
Molecular Targets of Minor Cannabinoids in Breast Cancer: In Silico and In Vitro Studies

 , , , ,
, , , ,  ,
,
Abstract

1. Introduction
2. Results
2.1. Molecular Docking Results
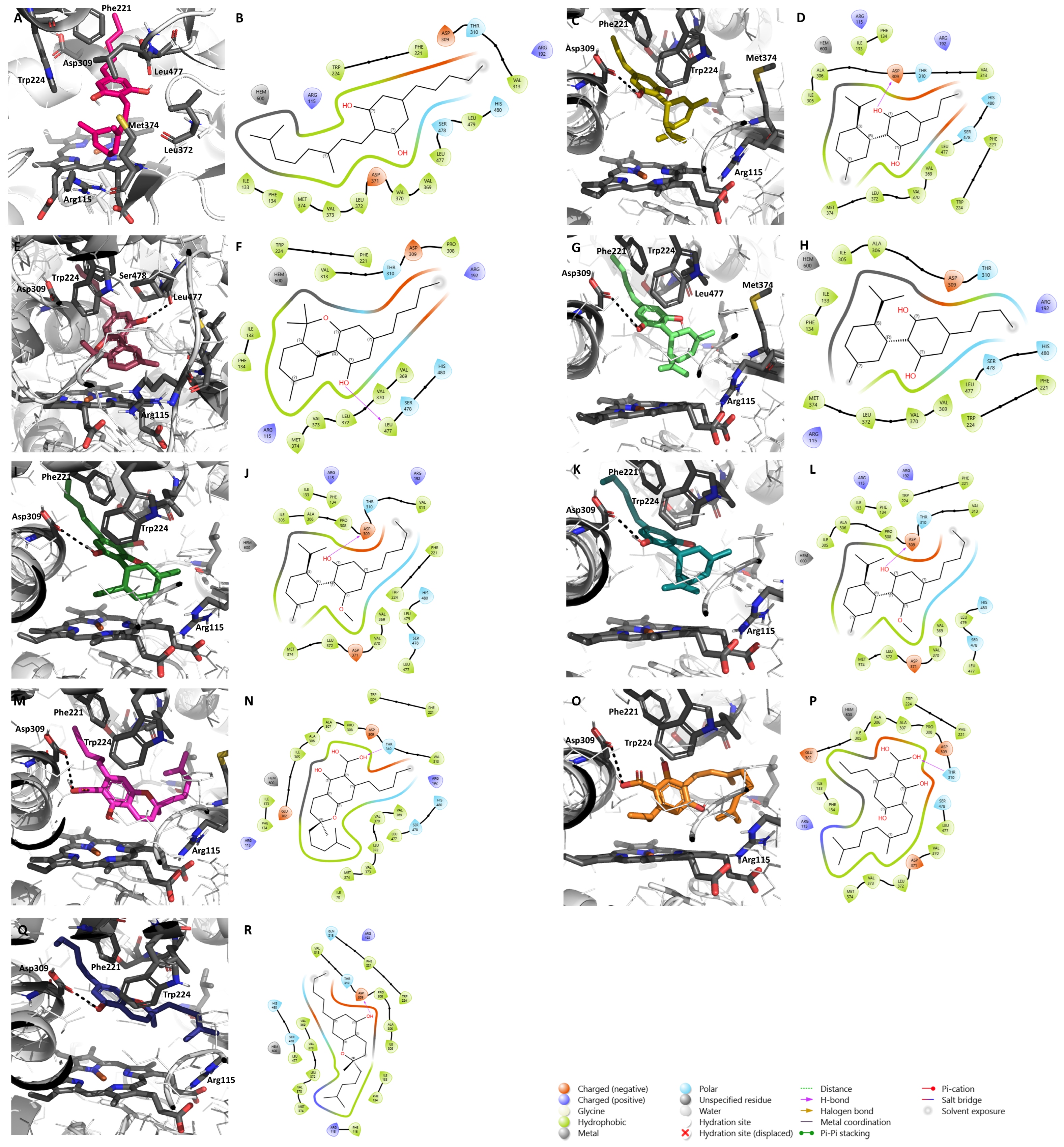


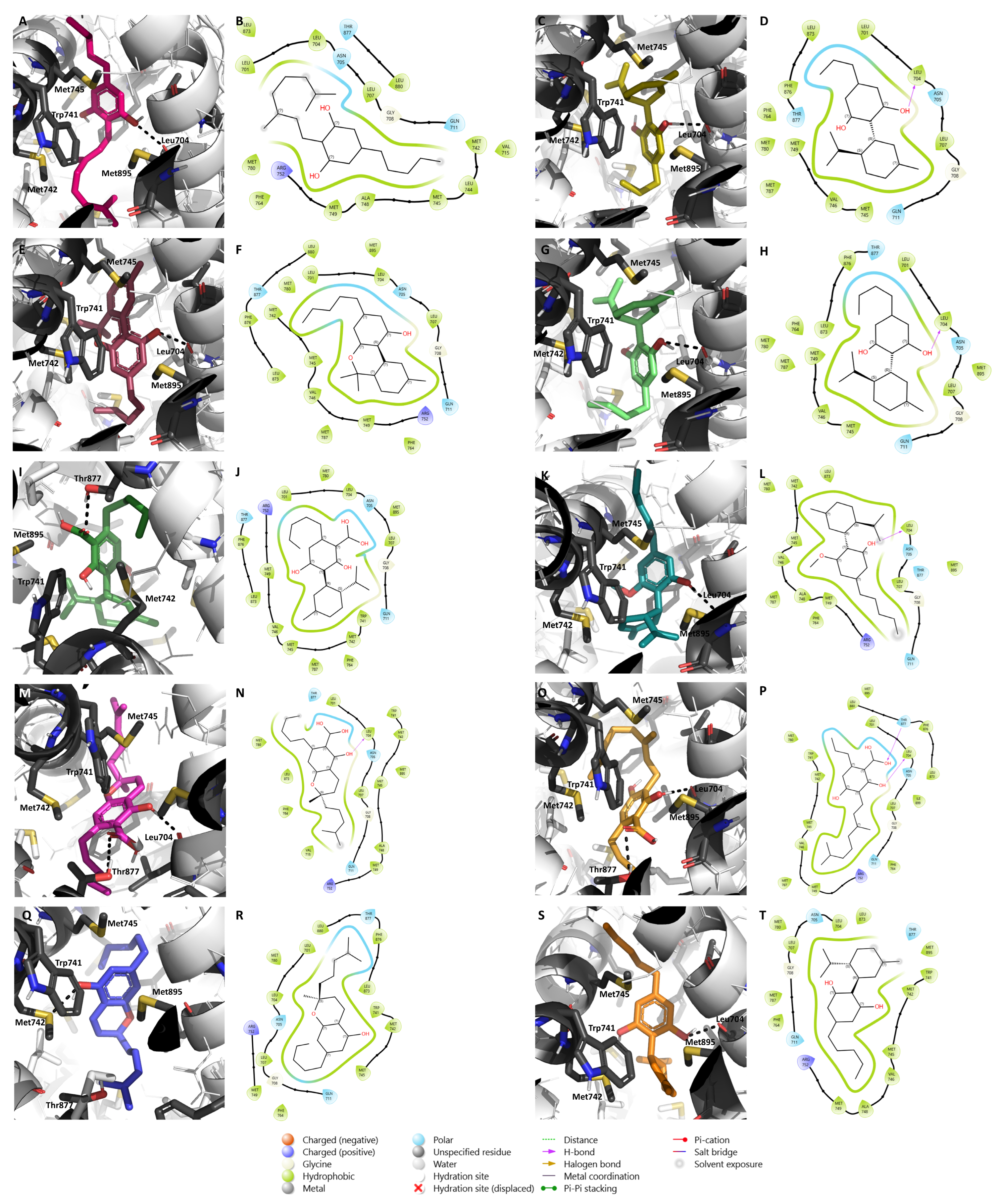
2.2. Effects of the Cannabinoids in Aromatase
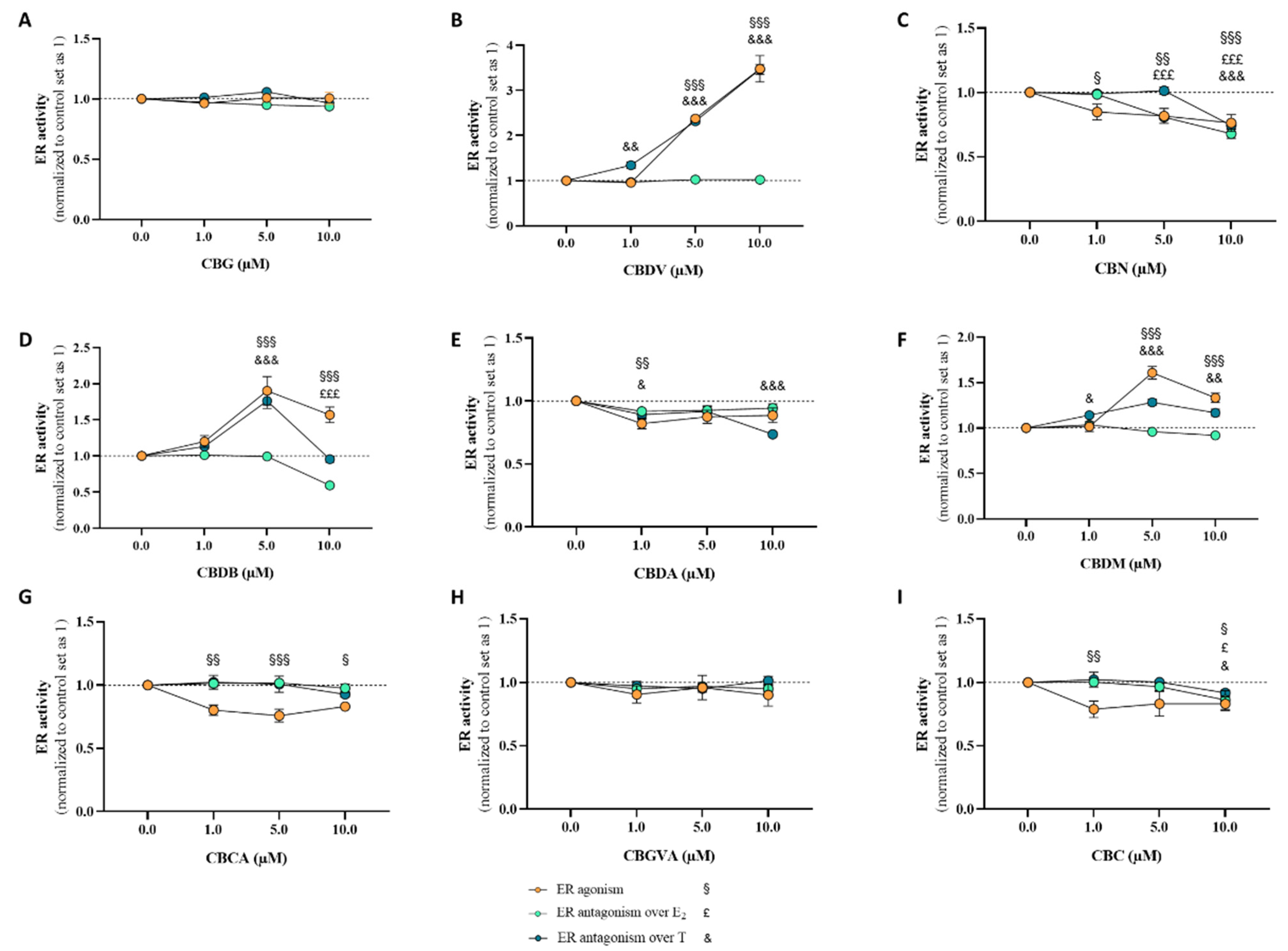
2.3. Effects of Cannabinoids in Estrogen Receptor (ER)
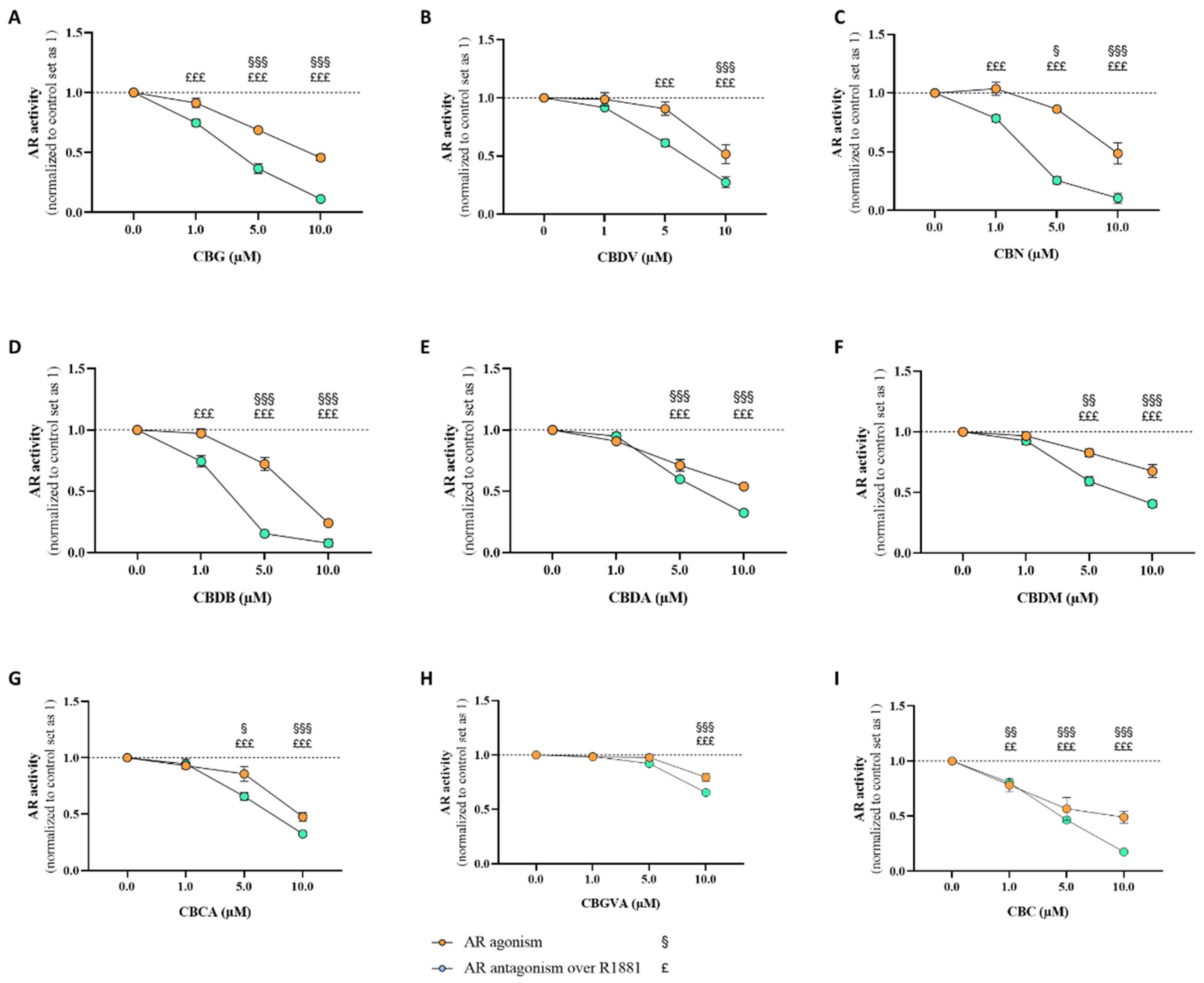
2.4. Effects of Cannabinoids in Androgen Receptor (AR)
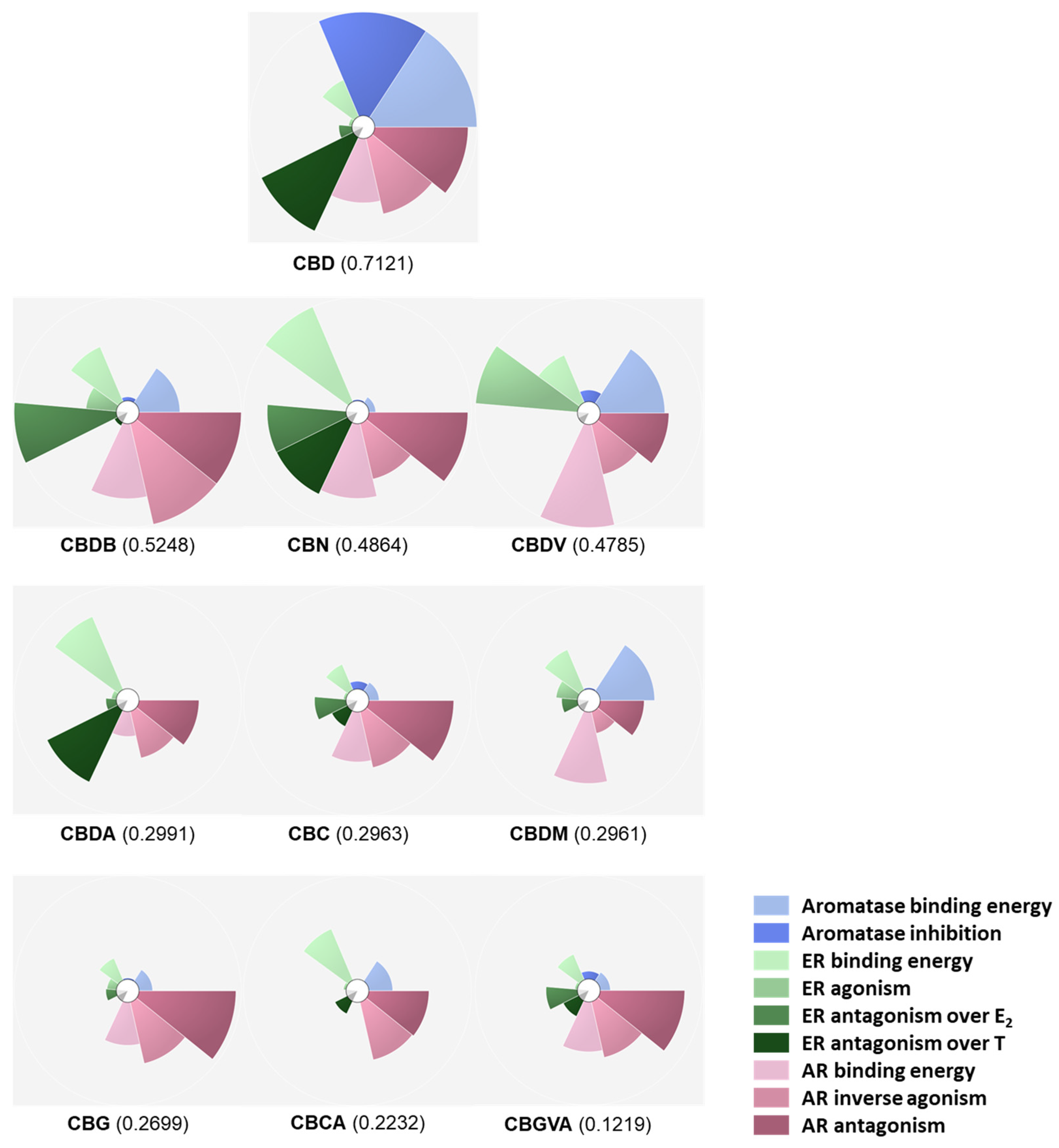
2.5. Scores and Ranking of the Cannabinoids Using ToxPi
3. Discussion
4. Materials and Methods
4.1. Molecular Docking
4.2. Molecular Dynamics Simulation
4.3. Anti-Aromatase Activity
4.4. ER and AR Transactivation Assays
4.5. ToxPi Construction
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Society, A.C. Breast Cancer Facts & Figures 2019–2020; American Cancer Society Inc.: Atlanta, GA, USA, 2019. [Google Scholar]
- Muller, K.; Jorns, J.M.; Tozbikian, G. What’s new in breast pathology 2022: WHO 5th edition and biomarker updates. J. Pathol. Transl. Med. 2022, 56, 170–171. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.S.; Conant, E.F.; Soo, M.S. Molecular Subtypes of Breast Cancer: A Review for Breast Radiologists. J. Breast Imaging 2021, 3, 12–24. [Google Scholar] [CrossRef]
- Ferreira Almeida, C.; Oliveira, A.; João Ramos, M.; Fernandes, P.A.; Teixeira, N.; Amaral, C. Estrogen receptor-positive (ER+) breast cancer treatment: Are multi-target compounds the next promising approach? Biochem. Pharmacol. 2020, 177, 113989. [Google Scholar] [CrossRef]
- Ghosh, D. Aromatase and steroid sulfatase from human placenta. Methods Enzymol. 2023, 689, 67–86. [Google Scholar] [CrossRef]
- Awan, A.; Esfahani, K. Endocrine therapy for breast cancer in the primary care setting. Curr. Oncol. 2018, 25, 285–291. [Google Scholar] [CrossRef]
- Cardoso, F.; Kyriakides, S.; Ohno, S.; Penault-Llorca, F.; Poortmans, P.; Rubio, I.T.; Zackrisson, S.; Senkus, E. Early breast cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up†. Ann. Oncol. 2019, 30, 1194–1220. [Google Scholar] [CrossRef]
- Paluch-Shimon, S.; Cardoso, F.; Partridge, A.H.; Abulkhair, O.; Azim, H.A.; Bianchi-Micheli, G.; Cardoso, M.J.; Curigliano, G.; Gelmon, K.A.; Gentilini, O.; et al. ESO-ESMO fifth international consensus guidelines for breast cancer in young women (BCY5). Ann. Oncol. 2022, 33, 1097–1118. [Google Scholar] [CrossRef]
- Gennari, A.; André, F.; Barrios, C.H.; Cortés, J.; de Azambuja, E.; DeMichele, A.; Dent, R.; Fenlon, D.; Gligorov, J.; Hurvitz, S.A.; et al. ESMO Clinical Practice Guideline for the diagnosis, staging and treatment of patients with metastatic breast cancer. Ann. Oncol. 2021, 32, 1475–1495. [Google Scholar] [CrossRef]
- Cardoso, F.; Paluch-Shimon, S.; Schumacher-Wulf, E.; Matos, L.; Gelmon, K.; Aapro, M.S.; Bajpai, J.; Barrios, C.H.; Bergh, J.; Bergsten-Nordström, E.; et al. 6th and 7th International consensus guidelines for the management of advanced breast cancer (ABC guidelines 6 and 7). Breast 2024, 76, 103756. [Google Scholar] [CrossRef] [PubMed]
- Augusto, T.V.; Correia-da-Silva, G.; Rodrigues, C.M.P.; Teixeira, N.; Amaral, C. Acquired resistance to aromatase inhibitors: Where we stand! Endocr. Relat. Cancer 2018, 25, R283–R301. [Google Scholar] [CrossRef] [PubMed]
- Saatci, O.; Huynh-Dam, K.T.; Sahin, O. Endocrine resistance in breast cancer: From molecular mechanisms to therapeutic strategies. J. Mol. Med. 2021, 99, 1691–1710. [Google Scholar] [CrossRef] [PubMed]
- Olson, E. Combination Therapies in Advanced, Hormone Receptor-Positive Breast Cancer. J. Adv. Pract. Oncol. 2018, 9, 43–54. [Google Scholar] [PubMed]
- Roberto, M.; Astone, A.; Botticelli, A.; Carbognin, L.; Cassano, A.; D’Auria, G.; Fabbri, A.; Fabi, A.; Gamucci, T.; Krasniqi, E.; et al. CDK4/6 Inhibitor Treatments in Patients with Hormone Receptor Positive, Her2 Negative Advanced Breast Cancer: Potential Molecular Mechanisms, Clinical Implications and Future Perspectives. Cancers 2021, 13, 332. [Google Scholar] [CrossRef]
- André, F.; Ciruelos, E.; Rubovszky, G.; Campone, M.; Loibl, S.; Rugo, H.S.; Iwata, H.; Conte, P.; Mayer, I.A.; Kaufman, B.; et al. Alpelisib for PIK3CA-Mutated, Hormone Receptor-Positive Advanced Breast Cancer. N. Engl. J. Med. 2019, 380, 1929–1940. [Google Scholar] [CrossRef]
- Kolyvas, E.A.; Caldas, C.; Kelly, K.; Ahmad, S.S. Androgen receptor function and targeted therapeutics across breast cancer subtypes. Breast Cancer Res. 2022, 24, 79. [Google Scholar] [CrossRef]
- García, X.; Elía, A.; Galizzi, L.; May, M.; Spengler, E.; Martínez Vázquez, P.; Burruchaga, J.; Gass, H.; Lanari, C.; Lamb, C.A. Increased androgen receptor expression in estrogen receptor-positive/progesterone receptor-negative breast cancer. Breast Cancer Res. Treat. 2020, 180, 257–263. [Google Scholar] [CrossRef]
- Michmerhuizen, A.R.; Spratt, D.E.; Pierce, L.J.; Speers, C.W. ARe we there yet? Understanding androgen receptor signaling in breast cancer. NPJ Breast Cancer 2020, 6, 47. [Google Scholar] [CrossRef]
