

Stereoselective Synthesis and Antimicrobial Studies of Allo-Gibberic Acid-Based 2,4-Diaminopyrimidine Chimeras



Abstract

1. Introduction
2. Results and Discussion
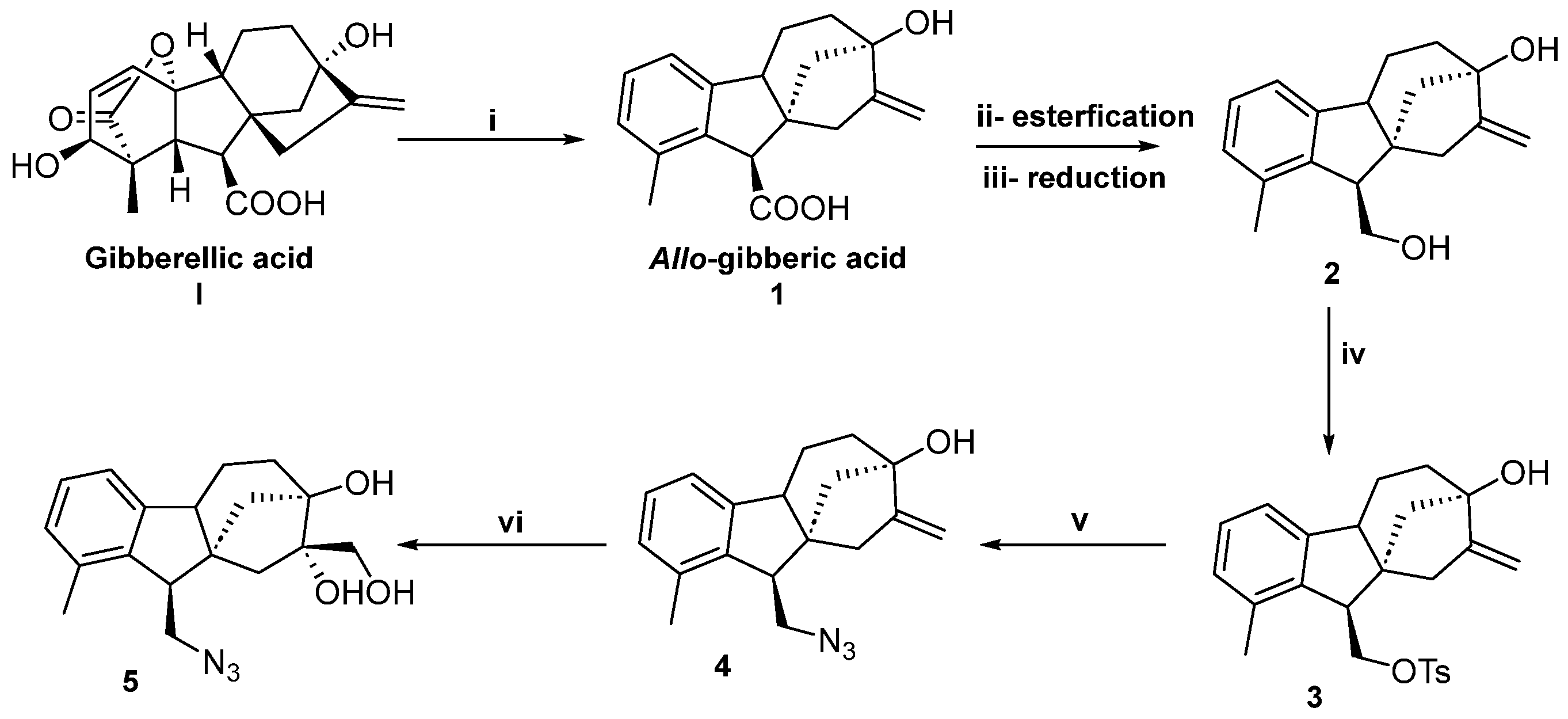
2.1. Synthesis of Allo-Gibberic Acid-Based Azides
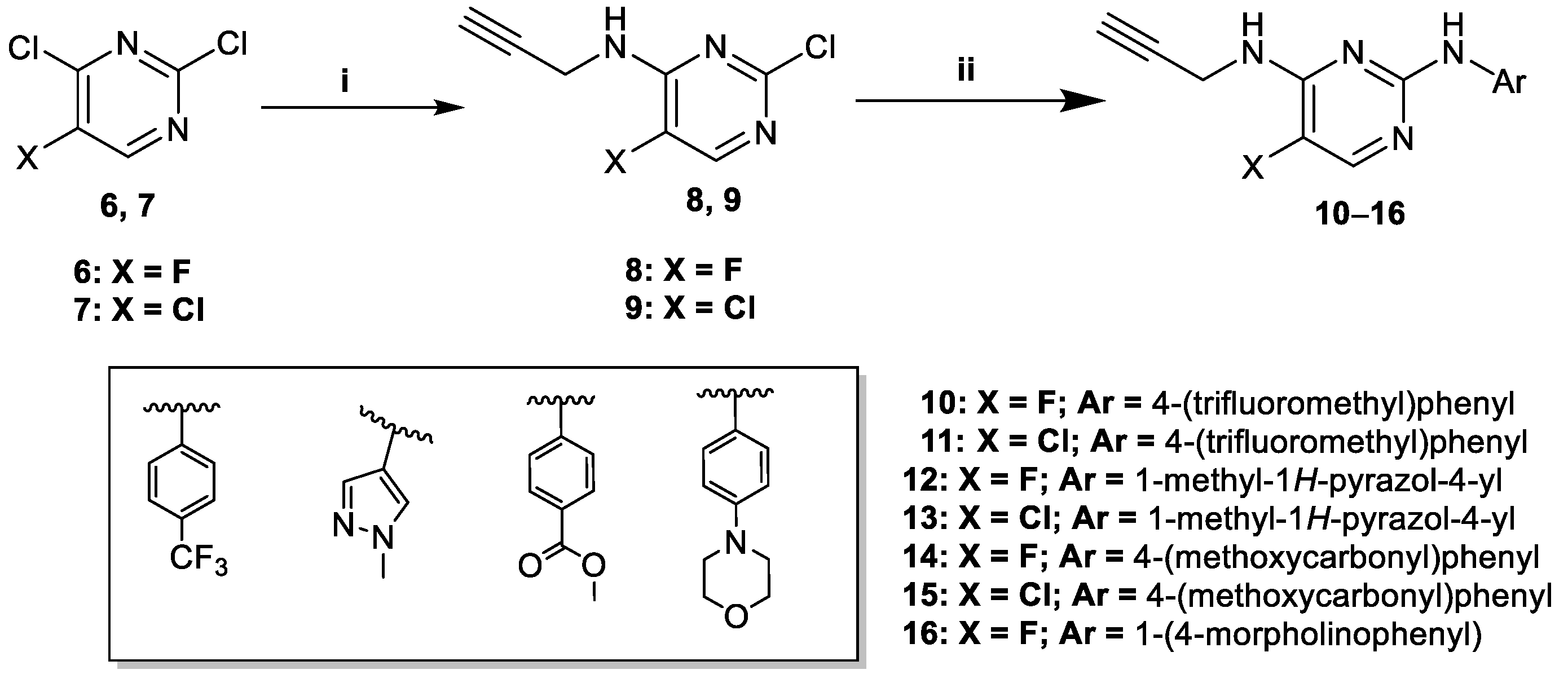
2.2. Synthesis of 2,4-Diaminopyrimidine-Derived Alkynes
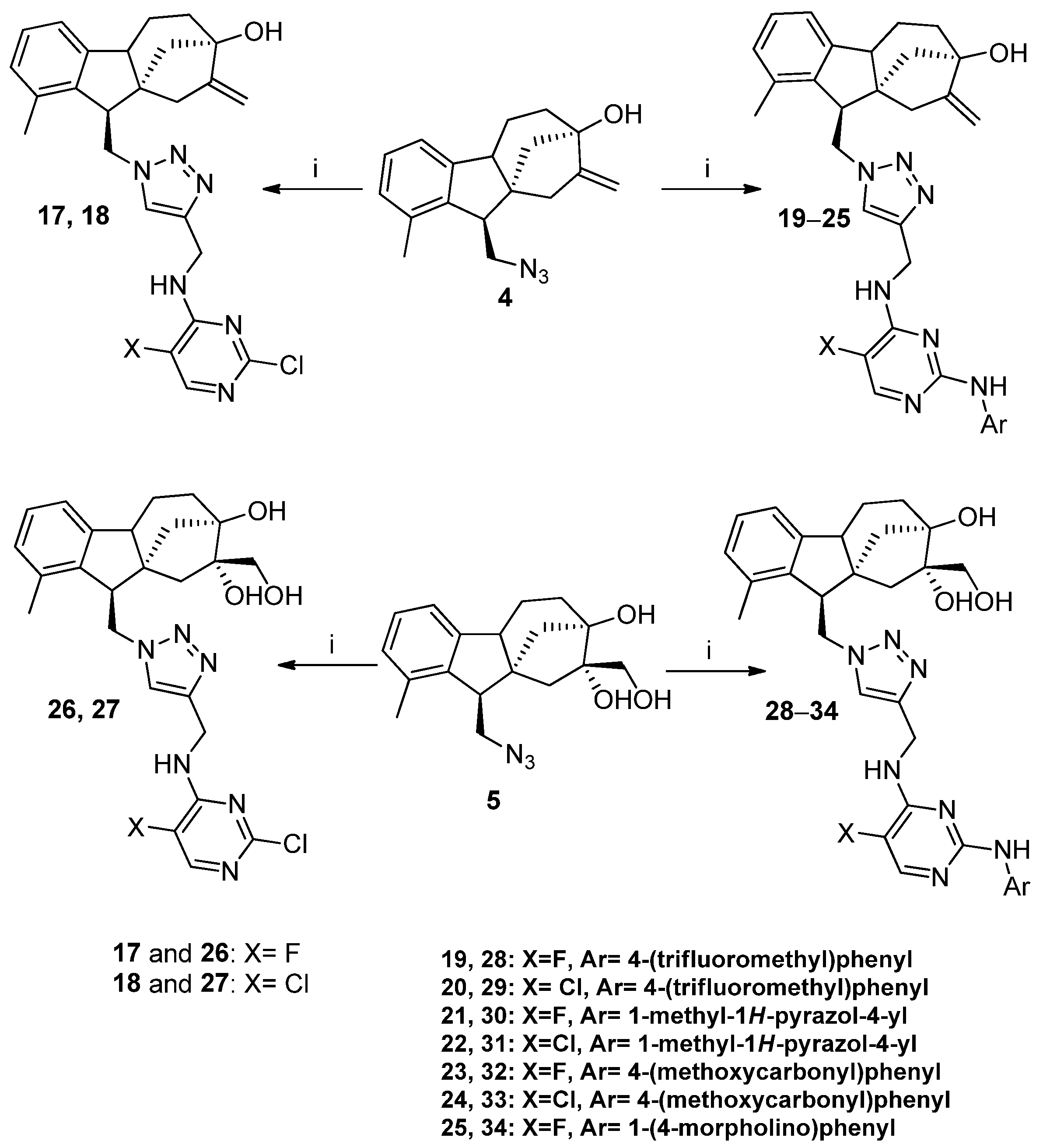
2.3. Coupling the Diterpene Skeleton with Monoamino- and Diaminopyrimidines via Click Reaction
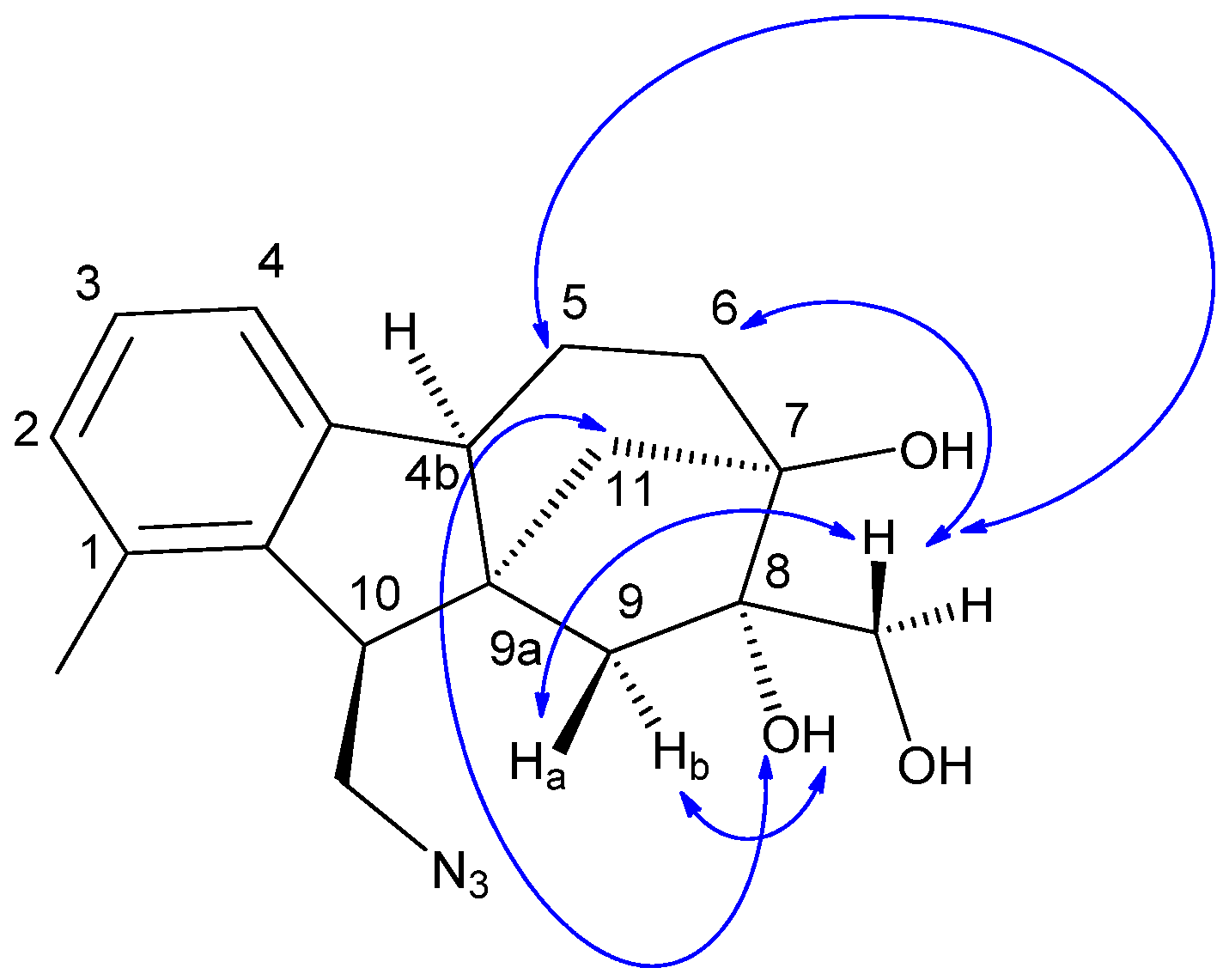
2.4. Determination of the Relative Configuration of Azide 5
2.5. In Vitro Studies of Antimicrobial Effects of 4-Amino- and 2,4-Diaminopyrimidine Derivatives and Their Structure–Activity Relationship (SAR)
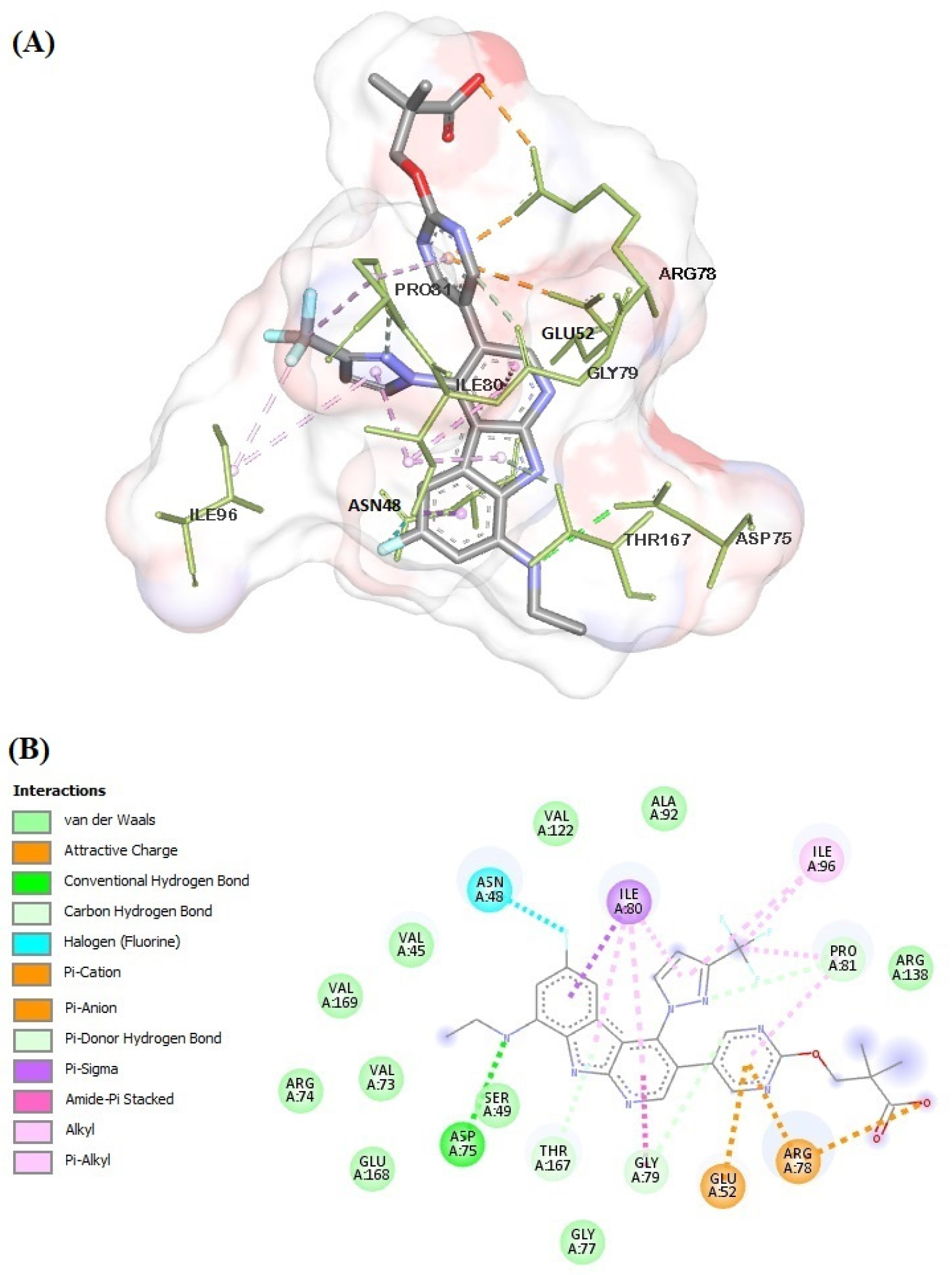
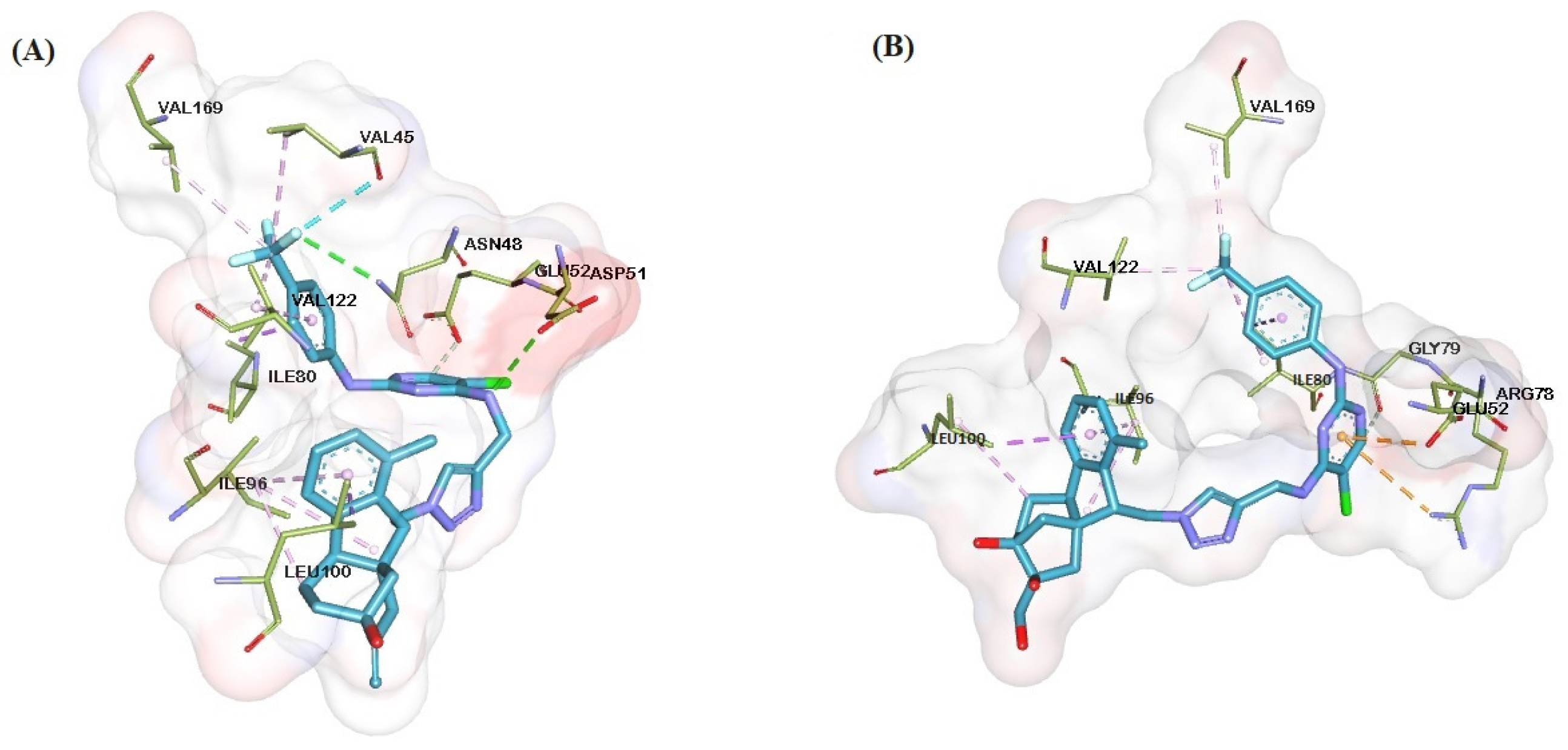
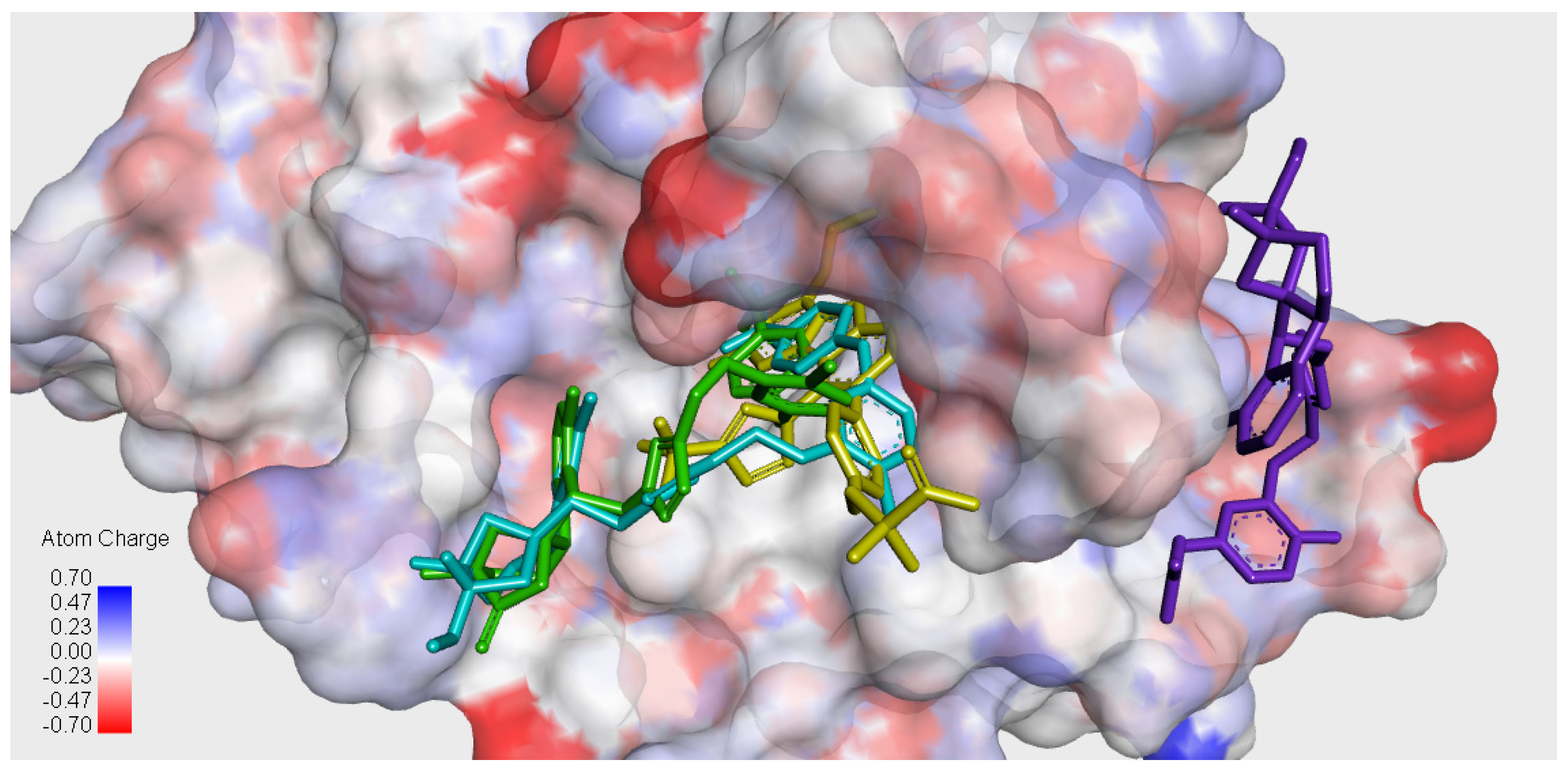
2.6. Molecular Docking
3. Materials and Methods
3.1. General Methods
3.2. Starting Materials
3.3. Synthesis of New Compounds
3.3.1. (7S,8S,9aR,10R)-10-(Azidomethyl)-8-(hydroxymethyl)-1-methyl-4b,5,6,8,9,10-hexahydro-7H-7,9a-methanobenzo[a]azulene-7,8-diol (5)
3.3.2. General Procedure for Preparing the N-Propargyl-Substituted Diaminopyrimidine Derivatives
Methyl 4-((5-Fluoro-4-(prop-2-yn-1-ylamino)pyrimidin-2-yl)amino)benzoate (14)
Methyl 4-((5-Chloro-4-(prop-2-yn-1-ylamino)pyrimidin-2-yl)amino)benzoate (15)
5-Fluoro-N2-(4-Morpholinophenyl)-N4-(prop-2-yn-1-yl)pyrimidine-2,4-diamine (16)
3.3.3. General Procedure for Preparation of 1,2,3-Triazols by Click Reaction
(7S,9aS,10R)-10-((4-(((2-Chloro-5-fluoropyrimidin-4-yl)amino)methyl)-1H-1,2,3-triazol-1-yl)methyl)-1-methyl-8-methylene-4b,5,6,8,9,10-hexahydro-7H-7,9a-methanobenzo[a]azulen-7-ol (17)
(7S,9aS,10R)-10-((4-(((2,5-Dichloropyrimidin-4-yl)amino)methyl)-1H-1,2,3-triazol-1-yl)methyl)-1-methyl-8-methylene-4b,5,6,8,9,10-hexahydro-7H-7,9a-methanobenzo[a]azulen-7-ol (18)
(7S,9aS,10R)-10-((4-(((5-Fluoro-2-((4-(trifluoromethyl)phenyl)amino)pyrimidin-4-yl)amino)methyl)-1H-1,2,3-triazol-1-yl)methyl)-1-methyl-8-methylene-4b,5,6,8,9,10-hexahydro-7H-7,9a-methanobenzo[a]azulen-7-ol (19)
(7S,9aS,10R)-10-((4-(((5-Chloro-2-((4-(trifluoromethyl)phenyl)amino)pyrimidin-4-yl)amino)methyl)-1H-1,2,3-triazol-1-yl)methyl)-1-methyl-8-methylene-4b,5,6,8,9,10-hexahydro-7H-7,9a-methanobenzo[a]azulen-7-ol (20)
(7S,9aS,10R)-10-((4-(((5-Fluoro-2-((1-methyl-1H-pyrazol-4-yl)amino)pyrimidin-4-yl)amino)methyl)-1H-1,2,3-triazol-1-yl)methyl)-1-methyl-8-methylene-4b,5,6,8,9,10-hexahydro-7H-7,9a-methanobenzo[a]azulen-7-ol (21)
(7S,9aS,10R)-10-((4-(((5-Chloro-2-((1-methyl-1H-pyrazol-4-yl)amino)pyrimidin-4-yl)amino)methyl)-1H-1,2,3-triazol-1-yl)methyl)-1-methyl-8-methylene-4b,5,6,8,9,10-hexahydro-7H-7,9a-methanobenzo[a]azulen-7-ol (22)
