
Design, Synthesis, and Computational Evaluation of 3,4-Dihydroquinolin-2(1H)-One Analogues as Potential VEGFR2 Inhibitors in Glioblastoma Multiforme

 , ,
, ,  , ,
, ,  and
and
Abstract
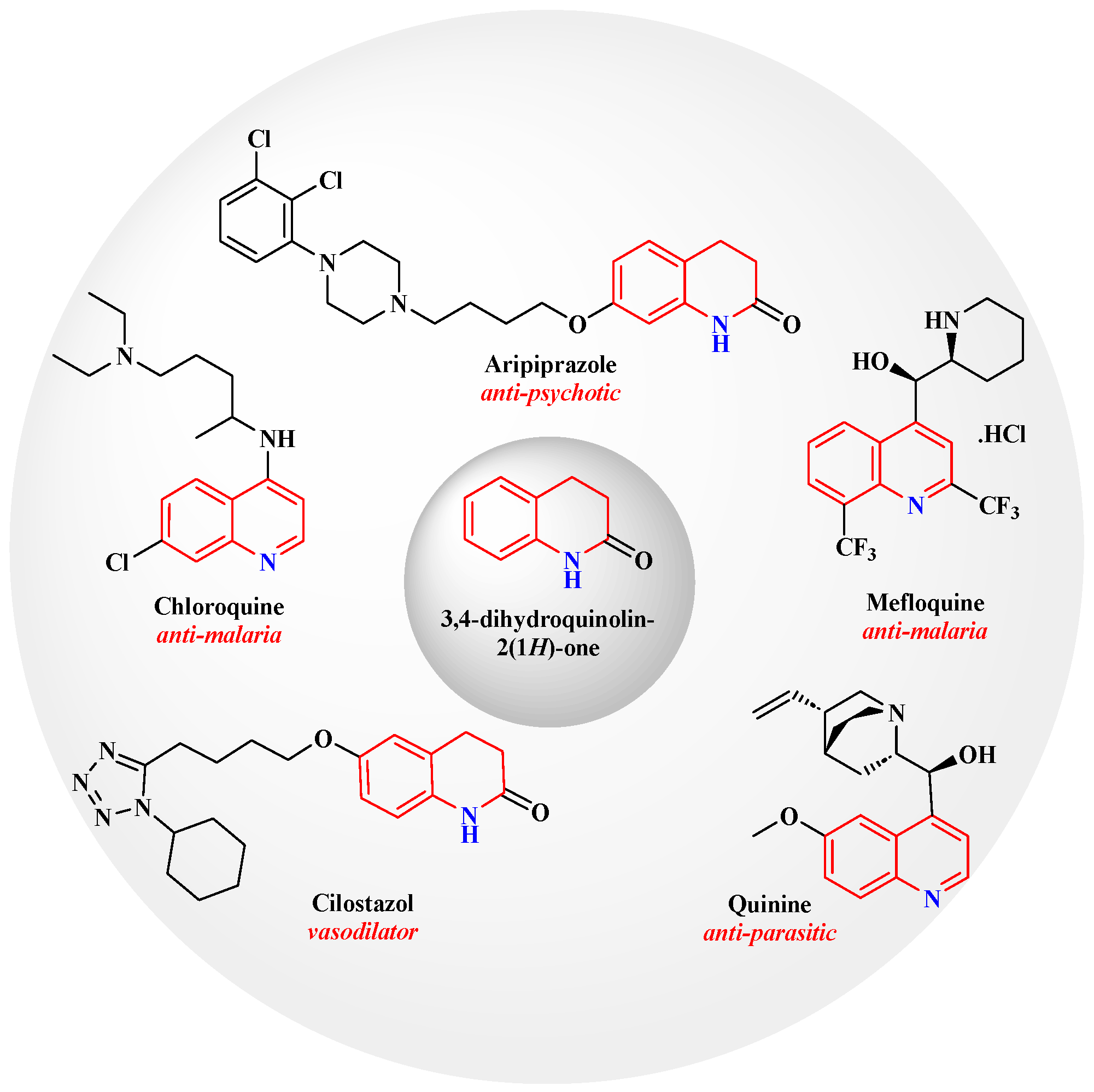
1. Introduction
2. Results and Discussion
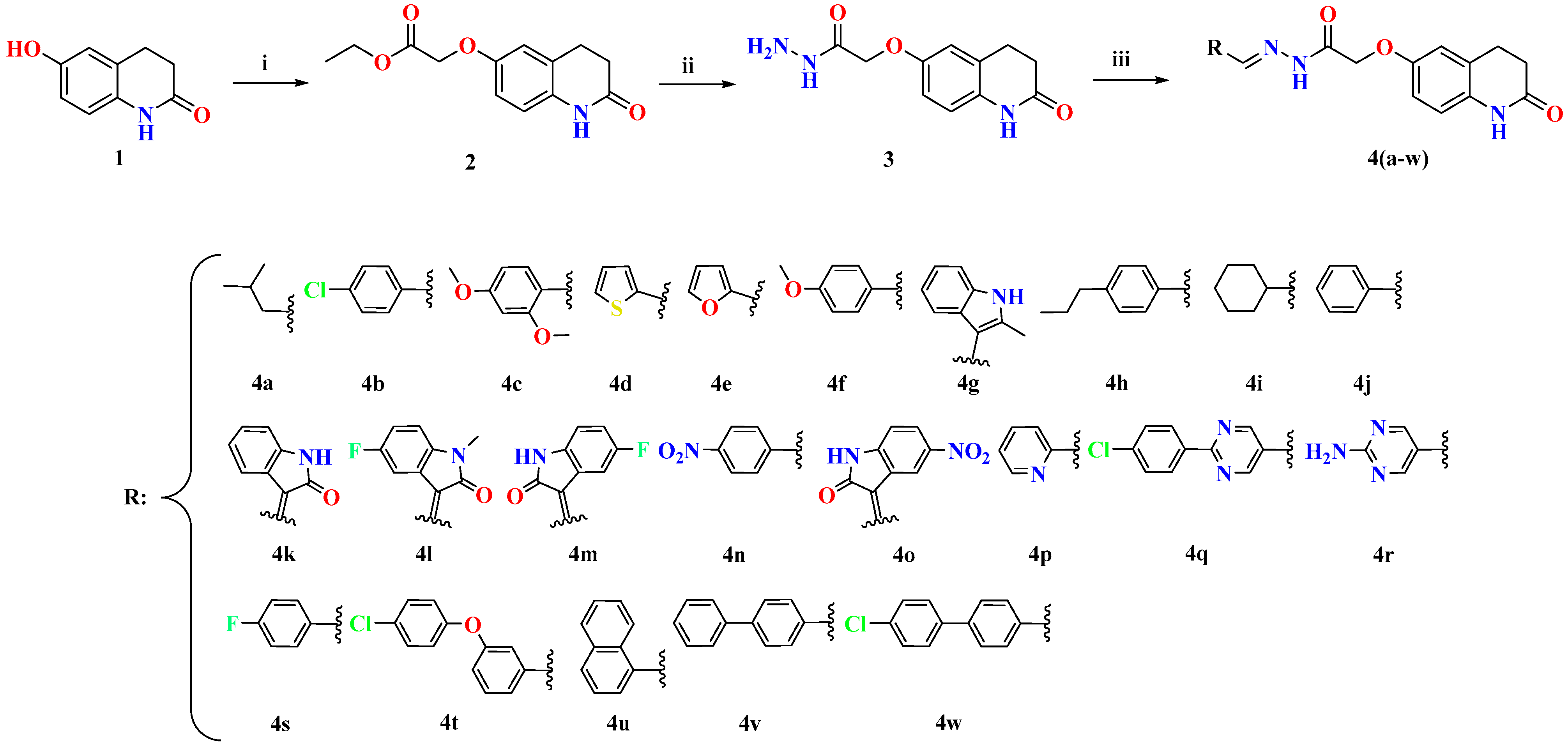
2.1. Chemistry
2.2. In Vitro Studies
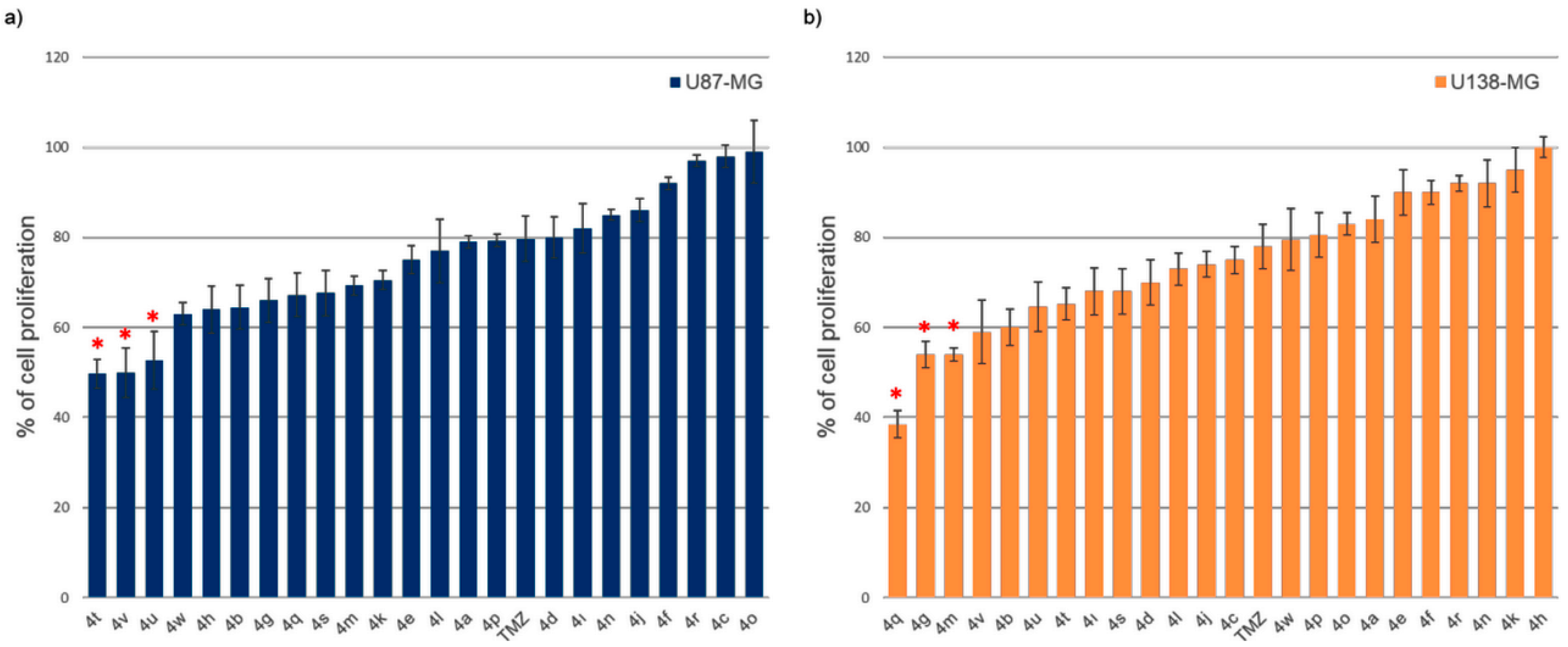
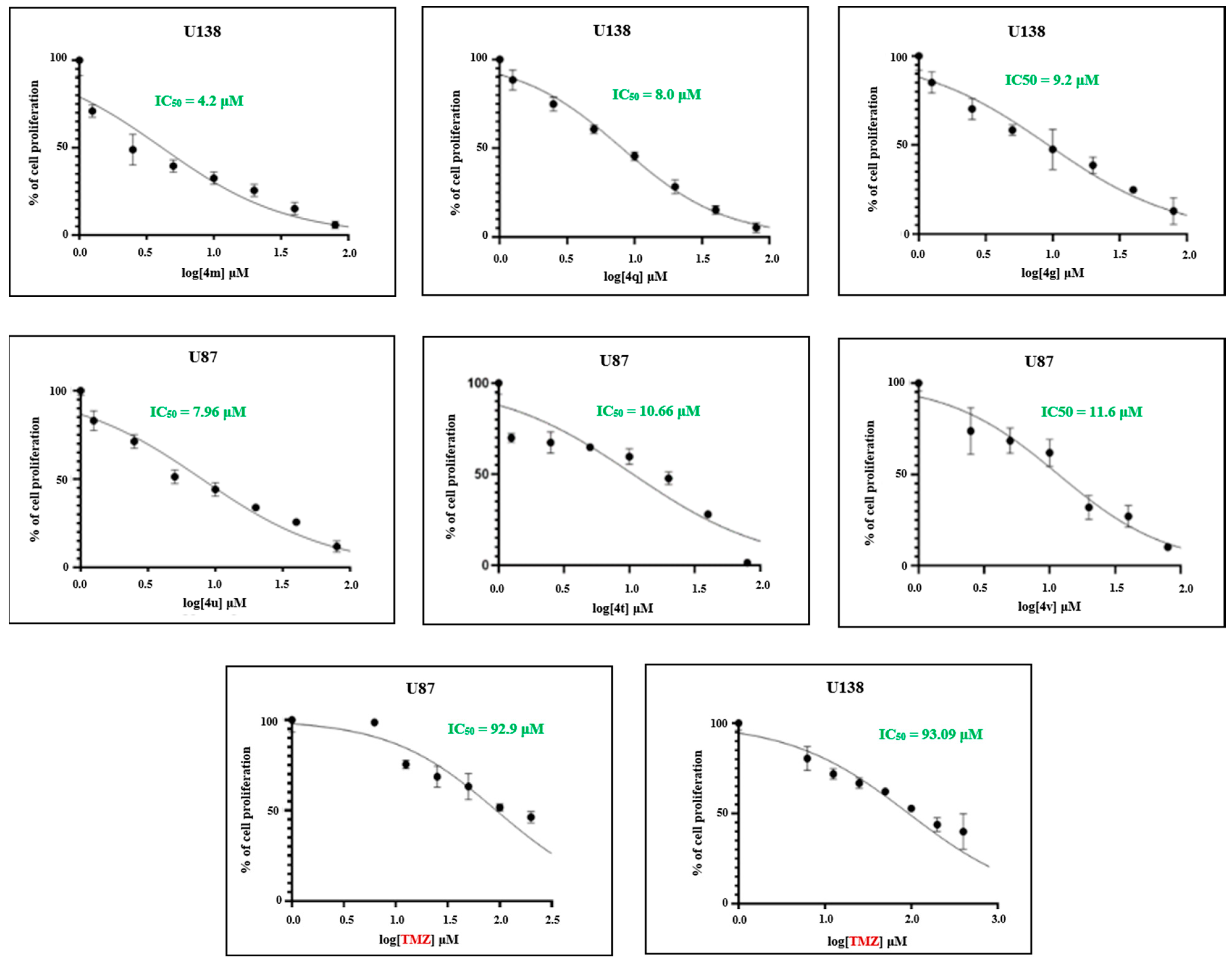
2.2.1. Efficacy of Novel Analogues on Cell Proliferation
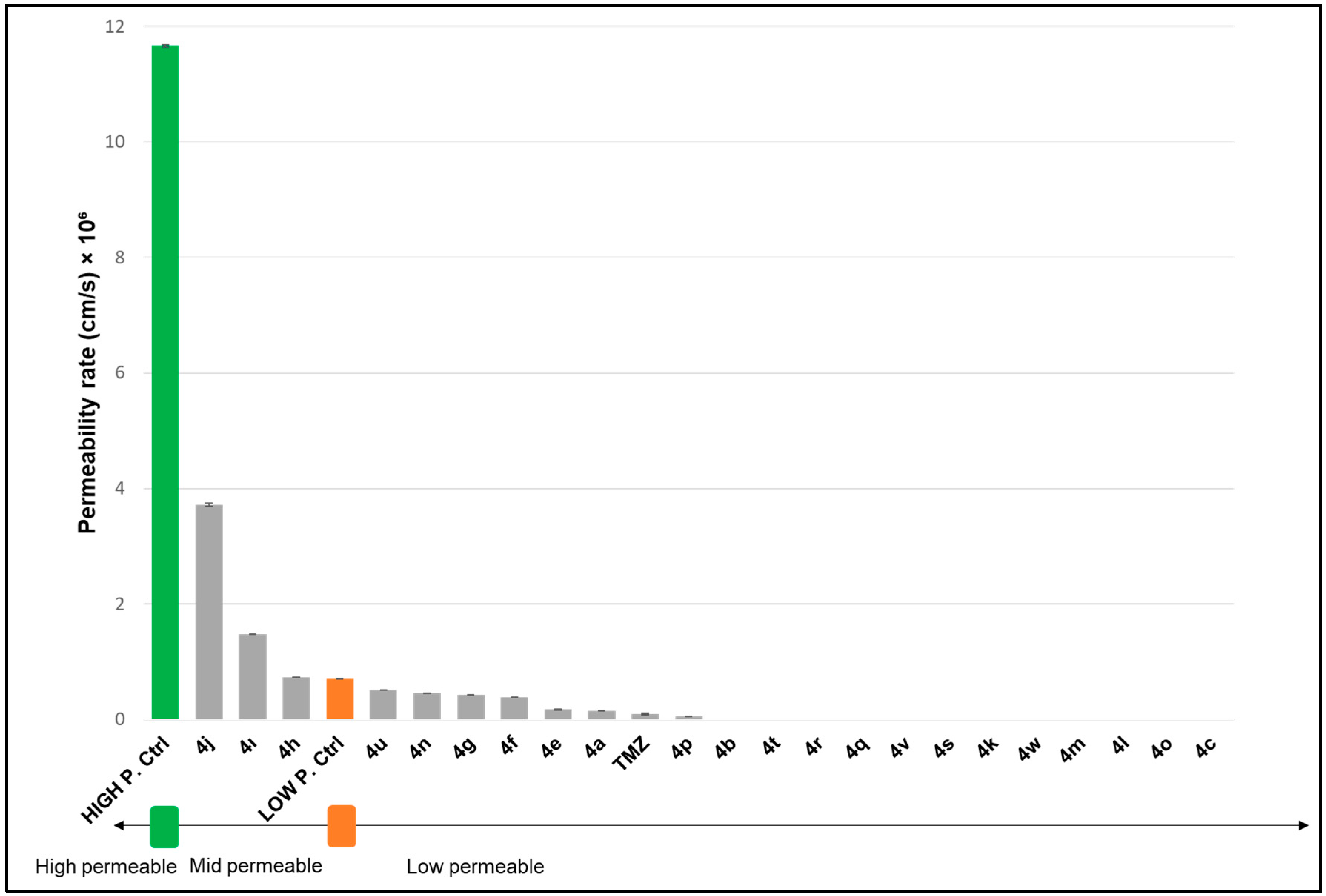
2.2.2. Permeability of Novel Analogues Through Biological Membrane Mimics of the Blood–Brain Barrier (BBB) and Gastrointestinal (GI) Membrane
2.3. In Silico Studies
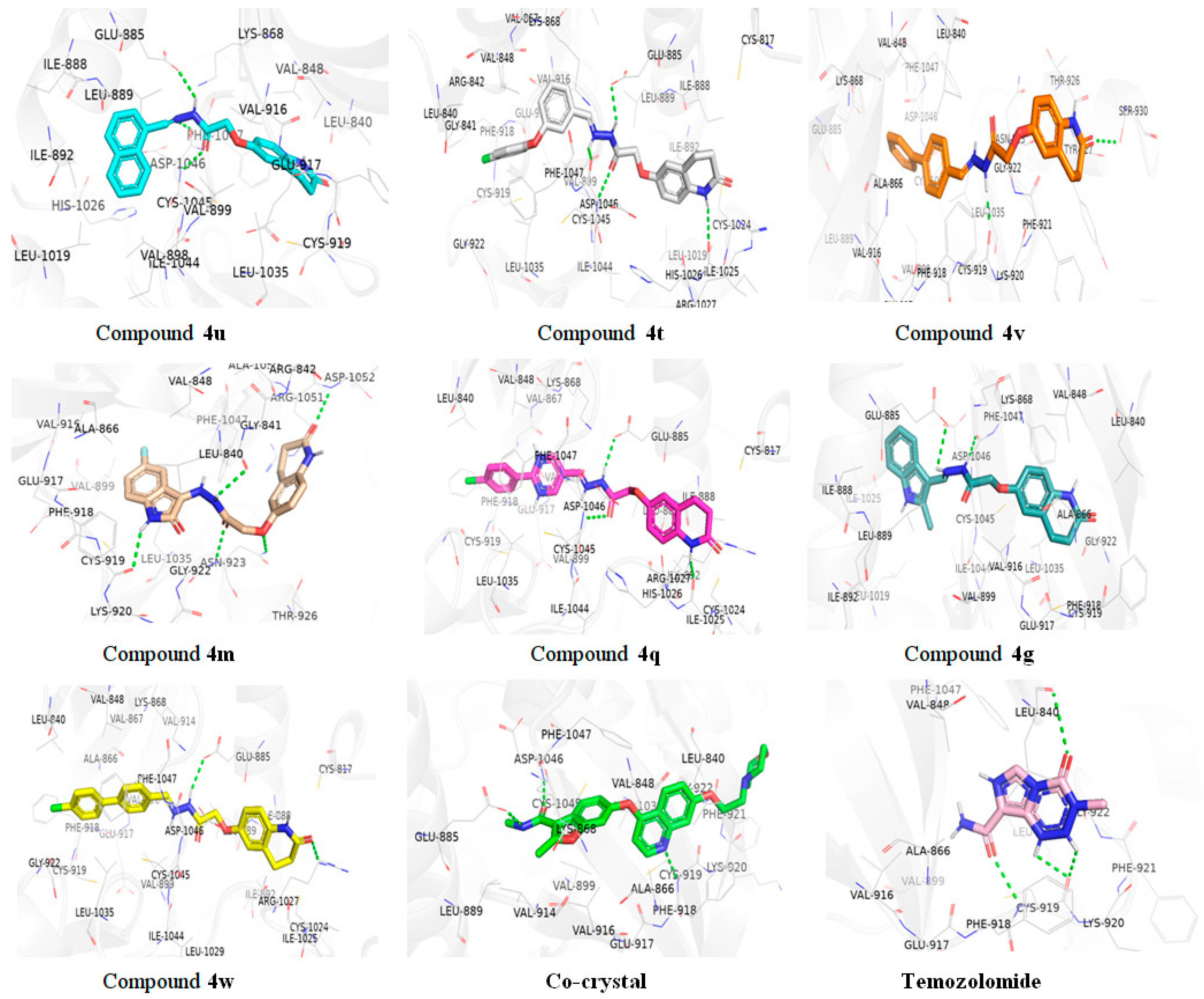
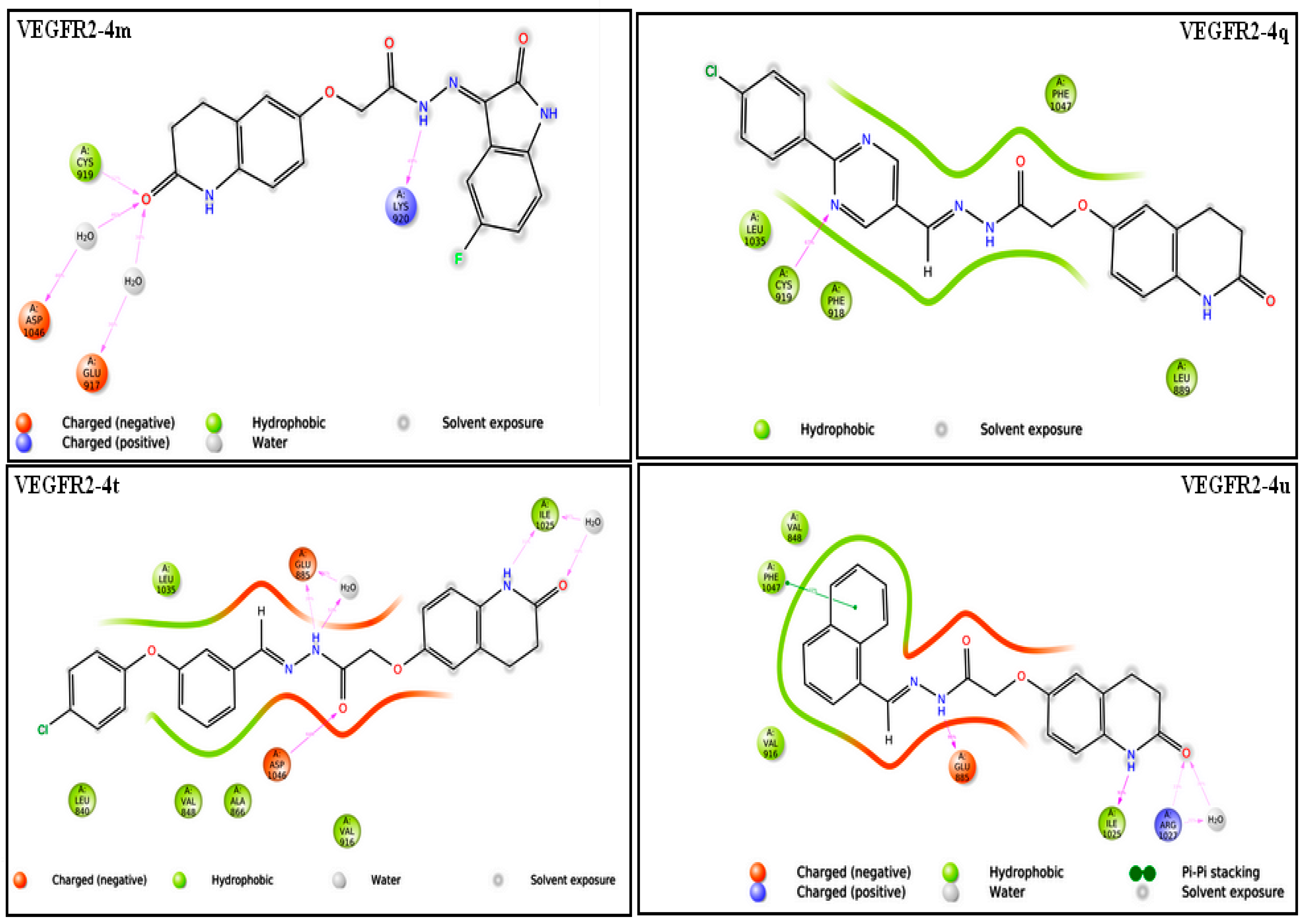
2.3.1. Molecular Docking Studies
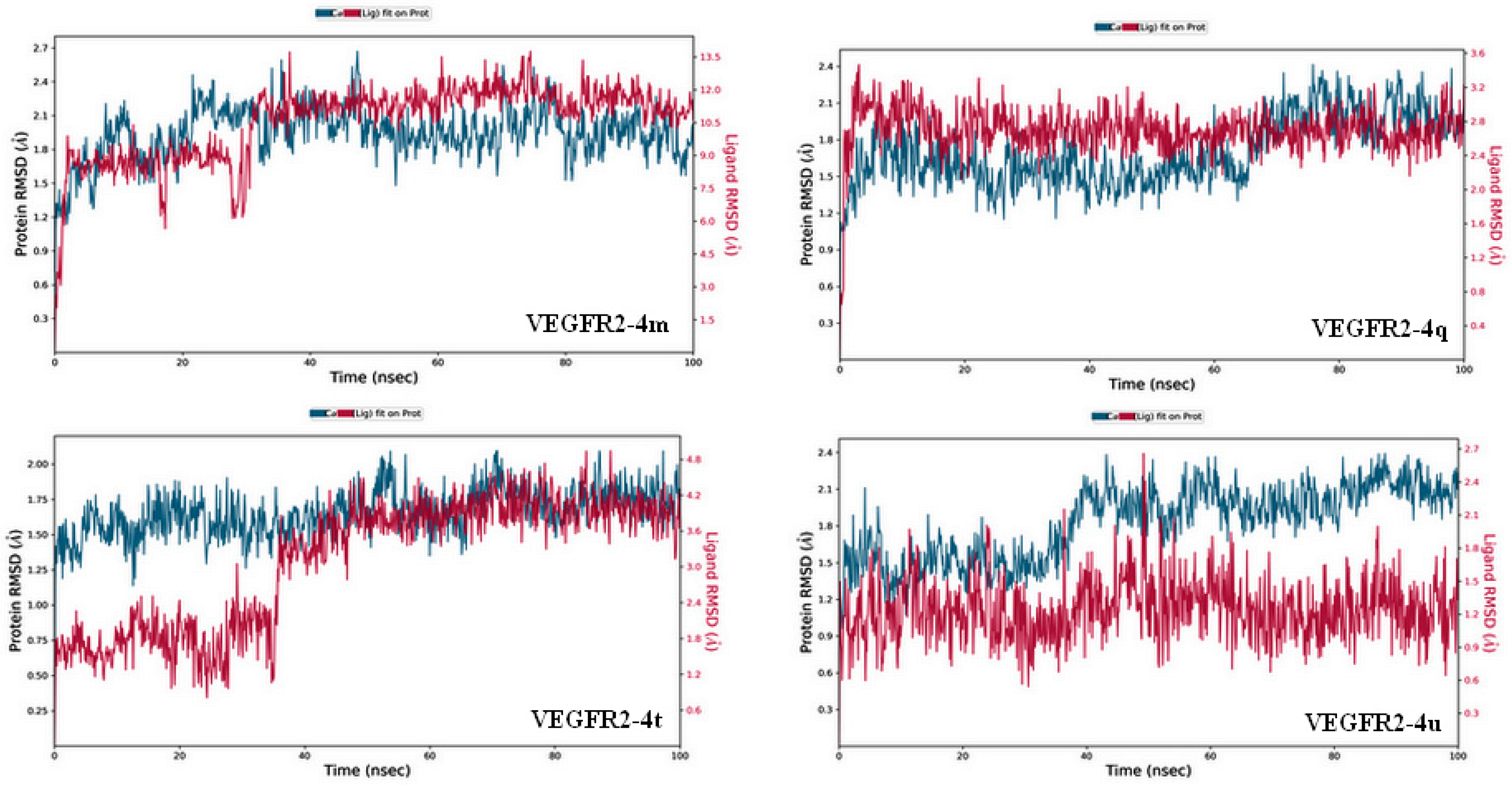
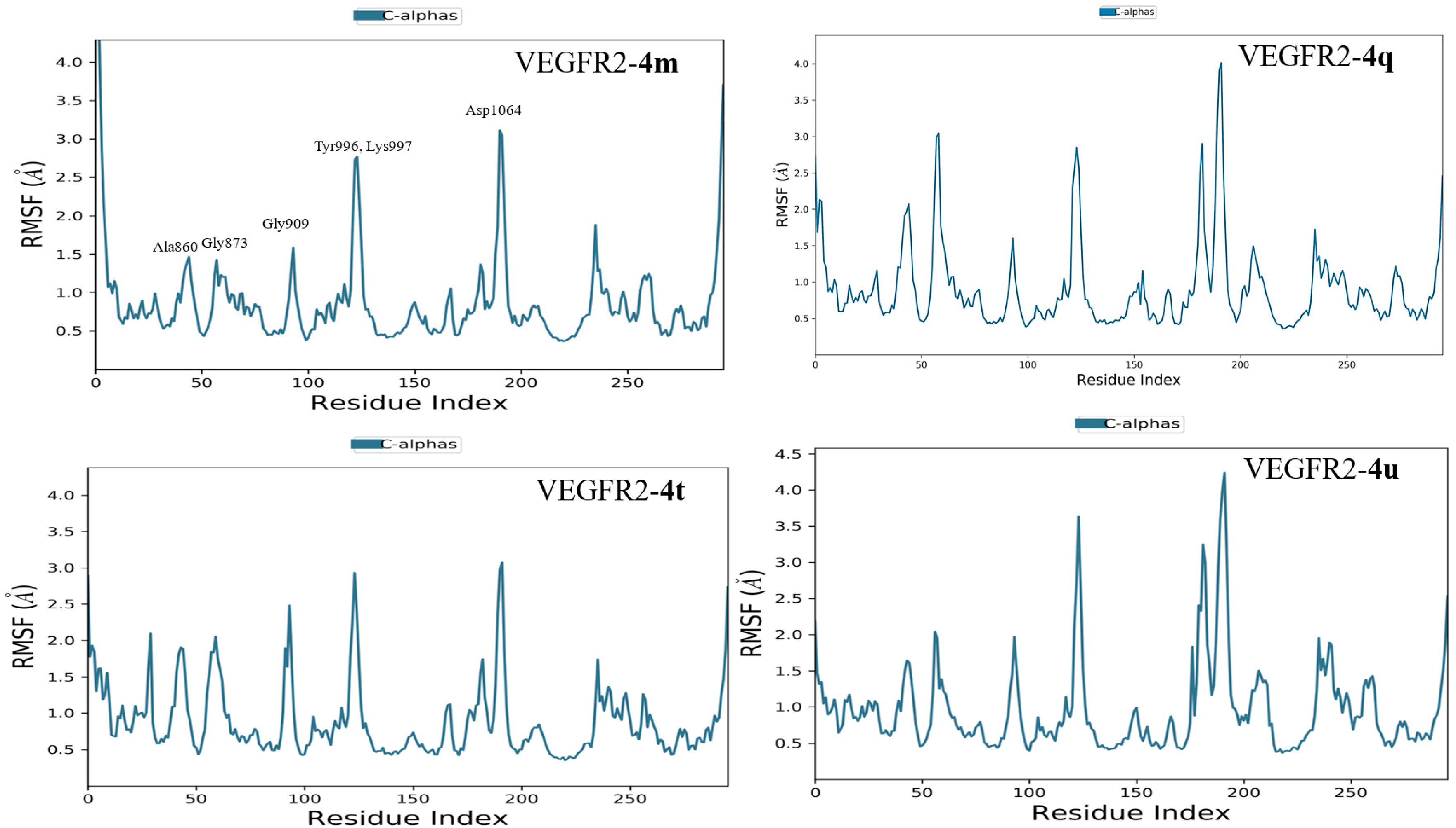
2.3.2. Molecular Dynamics Simulation
2.3.3. In Silico ADME Studies
2.4. Structure–Activity Relationship (SAR) Exploration of Compounds (4a–4w) Against U138-MG Glioblastoma Cells
3. Materials and Methods
3.1. Materials
3.2. General Procedures and Spectral Data
3.2.1. Synthesis of Ethyl 2-((2-Oxo-1,2,3,4-Tetrahydroquinolin-6-Yl) Oxy)Acetate (2)
3.2.2. Synthesis of 2-((2-Oxo-1,2,3,4-Tetrahydroquinolin-6-Yl)Oxy)Acetohydrazide (3)
3.2.3. Synthesis of (E)-N′-(3-Methylbutylidene)-2-((2-Oxo-1,2,3,4-Tetrahydroquinolin-6-Yl)Oxy)Acetohydrazide (4a)
3.2.4. Synthesis of (E)-N′-(4-Chlorobenzylidene)-2-((2-Oxo-1,2,3,4-Tetrahydroquinolin-6-Yl) Oxy) Acetohydrazide (4b)
3.2.5. Synthesis of N′-(2,4-Dimethoxybenzylidene)-2-((2-Oxo-1,2,3,4-Tetrahydroquinolin-6-Yl)Oxy) Acetohydrazide (4c)
3.2.6. Synthesis 2-((2-Oxo-1,2,3,4-Tetrahydroquinolin-6-Yl)Oxy)-N′-(Thiophen-2-Ylmethylene)Acetohydrazide (4d)
3.2.7. Synthesis (E)-N′-(Furan-2-Ylmethylene)-2-((2-Oxo-1,2,3,4-Tetrahydroquinolin-6-Yl)Oxy)Acetohydrazide (4e)
3.2.8. Synthesis (E)-N′-(4-Methoxybenzylidene)-2-((2-Oxo-1,2,3,4-Tetrahydroquinolin-6-Yl)Oxy)Acetohydrazide (4f)
3.2.9. Synthesis of (E)-N′-((2-Methyl-1H-Indol-3-Yl)Methylene)-2-((2-Oxo-1,2,3,4-Tetrahydroquinolin-6-Yl)Oxy)Acetohydrazide (4g)
3.2.10. Synthesis of (E)-2-((2-Oxo-1,2,3,4-Tetrahydroquinolin-6-Yl)Oxy)-N′-(4-Propylbenzylidene)Acetohydrazide (4h)