- Hanamura, T.; Hayashi, S.I. Overcoming aromatase inhibitor resistance in breast cancer: Possible mechanisms and clinical applications. Breast Cancer 2018, 25, 379–391. [Google Scholar] [CrossRef]
- Rechoum, Y.; Rovito, D.; Iacopetta, D.; Barone, I.; Andò, S.; Weigel, N.L.; O’Malley, B.W.; Brown, P.H.; Fuqua, S.A. AR collaborates with ERα in aromatase inhibitor-resistant breast cancer. Breast Cancer Res. Treat. 2014, 147, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Basile, D.; Cinausero, M.; Iacono, D.; Pelizzari, G.; Bonotto, M.; Vitale, M.G.; Gerratana, L.; Puglisi, F. Androgen receptor in estrogen receptor positive breast cancer: Beyond expression. Cancer Treat. Rev. 2017, 61, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Amaral, C.; Augusto, T.V.; Almada, M.; Cunha, S.C.; Correia-da-Silva, G.; Teixeira, N. The potential clinical benefit of targeting androgen receptor (AR) in estrogen-receptor positive breast cancer cells treated with Exemestane. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165661. [Google Scholar] [CrossRef] [PubMed]
- Augusto, T.V.; Cunha, S.C.; Amaral, C.; Fernandes, J.O.; da Silva, E.T.; Roleira, F.F.M.; Teixeira, N.; Correia-da-Silva, G. A novel GC-MS methodology to evaluate aromatase activity in human placental microsomes: A comparative study with the standard radiometric assay. Anal. Bioanal. Chem. 2019, 411, 7005–7013. [Google Scholar] [CrossRef]
- Roleira, F.M.F.; Varela, C.; Amaral, C.; Costa, S.C.; Correia-da-Silva, G.; Moraca, F.; Costa, G.; Alcaro, S.; Teixeira, N.A.A.; Tavares da Silva, E.J. C-6α- vs. C-7α-Substituted Steroidal Aromatase Inhibitors: Which Is Better? Synthesis, Biochemical Evaluation, Docking Studies, and Structure-Activity Relationships. J. Med. Chem. 2019, 62, 3636–3657. [Google Scholar] [CrossRef]
- Klumpers, L.E.; Thacker, D.L. A Brief Background on Cannabis: From Plant to Medical Indications. J. AOAC Int. 2019, 102, 412–420. [Google Scholar] [CrossRef]
- Alves, P.; Amaral, C.; Teixeira, N.; Correia-da-Silva, G. Cannabis sativa: Much more beyond Δ9-tetrahydrocannabinol. Pharmacol. Res. 2020, 157, 104822. [Google Scholar] [CrossRef]
- Pertwee, R.G. Cannabinoid pharmacology: The first 66 years. Br. J. Pharmacol. 2006, 147 (Suppl. 1), S163–S171. [Google Scholar] [CrossRef]
- Grimaldi, C.; Capasso, A. The endocannabinoid system in the cancer therapy: An overview. Curr. Med. Chem. 2011, 18, 1575–1583. [Google Scholar] [CrossRef]
- Velasco, G.; Sánchez, C.; Guzmán, M. Towards the use of cannabinoids as antitumour agents. Nat. Rev. Cancer 2012, 12, 436–444. [Google Scholar] [CrossRef]
- Caffarel, M.M.; Andradas, C.; Pérez-Gómez, E.; Guzmán, M.; Sánchez, C. Cannabinoids: A new hope for breast cancer therapy? Cancer Treat. Rev. 2012, 38, 911–918. [Google Scholar] [CrossRef] [PubMed]
- Almeida, C.F.; Teixeira, N.; Correia-da-Silva, G.; Amaral, C. Cannabinoids in Breast Cancer: Differential Susceptibility According to Subtype. Molecules 2021, 27, 156. [Google Scholar] [CrossRef] [PubMed]
- Velasco, G.; Sánchez, C.; Guzmán, M. Anticancer mechanisms of cannabinoids. Curr. Oncol. 2016, 23, S23–S32. [Google Scholar] [CrossRef]
- Hinz, B.; Ramer, R. Anti-tumour actions of cannabinoids. Br. J. Pharmacol. 2019, 176, 1384–1394. [Google Scholar] [CrossRef]
- Sledzinski, P.; Zeyland, J.; Slomski, R.; Nowak, A. The current state and future perspectives of cannabinoids in cancer biology. Cancer Med. 2018, 7, 765–775. [Google Scholar] [CrossRef] [PubMed]
- Fraguas-Sánchez, A.I.; Martín-Sabroso, C.; Torres-Suárez, A.I. Insights into the effects of the endocannabinoid system in cancer: A review. Br. J. Pharmacol. 2018, 175, 2566–2580. [Google Scholar] [CrossRef]
- Seltzer, E.S.; Watters, A.K.; MacKenzie, D., Jr.; Granat, L.M.; Zhang, D. Cannabidiol (CBD) as a Promising Anti-Cancer Drug. Cancers 2020, 12, 3203. [Google Scholar] [CrossRef]
- Munson, A.E.; Harris, L.S.; Friedman, M.A.; Dewey, W.L.; Carchman, R.A. Antineoplastic activity of cannabinoids. J. Natl. Cancer Inst. 1975, 55, 597–602. [Google Scholar] [CrossRef]
- Bifulco, M.; Laezza, C.; Pisanti, S.; Gazzerro, P. Cannabinoids and cancer: Pros and cons of an antitumour strategy. Br. J. Pharmacol. 2006, 148, 123–135. [Google Scholar] [CrossRef]
- Ramer, R.; Hinz, B. Cannabinoids as Anticancer Drugs. Adv. Pharmacol. 2017, 80, 397–436. [Google Scholar] [CrossRef]
- Amaral, C.; Trouille, F.M.; Almeida, C.F.; Correia-da-Silva, G.; Teixeira, N. Unveiling the mechanism of action behind the anti-cancer properties of cannabinoids in ER+ breast cancer cells: Impact on aromatase and steroid receptors. J. Steroid Biochem. Mol. Biol. 2021, 210, 105876. [Google Scholar] [CrossRef] [PubMed]
- Mokoena, D.; George, B.P.; Abrahamse, H. Cannabidiol Combination Enhances Photodynamic Therapy Effects on MCF-7 Breast Cancer Cells. Cells 2024, 13, 187. [Google Scholar] [CrossRef] [PubMed]
- Almeida, C.F.; Teixeira, N.; Valente, M.J.; Vinggaard, A.M.; Correia-da-Silva, G.; Amaral, C. Cannabidiol as a Promising Adjuvant Therapy for Estrogen Receptor-Positive Breast Tumors: Unveiling Its Benefits with Aromatase Inhibitors. Cancers 2023, 15, 2517. [Google Scholar] [CrossRef]
- Schoeman, R.; Beukes, N.; Frost, C. Cannabinoid Combination Induces Cytoplasmic Vacuolation in MCF-7 Breast Cancer Cells. Molecules 2020, 25, 4682. [Google Scholar] [CrossRef]