Methyl 4-((5-Fluoro-4-(((1-(((7S,9aS,10R)-7-hydroxy-1-methyl-8-methylene-5,6,7,8,9,10-hexahydro-4bH-7,9a-methanobenzo[a]azulen-10-yl)methyl)-1H-1,2,3-triazol-4-yl)methyl)amino)pyrimidin-2-yl)amino)benzoate (23)
Methyl 4-((5-Chloro-4-(((1-(((7S,9aS,10R)-7-hydroxy-1-methyl-8-methylene-4b,6,7,8,9,10-hexahydro-5H-7,9a-methanobenzo[a]azulen-10-yl)methyl)-1H-1,2,3-triazol-4-yl)methyl)amino)pyrimidin-2-yl)amino)benzoate (24)
(7S,9aS,10R)-10-((4-(((5-Fluoro-2-((4-morpholinophenyl)amino)pyrimidin-4-yl)amino)methyl)-1H-1,2,3-triazol-1-yl)methyl)-1-methyl-8-methylene-4b,5,6,8,9,10-hexahydro-7H-7,9a-methanobenzo[a]azulen-7-ol (25)
(7S,8S,9aR,10R)-10-((4-(((2-Chloro-5-fluoropyrimidin-4-yl)amino)methyl)-1H-1,2,3-triazol-1-yl)methyl)-8-(hydroxymethyl)-1-methyl-4b,5,6,8,9,10-hexahydro-7H-7,9a-methanobenzo[a]azulene-7,8-diol (26)
(7S,8S,9aR,10R)-10-((4-(((2,5-Dichloropyrimidin-4-yl)amino)methyl)-1H-1,2,3-triazol-1-yl)methyl)-8-(hydroxymethyl)-1-methyl-4b,5,6,8,9,10-hexahydro-7H-7,9a-methanobenzo[a]azulene-7,8-diol (27)
(7S,8S,9aR,10R)-10-((4-(((5-Fluoro-2-((4-(trifluoromethyl)phenyl)amino)pyrimidin-4-yl)amino)methyl)-1H-1,2,3-triazol-1-yl)methyl)-8-(hydroxymethyl)-1-methyl-4b,5,6,8,9,10-hexahydro-7H-7,9a-methanobenzo[a]azulene-7,8-diol (28)
(7S,8S,9aR,10R)-10-((4-(((5-Chloro-2-((4-(trifluoromethyl)phenyl)amino)pyrimidin-4-yl)amino)methyl)-1H-1,2,3-triazol-1-yl)methyl)-8-(hydroxymethyl)-1-methyl-4b,5,6,8,9,10-hexahydro-7H-7,9a-methanobenzo[a]azulene-7,8-diol (29)
(7S,8S,9aR,10R)-10-((4-(((5-Fluoro-2-((1-methyl-1H-pyrazol-4-yl)amino)pyrimidin-4-yl)amino)methyl)-1H-1,2,3-triazol-1-yl)methyl)-8-(hydroxymethyl)-1-methyl-4b,5,6,8,9,10-hexahydro-7H-7,9a-methanobenzo[a]azulene-7,8-diol (30)
(7S,8S,9aR,10R)-10-((4-(((5-Chloro-2-((1-methyl-1′H-pyrazol-4-yl)amino)pyrimidin-4-yl)amino)methyl)-1H-1,2,3-triazol-1-yl)methyl)-8-(hydroxymethyl)-1-methyl-4b,5,6,8,9,10-hexahydro-7H-7,9a-methanobenzo[a]azulene-7,8-diol (31)
Methyl 4-((4-(((1-(((7S,8S,9aR,10R)-7,8-Dihydroxy-8-(hydroxymethyl)-1-methyl-4b,6,7,8,9,10-hexahydro-5H-7,9a-methanobenzo[a]azulen-10-yl)methyl)-1H-1,2,3-triazol-4-yl)methyl)amino)-5-fluoropyrimidin-2-yl)amino)benzoate (32)
Methyl 4-((5-Chloro-4-(((1-(((7S,8S,9aR,10R)-7,8-dihydroxy-8-(hydroxymethyl)-1-methyl-4b,6,7,8,9,10-hexahydro-5H-7,9a-methanobenzo[a]azulen-10-yl)methyl)-1H-1,2,3-triazol-4-yl)methyl)amino)pyrimidin-2-yl)amino)benzoate (33)
(7S,8S,9aR,10R)-10-((4-(((5-Fluoro-2-((4-morpholinophenyl)amino)pyrimidin-4-yl)amino)methyl)-1H-1,2,3-triazol-1-yl)methyl)-8-(hydroxymethyl)-1-methyl-4b,5,6,8,9,10-hexahydro-7H-7,9a-methanobenzo[a]azulene-7,8-diol (34)
3.4. Antimicrobial Analyses