3.2.11. Synthesis of (E)-N′-(Cyclohexylmethylene)-2-((2-Oxo-1,2,3,4-Tetrahydroquinolin-6-Yl)Oxy)Acetohydrazide (4i)
3.2.12. Synthesis of (E)-N′-Benzylidene-2-((2-Oxo-1,2,3,4-Tetrahydroquinolin-6-Yl)Oxy)Acetohydrazide (4j)
3.2.13. Synthesis of (Z)-2-((2-Oxo-1,2,3,4-Tetrahydroquinolin-6-Yl)Oxy)-N′-(2-Oxoindolin-3-Ylidene)Acetohydrazide (4k)
3.2.14. Synthesis of (Z)-N′-(5-Fluoro-1-Methyl-2-Oxoindolin-3-Ylidene)-2-((2-Oxo-1,2,3,4-Tetrahydroquinolin-6-Yl)Oxy)Acetohydrazide (4l)
3.2.15. Synthesis of (Z)-N′-(5-Fluoro-2-Oxoindolin-3-Ylidene)-2-((2-Oxo-1,2,3,4-Tetrahydroquinolin-6-Yl)Oxy)Acetohydrazide (4m)
3.2.16. Synthesis of (E)-N′-(4-Nitrobenzylidene)-2-((2-Oxo-1,2,3,4-Tetrahydroquinolin-6-Yl)Oxy)Acetohydrazide (4n)
3.2.17. Synthesis of (Z)-N′-(5-Nitro-2-Oxoindolin-3-Ylidene)-2-((2-Oxo-1,2,3,4-Tetrahydroquinolin-6-Yl)Oxy)Acetohydrazide (4o)
3.2.18. Synthesis of (E)-2-((2-Oxo-1,2,3,4-Tetrahydroquinolin-6-Yl)Oxy)-N′-(Pyridin-2-Ylmethylene)Acetohydrazide (4p)
3.2.19. Synthesis of (E)-N′-((2-(4-Chlorophenyl)Pyrimidin-5-Yl)Methylene)-2-((2-Oxo-1,2,3,4-Tetrahydroquinolin-6-Yl)Oxy)Acetohydrazide (4q)
3.2.20. Synthesis of (E)-N′-((2-Aminopyrimidin-5-Yl)Methylene)-2-((2-Oxo-1,2,3,4-Tetrahydroquinolin-6-Yl)Oxy)Acetohydrazide (4r)
3.2.21. Synthesis of (E)-N′-(4-Fluorobenzylidene)-2-((2-Oxo-1,2,3,4-Tetrahydroquinolin-6-Yl)Oxy)Acetohydrazide (4s)
3.2.22. Synthesis of (E)-N′-(3-(4-Chlorophenoxy)Benzylidene)-2-((2-Oxo-1,2,3,4-Tetrahydroquinolin-6-Yl)Oxy)Acetohydrazide (4t)
3.2.23. Synthesis of (E)-N′-(Naphthalen-1-Ylmethylene)-2-((2-Oxo-1,2,3,4-Tetrahydroquinolin-6-Yl)Oxy)Acetohydrazide (4u)
3.2.24. Synthesis of (E)-N′-([1,1′-Biphenyl]-4-Ylmethylene)-2-((2-Oxo-1,2,3,4-Tetrahydroquinolin-6-Yl)Oxy)Acetohydrazide (4v)
3.2.25. Synthesis of (E)-N′-((4′-Chloro-[1,1′-Biphenyl]-4-Yl)Methylene)-2-((2-Oxo-1,2,3,4-Tetrahydroquinolin-6-Yl)Oxy)Acetohydrazide (4w)
3.3. Molecular Docking
3.4. Molecular Dynamics Simulation
3.5. In Silico ADME Study
3.6. In Vitro Experiments on GBM Models
3.6.1. Cell Cultures
3.6.2. MTT Assay
3.6.3. PAMPA Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sucu, B.O.; Koc, E.B.; Ipek, O.S.; Mirat, A.; Almas, F.; Guzel, M.A.; Dogan, B.; Uludag, D.; Karakas, N.; Durdagi, S.; et al. Design and synthesis of novel caffeic acid phenethyl ester (CAPE) derivatives and their biological activity studies in glioblastoma multiforme (GBM) cancer cell lines. J. Mol. Graph. Model. 2022, 113, 108160. [Google Scholar] [CrossRef]