- Shrivastava, A.; Kuzontkoski, P.M.; Groopman, J.E.; Prasad, A. Cannabidiol induces programmed cell death in breast cancer cells by coordinating the cross-talk between apoptosis and autophagy. Mol. Cancer Ther. 2011, 10, 1161–1172. [Google Scholar] [CrossRef]
- Sultan, A.S.; Marie, M.A.; Sheweita, S.A. Novel mechanism of cannabidiol-induced apoptosis in breast cancer cell lines. Breast 2018, 41, 34–41. [Google Scholar] [CrossRef]
- Suttithumsatid, W.; Sukketsiri, W.; Panichayupakaranant, P. Cannabinoids and standardized cannabis extracts inhibit migration, invasion, and induce apoptosis in MCF-7 cells through FAK/MAPK/Akt/NF-κB signaling. Toxicol. In Vitro 2023, 93, 105667. [Google Scholar] [CrossRef] [PubMed]
- Pino, S.; Espinoza, L.; Jara-Gutiérrez, C.; Villena, J.; Olea, A.F.; Díaz, K. Study of Cannabis Oils Obtained from Three Varieties of C. sativa and by Two Different Extraction Methods: Phytochemical Characterization and Biological Activities. Plants 2023, 12, 1772. [Google Scholar] [CrossRef]
- Schoeman, R.; de la Harpe, A.; Beukes, N.; Frost, C.L. Cannabis with breast cancer treatment: Propitious or pernicious? 3 Biotech. 2022, 12, 54. [Google Scholar] [CrossRef]
- García-Morales, L.; Mendoza-Rodríguez, M.G.; Tapia Ramírez, J.; Meza, I. CBD Inhibits In Vivo Development of Human Breast Cancer Tumors. Int. J. Mol. Sci. 2023, 24, 13235. [Google Scholar] [CrossRef]
- Oliveira, H.A.; Somvanshi, R.K.; Kumar, U. Comparative changes in breast cancer cell proliferation and signalling following somatostatin and cannabidiol treatment. Biochem. Biophys. Res. Commun. 2023, 643, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Alsherbiny, M.A.; Bhuyan, D.J.; Low, M.N.; Chang, D.; Li, C.G. Synergistic Interactions of Cannabidiol with Chemotherapeutic Drugs in MCF7 Cells: Mode of Interaction and Proteomics Analysis of Mechanisms. Int. J. Mol. Sci. 2021, 22, 10103. [Google Scholar] [CrossRef] [PubMed]
- García-Morales, L.; Castillo, A.M.; Tapia Ramírez, J.; Zamudio-Meza, H.; Domínguez-Robles, M.D.C.; Meza, I. CBD Reverts the Mesenchymal Invasive Phenotype of Breast Cancer Cells Induced by the Inflammatory Cytokine IL-1β. Int. J. Mol. Sci. 2020, 21, 2429. [Google Scholar] [CrossRef]
- Takeda, S.; Yoshida, K.; Nishimura, H.; Harada, M.; Okajima, S.; Miyoshi, H.; Okamoto, Y.; Amamoto, T.; Watanabe, K.; Omiecinski, C.J.; et al. Δ9-Tetrahydrocannabinol disrupts estrogen-signaling through up-regulation of estrogen receptor β (ERβ). Chem. Res. Toxicol. 2013, 26, 1073–1079. [Google Scholar] [CrossRef]
- Takeda, S.; Yamaori, S.; Motoya, E.; Matsunaga, T.; Kimura, T.; Yamamoto, I.; Watanabe, K. Δ9-Tetrahydrocannabinol enhances MCF-7 cell proliferation via cannabinoid receptor-independent signaling. Toxicology 2008, 245, 141–146. [Google Scholar] [CrossRef]
- Takeda, S.; Yamamoto, I.; Watanabe, K. Modulation of Δ9-tetrahydrocannabinol-induced MCF-7 breast cancer cell growth by cyclooxygenase and aromatase. Toxicology 2009, 259, 25–32. [Google Scholar] [CrossRef]
- Morin-Buote, J.; Ennour-Idrissi, K.; Poirier, É.; Lemieux, J.; Furrer, D.; Burguin, A.; Durocher, F.; Diorio, C. Association of Breast Tumour Expression of Cannabinoid Receptors CBR1 and CBR2 with Prognostic Factors and Survival in Breast Cancer Patients. J. Pers. Med. 2021, 11, 852. [Google Scholar] [CrossRef] [PubMed]
- Kisková, T.; Mungenast, F.; Suváková, M.; Jäger, W.; Thalhammer, T. Future Aspects for Cannabinoids in Breast Cancer Therapy. Int. J. Mol. Sci. 2019, 20, 1673. [Google Scholar] [CrossRef]
- Dobovišek, L.; Krstanović, F.; Borštnar, S.; Debeljak, N. Cannabinoids and Hormone Receptor-Positive Breast Cancer Treatment. Cancers 2020, 12, 525. [Google Scholar] [CrossRef]
- Dobovišek, L.; Hojnik, M.; Ferk, P. Overlapping molecular pathways between cannabinoid receptors type 1 and 2 and estrogens/androgens on the periphery and their involvement in the pathogenesis of common diseases (Review). Int. J. Mol. Med. 2016, 38, 1642–1651. [Google Scholar] [CrossRef]
- Ruh, M.F.; Taylor, J.A.; Howlett, A.C.; Welshons, W.V. Failure of cannabinoid compounds to stimulate estrogen receptors. Biochem. Pharmacol. 1997, 53, 35–41. [Google Scholar] [CrossRef] [PubMed]
- von Bueren, A.O.; Schlumpf, M.; Lichtensteiger, W. Δ9-Tetrahydrocannabinol inhibits 17beta-estradiol-induced proliferation and fails to activate androgen and estrogen receptors in MCF7 human breast cancer cells. Anticancer. Res. 2008, 28, 85–89. [Google Scholar] [PubMed]
- Almada, M.; Oliveira, A.; Amaral, C.; Fernandes, P.A.; Ramos, M.J.; Fonseca, B.; Correia-da-Silva, G.; Teixeira, N. Anandamide targets aromatase: A breakthrough on human decidualization. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 158512. [Google Scholar] [CrossRef]
- Almada, M.; Amaral, C.; Oliveira, A.; Fernandes, P.A.; Ramos, M.J.; Fonseca, B.M.; Correia-da-Silva, G.; Teixeira, N. Cannabidiol (CBD) but not tetrahydrocannabinol (THC) dysregulate in vitro decidualization of human endometrial stromal cells by disruption of estrogen signaling. Reprod. Toxicol. 2020, 93, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Pertwee, R.G. Handbook of Cannabis; Oxford University Press: Oxford, UK, 2016. [Google Scholar]
