular Remodeling through Apelin/Angiotensin-Converting Enzyme 2 Signaling in Heart Failure with Preserved Ejection Fraction Rats. Pharmacology 2023, 108, 478–491. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; He, L.L.; Zhang, G.R.; Zuo, Q.J.; Wang, Z.L.; Zhai, J.L.; Zhang, T.T.; Wang, Y.; Ma, H.J.; Guo, Y.F. Canagliflozin mitigates ferroptosis and ameliorates heart failure in rats with preserved ejection fraction. Naunyn Schmiedebergs Arch. Pharmacol. 2022, 395, 945–962. [Google Scholar] [CrossRef] [PubMed]
- Osaka, N.; Mori, Y.; Terasaki, M.; Hiromura, M.; Saito, T.; Yashima, H.; Shiraga, Y.; Kawakami, R.; Ohara, M.; Fukui, T.; et al. Luseogliflozin inhibits high glucose-induced TGF- β 2 expression in mouse cardiomyocytes by suppressing NHE-1 activity. J. Int. Med. Res. 2022, 50, 3000605221097490. [Google Scholar] [CrossRef]
- Solomon, S.D.; McMurray, J.J.V.; Claggett, B.; Boer, R.A.; de DeMets, D.; Hernandez, A.F.; Inzucchi, S.E.; Kosiborod, M.N.; Lam, C.S.P.; Martinez, F.; et al. Dapagliflozin in Heart Failure with Mildly Reduced or Preserved Ejection Fraction. N. Engl. J. Med. 2022, 387, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ding, L.; Tian, L.; Tian, Y.; Liao, L.; Zhao, J. Empagliflozin reduces diffuse myocardial fibrosis by extracellular volume mapping: A meta-analysis of clinical studies. Front. Endocrinol. 2022, 13, 917761. [Google Scholar] [CrossRef] [PubMed]
- Hawley, S.A.; Ford, R.J.; Smith, B.K.; Gowans, G.J.; Mancini, S.J.; Pitt, R.D.; Day, E.A.; Salt, I.P.; Steinberg, G.R.; Hardie, D.G. The Na+/Glucose Cotransporter Inhibitor Canagliflozin Activates AMPK by Inhibiting Mitochondrial Function and Increasing Cellular AMP Levels. Diabetes 2016, 65, 2784–2794. [Google Scholar] [CrossRef]
- Kang, S.; Verma, S.; Hassanabad, A.F.; Teng, G.; Belke, D.D.; Dundas, J.A.; Guzzardi, D.G.; Svystonyuk, D.A.; Pattar, S.S.; Park, D.S.J.; et al. Direct Effects of Empagliflozin on Extracellular Matrix Remodelling in Human Cardiac Myofibroblasts: Novel Translational Clues to Explain EMPA-REG OUTCOME Results. Can. J. Cardiol. 2020, 36, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Daud, E.; Ertracht, O.; Bandel, N.; Moady, G.; Shehadeh, M.; Reuveni, T.; Atar, S. The impact of empagliflozin on cardiac physiology and fibrosis early after myocardial infarction in non-diabetic rats. Cardiovasc. Diabetol. 2021, 20, 132. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Paz, L.; Cristóbal, H.; Tomás Ortiz-Perez, J.; García De Frutos, P.; Mendieta, G.; Sandoval, E.; Rodriguez, J.J.; Ortega, E.; García-Álvarez, A.; Brugaletta, S.; et al. Direct actions of dapagliflozin and interactions with LCZ696 and spironolactone on cardiac fibroblasts of patients with heart failure and reduced ejection fraction. ESC Heart Fail. 2022, 10, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, K.; Schmidt, A.; Groß, S.; Just, A.; Pfanne, A.; Fuchs, M.; Jordan, M.; Mohr, E.; Pich, A.; Fiedler, J.; et al. SGLT2 inhibitors attenuate endothelial to mesenchymal transition and cardiac fibroblast activation. Sci. Rep. 2024, 14, 16459. [Google Scholar] [CrossRef]