- Stackhouse, C.T.; Gillespie, G.Y.; Willey, C.D. Exploring the Roles of lncRNAs in GBM Pathophysiology and Their Therapeutic Potential. Cells 2020, 9, 2369. [Google Scholar] [CrossRef]
- Ajay Bemis, G.W.; Murcko, M.A. Designing libraries with CNS activity. J. Med. Chem. 1999, 42, 4942–4951. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. Drug transport across the blood-brain barrier. J. Cereb. Blood Flow Metab. 2012, 32, 1959–1972. [Google Scholar] [CrossRef]
- Shergalis, A.; Bankhead, A.; Luesakul, U.; Muangsin, N.; Neamati, N. Current challenges and opportunities in treating glio-blastomas. Pharmacol. Rev. 2018, 70, 412–445. [Google Scholar] [CrossRef]
- Bellettato, C.M.; Scarpa, M. Possible strategies to cross the blood–brain barrier. Ital. J. Pediatr. 2018, 44, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [Google Scholar] [CrossRef]
- Cornelissen, F.M.; Markert, G.; Deutsch, G.; Antonara, M.; Faaij, N.; Bartelink, I.; Noske, D.; Vandertop, W.P.; Bender, A.; Westerman, B.A. Explaining Blood–Brain Barrier Permeability of Small Molecules by Integrated Analysis of Different Transport Mechanisms. J. Med. Chem. 2023, 66, 7253–7267. [Google Scholar] [CrossRef]
- Thakkar, J.P.; Dolecek, T.A.; Horbinski, C.; Ostrom, Q.T.; Lightner, D.D.; Barnholtz-Sloan, J.S.; Villano, J.L. Epidemiologic and Molecular Prognostic Review of Glioblastoma. Cancer Epidemiol. Biomark. Prev. 2014, 23, 1985–1996. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; Van Den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma in elderly patients. J. Neurooncol. 2005, 352, 987–996. [Google Scholar]
- Brown, D.; Chen, Z.-P.; Guo, C.; Yang, Q.; Li, J.; Wu, S.; Deng, M.; Du, X.; Sai, K.; Jiang, X.; et al. Phase 2 clinical trial of VAL-083 as first-line treatment in newly-diagnosed MGMT-unmethylated glioblastoma multiforme (GBM): Halfway report. Glioma 2019, 2, 167. [Google Scholar] [CrossRef]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; De Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT Gene Silencing and Benefit from Temozolomide in Glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef]