- Appendino, G.; Chianese, G.; Taglialatela-Scafati, O. Cannabinoids: Occurrence and medicinal chemistry. Curr. Med. Chem. 2011, 18, 1085–1099. [Google Scholar] [CrossRef]
- Gould, J. The cannabis crop. Nature 2015, 525, S2–S3. [Google Scholar] [CrossRef]
- Procaccia, S.; Lewitus, G.M.; Lipson Feder, C.; Shapira, A.; Berman, P.; Meiri, D. Cannabis for Medical Use: Versatile Plant Rather Than a Single Drug. Front. Pharmacol. 2022, 13, 894960. [Google Scholar] [CrossRef]
- Sampson, P.B. Phytocannabinoid Pharmacology: Medicinal Properties of Cannabis sativa Constituents Aside from the “Big Two”. J. Nat. Prod. 2021, 84, 142–160. [Google Scholar] [CrossRef] [PubMed]
- Welling, M.T.; Deseo, M.A.; Bacic, A.; Doblin, M.S. Biosynthetic origins of unusual cannabimimetic phytocannabinoids in Cannabis sativa L: A review. Phytochemistry 2022, 201, 113282. [Google Scholar] [CrossRef]
- Nachnani, R.; Raup-Konsavage, W.M.; Vrana, K.E. The Pharmacological Case for Cannabigerol. J. Pharmacol. Exp. Ther. 2021, 376, 204–212. [Google Scholar] [CrossRef]
- Lah, T.T.; Novak, M.; Pena Almidon, M.A.; Marinelli, O.; Žvar Baškovič, B.; Majc, B.; Mlinar, M.; Bošnjak, R.; Breznik, B.; Zomer, R.; et al. Cannabigerol Is a Potential Therapeutic Agent in a Novel Combined Therapy for Glioblastoma. Cells 2021, 10, 340. [Google Scholar] [CrossRef] [PubMed]
- Lah, T.T.; Majc, B.; Novak, M.; Sušnik, A.; Breznik, B.; Porčnik, A.; Bošnjak, R.; Sadikov, A.; Malavolta, M.; Halilčević, S.; et al. The Cytotoxic Effects of Cannabidiol and Cannabigerol on Glioblastoma Stem Cells May Mostly Involve GPR55 and TRPV1 Signalling. Cancers 2022, 14, 5918. [Google Scholar] [CrossRef] [PubMed]
- Ligresti, A.; Moriello, A.S.; Starowicz, K.; Matias, I.; Pisanti, S.; De Petrocellis, L.; Laezza, C.; Portella, G.; Bifulco, M.; Di Marzo, V. Antitumor activity of plant cannabinoids with emphasis on the effect of cannabidiol on human breast carcinoma. J. Pharmacol. Exp. Ther. 2006, 318, 1375–1387. [Google Scholar] [CrossRef] [PubMed]
- McAllister, S.D.; Christian, R.T.; Horowitz, M.P.; Garcia, A.; Desprez, P.Y. Cannabidiol as a novel inhibitor of Id-1 gene expression in aggressive breast cancer cells. Mol. Cancer Ther. 2007, 6, 2921–2927. [Google Scholar] [CrossRef]
- Silva-Reis, R.; Silva, A.M.S.; Oliveira, P.A.; Cardoso, S.M. Antitumor Effects of Cannabis sativa Bioactive Compounds on Colorectal Carcinogenesis. Biomolecules 2023, 13, 764. [Google Scholar] [CrossRef]
- Anis, O.; Vinayaka, A.C.; Shalev, N.; Namdar, D.; Nadarajan, S.; Anil, S.M.; Cohen, O.; Belausov, E.; Ramon, J.; Mayzlish Gati, E.; et al. Cannabis-Derived Compounds Cannabichromene and Δ9-Tetrahydrocannabinol Interact and Exhibit Cytotoxic Activity against Urothelial Cell Carcinoma Correlated with Inhibition of Cell Migration and Cytoskeleton Organization. Molecules 2021, 26, 465. [Google Scholar] [CrossRef]
- Nahler, G. Treatment of malignant diseases with phytocannabinoids: Promising observations in animal models and patients. Explor. Med. 2023, 4, 847–877. [Google Scholar] [CrossRef]
- Pagano, C.; Navarra, G.; Coppola, L.; Bifulco, M.; Laezza, C. Molecular Mechanism of Cannabinoids in Cancer Progression. Int. J. Mol. Sci. 2021, 22, 3680. [Google Scholar] [CrossRef]
- Salbini, M.; Quarta, A.; Russo, F.; Giudetti, A.M.; Citti, C.; Cannazza, G.; Gigli, G.; Vergara, D.; Gaballo, A. Oxidative Stress and Multi-Organel Damage Induced by Two Novel Phytocannabinoids, CBDB and CBDP, in Breast Cancer Cells. Molecules 2021, 26, 5576. [Google Scholar] [CrossRef]
- Chan, H.J.; Petrossian, K.; Chen, S. Structural and functional characterization of aromatase, estrogen receptor, and their genes in endocrine-responsive and -resistant breast cancer cells. J. Steroid Biochem. Mol. Biol. 2016, 161, 73–83. [Google Scholar] [CrossRef]
- Hong, Y.; Rashid, R.; Chen, S. Binding features of steroidal and nonsteroidal inhibitors. Steroids 2011, 76, 802–806. [Google Scholar] [CrossRef] [PubMed]
- Di Nardo, G.; Breitner, M.; Bandino, A.; Ghosh, D.; Jennings, G.K.; Hackett, J.C.; Gilardi, G. Evidence for an elevated aspartate pKa in the active site of human aromatase. J. Biol. Chem. 2015, 290, 1186–1196. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Czapla, L.; Amaro, R.E. Molecular simulations of aromatase reveal new insights into the mechanism of ligand binding. J. Chem. Inf. Model. 2013, 53, 2047–2056. [Google Scholar] [CrossRef] [PubMed]
- Cochrane, D.R.; Bernales, S.; Jacobsen, B.M.; Cittelly, D.M.; Howe, E.N.; D’Amato, N.C.; Spoelstra, N.S.; Edgerton, S.M.; Jean, A.; Guerrero, J.; et al. Role of the androgen receptor in breast cancer and preclinical analysis of enzalutamide. Breast Cancer Res. 2014, 16, R7. [Google Scholar] [CrossRef] [PubMed]
- Caswell-Jin, J.L.; Curtis, C. Androgen receptor agonists as breast cancer therapeutics. Nat. Med. 2021, 27, 198–199. [Google Scholar] [CrossRef]
- Li, D.; Zhou, W.; Pang, J.; Tang, Q.; Zhong, B.; Shen, C.; Xiao, L.; Hou, T. A magic drug target: Androgen receptor. Med. Res. Rev. 2019, 39, 1485–1514. [Google Scholar] [CrossRef]