3.5. General Procedure for Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| MDPI | Multidisciplinary Digital Publishing Institute |
| DOAJ | Directory of open access journals |
| TLA | Three-letter acronym |
| LD | Linear dichroism |
| CuAAC reaction | Copper-catalyzed azide–alkyne cycloaddition |
| DCM | Dichloromethane |
| DIPEA | N,N-Diisopropylethylamine |
| DMAP | 4-(Dimethylamino)pyridine |
| DMSO | Dimethyl sulfoxide |
| MW | Microwave |
| NMO | 4-Methylmorpholine N-oxide |
| NOE | Nuclear Overhauser effect |
| SZMC | Szeged Microbiology Collection |
| TEA | Triethylamine |
References
- Murray, C.J.L.; Ikuta, K.S.; Sharara, F.; Swetschinski, L.; Robles Aguilar, G.; Gray, A.; Han, C.; Bisignano, C.; Rao, P.; Wool, E.; et al. Global Burden of Bacterial Antimicrobial Resistance in 2019: A Systematic Analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef] [PubMed]
- European Centre for Disease Prevention and Control. Antimicrobial Resistance in the EU/EEA (EARS-Net)—Annual Epidemiological Report 2023; ECDC: Stockholm, Sweden, 2024. [Google Scholar]
- Farha, M.A.; Brown, E.D. Drug Repurposing for Antimicrobial Discovery. Nat. Microbiol. 2019, 4, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Vaou, N.; Stavropoulou, E.; Voidarou, C.; Tsigalou, C.; Bezirtzoglou, E. Towards Advances in Medicinal Plant Antimicrobial Activity: A Review Study on Challenges and Future Perspectives. Microorganisms 2021, 9, 2041. [Google Scholar] [CrossRef] [PubMed]
- Plant Bioactives and Drug Discovery: Principles, Practice, and Perspectives; Cechinel-Filho, V., Ed.; Wiley Series in Drug Discovery and Development; John Wiley & Sons: Hoboken, NJ, USA, 2012; ISBN 978-0-470-58226-8. [Google Scholar]
- Studies in Natural Products Chemistry; Rahman, A., Ed.; Elsevier: Amsterdam, The Netherlands; New York, NY, USA, 1988; ISBN 978-0-444-42970-4. [Google Scholar]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef]
- Schreiber, K.; Schneider, G.; Sembdner, G. Gibberelline—XII. Tetrahedron 1968, 24, 73–78. [Google Scholar] [CrossRef]
- Hanson, J.R. The Chemistry of the Gibberellins. Nat. Prod. Rep. 1990, 7, 41. [Google Scholar] [CrossRef]
- Mander, L.N. The Chemistry of Gibberellins: An Overview. Chem. Rev. 1992, 92, 573–612. [Google Scholar] [CrossRef]
- Chen, S.-Y.; Kuo, S.-R.; Chien, C.-T. Roles of Gibberellins and Abscisic Acid in Dormancy and Germination of Red Bayberry (Myrica Rubra) Seeds. Tree Physiol. 2008, 28, 1431–1439. [Google Scholar] [CrossRef]
- Alabdeen Khdar, Z.; Le, T.M.; Szakonyi, Z. Gibberellic Acid (GA3): A Versatile Chiral Building Block for Syntheses of Pharmaceutical Agents. Chem. Biodivers. 2024, 22, e202401823. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, H.; Chen, J.; Zhao, H.; Zeng, X.; Zhang, H.; Qing, C. Antitumor and Antiangiogenic Effects of GA-13315, a Gibberellin Derivative. Investig. New Drug. 2012, 30, 8–16. [Google Scholar] [CrossRef]