- Fan, C.-H.; Liu, W.-L.; Cao, H.; Wen, C.; Chen, L.; Jiang, G. O6-methylguanine DNA methyltransferase as a promising target for the treatment of temozolomide-resistant gliomas. Cell Death Dis. 2013, 4, e876. [Google Scholar] [CrossRef] [PubMed]
- Hanashalshahaby, E.H.A.; Unaleroglu, C. Mannich bases as enone precursors for water-mediated efficient synthesis of 2,3,6-trisubstituted pyridines and 5,6,7,8-tetrahydroquinolines. ACS Comb. Sci. 2015, 17, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Hanashalshahaby, E.H.A.; Ünaleroğlu, C.; Ak Can, A.; Özgün, A.; Garipcan, B. Design, synthesis, and antitumor evaluation of novel methylene moiety-tethered tetrahydroquinoline derivatives. Turkish J. Chem. 2019, 43, 1552–1569. [Google Scholar] [CrossRef]
- Trawally, M. Beyond the heart—Exploring the therapeutic potential of PDE3 inhibitors. J. Res. Pharm. 2023, 27, 2218–2241. [Google Scholar] [CrossRef]
- Mardinoglu, A.; Palsson, B.Ø. Genome-scale models in human metabologenomics. Nat. Rev. Genet. 2025, 26, 123–140. [Google Scholar] [CrossRef]
- Xu, X.; Wang, J.; Han, K.; Li, S.; Xu, F.; Yang, Y. Antimalarial drug mefloquine inhibits nuclear factor kappa B signaling and induces apoptosis in colorectal cancer cells. Cancer Sci. 2018, 109, 1220–1229. [Google Scholar] [CrossRef]
- Baudry, S.; Pham, Y.T.; Baune, B.; Vidrequin, S.; Crevoisier, C.; Gimenez, F.; Farinotti, R. Stereoselective passage of mefloquine through the blood-brain barrier in the rat. J. Pharm. Pharmacol. 1997, 49, 1086–1090. [Google Scholar] [CrossRef] [PubMed]
- Golden, E.B.; Cho, H.-Y.; Hofman, F.M.; Louie, S.G.; Schönthal, A.H.; Chen, T.C. Quinoline-based antimalarial drugs: A novel class of autophagy inhibitors. Neurosurg. Focus 2015, 38, E12. [Google Scholar] [CrossRef]
- Kim, M.S.; Yoo, B.C.; Yang, W.S.; Han, S.Y.; Jeong, D.; Song, J.M.; Kim, K.H.; Aravinthan, A.; Kim, J.H.; Kim, J.-H.; et al. Src is the primary target of aripiprazole, an atypical antipsychotic drug, in its anti-tumor action. Oncotarget 2018, 9, 5979–5992. [Google Scholar] [CrossRef]