- Liu, N.; Zhou, W.; Guo, Y.; Wang, J.; Fu, W.; Sun, H.; Li, D.; Duan, M.; Hou, T. Molecular Dynamics Simulations Revealed the Regulation of Ligands to the Interactions between Androgen Receptor and Its Coactivator. J. Chem. Inf. Model. 2018, 58, 1652–1661. [Google Scholar] [CrossRef]
- Sakkiah, S.; Kusko, R.; Pan, B.; Guo, W.; Ge, W.; Tong, W.; Hong, H. Structural Changes Due to Antagonist Binding in Ligand Binding Pocket of Androgen Receptor Elucidated Through Molecular Dynamics Simulations. Front. Pharmacol. 2018, 9, 492. [Google Scholar] [CrossRef]
- Almeida, C.F.; Teixeira, N.; Oliveira, A.; Augusto, T.V.; Correia-da-Silva, G.; Ramos, M.J.; Fernandes, P.A.; Amaral, C. Discovery of a multi-target compound for estrogen receptor-positive (ER+) breast cancer: Involvement of aromatase and ERs. Biochimie 2021, 181, 65–76. [Google Scholar] [CrossRef]
- Varela, C.L.; Amaral, C.; Correia-da-Silva, G.; Costa, S.C.; Carvalho, R.A.; Costa, G.; Alcaro, S.; Teixeira, N.A.; Tavares-da-Silva, E.J.; Roleira, F.M. Exploring new chemical functionalities to improve aromatase inhibition of steroids. Bioorg. Med. Chem. 2016, 24, 2823–2831. [Google Scholar] [CrossRef]
- Roleira, F.M.F.; Costa, S.C.; Gomes, A.R.; Varela, C.L.; Amaral, C.; Augusto, T.V.; Correia-da-Silva, G.; Romeo, I.; Costa, G.; Alcaro, S.; et al. Design, synthesis, biological activity evaluation and structure-activity relationships of new steroidal aromatase inhibitors. The case of C-ring and 7β substituted steroids. Bioorg. Chem. 2023, 131, 106286. [Google Scholar] [CrossRef] [PubMed]
- Rasha, F.; Sharma, M.; Pruitt, K. Mechanisms of endocrine therapy resistance in breast cancer. Mol. Cell. Endocrinol. 2021, 532, 111322. [Google Scholar] [CrossRef]
- Portman, N.; Alexandrou, S.; Carson, E.; Wang, S.; Lim, E.; Caldon, C.E. Overcoming CDK4/6 inhibitor resistance in ER-positive breast cancer. Endocr. Relat. Cancer 2019, 26, R15–R30. [Google Scholar] [CrossRef]
- Papadimitriou, M.C.; Pazaiti, A.; Iliakopoulos, K.; Markouli, M.; Michalaki, V.; Papadimitriou, C.A. Resistance to CDK4/6 inhibition: Mechanisms and strategies to overcome a therapeutic problem in the treatment of hormone receptor-positive metastatic breast cancer. Biochim. Biophys. Acta Mol. Cell Res. 2022, 1869, 119346. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Yu, B.; Sherman, M.; Yuan, Y.C.; Zhou, D.; Chen, S. Molecular basis for the aromatization reaction and exemestane-mediated irreversible inhibition of human aromatase. Mol. Endocrinol. 2007, 21, 401–414. [Google Scholar] [CrossRef]
- Suvannang, N.; Nantasenamat, C.; Isarankura-Na-Ayudhya, C.; Prachayasittikul, V. Molecular Docking of Aromatase Inhibitors. Molecules 2011, 16, 3597–3617. [Google Scholar] [CrossRef]
- Pavlin, M.; Spinello, A.; Pennati, M.; Zaffaroni, N.; Gobbi, S.; Bisi, A.; Colombo, G.; Magistrato, A. A Computational Assay of Estrogen Receptor alpha Antagonists Reveals the Key Common Structural Traits of Drugs Effectively Fighting Refractory Breast Cancers. Sci. Rep. 2018, 8, 649. [Google Scholar] [CrossRef]
- Amaral, C.; Correia-da-Silva, G.; Almeida, C.F.; Valente, M.J.; Varela, C.; Tavares-da-Silva, E.; Vinggaard, A.M.; Teixeira, N.; Roleira, F.M.F. An Exemestane Derivative, Oxymestane-D1, as a New Multi-Target Steroidal Aromatase Inhibitor for Estrogen Receptor-Positive (ER+) Breast Cancer: Effects on Sensitive and Resistant Cell Lines. Molecules 2023, 28, 789. [Google Scholar] [CrossRef]
- Kucuksayan, E.; Ozben, T. Hybrid Compounds as Multitarget Directed Anticancer Agents. Curr. Top. Med. Chem. 2017, 17, 907–918. [Google Scholar] [CrossRef]
- Li, X.; Wu, C.; Lin, X.; Cai, X.; Liu, L.; Luo, G.; You, Q.; Xiang, H. Synthesis and biological evaluation of 3-aryl-quinolin derivatives as anti-breast cancer agents targeting ERα and VEGFR-2. Eur. J. Med. Chem. 2019, 161, 445–455. [Google Scholar] [CrossRef]
- Petrelli, A.; Giordano, S. From single- to multi-target drugs in cancer therapy: When aspecificity becomes an advantage. Curr. Med. Chem. 2008, 15, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Amaral, C.; Varela, C.L.; Mauricio, J.; Sobral, A.F.; Costa, S.C.; Roleira, F.M.F.; Tavares-da-Silva, E.J.; Correia-da-Silva, G.; Teixeira, N. Anti-tumor efficacy of new 7α-substituted androstanes as aromatase inhibitors in hormone-sensitive and resistant breast cancer cells. J. Steroid Biochem. Mol. Biol. 2017, 171, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Berg, K.A.; Clarke, W.P. Making Sense of Pharmacology: Inverse Agonism and Functional Selectivity. Int. J. Neuropsychopharmacol. 2018, 21, 962–977. [Google Scholar] [CrossRef]
- Paterni, I.; Granchi, C.; Katzenellenbogen, J.A.; Minutolo, F. Estrogen receptors alpha (ERα) and beta (ERβ): Subtype-selective ligands and clinical potential. Steroids 2014, 90, 13–29. [Google Scholar] [CrossRef]
- Li, J.; Yu, J.; Zou, H.; Zhang, J.; Ren, L. Estrogen receptor-mediated health benefits of phytochemicals: A review. Food Funct. 2023, 14, 10681–10699. [Google Scholar] [CrossRef] [PubMed]
- Martinkovich, S.; Shah, D.; Planey, S.L.; Arnott, J.A. Selective estrogen receptor modulators: Tissue specificity and clinical utility. Clin. Interv. Aging 2014, 9, 1437–1452. [Google Scholar] [CrossRef] [PubMed]