- Zhu, S.; Luo, F.; Sun, P.-H. Synthesis and Antitumor Activity of Novel Gibberellin Derivatives with Tetracyclic Diterpenoid Skeletons. Med. Chem. Res. 2020, 29, 1341–1354. [Google Scholar] [CrossRef]
- Annand, J.R.; Henderson, A.R.; Cole, K.S.; Maurais, A.J.; Becerra, J.; Liu, Y.; Weerapana, E.; Koehler, A.N.; Mapp, A.K.; Schindler, C.S. Gibberellin JRA-003: A Selective Inhibitor of Nuclear Translocation of IKKα. ACS Med. Chem. Lett. 2020, 11, 1913–1918. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F.; Mantovani, A. Inflammation and Cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Werb, Z. Inflammation and Cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Khdar, Z.A.; Le, T.M.; Schelz, Z.; Zupkó, I.; Szakonyi, Z. Stereoselective Synthesis and Antiproliferative Activity of Allo -Gibberic Acid-Based 1,3-Aminoalcohol Regioisomers. RSC Med. Chem. 2024, 15, 874–887. [Google Scholar] [CrossRef]
- Khdar, Z.A.; Le, T.M.; Schelz, Z.; Zupkó, I.; Szakonyi, Z. Stereoselective Synthesis and Application of Gibberellic Acid-Derived Aminodiols. Int. J. Mol. Sci. 2022, 23, 10366. [Google Scholar] [CrossRef]
- Khdar, Z.A.; Le, T.M.; Schelz, Z.; Zupkó, I.; Szakonyi, Z. Aminodiols, Aminotetraols and 1,2,3-Triazoles Based on Allo -Gibberic Acid: Stereoselective Syntheses and Antiproliferative Activities. RSC Adv. 2024, 14, 36698–36712. [Google Scholar] [CrossRef]
- Wu, M.-J.; Wu, D.-M.; Chen, J.-B.; Zhao, J.-F.; Gong, L.; Gong, Y.-X.; Li, Y.; Yang, X.-D.; Zhang, H. Synthesis and Anti-Proliferative Activity of Allogibberic Acid Derivatives Containing 1,2,3-Triazole Pharmacophore. Bioorg. Med. Chem. Lett. 2018, 28, 2543–2549. [Google Scholar] [CrossRef]
- Richter, M.F.; Drown, B.S.; Riley, A.P.; Garcia, A.; Shirai, T.; Svec, R.L.; Hergenrother, P.J. Predictive Compound Accumulation Rules Yield a Broad-Spectrum Antibiotic. Nature 2017, 545, 299–304. [Google Scholar] [CrossRef]
- El-Sayed, M.Y.; Fetooh, H.; Refat, M.S.; Eldaroti, H.H.; Adam, A.M.A.; Saad, H.A. Complexes of the Plant Hormone Gibberellic Acid with the Pt(II), Au(III), Ru(III), V(III), and Se(IV) Ions: Preparation, Characterization, and in Vitro Evaluation of Biological Activity. J. Mol. Liq. 2019, 296, 111895. [Google Scholar] [CrossRef]
- Toner, P.; Nelson, D.; Rao, J.R.; Ennis, M.; Moore, J.E.; Schock, B. Antimicrobial Properties of Phytohormone (Gibberellins) against Phytopathogens and Clinical Pathogens. Access Microbiol. 2021, 3, 000278. [Google Scholar] [CrossRef] [PubMed]