- Zhuo, C.; Xun, Z.; Hou, W.; Ji, F.; Lin, X.; Tian, H.; Zheng, W.; Chen, M.; Liu, C.; Wang, W.; et al. Surprising anticancer activities of psychiatric medications: Old drugs offer new hope for patients with brain cancer. Front. Pharmacol. 2019, 10, 1262. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.K.; Jermakowicz, A.; Mookhtiar, A.K.; Nemeroff, C.B.; Schürer, S.C.; Ayad, N.G. Drug repositioning in glioblastoma: A pathway perspective. Front. Pharmacol. 2018, 9, 218. [Google Scholar] [CrossRef] [PubMed]
- Forno, F.; Maatuf, Y.; Boukeileh, S.; Dipta, P.; Mahameed, M.; Darawshi, O.; Ferreira, V.; Rada, P.; García-Martinez, I.; Gross, E.; et al. Aripiprazole cytotoxicity coincides with activation of the unfolded protein response in human hepatic cells. J. Pharmacol. Exp. Ther. 2020, 374, 452–461. [Google Scholar] [CrossRef]
- Liu, C.-C.; Wu, C.-L.; Yeh, I.-C.; Wu, S.-N.; Sze, C.; Gean, P.-W. Cilostazol eliminates radiation-resistant glioblastoma by re-evoking big conductance calcium-activated potassium channel activity. Am. J. Cancer Res. 2021, 11, 1148–1169. [Google Scholar]
- Farzaneh Behelgardi, M.; Zahri, S.; Mashayekhi, F.; Mansouri, K.; Asghari, S.M. A peptide mimicking the binding sites of VEGF-A and VEGF-B inhibits VEGFR-1/-2 driven angiogenesis, tumor growth and metastasis. Sci. Rep. 2018, 8, 17924. [Google Scholar] [CrossRef]
- Batchelor, T.T.; Duda, D.G.; di Tomaso, E.; Ancukiewicz, M.; Plotkin, S.R.; Gerstner, E.; Eichler, A.F.; Drappatz, J.; Hochberg, F.H.; Benner, T.; et al. Phase II Study of Cediranib, an Oral Pan–Vascular Endothelial Growth Factor Receptor Tyrosine Kinase Inhibitor, in Patients With Recurrent Glioblastoma. J. Clin. Oncol. 2010, 28, 2817–2823. [Google Scholar] [CrossRef]
- Bai, R.-Y.; Staedtke, V.; Riggins, G.J. Molecular targeting of glioblastoma: Drug discovery and therapies. Trends Mol. Med. 2011, 17, 301–312. [Google Scholar] [CrossRef]
- Brandes, A.A.; Bartolotti, M.; Tosoni, A.; Poggi, R.; Franceschi, E. Practical Management of Bevacizumab-Related Toxicities in Glioblastoma. Oncologist 2015, 20, 166–175. [Google Scholar] [CrossRef]
- Friedman, H.S.; Prados, M.D.; Wen, P.Y.; Mikkelsen, T.; Schiff, D.; Abrey, L.E.; Yung, W.A.; Paleologos, N.; Nicholas, M.K.; Jensen, R.; et al. Bevacizumab Alone and in Combination With Irinotecan in Recurrent Glioblastoma. J. Clin. Oncol. 2009, 27, 4733–4740. [Google Scholar] [CrossRef]