- Kono, M.; Fujii, T.; Lim, B.; Karuturi, M.S.; Tripathy, D.; Ueno, N.T. Androgen Receptor Function and Androgen Receptor-Targeted Therapies in Breast Cancer: A Review. JAMA Oncol. 2017, 3, 1266–1273. [Google Scholar] [CrossRef] [PubMed]
- Purohit, V.; Ahluwahlia, B.S.; Vigersky, R.A. Marihuana inhibits dihydrotestosterone binding to the androgen receptor. Endocrinology 1980, 107, 848–850. [Google Scholar] [CrossRef]
- Mobisson, S.K.; Ikpi, D.E.; Wopara, I.; Obembe, A.O.; Omotuyi, O. Inhibition of human androgen receptor by Δ9-tetrahydro-cannabinol and cannabidiol related to reproductive dysfunction: A computational study. Andrologia 2022, 54, e14454. [Google Scholar] [CrossRef]
- Bleach, R.; McIlroy, M. The Divergent Function of Androgen Receptor in Breast Cancer; Analysis of Steroid Mediators and Tumor Intracrinology. Front. Endocrinol. 2018, 9, 594. [Google Scholar] [CrossRef]
- Krop, I.; Abramson, V.; Colleoni, M.; Traina, T.; Holmes, F.; Garcia-Estevez, L.; Hart, L.; Awada, A.; Zamagni, C.; Morris, P.G.; et al. A Randomized Placebo Controlled Phase II Trial Evaluating Exemestane with or without Enzalutamide in Patients with Hormone Receptor-Positive Breast Cancer. Clin. Cancer Res. 2020, 26, 6149–6157. [Google Scholar] [CrossRef] [PubMed]
- Schwartzberg, L.S.; Yardley, D.A.; Elias, A.D.; Patel, M.; LoRusso, P.; Burris, H.A.; Gucalp, A.; Peterson, A.C.; Blaney, M.E.; Steinberg, J.L.; et al. A Phase I/Ib Study of Enzalutamide Alone and in Combination with Endocrine Therapies in Women with Advanced Breast Cancer. Clin. Cancer Res. 2017, 23, 4046–4054. [Google Scholar] [CrossRef] [PubMed]
- Madersbacher, S.; Sampson, N.; Culig, Z. Pathophysiology of Benign Prostatic Hyperplasia and Benign Prostatic Enlargement: A Mini-Review. Gerontology 2019, 65, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Vickman, R.E.; Franco, O.E.; Moline, D.C.; Vander Griend, D.J.; Thumbikat, P.; Hayward, S.W. The role of the androgen receptor in prostate development and benign prostatic hyperplasia: A review. Asian J. Urol. 2020, 7, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Gerratana, L.; Basile, D.; Buono, G.; De Placido, S.; Giuliano, M.; Minichillo, S.; Coinu, A.; Martorana, F.; De Santo, I.; Del Mastro, L.; et al. Androgen receptor in triple negative breast cancer: A potential target for the targetless subtype. Cancer Treat. Rev. 2018, 68, 102–110. [Google Scholar] [CrossRef]
- Traina, T.A.; Miller, K.; Yardley, D.A.; Eakle, J.; Schwartzberg, L.S.; O’Shaughnessy, J.; Gradishar, W.; Schmid, P.; Winer, E.; Kelly, C.; et al. Enzalutamide for the Treatment of Androgen Receptor-Expressing Triple-Negative Breast Cancer. J. Clin. Oncol. 2018, 36, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Gucalp, A.; Tolaney, S.; Isakoff, S.J.; Ingle, J.N.; Liu, M.C.; Carey, L.A.; Blackwell, K.; Rugo, H.; Nabell, L.; Forero, A.; et al. Phase II trial of bicalutamide in patients with androgen receptor-positive, estrogen receptor-negative metastatic Breast Cancer. Clin. Cancer Res. 2013, 19, 5505–5512. [Google Scholar] [CrossRef]
- Traina, T.A.; Miller, K.; Yardley, D.A.; O’Shaughnessy, J.; Cortes, J.; Awada, A.; Kelly, C.M.; Trudeau, M.E.; Schmid, P.; Gianni, L.; et al. Results from a phase 2 study of enzalutamide (ENZA), an androgen receptor (AR) inhibitor, in advanced AR+ triple-negative breast cancer (TNBC). J. Clin. Oncol. 2015, 33 (Supp. 15), 1003. [Google Scholar] [CrossRef]
- Ghosh, D.; Lo, J.; Morton, D.; Valette, D.; Xi, J.; Griswold, J.; Hubbell, S.; Egbuta, C.; Jiang, W.; An, J.; et al. Novel aromatase inhibitors by structure-guided design. J. Med. Chem. 2012, 55, 8464–8476. [Google Scholar] [CrossRef]
- Pereira de Jésus-Tran, K.; Côté, P.L.; Cantin, L.; Blanchet, J.; Labrie, F.; Breton, R. Comparison of crystal structures of human androgen receptor ligand-binding domain complexed with various agonists reveals molecular determinants responsible for binding affinity. Protein Sci. 2006, 15, 987–999. [Google Scholar] [CrossRef]
- Spinello, A.; Pavlin, M.; Casalino, L.; Magistrato, A. A Dehydrogenase Dual Hydrogen Abstraction Mechanism Promotes Estrogen Biosynthesis: Can We Expand the Functional Annotation of the Aromatase Enzyme? Chemistry 2018, 24, 10840–10849. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B.; et al. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, D1100–D1107. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Dallakyan, S.; Olson, A.J. Small-molecule library screening by docking with PyRx. Methods Mol. Biol. 2015, 1263, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Thompson, E.A., Jr.; Siiteri, P.K. The involvement of human placental microsomal cytochrome P-450 in aromatization. J. Biol. Chem. 1974, 249, 5373–5378. [Google Scholar] [CrossRef]
- Heldring, N.; Pawson, T.; McDonnell, D.; Treuter, E.; Gustafsson, J.A.; Pike, A.C. Structural insights into corepressor recognition by antagonist-bound estrogen receptors. J. Biol. Chem. 2007, 282, 10449–10455. [Google Scholar] [CrossRef]
- EDSP-USEP Agency. Aromatase Assay (Human Recombinant) OCSPP Guideline 890.1200; U.S. Environmental Protection Agency: Washington, DC, USA, 2011.