- Then, R.L. History and Future of Antimicrobial Diaminopyrimidines. J. Chemother. 1993, 5, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Landi, G.; Linciano, P.; Tassone, G.; Costi, M.P.; Mangani, S.; Pozzi, C. High-Resolution Crystal Structure of Trypanosoma Brucei Pteridine Reductase 1 in Complex with an Innovative Tricyclic-Based Inhibitor. Acta Crystallogr. D Struct. Biol. 2020, 76, 558–564. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, M.; Sankhe, K.; Bhandare, R.R.; Edis, Z.; Bloukh, S.H.; Khan, T.A. Synthetic Strategies of Pyrimidine-Based Scaffolds as Aurora Kinase and Polo-like Kinase Inhibitors. Molecules 2021, 26, 5170. [Google Scholar] [CrossRef]
- Mahapatra, A.; Prasad, T.; Sharma, T. Pyrimidine: A Review on Anticancer Activity with Key Emphasis on SAR. Futur. J. Pharm. Sci. 2021, 7, 123. [Google Scholar] [CrossRef]
- Kaur, R.; Kaur, P.; Sharma, S.; Singh, G.; Mehndiratta, S.; Bedi, P.; Nepali, K. Anti-Cancer Pyrimidines in Diverse Scaffolds: A Review of Patent Literature. Recent Pat. Anti-Canc. 2014, 10, 23–71. [Google Scholar] [CrossRef]
- VanRheenen, V.; Cha, D.Y.; Hartley, W.M. Catalytic Osmium Tetroxide Oxidation of Olefins: Cis -1,2-Cyclohexanediol. In Organic Syntheses; Kende, A.S., Freeman, J.P., Eds.; Wiley: Hoboken, NJ, USA, 2003; p. 43. ISBN 978-0-471-26422-4. [Google Scholar]
- Lagoja, I. Pyrimidine as Constituent of Natural Biologically Active Compounds. Chem. Biodivers. 2005, 2, 1–50. [Google Scholar] [CrossRef]
- Bamou, F.Z.; Le, T.M.; Tayeb, B.A.; Tahaei, S.A.S.; Minorics, R.; Zupkó, I.; Szakonyi, Z. Antiproliferative Activity of (−)-Isopulegol-based 1,3-Oxazine, 1,3-Thiazine and 2,4-Diaminopyrimidine Derivatives. ChemistryOpen 2022, 11, e202200169. [Google Scholar] [CrossRef]
- Nammalwar, B.; Bunce, R.A. Recent Advances in Pyrimidine-Based Drugs. Pharmaceuticals 2024, 17, 104. [Google Scholar] [CrossRef]
- Rauckman, B.S.; Tidwell, M.Y.; Johnson, J.V.; Roth, B. 2,4-Diamino-5-Benzylpyrimidines and Analogs as Antibacterial Agents. 10. 2,4-Diamino-5-(6-Quinolylmethyl)- and -[(Tetrahydro-6-Quinolyl)Methyl]Pyrimidine Derivatives. Further Specificity Studies. J. Med. Chem. 1989, 32, 1927–1935. [Google Scholar] [CrossRef]
- Perales, J.B.; Freeman, J.; Bacchi, C.J.; Bowling, T.; Don, R.; Gaukel, E.; Mercer, L.; Moore Iii, J.A.; Nare, B.; Nguyen, T.M.; et al. SAR of 2-Amino and 2,4-Diamino Pyrimidines with in Vivo Efficacy against Trypanosoma Brucei. Bioorg. Med. Chem. Lett. 2011, 21, 2816–2819. [Google Scholar] [CrossRef] [PubMed]