- Keunen, O.; Johansson, M.; Oudin, A.; Sanzey, M.; Rahim, S.A.A.; Fack, F.; Thorsen, F.; Taxt, T.; Bartos, M.; Jirik, R.; et al. Anti-VEGF treatment reduces blood supply and increases tumor cell invasion in glioblastoma. Proc. Natl. Acad. Sci. USA 2011, 108, 3749–3754. [Google Scholar] [CrossRef]
- Ahmed, S.I.; Thomas, A.L.; Steward, W.P. Vascular endothelial growth factor (VEGF) inhibition by small molecules. J. Chemother. 2004, 4, 59–63. [Google Scholar] [CrossRef]
- Wilhelm, S.M.; Adnane, L.; Newell, P.; Villanueva, A.; Llovet, J.M.; Lynch, M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol. Cancer Ther. 2008, 7, 3129–3140. [Google Scholar] [CrossRef] [PubMed]
- Aly, R.M.; Serya, R.A.; Amira, M.; Al-Ansary, G.H.; Abou El Ella, D.A. Quinoline-based small molecules as effective protein kinases inhibitors (Review). J. Am. Sci. 2016, 12, 10–32. [Google Scholar]
- Elkaeed, E.B.; Yousef, R.G.; Khalifa, M.M.; Ibrahim, A.; Mehany, A.B.M.; Gobaara, I.M.M.; Alsfouk, B.A.; Eldehna, W.M.; Metwaly, A.M.; Eissa, I.H.; et al. Discovery of New VEGFR-2 Inhibitors: Design, Synthesis, Anti-Proliferative Evaluation, Docking, and MD Simulation Studies. Molecules 2022, 27, 6203. [Google Scholar] [CrossRef] [PubMed]
- Alafeefy, A.M.; Ashour, A.E.; Prasad, O.; Sinha, L.; Pathak, S.; Alasmari, F.A.; Rishi, A.K.; Abdel-Aziz, H.A. Development of certain novel N-(2-(2-(2-oxoindolin-3-ylidene) hydrazinecarbonyl)phenyl)-benzamides and 3-(2-oxoindolin-3-ylideneamino)-2-substituted quinazolin-4(3H)-ones as CFM-1 analogs: Design, synthesis, QSAR analysis and anticancer activity. Eur. J. Med. Chem. 2015, 92, 191–201. [Google Scholar] [CrossRef]
- Liao, W.; Xu, C.; Ji, X.; Hu, G.; Ren, L.; Liu, Y.; Li, R.; Gong, P.; Sun, T. Design and optimization of novel 4-(2-fluorophenoxy)quinoline derivatives bearing a hydrazone moiety as c-Met kinase inhibitors. Eur. J. Med. Chem. 2014, 87, 508–518. [Google Scholar] [CrossRef]
- McTigue, M.; Wickersham, J.; Pinko, C.; Hong, Y.; Marrone, T. Crystal Structure of the VEGFR2 Kinase Domain in Complex with PF-00337210 (N,2-dimethyl-6-(7-(2-morpholinoethoxy)quinolin-4-yloxy) Benzofuran-3-carb Oxamide). wwpdb 2023. Available online: https://www.wwpdb.org/pdb?id=pdb_00002xir (accessed on 20 December 2023).