- Reif, D.M.; Sypa, M.; Lock, E.F.; Wright, F.A.; Wilson, A.; Cathey, T.; Judson, R.R.; Rusyn, I. ToxPi GUI: An interactive visualization tool for transparent integration of data from diverse sources of evidence. Bioinformatics 2013, 29, 402–403. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cannabinoid Name | Cannabinoid Class | Binding Affinity (kcal/mol) | Relevant Interactions |
|---|---|---|---|
| 8-Oxo-(-)-Δ9-trans-tetrahydrocannabinol | Δ9-THC | −9.2 | Hydrogen bonds with Arg115 and Leu477; hydrophobic interactions with Trp224 and Phe221 |
| 11-Acetoxy-(-)-Δ9-trans-tetrahydrocannabinolic acid A | Δ9-THC | −8.3 | Hydrogen bonds with Arg115, Met374, Asp309, and Leu477; hydrophobic interactions with Phe221 |
| (-)-Δ9-trans-Tetrahydrocannabivarinic acid | Δ9-THC | −7.1 | Hydrogen bonds with the heme group, Leu372, and Leu477; hydrophobic interactions with Trp224 and Phe221 |
| (-)-Δ9-trans-Tetrahydrocannabinolic acid A-C4 | Δ9-THC | −6.7 | Hydrogen bonds with the heme group, Leu372, Met374, and Leu477; hydrophobic interactions with Trp224 and Phe221 |
| (-)-Δ9-trans-Tetrahydrocannabinolic acid B | Δ9-THC | −6.0 | Hydrogen bonds with Asp309 and Leu477; hydrophobic interactions with Trp224 and Phe221 |
| Δ8-trans-Tetrahydrocannabinolic acid A | Δ9-THC | −4.4 | Hydrogen bond with Asp309; hydrophobic interactions with Phe221 |
| (-)-Δ9-trans-Tetrahydrocannabinal | Δ9-THC | −3.8 | Hydrogen bond with Asp309; hydrophobic interactions with Trp224 and Phe221 |
| Cannabidiol (CBD) | Cannabidiol | −8.7 | Hydrogen bond with Asp309; hydrophobic interactions with Phe221 |
| Cannabidiorcol | Cannabidiol | −7.8 | Hydrogen bonds with Asp309 and Leu477; hydrophobic interactions with Phe221 |
| (-)-Cannabidivarin (CBDV) | Cannabidiol | −7.6 | Hydrogen bonds with Asp309 and Leu477; hydrophobic interactions with Phe221 |
| Cannabidiol monomethylether (CBDM) | Cannabidiol | −7.3 | Hydrogen bonds with Asp309 and Ser478; hydrophobic interactions with Phe221 |
| Cannabidiol-C4 (CBDB) | Cannabidiol | −6.9 | Hydrogen bonds with Asp309 and Leu477; hydrophobic interactions with Phe221 |
| Cannabidivarinic acid | Cannabidiol | −6.6 | Hydrogen bonds with Asp309 and Leu477; hydrophobic interactions with Phe221 |
| Cannabidiolic acid (CBDA) | Cannabidiol | −5.7 | Hydrogen bonds with Asp309 and Leu477; hydrophobic interactions with Phe221 |
| Cannabigerol (CBG) | Cannabigerol | −6.1 | Hydrophobic interactions with Phe221 and Trp224 |
| Cannabinerolic acid | Cannabigerol | −6.7 | Hydrogen bond with Asp309; hydrophobic interactions with Trp224 and Phe221 |
| Camagerol | Cannabigerol | −6.6 | Hydrogen bonds with Asp309, Arg115, and Met374 |
| (-)-6,7-cis-Epoxycannabigerolic acid | Cannabigerol | −6.3 | Hydrogen bonds with Asp309, Leu477, and Arg115; hydrophobic interactions with Trp224 |
| Cannabigerovarinic acid (CBGVA) | Cannabigerol | −5.8 | Hydrogen bond with Asp309 |
| 5-Acetyl-4-hydroxy cannabigerol | Cannabigerol | −5.1 | Hydrogen bond with Asp309; hydrophobic interactions with Phe221 |
| (-)-6,7-trans-Epoxycannabigerolic acid | Cannabigerol | −5.4 | Hydrogen bonds with Asp309 and Leu477; hydrophobic interactions with Trp224 |
| Cannabielsoic acid B-C3 | Cannabielsoin | −6.0 | Hydrogen bonds with the heme group and Asp309 |
| Cannabielsoin acid A | Cannabielsoin | −5.9 | Hydrogen bonds with Asp309 and Arg115 |
| Cannabielsoin acid B | Cannabielsoin | −5.1 | Hydrogen bond with Asp309; hydrophobic interactions with Trp224 and Phe221 |
| Cannabicyclol | Cannabicyclol | −9.8 | Hydrogen bond with Asp309; hydrophobic interactions with Phe221 |
| Cannabicyclovarin | Cannabicyclol | −9.3 | Hydrogen bond with Asp309; hydrophobic interactions with Phe221 |
| Cannabicyclolic acid | Cannabicyclol | −5.5 | Hydrogen bond with Leu477 |
| Cannabichromevarin | Cannabichromene | −7.1 | Hydrogen bond with Asp309 |
| Cannabichromenic acid (CBCA) | Cannabichromene | −6.4 | Hydrogen bond with Asp309; hydrophobic interactions with Trp224 |
| Cannabichromene (CBC) | Cannabichromene | −6.0 | Hydrogen bond with Asp309; hydrophobic interactions with Phe221 |
| (-)-7-Hydroxycannabichromene | Cannabichromene | −5.5 | Hydrogen bonds with the heme group and Asp309; hydrophobic interactions with Phe221 |
| Cannabichromevarinic acid | Cannabichromene | −5.2 | Hydrogen bond with Arg115 |
| Cannabinol (CBN) | Cannabinol | −5.9 | Hydrogen bond with Leu477; hydrophobic interactions with Phe221 and Trp224 |
| 8-Hydroxycannabinol | Cannabinol | −7.9 | Hydrogen bonds with Asp309 and Arg115 |
| Cannabiorcol-C1 | Cannabinol | −6.6 | Hydrogen bond with Asp309; hydrophobic interactions with Trp224 and Phe221 |
| 8-Hydroxycannabinolic acid A | Cannabinol | −4.8 | Hydrogen bonds with Asp309 and Arg115; hydrophobic interactions with Trp224 and Phe221 |
| (-)-trans-Cannabitriol-OEt-C5 * | Cannabitriol | −3.9 | Hydrogen bonds with Asp309, Arg115, and Leu477; hydrophobic interactions with Phe221 |
| (-)-trans-Cannabitriol-OEt-C3 * | Cannabitriol | −3.5 | Hydrogen bonds with Asp309 and Ser478; hydrophobic interactions with Phe221 |
| (-)-Δ7-trans-(1R,3R,6R)-isotetrahydrocannabinol-C5 | Miscellaneous | −9.8 | Hydrogen bonds with the heme group and Asp309; hydrophobic interactions with Trp224 and Phe221 |
| Cannabimovone | Miscellaneous | −8.4 | Hydrogen bond with Asp309; hydrophobic interactions with Phe221 |
| Cannabichromanone C | Miscellaneous | −7.8 | Hydrogen bonds with Asp309 and Arg115; hydrophobic interactions with Phe221 |
| Dehydrocannabifuran | Miscellaneous | −7.3 | Hydrogen bond with Asp309; hydrophobic interactions with Phe221 |
| Cannabichromanone B | Miscellaneous | −7.0 | Hydrogen bonds with the heme group and Asp309; hydrophobic interactions with Phe221 |
| (-)-(7R)-Cannabicoumarononic acid | Miscellaneous | −6.5 | Hydrogen bonds with Asp309 and Met374; hydrophobic interactions with Trp224 and Phe221 |
| 8-Hydroxy-isohexahydrocannabivarin | Miscellaneous | −6.0 | Hydrogen bonds with the heme group and Asp309; hydrophobic interactions with Trp224 and Phe221 |
| Cannabicoumaronome-C5 | Miscellaneous | −6.0 | Hydrogen bonds with the heme group and Asp309; hydrophobic interactions with Phe221 |
| Cannabichromanone-C3 | Miscellaneous | −5.4 | Hydrogen bonds with the heme group and Asp309; hydrophobic interactions with Phe221 |
| Cannabinoid Name | Cannabinoid Class | Binding Affinity (kcal/mol) | Relevant Interactions |
|---|---|---|---|
| (-)-Δ9-trans-Tetrahydrocannabihexol | Δ9-THC | −8.5 | Close to Asp351; hydrophobic interactions with Trp383 |
| α-Terpenyl (-)-Δ9-trans- tetrahydrocannabinolate | Δ9-THC | −8.0 | Close to Asp351; hydrophobic interactions with Phe404 |
| 11-Acetoxy-(-)-Δ9-trans-tetrahydrocannabinolic acid A | Δ9-THC | −7.8 | Hydrogen bonds with Glu353 and Arg394; close to Asp351; hydrophobic interactions with Phe404 |
| 8-oxo-(-)-Δ9-trans-Tetrahydrocannabinol | Δ9-THC | −7.8 | Close to Asp351; hydrophobic interactions with Trp383 |
| (-)-Δ9-trans-Tetrahydrocannabiphorol | Δ9-THC | −7.8 | Close to Asp351; hydrophobic interactions with Trp383 |
| 4-Terpenyl (-)-Δ9-trans-tetrahydrocannabinolate | Δ9-THC | −7.3 | Hydrogen bond with Asp351; hydrophobic interactions with Trp383 |
| Δ9-Tetrahydrocannabinol (THC) | Δ9-THC | −7.1 | Close to Asp351; hydrophobic interactions with Trp383 |
| (-)-Δ9-trans-Tetrahydrocannabinol-C4 | Δ9-THC | −7.0 | Close to Asp351; hydrophobic interactions with Trp383 |
| (-)-Δ9-trans-Tetrahydrocannabinal | Δ9-THC | −6.5 | Hydrogen bond with Asp351; hydrophobic interactions with Trp383 |
| (-)-Δ9-trans-Tetrahydrocannabinolic acid A-C4 | Δ9-THC | −5.4 | Hydrogen bonds with Val534 and Asp351; hydrophobic interactions with Trp383 |
| (-)-Δ8-trans-Tetrahydrocannabinol | Δ8-THC | −8.4 | Close to Asp351; hydrophobic interactions with Trp383 |
| 10β-Hydroxy-Δ8-tetrahydrocannabinol | Δ8-THC | −8.1 | Hydrogen bond with Glu353; close to Asp351; hydrophobic interactions with Trp383 |
| 10α-Hydroxy-Δ8-tetrahydrocannabinol | Δ8-THC | −7.4 | Close to Asp351; hydrophobic interactions with Trp383 |
| 10a-α-Hydroxy-o-oxo-Δ8-tetrahydrocannabinol | Δ8-THC | −6.4 | Close to Asp351; hydrophobic interactions with Trp383 |
| Cannabidiolic acid | Cannabidiol | −8.3 | Close to Asp351; hydrophobic interactions with Trp383 |
| Cannabidiol-C4 | Cannabidiol | −7.8 | Close to Asp351; hydrophobic interactions with Trp383 |
| (-)-Cannabidivarin | Cannabidiol | −7.6 | Close to Asp351; hydrophobic interactions with Trp383 |
| Cannabidiol monomethyl ether | Cannabidiol | −7.4 | Close to Asp351; hydrophobic interactions with Trp383 |