- Mercer, L.; Bowling, T.; Perales, J.; Freeman, J.; Nguyen, T.; Bacchi, C.; Yarlett, N.; Don, R.; Jacobs, R.; Nare, B. 2,4-Diaminopyrimidines as Potent Inhibitors of Trypanosoma Brucei and Identification of Molecular Targets by a Chemical Proteomics Approach. PLoS Negl. Trop. Dis. 2011, 5, e956. [Google Scholar] [CrossRef] [PubMed]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chem. 2002, 114, 2708–2711. [Google Scholar] [CrossRef]
- Zhu, L.; Brassard, C.J.; Zhang, X.; Guha, P.M.; Clark, R.J. On the Mechanism of Copper(I)-Catalyzed Azide-Alkyne Cycloaddition. Chem. Rec. 2016, 16, 1501–1517. [Google Scholar] [CrossRef]
- Weinstein, M.P. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically. In Clinical and Laboratory, 11th ed.; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2018. [Google Scholar]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Rajakumari, K.; Aravind, K.; Balamugundhan, M.; Jagadeesan, M.; Somasundaram, A.; Brindha Devi, P.; Ramasamy, P. Comprehensive Review of DNA Gyrase as Enzymatic Target for Drug Discovery and Development. Eur. J. Med. Chem. Res. 2024, 12, 100233. [Google Scholar] [CrossRef]
- Shahul, H.P.; Solapure, S.; Mukherjee, K.; Nandi, V.; Waterson, D.; Shandil, R.; Balganesh, M.; Sambandamurthy, V.K.; Raichurkar, A.K.; Deshpande, A.; et al. Optimization of Pyrrolamides as Mycobacterial GyrB ATPase Inhibitors: Structure-Activity Relationship and In Vivo Efficacy in a Mouse Model of Tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 61–70. [Google Scholar] [CrossRef]
- Sharma, V.; Gupta, M. Designing of Kinase Hinge Binders: A Medicinal Chemistry Perspective. Chem. Biol. Drug Des. 2022, 100, 968–980. [Google Scholar] [CrossRef]
- Hu, Y.; Shi, H.; Zhou, M.; Ren, Q.; Zhu, W.; Zhang, W.; Zhang, Z.; Zhou, C.; Liu, Y.; Ding, X.; et al. Discovery of Pyrido [2,3-b] Indole Derivatives with Gram-Negative Activity Targeting Both DNA Gyrase and Topoisomerase IV. J. Med. Chem. 2020, 63, 9623–9649. [Google Scholar] [CrossRef]
- Novais, J.S.; Campos, V.R.; Silva, A.C.J.A.; De Souza, M.C.B.V.; Ferreira, V.F.; Keller, V.G.L.; Ferreira, M.O.; Dias, F.R.F.; Vitorino, M.I.; Sathler, P.C.; et al. Synthesis and Antimicrobial Evaluation of Promising 7-Arylamino-5,8-Dioxo-5,8-Dihydroisoquinoline-4-Carboxylates and Their Halogenated Amino Compounds for Treating Gram-Negative Bacterial Infections. RSC Adv. 2017, 7, 18311–18320. [Google Scholar] [CrossRef]
- Cao, M.; Chen, Y.; Zhao, T.; Wei, S.; Guo, M.; Zhai, X. Pyrroformyl-Containing 2,4-Diaminopyrimidine Derivatives as a New Optimization Strategy of ALK Inhibitors Combating Mutations. Bioorg. Med. Chem. 2020, 28, 115715. [Google Scholar] [CrossRef] [PubMed]
- Brocksom, T.J.; dos Santos, R.B.; Varanda, N.A.; Brocksom, U. An Efficient Synthesis of Monoterpene α-Methylene-γ-Butyrolactones. Synth. Commun. 1988, 18, 1403–1410. [Google Scholar] [CrossRef]