- Peng, Q.; Shan, D.; Cui, K.; Li, K.; Zhu, B.; Wu, H.; Wang, B.; Wong, S.; Norton, V.; Dong, Y.; et al. The Role of Endothelial-to-Mesenchymal Transition in Cardiovascular Disease. Cells 2022, 11, 1834. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.J.; Nam, J.K.; Kim, J.H.; Choi, S.H.; Lee, Y.J. Endothelial-to-mesenchymal transition in anticancer therapy and normal tissue damage. Exp. Mol. Med. 2020, 52, 781. [Google Scholar] [CrossRef] [PubMed]
- Anbara, T.; Sharifi, M.; Aboutaleb, N. Endothelial to Mesenchymal Transition in the Cardiogenesis and Cardiovascular Diseases. Curr. Cardiol. Rev. 2020, 16, 306. [Google Scholar] [CrossRef]
- Zhang, W.; Li, X.; Li, M.; He, H.; Yang, C.; Wang, M.; Liu, D.; Zhang, L.; Shu, C. Empagliflozin inhibits neointimal hyperplasia through attenuating endothelial-to-mesenchymal transition via TAK-1/NF-κB pathway. Eur. J. Pharmacol. 2023, 954, 175826. [Google Scholar] [CrossRef]
- Smolgovsky, S.; Ibeh, U.; Tamayo, T.P.; Alcaide, P. Adding insult to injury—Inflammation at the heart of cardiac fibrosis. Cell. Signal. 2021, 77, 109828. [Google Scholar] [CrossRef] [PubMed]
- Sheng, W.; Yu, J.; Zhang, H.; Zhang, J. Empagliflozin attenuates inflammation levels in autoimmune myocarditis through the STAT3 pathway and macrophage phenotype transformation. Mol. Immunol. 2024, 167, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Koyani, C.N.; Plastira, I.; Sourij, H.; Hallström, S.; Schmidt, A.; Rainer, P.P.; Bugger, H.; Frank, S.; Malle, E.; von Lewinski, D. Empagliflozin protects heart from inflammation and energy depletion via AMPK activation. Pharmacol. Res. 2020, 158, 104870. [Google Scholar] [CrossRef] [PubMed]
- Quagliariello, V.; De Laurentiis, M.; Rea, D.; Barbieri, A.; Monti, M.G.; Carbone, A.; Paccone, A.; Altucci, L.; Conte, M.; Canale, M.L.; et al. The SGLT-2 inhibitor empagliflozin improves myocardial strain, reduces cardiac fibrosis and pro-inflammatory cytokines in non-diabetic mice treated with doxorubicin. Cardiovasc. Diabetol. 2021, 20, 150. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Xu, J.; Tan, X.; Li, D.; Yao, D.; Xu, B.; Lei, Y. Dapagliflozin protects against dilated cardiomyopathy progression by targeting NLRP3 inflammasome activation. Naunyn Schmiedebergs Arch. Pharmacol. 2023, 396, 1461–1470. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.M.; Chang, N.C.; Lin, S.Z. Dapagliflozin, a selective SGLT2 Inhibitor, attenuated cardiac fibrosis by regulating the macrophage polarization via STAT3 signaling in infarcted rat hearts. Free Radic. Biol. Med. 2017, 104, 298–310. [Google Scholar] [CrossRef]
- Yan, P.; Song, X.; Tran, J.; Zhou, R.; Cao, X.; Zhao, G.; Yuan, H. Dapagliflozin Alleviates Coxsackievirus B3-induced Acute Viral Myocarditis by Regulating the Macrophage Polarization Through Stat3-related Pathways. Inflammation 2022, 45, 2078–2090. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.T.; Shih, J.Y.; Lin, Y.W.; Chen, Z.C.; Kan, W.C.; Lin, T.H.; Hong, C.S. Dapagliflozin protects against doxorubicin-induced cardiotoxicity by restoring STAT3. Arch. Toxicol. 2022, 96, 2021–2032. [Google Scholar] [CrossRef] [PubMed]