- Yao, X.; Ping, Y.; Liu, Y.; Chen, K.; Yoshimura, T.; Liu, M.; Gong, W.; Chen, C.; Niu, Q.; Guo, D.; et al. Vascular Endothelial Growth Factor Receptor 2 (VEGFR-2) Plays a Key Role in Vasculogenic Mimicry Formation, Neovascularization and Tumor Initiation by Glioma Stem-like Cells. PLoS ONE 2013, 8, e57188. [Google Scholar] [CrossRef]
- Abdelgawad, M.A.; Hayallah, A.M.; Bukhari, S.N.A.; Musa, A.; Elmowafy, M.; Abdel-Rahman, H.M.; El-Gaber, M.K.A. Design, Synthesis, Molecular Modeling, and Anticancer Evaluation of New VEGFR-2 Inhibitors Based on the Indolin-2-One Scaffold. Pharmaceuticals 2022, 15, 1416. [Google Scholar] [CrossRef]
- Nguyen, H.-M.; Guz-Montgomery, K.; Lowe, D.B.; Saha, D. Pathogenetic features and current management of glioblastoma. Cancers 2021, 13, 856. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2012, 64, 4–17. [Google Scholar] [CrossRef]
- Athanasiou, C.; Cournia, Z. From Computers to Bedside: Computational Chemistry Contributing to FDA Approval. In Biomolecular Simulations in Structure-Based Drug Discovery; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2018; pp. 163–203. [Google Scholar] [CrossRef]
- Abad, N.; Sallam, H.H.; Al-Ostoot, F.H.; Khamees, H.A.; Al-Horaibi, S.A.; Khanum, S.A.; Madegowda, M.; El Hafi, M.; Mague, J.T.; Essassi, E.M.; et al. Synthesis, crystal structure, DFT calculations, Hirshfeld surface analysis, energy frameworks, molecular dynamics and docking studies of novel isoxazolequinoxaline derivative (IZQ) as anti-cancer drug. J. Mol. Struct. 2021, 1232, 130004. [Google Scholar] [CrossRef]
- Dallakyan, S.; Olson, A.J. Small-molecule library screening by docking with PyRx. Methods Mol. Biol. 2015, 1263, 243–250. [Google Scholar] [PubMed]
- Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No | Code | R | Cell Proliferation ± Std dev. (%) | No | Code | R | Cell Proliferation ± Std dev. (%) | ||
|---|---|---|---|---|---|---|---|---|---|
| U87 | U138 | U87 | U138 | ||||||
| 1 | 4a | 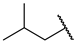 | 79 ± 1.3 | 84 ± 5.2 | 13 | 4m | 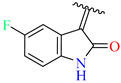 | 69 ± 2.1 | 54 ± 1.4 |
| 2 | 4b | 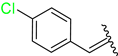 | 65 ± 4.9 | 60 ± 4.0 | 14 | 4n | 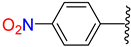 | 85 ± 1.2 | 92 ± 5.2 |
| 3 | 4c |  | 98 ± 2.5 | 75 ± 3.0 | 15 | 4o | 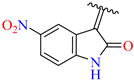 | 99 ± 7.0 | 83 ± 2.5 |
| 4 | 4d |  | 80 ± 4.5 | 70 ± 5.0 | 16 | 4p | 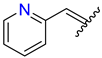 | 79 ± 1.4 | 81 ± 4.9 |
| 5 | 4e | 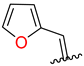 | 75 ± 3.1 | 90 ± 5.0 | 17 | 4q |  | 67 ± 4.9 | 39 ± 3.0 |
| 6 | 4f | 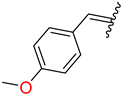 | 92 ± 1.4 | 90 ± 2.6 | 18 | 4r | 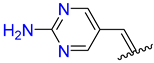 | 97 ± 1.3 | 92 ± 1.8 |
| 7 | 4g |  | 66 ± 4.9 | 54 ± 3.0 | 19 | 4s | 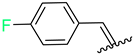 | 68 ± 5.0 | 68 ± 5.0 |
| 8 | 4h | 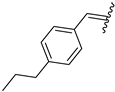 | 64 ± 5.2 | 100 ± 2.3 | 20 | 4t |  | 50 ± 3.2 | 65 ± 3.5 |
| 9 | 4ı | 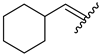 | 82 ± 5.5 | 68 ± 5.2 | 21 | 4u |  | 53 ± 6.4 | 65 ± 5.5 |
| 10 | 4j | 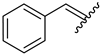 | 86 ± 2.6 | 74 ± 2.8 | 22 | 4v | 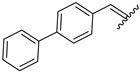 | 49 ± 5.5 | 59 ± 7.0 |
| 11 | 4k | 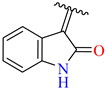 | 71± 2.1 | 95 ± 5 | 23 | 4w | 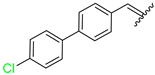 | 63 ± 2.5 | 80 ± 6.9 |
| 12 | 4l | 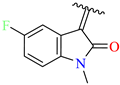 | 77 ± 7.0 | 73 ± 3.6 | 24 | TMZ | 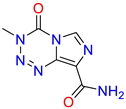 | 80 ± 5 | 78 ± 5 |
| Molecules | VEGF Receptor (PDB ID: 2XIR) | ||
|---|---|---|---|
| Binding Affinity (kcal/mol) | Number of H-Bonds | H-Bonding Residues | |
| 4a | −9.0 | 2 | Asp1046 |
| 4b | −10.3 | 1 | Glu885 |
| 4c | −9.9 | 2 | Glu885, Arg1027 |
| 4d | −8.4 | 3 | Leu840, Asn923 |
| 4e | −9.2 | 2 | Asp1046, Glu885 |
| 4f | −8.8 | 2 | Asp1046, Glu885, |
| 4g | −10.3 | 2 | Asp1046, Glu885 |
| 4h | −10.2 | 2 | Glu885, Asp1046 |
| 4i | −9.2 | 5 | Asp1046, Ile1025, Arg1027, Glu885 |
| 4j | −10.1 | 1 | Glu885 |
| 4k | −10.7 | 5 | Asp1046, Arg1027, Glu885 |
| 4l | −9.0 | 4 | Ala881, His1026, Arg1027, Ile1025 |
| 4m | −9.9 | 5 | Asp1052, Leu840, Asn923, Cys919 |
| 4n | −10.2 | 1 | Asp1046 |
| 4o | −9.3 | 1 | Asp1046 |
| 4p | −9.9 | 2 | Asp1046, Glu885 |
| 4q | −11.1 | 3 | Asp1046, Glu885, Ile1025 |
| 4r | −9.4 | 2 | Asp1046, Glu885 |
| 4s | −9.6 | 2 | Asp1046, Glu885 |
| 4t | −11.4 | 4 | Asp1046, Glu885, Ile1025 |
| 4u | −11.4 | 3 | Asp1046, Glu885 |
| 4v | −10.4 | 2 | Ser930, Cys919 |
| 4w | −12.2 | 1 | Glu885 |
| TMZ | −6.2 | 4 | Leu840, Cys919 |
| Comp. | CNS (−2 to +2) | Mw < 500 | HBD (0–6) | HBA (2.0–20) | QPlogPo/w (−2.0–6.5) | QPlogS (−6.5–0.5) | QPPCaco <25 Poor >500 High | Metabsm (1–8) | %Human Oral Ab (>80% High <25% Poor) | N&O (<2 – 15>) | Rule of 5 (Max 4) | Rule of 3 (Max 3) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 4a | −2 | 303.36 | 2 | 5.75 | 2.431 | −4.886 | 209.18 | 5 | 82.715 | 6 | 0 | 0 |
| 4b | −2 | 357.796 | 2 | 5.75 | 3.319 | −5.642 | 313.438 | 4 | 91.058 | 6 | 0 | 0 |