- Béni, Z.; Dékány, M.; Kovács, B.; Csupor-Löffler, B.; Zomborszki, Z.P.; Kerekes, E.; Szekeres, A.; Urbán, E.; Hohmann, J.; Ványolós, A. Bioactivity-Guided Isolation of Antimicrobial and Antioxidant Metabolites from the Mushroom Tapinella Atrotomentosa. Molecules 2018, 23, 1082. [Google Scholar] [CrossRef] [PubMed]
- De Morais Oliveira-Tintino, C.D.; Da Silva, F.E.F.; Santiago, G.M.P.; Das, C.L.; Pinto, F.; Pessoa, O.D.L.; Da Fonseca, A.M.; Paulo, C.L.R.; Dos Santos, H.S.; Marinho, M.M.; et al. Molecular Docking and Antibacterial Activity of Campesterol Derivatives Against Staphylococcus aureus, Escherichia coli and Pseudomonas aeruginosa Multiresistant Strains. Chem. Biodivers. 2024, 22, e202401073. [Google Scholar] [CrossRef] [PubMed]
- Yusuf, D.; Davis, A.M.; Kleywegt, G.J.; Schmitt, S. An Alternative Method for the Evaluation of Docking Performance: RSR vs RMSD. J. Chem. Inf. Model. 2008, 48, 1411–1422. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitory Effect (%) ± RSD (%) | |||||||
|---|---|---|---|---|---|---|---|
| Gram-Positive | Gram-Negative | Yeast | |||||
| Compound | Conc. (μg/mL) | B. subtilis | S. aureus | E. coli | P. aeruginosa | C. krusei | C. albicans |
| Ampicillin | 50 | 99.7 ± 2.2 | 99.1 ± 1.2 | 99.1 ± 1.2 | 99.1 ± 1.2 | - | - |
| 5 | 100.3 ± 0.2 | 100.0 ± 2.1 | 100.0 ± 2.1 | 100.0 ± 2.1 | - | - | |
| Nystatine | 50 | - | - | - | - | 100.0 ± 4.6 | 100.0 ± 1.0 |
| 5 | - | - | - | - | 77.5 ± 6.4 | 100.0 ± 2.0 | |
| 4 | 50 | 100.0 ± 5.9 | 100.0 ± 1.9 | 100.0 ± 2.6 | 44.5 ± 1.6 | 86.6 ± 22.0 | 48.3 ± 5.5 |
| 5 | 28.7 ± 9.4 | 19.8 ± 7.9 | - | 18.3 ± 3.8 | 95.0 ± 6.3 | 32.5 ± 2.5 | |
| 5 | 50 | - | 3.2 ± 3.0 | 15.4 ± 1.2 | 15.8 ± 1.9 | - | 4.6 ± 7.1 |
| 5 | 15.6 ± 5.3 | 8.5 ± 4.4 | - | 14.9 ± 3.9 | 62.1 ± 2.2 | 3.9 ± 2.2 | |
| 17 | 50 | 58.7 ± 3.1 | 12.3 ± 6.0 | 82.6 ± 2.1 | 22.8 ± 5.5 | 59.3 ± 4.6 | 7.7 ± 2.2 |
| 5 | 69.3 ± 9.4 | 45.1 ± 6.7 | 42.4 ± 4.8 | 50.0 ± 7.7 | 41.8 ± 4.0 | - | |
| 18 | 50 | - | 43.4 ± 5.3 | - | 31.0 ± 5.0 | 58.8 ± 9.3 | 13.8 ± 3.1 |
| 5 | 11.7 ± 7.2 | 14.2 ± 4.0 | - | 49.4 ± 3.0 | 50.9 ± 17.8 | 10.6 ± 2.2 | |
| 19 | 50 | 82.2 ± 4.8 | 50.6 ± 3.3 | 18.8 ± 2.3 | 19.8 ± 3.8 | 46.0 ± 8.5 | - |
| 5 | - | 17.9 ± 1.9 | 13.3 ± 1.4 | 18.5 ± 2.0 | - | - | |
| 20 | 50 | 23.4 ± 3.7 | 26.1 ± 4.6 | 17.2 ± 4.9 | 81.6 ± 4.2 | 12.4 ± 3.0 | 4.9 ± 2.9 |
| 5 | - | 15.8 ± 4.0 | 9.5 ± 2.2 | 100.0 ± 1.4 | 12.9 ± 2.8 | 3.2 ± 3.3 | |
| 21 | 50 | - | 23.6 ± 2.8 | 27.2 ± 5.6 | 7.6 ± 1.2 | 79.2 ± 1.6 | 71.8 ± 1.1 |
| 5 | - | 17.0 ± 4.5 | - | 12.0 ± 4.1 | 39.6 ± 8.5 | 37.3 ± 1.1 | |
| 22 | 50 | - | 9.5 ± 1.9 | 32.1 ± 2.4 | - | 58.6 ± 3.2 | 47.1 ± 0.6 |
| 5 | - | 2.4 ± 2.5 | - | 9.3 ± 8.7 | 29.2 ± 10.3 | 32.1 ± 0.6 | |