- Kolijn, D.; Pabel, S.; Tian, Y.; Lódi, M.; Herwig, M.; Carrizzo, A.; Zhazykbayeva, S.; Kovács, Á.; Fülöp, G.; Falcão-Pires, I.; et al. Empagliflozin improves endothelial and cardiomyocyte function in human heart failure with preserved ejection fraction via reduced pro-inflammatory-oxidative pathways and protein kinase Gα oxidation. Cardiovasc. Res. 2021, 117, 495–507. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Liu, X.; Li, T.; Fang, T.; Cheng, Y.; Han, L.; Sun, B.; Chen, L. The SGLT2 inhibitor empagliflozin negatively regulates IL-17/IL-23 axis-mediated inflammatory responses in T2DM with NAFLD via the AMPK/mTOR/autophagy pathway. Int. Immunopharmacol. 2021, 94, 107492. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhang, Z.; Si, Z.; Yang, Y.; Li, S.; Xue, Y. Dapagliflozin reverses the imbalance of T helper 17 and T regulatory cells by inhibiting SGK1 in a mouse model of diabetic kidney disease. FEBS Open Bio 2021, 11, 1395. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Liu, Q.; Liu, A.; Leng, S.; Wang, S.; Li, C.; Ma, J.; Peng, J.; Xu, M. Empagliflozin modulates CD4+ T-cell differentiation via metabolic reprogramming in immune thrombocytopenia. Br. J. Haematol. 2022, 198, 765–775. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Li, W.; Yang, N.; Song, Y. IL-17 RA PLAD-Ig Reduces Cardiac Fibrosis in Experimental Autoimmune Myocarditis Rats. J. Card. Fail. 2011, 17, S29–S30. [Google Scholar] [CrossRef]
- Sisto, M.; Lisi, S. Targeting Interleukin-17 as a Novel Treatment Option for Fibrotic Diseases. J. Clin. Med. 2023, 13, 164. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, Y.Y.; Li, T.T.; Wang, J.; Jiang, Y.; Zhao, Y.; Jin, X.X.; Xue, G.L.; Yang, Y.; Zhang, X.F.; et al. Ablation of interleukin-17 alleviated cardiac interstitial fibrosis and improved cardiac function via inhibiting long non-coding RNA-AK081284 in diabetic mice. J. Mol. Cell Cardiol. 2018, 115, 64–72. [Google Scholar] [CrossRef]
- Jenkins, B.J.; Blagih, J.; Ponce-Garcia, F.M.; Canavan, M.; Gudgeon, N.; Eastham, S.; Hill, D.; Hanlon, M.M.; Ma, E.H.; Bishop, E.L.; et al. Canagliflozin impairs T cell effector function via metabolic suppression in autoimmunity. Cell Metab. 2023, 35, 1132–1146.e9. [Google Scholar] [CrossRef]
- Borzouei, S.; Moghimi, H.; Zamani, A.; Behzad, M. Changes in T helper cell-related factors in patients with type 2 diabetes mellitus after empagliflozin therapy. Hum. Immunol. 2021, 82, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Kabłak-Ziembicka, A.; Badacz, R.; Przewłocki, T. Clinical Application of Serum microRNAs in Atherosclerotic Coronary Artery Disease. J. Clin. Med. 2022, 11, 6849. [Google Scholar] [CrossRef] [PubMed]
- Gocer, Z.; Elek, A.; Caska, H.; Bozgeyik, I. MicroRNAs and cardiac fibrosis: A comprehensive update on mechanisms and consequences. Pathol. Res. Pract. 2023, 251, 154853. [Google Scholar] [CrossRef]
- Mone, P.; Lombardi, A.; Kansakar, U.; Varzideh, F.; Jankauskas, S.S.; Pansini, A.; Marzocco, S.; De Gennaro, S.; Famiglietti, M.; Macina, G.; et al. Empagliflozin Improves the MicroRNA Signature of Endothelial Dysfunction in Patients with Heart Failure with Preserved Ejection Fraction and Diabetes. J. Pharmacol. Exp. Ther. 2023, 384, 116. [Google Scholar] [CrossRef] [PubMed]
- Bellera, N.; Barba, I.; Rodriguez-Sinovas, A.; Ferret, E.; Asín, M.A.; Gonzalez-Alujas, M.T.; Pérez-Rodon, J.; Esteves, M.; Fonseca, C.; Toran, N.; et al. Single Intracoronary Injection of Encapsulated Antagomir-92a Promotes Angiogenesis and Prevents Adverse Infarct Remodeling. J. Am. Heart Assoc. Cardiovasc. Cerebrovasc. Dis. 2014, 3, e000946. [Google Scholar] [CrossRef] [PubMed]