| 4c | −2 | 383.403 | 2 | 7.25 | 3.075 | −5.791 | 296.37 | 6 | 89.191 | 8 | 0 | 1 |
| 4d | −2 | 329.373 | 2 | 5.75 | 2.765 | −4.901 | 308.989 | 5 | 87.702 | 6 | 0 | 0 |
| 4e | −2 | 313.312 | 2 | 6.25 | 2.163 | −4.157 | 301.102 | 5 | 83.972 | 7 | 0 | 0 |
| 4f | −2 | 353.377 | 2 | 6.5 | 2.938 | −5.185 | 309.992 | 5 | 88.736 | 7 | 0 | 0 |
| 4g | −2 | 376.414 | 3 | 5.75 | 3.386 | −5.97 | 228.303 | 5 | 88.981 | 7 | 0 | 1 |
| 4h | −2 | 365.431 | 2 | 5.75 | 3.849 | −6.233 | 312.955 | 5 | 94.148 | 6 | 0 | 1 |
| 4i | −2 | 329.398 | 2 | 5.75 | 2.831 | −5.617 | 203.198 | 5 | 84.828 | 6 | 0 | 0 |
| 4j | −2 | 323.351 | 2 | 5.75 | 2.831 | −4.93 | 310.285 | 4 | 88.119 | 6 | 0 | 0 |
| 4k | −2 | 364.36 | 2 | 6.75 | 2.144 | −5.335 | 49.12 | 4 | 69.77 | 8 | 0 | 0 |
| 4l | −2 | 396.377 | 1 | 7.25 | 2.775 | −5.346 | 102.083 | 4 | 79.152 | 8 | 0 | 0 |
| 4m | −2 | 382.35 | 2 | 6.75 | 2.404 | −5.603 | 54.475 | 4 | 72.098 | 8 | 0 | 0 |
| 4n | −2 | 368.348 | 2 | 6.75 | 2.135 | −5.132 | 35.954 | 5 | 67.293 | 9 | 0 | 0 |
| 4o | −2 | 423.384 | 1 | 8.25 | 1.733 | −4.265 | 27.701 | 5 | 49.954 | 11 | 1 | 0 |
| 4p | −2 | 323.351 | 2 | 5.75 | 2.828 | −4.948 | 303.005 | 4 | 87.915 | 6 | 0 | 0 |
| 4q | −2 | 435.869 | 2 | 7.75 | 3.732 | −6.758 | 171.738 | 6 | 88.794 | 8 | 0 | 1 |
| 4r | −2 | 340.341 | 4 | 8.25 | 0.686 | −4.007 | 25.356 | 6 | 56.094 | 9 | 0 | 0 |
| 4s | −2 | 341.341 | 2 | 5.75 | 3.062 | −5.289 | 310.103 | 4 | 89.47 | 6 | 0 | 0 |
| 4t | −2 | 449.893 | 2 | 6.25 | 4.882 | −7.62 | 306.659 | 4 | 100 | 7 | 0 | 1 |
| 4u | −2 | 373.41 | 2 | 5.75 | 3.724 | −6.051 | 304.844 | 4 | 93.21 | 6 | 0 | 1 |
| 4v | −2 | 399.448 | 2 | 5.75 | 4.409 | −6.883 | 305.004 | 4 | 100 | 6 | 0 | 1 |
| 4w | −2 | 433.893 | 2 | 5.75 | 4.89 | −7.596 | 303.939 | 4 | 100 | 6 | 0 | 1 |
| TMZ | −2 | 194.152 | 2 | 7 | −1.207 | −1.379 | 59.807 | 1 | 51.677 | 8 | 0 | 0 |
| Molecules | BBB | |||
|---|---|---|---|---|
| PreADME | SwissADME | QikProp Schrödinger | BBB | |
| 4a | 0.097/Low | No | Yes | Yes/Low |
| 4b | 0.149/Med | No | Yes | Yes/Low |
| 4c | 0.030/Low | No | Yes | Yes/Low |
| 4d | 0.0220/Low | No | Yes | Yes/Low |
| 4e | 0.0236/Low | No | Yes | Yes/Low |
| 4f | 0.0375/Low | No | Yes | Yes/Low |
| 4g | 0.314/Medl | No | Yes | Yes/Med |
| 4h | 0.394/Medl | No | Yes | Yes/Med |
| 4i | 0.258/Medl | No | Yes | Yes/Med |
| 4j | 0.062/Low | No | Yes | Yes/Low |
| 4k | 0.025/Low | No | Yes | Yes/Low |
| 4l | 0.0212/Low | No | Yes | Yes/Low |
| 4m | 0.024/Low | No | No | Yes/Low |
| 4n | 0.024/Low | No | No | Yes/Low |
| 4o | 1.061/Medl | Yes | No | Yes/Medl |
| 4p | 0.020/Low | No | Yes | Yes/Low |
| 4q | 0.015/Low | No | Yes | Yes/Low |
| 4r | 0.028/Low | No | No | Yes/Low |
| 4s | 0.082/Low | No | Yes | Yes/Low |
| 4t | 0.184/Medl | No | Yes | Yes/Medl |
| 4u | 0.090/Low | No | Yes | Yes/Low |
| 4v | 0.156/Medl | No | Yes | Yes/Medl |
| 4w | 0.450/Med | No | Yes | Yes/Medl |
| TMZ | 0.017/Low | No | No | Yes/Low |
| No. | Binding Affinity for VEGFR2 (kcal/mol) | IC50 Values (μM) U138 | ADME | ||||||
|---|---|---|---|---|---|---|---|---|---|
| BBB | CNS (−2 to +2) | MolWt < 500 | QPlogS (−6.5–0.5) | QPPCaco <25 Poor >500 High | Metabsm (1–8) | %Human OralAb >80% High <25% Poor | |||
| 4g | −10.3 | 9.2 | Yes | −2 | 376.41 | −5.97 | 228.303 | 5 | 88.981 |
| 4m | −9.9 | 4.2 | No | −2 | 382.35 | −5.603 | 54.475 | 4 | 72.098 |
| 4q | −11.1 | 8.0 | Yes | −2 | 435.86 | −6.758 | 171.738 | 6 | 88.794 |
| TMZ | −6.2 | 93.09 | Yes | −2 | 194.15 | −1.379 | 59.807 | 1 | 51.677 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buhlak, S.; Abad, N.; Akachar, J.; Saffour, S.; Kesgun, Y.; Dik, S.; Yasin, B.; Bati-Ayaz, G.; Hanashalshahaby, E.; Türkez, H.; et al. Design, Synthesis, and Computational Evaluation of 3,4-Dihydroquinolin-2(1H)-One Analogues as Potential VEGFR2 Inhibitors in Glioblastoma Multiforme. Pharmaceuticals 2025, 18, 233. https://doi.org/10.3390/ph18020233
Buhlak S, Abad N, Akachar J, Saffour S, Kesgun Y, Dik S, Yasin B, Bati-Ayaz G, Hanashalshahaby E, Türkez H, et al. Design, Synthesis, and Computational Evaluation of 3,4-Dihydroquinolin-2(1H)-One Analogues as Potential VEGFR2 Inhibitors in Glioblastoma Multiforme. Pharmaceuticals. 2025; 18(2):233. https://doi.org/10.3390/ph18020233
Chicago/Turabian StyleBuhlak, Shafeek, Nadeem Abad, Jihane Akachar, Sana Saffour, Yunus Kesgun, Sevval Dik, Betul Yasin, Gizem Bati-Ayaz, Essam Hanashalshahaby, Hasan Türkez, and et al. 2025. "Design, Synthesis, and Computational Evaluation of 3,4-Dihydroquinolin-2(1H)-One Analogues as Potential VEGFR2 Inhibitors in Glioblastoma Multiforme" Pharmaceuticals 18, no. 2: 233. https://doi.org/10.3390/ph18020233
APA StyleBuhlak, S., Abad, N., Akachar, J., Saffour, S., Kesgun, Y., Dik, S., Yasin, B., Bati-Ayaz, G., Hanashalshahaby, E., Türkez, H., & Mardinoglu, A. (2025). Design, Synthesis, and Computational Evaluation of 3,4-Dihydroquinolin-2(1H)-One Analogues as Potential VEGFR2 Inhibitors in Glioblastoma Multiforme. Pharmaceuticals, 18(2), 233. https://doi.org/10.3390/ph18020233