| 23 | 50 | - | - | 48.1 ± 2.5 | - | 57.4 ± 5.7 | 49.8 ± 1.1 |
| 5 | - | 20.1 ± 6.8 | - | 4.6 ± 1.9 | 20.2 ± 2.3 | 21.4 ± 1.3 | |
| 24 | 50 | - | - | - | - | 56.8 ± 9.0 | 58.1 ± 1.1 |
| 5 | - | 5.9 ± 1.1 | - | 10.6 ± 1.7 | - | 35.3 ± 0.5 | |
| 25 | 50 | - | - | - | 10.1 ± 2.2 | 42.3 ± 0.7 | 27.0 ± 0.3 |
| 5 | - | 24.1 ± 5.4 | - | 3.4 ± 0.7 | - | 22.5 ± 0.3 | |
| 26 | 50 | - | - | 17.5 ± 6.5 | 15.5 ± 3.1 | 18.5 ± 13.6 | 5.2 ± 1.3 |
| 5 | - | 16.7 ± 6.4 | - | 42.3 ± 12.9 | - | - | |
| 27 | 50 | - | - | 22.7 ± 1.3 | 30.2 ± 2.5 | 61.3 ± 10.8 | - |
| 5 | 39.5 ± 7.7 | 19.8 ± 6.6 | - | 21.9 ± 5.5 | 100.0 ± 21.3 | 56.3 ± 2.0 | |
| 28 | 50 | 96.5 ± 5.2 | 94.7 ± 1.6 | 97.5 ± 1.4 | 33.6 ± 3.9 | 26.7 ± 8.1 | 7.6 ± 1.8 |
| 5 | - | - | - | 21.9 ± 2.5 | 100.0 ± 3.0 | 56.1 ± 0.7 | |
| 29 | 50 | 100.7 ± 1.8 | 50.1 ± 5.5 | 32.4 ± 12.7 | 93.2 ± 3.4 | 84.0 ± 5.2 | 19.1 ± 0.6 |
| 5 | - | - | 5.7 ± 5.1 | 100.0 ± 1.4 | 43.7 ± 18.9 | - | |
| 30 | 50 | - | 17.0 ± 8.3 | - | - | 41.7 ± 2.7 | 48.1 ± 0.9 |
| 5 | - | - | - | - | 17.7 ± 6.5 | 34.5 ± 2.3 | |
| 31 | 50 | - | 100.0 ± 8.1 | 61.2 ± 3.3 | 49.3 ± 2.5 | 54.5 ± 4.4 | 60.2 ± 0.4 |
| 5 | - | 17.0 ± 2.8 | 27.5 ± 13.9 | - | 27.0 ± 4.2 | 18.9 ± 0.5 | |
| 32 | 50 | - | 45.9 ± 6.1 | 40.1 ± 7.8 | 15.9 ± 0.6 | 53.8 ± 2.7 | 60.1 ± 2.1 |
| 5 | - | 2.7 ± 2.2 | 12.3 ± 2.1 | 7.4 ± 4.9 | 29.2 ± 10.3 | 28.6 ± 0.8 | |
| 33 | 50 | - | 100.0 ± 2.8 | 60.7 ± 18 | 38.4 ± 0.8 | 57.0 ± 6.9 | 67.3 ± 0.9 |
| 5 | - | 11.1 ± 4.0 | 22.3 ± 1.3 | 12.4 ± 3.1 | - | 27.0 ± 0.6 | |
| 34 | 50 | - | 21.7 ± 1.4 | 18.4 ± 2.1 | - | 50.7 ± 3.2 | 29.6 ± 0.3 |
| 5 | 13.1 ± 7.6 | 25.8 ± 3.0 | - | - | 26.1 ± 9.4 | 31.6 ± 0.3 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Depp, D.; Sebők, N.R.; Szekeres, A.; Szakonyi, Z. Stereoselective Synthesis and Antimicrobial Studies of Allo-Gibberic Acid-Based 2,4-Diaminopyrimidine Chimeras. Pharmaceuticals 2025, 18, 168. https://doi.org/10.3390/ph18020168
Depp D, Sebők NR, Szekeres A, Szakonyi Z. Stereoselective Synthesis and Antimicrobial Studies of Allo-Gibberic Acid-Based 2,4-Diaminopyrimidine Chimeras. Pharmaceuticals. 2025; 18(2):168. https://doi.org/10.3390/ph18020168
Chicago/Turabian StyleDepp, Dima, Noémi Regina Sebők, András Szekeres, and Zsolt Szakonyi. 2025. "Stereoselective Synthesis and Antimicrobial Studies of Allo-Gibberic Acid-Based 2,4-Diaminopyrimidine Chimeras" Pharmaceuticals 18, no. 2: 168. https://doi.org/10.3390/ph18020168
APA StyleDepp, D., Sebők, N. R., Szekeres, A., & Szakonyi, Z. (2025). Stereoselective Synthesis and Antimicrobial Studies of Allo-Gibberic Acid-Based 2,4-Diaminopyrimidine Chimeras. Pharmaceuticals, 18(2), 168. https://doi.org/10.3390/ph18020168