- Ridwan, M.; Dimiati, H.; Syukri, M.; Lesmana, R.; Zaini, L.M. Effects of SGLT2-inhibitor on The Expression of MicroRNA-21, Transforming Growth Factor-β1, and Matrix Metalloproteinase-2 in The Process of Cardiac Fibrosis in Hyperglycemic Model Rats. Indones. Biomed. J. 2024, 16, 48–55. [Google Scholar] [CrossRef]
- Takasu, T. The Role of SGLT2 Inhibitor Ipragliflozin on Cardiac Hypertrophy and microRNA Expression Profiles in a Non-diabetic Rat Model of Cardiomyopathy. Biol. Pharm. Bull. 2022, 45, 1321–1331. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, R.N.; Ahmed, L.A.; Abdul Salam, R.M.; Ahmed, K.A.; Attia, A.S. Crosstalk Among NLRP3 Inflammasome, ETBR Signaling, and miRNAs in Stress-Induced Depression-Like Behavior: A Modulatory Role for SGLT2 Inhibitors. Neurotherapeutics 2021, 18, 2664–2681. [Google Scholar] [CrossRef] [PubMed]
- Monami, M.; Nardini, C.; Mannucci, E. Efficacy and safety of sodium glucose co-transport-2 inhibitors in type 2 diabetes: A meta-analysis of randomized clinical trials. Diabetes Obes. Metab. 2014, 16, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Peters, A.L.; Buschur, E.O.; Buse, J.B.; Cohan, P.; Diner, J.C.; Hirsch, I.B. Euglycemic Diabetic Ketoacidosis: A Potential Complication of Treatment with Sodium-Glucose Cotransporter 2 Inhibition. Diabetes Care 2015, 38, 1687–1693. [Google Scholar] [CrossRef]
- Heerspink, H.J.L.; Eickhoff, M.K.; Dekkers, C.C.J.; Kramers, B.J.; Laverman, G.D.; Frimodt-Møller, M.; Jørgensen, N.R.; Faber, J.; Danser, A.H.J.; Gansevoort, R.T.; et al. Effects of Dapagliflozin on Volume Status When Added to Renin–Angiotensin System Inhibitors. J. Clin. Med. 2019, 8, 779. [Google Scholar] [CrossRef] [PubMed]
- Kolwicz, S.C. Ketone Body Metabolism in the Ischemic Heart. Front. Cardiovasc. Med. 2021, 8, 789458. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.; Wilcox, C.S.; Testani, J.M. Critical Analysis of the Effects of SGLT2 Inhibitors on Renal Tubular Sodium, Water and Chloride Homeostasis and Their Role in Influencing Heart Failure Outcomes. Circulation 2023, 148, 354–372. [Google Scholar] [CrossRef]
- Vallon, V.; Verma, S. Effects of SGLT2 Inhibitors on Kidney and Cardiovascular Function. Annu. Rev. Physiol. 2021, 83, 503. [Google Scholar] [CrossRef]
- Mone, P.; Varzideh, F.; Jankauskas, S.S.; Pansini, A.; Lombardi, A.; Frullone, S.; Santulli, G. SGLT2 Inhibition via Empagliflozin Improves Endothelial Function and Reduces Mitochondrial Oxidative Stress: Insights from Frail Hypertensive and Diabetic Patients. Hypertension 2022, 79, 1633–1643. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rolski, F.; Mączewski, M. Cardiac Fibrosis: Mechanistic Discoveries Linked to SGLT2 Inhibitors. Pharmaceuticals 2025, 18, 313. https://doi.org/10.3390/ph18030313
Rolski F, Mączewski M. Cardiac Fibrosis: Mechanistic Discoveries Linked to SGLT2 Inhibitors. Pharmaceuticals. 2025; 18(3):313. https://doi.org/10.3390/ph18030313
Chicago/Turabian StyleRolski, Filip, and Michał Mączewski. 2025. "Cardiac Fibrosis: Mechanistic Discoveries Linked to SGLT2 Inhibitors" Pharmaceuticals 18, no. 3: 313. https://doi.org/10.3390/ph18030313
APA StyleRolski, F., & Mączewski, M. (2025). Cardiac Fibrosis: Mechanistic Discoveries Linked to SGLT2 Inhibitors. Pharmaceuticals, 18(3), 313. https://doi.org/10.3390/ph18030313