| Cannabidiol (CBD) | Cannabidiol | −7.3 | Close to Asp351; hydrophobic interactions with Trp383 |
| Cannabidihexol | Cannabidiol | −6.8 | Close to Asp351; hydrophobic interactions with Trp383 |
| Cannabigerol | Cannabigerol | −6.9 | Possible hydrogen bond with His 524; close to Asp351 |
| ᵞ-Eudesmyl-cannabigerolate | Cannabigerol | −7.7 | Close to Asp351; hydrophobic interactions with Trp383 |
| Cannabinerolic acid | Cannabigerol | −7.7 | Hydrogen bonds with His524 and Glu353; close to Asp351; hydrophobic interactions with Trp383 |
| 5-Acetyl-4-hydroxy-cannabigerol | Cannabigerol | −7.0 | Hydrogen bonds with His524 and Glu353; close to Asp351; hydrophobic interactions with Trp383 and Phe404 |
| (-)-6,7-trans-Epoxycannabigerolic acid | Cannabigerol | −6.9 | Hydrogen bonds with His524 and Ala350; close to Asp351; hydrophobic interactions with Trp383 and Phe404 |
| (-)-6,7-cis-Epoxycannabigerolic acid | Cannabigerol | −6.9 | Hydrogen bonds with His524 and Ala350; close to Asp351; hydrophobic interactions with Trp383 and Phe404 |
| Monomethyl ether of (E)-cannabigerol | Cannabigerol | −6.8 | Close to Asp351; hydrophobic interactions with Trp383 |
| Sesquicannabigerol | Cannabigerol | −6.5 | Hydrogen bonds with Asp351, Asn532, and Val534; hydrophobic interactions with Trp383 |
| (-)-6,7-trans-Epoxycannabigerol | Cannabigerol | −6.4 | Close to Asp351; hydrophobic interactions with Trp383 |
| Cannabigerolic acid | Cannabigerol | −6.3 | Close to Asp351; hydrophobic interactions with Trp383 |
| Cannabigerovarinic acid | Cannabigerol | −6.3 | Hydrogen bonds with Asp351 and Val534; hydrophobic interactions with Trp383 |
| Camagerol | Cannabigerol | −6.0 | Hydrogen bonds with Asp351 and Asn532 |
| Monomethyl ether of cannabigerolic acid | Cannabigerol | −5.9 | Close to Asp351; hydrophobic interactions with Trp383 |
| (-)-6,7-cis-Epoxycannabigerol | Cannabigerol | −5.1 | Close to Asp351; hydrophobic interactions with Trp383 |
| Cannabinodiol | Cannabinodiol | −8.1 | Close to Asp351; hydrophobic interactions with Trp383 and Phe404 |
| Cannabinodivirin | Cannabinodiol | −7.8 | Close to Asp351; hydrophobic interactions with Trp383 and Phe404 |
| Cannabielsoin | Cannabielsoin | −8.1 | Close to Asp351; hydrophobic interactions with Trp383 |
| Cannabielsoin acid B | Cannabielsoin | −6.0 | Hydrogen bond with His524; close to Asp351; hydrophobic interactions with Phe404 |
| Cannabielsoin acid A | Cannabielsoin | −5.2 | Hydrogen bonds with His524 and Ala350; close to Asp351; hydrophobic interactions with Trp383 |
| Cannabicyclol | Cannabicyclol | −8.9 | Close to Asp351; hydrophobic interactions with Trp383 |
| Cannabicyclolic acid | Cannabicyclol | −8.7 | Hydrogen bond with His524; hydrophobic interactions with Trp383 |
| Cannabicyclovarin | Cannabicyclol | −7.8 | Close to Asp351; hydrophobic interactions with Trp383 |
| (-)-7-Hydroxycannabichromene | Cannabichromene | −8.5 | Close to Asp351; hydrophobic interactions with Phe404 |
| ( ± )-4-Acetoxycannabichromene | Cannabichromene | −7.8 | Hydrogen bond with Met421; close to Asp351; hydrophobic interactions with Trp383 and Phe404 |
| Cannabichromenic acid | Cannabichromene | −7.7 | Close to Asp351; hydrophobic interactions with Trp383 |
| 2-Methyl-2-(4-methyl-2-pentyl)-7-propyl-2H-1-benzopyran-5-ol | Cannabichromene | −7.6 | Close to Asp351; hydrophobic interactions with Phe404 |
| Cannabichromevarin | Cannabichromene | −7.6 | Close to Asp351; hydrophobic interactions with Phe404 |
| Cannabichromevarinic acid | Cannabichromene | −7.6 | Hydrogen bond with Asp351; hydrophobic interactions with Trp383 |
| (-)-3′′-Hydroxy-Δ4′′-cannabichromene | Cannabichromene | −7.2 | Close to Asp351; hydrophobic interactions with Trp383 |
| Cannabichromene | Cannabichromene | −7.0 | Close to Asp351; hydrophobic interactions with Trp383 |
| Cannabinol | Cannabinol | −8.9 | Hydrophobic interactions with Trp383 and Phe404 |
| Cannabinol methyl ether | Cannabinol | −8.4 | Close to Asp351; hydrophobic interactions with Trp383 and Phe404 |
| Cannabinol-C4 | Cannabinol | −6.6 | Close to Asp351; hydrophobic interactions with Trp383 and Phe404 |
| (10S)-Hydroxycannabinol | Cannabinol | −6.4 | Hydrogen bond with Val534; close to Asp351; hydrophobic interactions with Trp383 and Phe404 |
| 8-Hydroxycannabinol | Cannabinol | −6.0 | Close to Asp351; hydrophobic interactions with Trp383 and Phe404 |
| 8-Hydroxycannabinolic acid A | Cannabinol | −5.4 | Hydrogen bond with Val524; hydrophobic interaction with Phe404 |
| (+)-trans-Cannabitriol-C3 | Cannabitriol | −7.7 | Close to Asp351; hydrophobic interactions with Trp383 |
| (+)-cis-Cannabitriol-C5 | Cannabitriol | −7.6 | Close to Asp351; hydrophobic interactions with Trp383 |
| (-)-cis-Cannabitriol-C5 | Cannabitriol | −7.6 | Close to Asp351; hydrophobic interactions with Trp383 |
| (+)-trans-Cannabitriol-C5 | Cannabitriol | −7.5 | Close to Asp351; hydrophobic interactions with Trp383 |
| (-)-trans-Cannabitriol-OEt-C3 * | Cannabitriol | −7.5 | Hydrogen bond with His524; close to Asp351; hydrophobic interactions with Trp383 |
| (-)-trans-Cannabitriol-C5 | Cannabitriol | −6.8 | Hydrogen bond with Met421; close to Asp351; hydrophobic interactions with Trp383 |
| Cannabitriol | Cannabitriol | −6.5 | Close to Asp351; hydrophobic interactions with Trp383 |
| Cannabioxepane | Miscellaneous | −9.2 | Close to Asp351; hydrophobic interactions with Trp383 and Phe404 |
| (-)-Δ9-cis-(6aS,10aR)-Tetrahydrocannabinol | Miscellaneous | −8.7 | Close to Asp351; hydrophobic interactions with Trp383 |
| Dehydrocannabifuran | Miscellaneous | −8.5 | Close to Asp351; hydrophobic interactions with Trp383 and Phe404 |
| Cannabifuran | Miscellaneous | −8.1 | Close to Asp351; hydrophobic interactions with Trp383 and Phe404 |
| 10α-Hydroxyhexahydrocannabinol | Miscellaneous | −8.0 | Close to Asp351; hydrophobic interactions with Trp383 |
| 2-Geranyl-5-hydroxy-3-n-pentyl-1,4-benzoquinone | Miscellaneous | −7.9 | Close to Asp351; hydrophobic interactions with Trp383 |
| 9α-Hydroxyhexahydrocannabinol | Miscellaneous | −7.9 | Close to Asp351; hydrophobic interactions with Trp383 |
| 10-Oxo-Δ6a(10a)-tetrahydrocannabinol | Miscellaneous | −7.7 | Close to Asp351; hydrophobic interactions with Trp383 |
| Cannabichromanone D | Miscellaneous | −7.7 | Close to Asp351; hydrophobic interactions with Trp383 |
| Cannabichromanone-C3 | Miscellaneous | −7.5 | Hydrogen bond with His524; close to Asp351; hydrophobic interactions with Trp383 |
| Cannabicoumaronome-C5 | Miscellaneous | −7.2 | Close to Asp351; hydrophobic interactions with Trp383 |
| Cannabimovone | Miscellaneous | −7.2 | Close to Asp351; hydrophobic interactions with Trp383 |
| 9β, 10β-Epoxyhexahydrocannabinol | Miscellaneous | −7.0 | Close to Asp351; hydrophobic interactions with Trp383 |
| 10α-Hydroxy-Δ9,11-hexahydrocannabinol | Miscellaneous | −7.0 | Close to Asp351; hydrophobic interactions with Trp383 |
| 10αR-Hydroxyhexahydrocannabinol | Miscellaneous | −6.7 | Close to Asp351; hydrophobic interactions with Trp383 |
| Cannabichromanone C | Miscellaneous | −6.7 | Hydrogen bond with Val534; close to Asp351; hydrophobic interactions with Phe404 |
| (-)-Δ7-trans-(1R,3R,6R)-Isotetrahydrocannabinol-C5 | Miscellaneous | −6.6 | Hydrogen bond with His524; close to Asp351 |
| 9α-Hydroxy-10-oxo-Δ6a,10a-tetrahydrocannabinol | Miscellaneous | −6.6 | Close to Asp351; hydrophobic interactions with Trp383 |
| 4-Acetoxy-2-geranyl-5-hydroxy-3-n-pentylphenol | Miscellaneous | −6.4 | Close to Asp351; hydrophobic interactions with Phe404 |
| 7-Oxo-9α-hydroxyhexahydrocannabinol | Miscellaneous | −6.4 | Close to Asp351; hydrophobic interactions with Trp383 |
| (-)-Cannabitetrol | Miscellaneous | −6.4 | Close to Asp351; hydrophobic interactions with Trp383 |
| (-)-(7R)-Cannabicoumaronic acid | Miscellaneous | −6.4 | Close to Asp351; hydrophobic interactions with Trp383 |
| Cannabichromanone B | Miscellaneous | −6.4 | Close to Asp351; hydrophobic interactions with Phe404 |
| (-)-Cannabiripsol-C5 | Miscellaneous | −6.2 | Close to Asp351; hydrophobic interactions with Trp383 |
| Cannabinoid Name | Cannabinoid Class | Binding Affinity—Aromatase (kcal/mol) | Binding Affinity—ERα (kcak/mol) |
|---|---|---|---|
| 8-Oxo-(-)-Δ9-trans-tetrahydrocannabinol | Δ9-THC | −9.2 | −7.8 |
| 11-Acetoxy-(-)-Δ9-trans-tetrahydrocannabinolic acid A | Δ9-THC | −8.3 | −7.8 |
| (-)-Δ9-trans-Tetrahydrocannabinolic acid A-C4 | Δ9-THC | −6.7 | −5.4 |
| (-)-Δ9-trans-Tetrahydrocannabinal | Δ9-THC | −3.8 | −6.5 |
| Cannabidiol (CBD) | Cannabidiol | −8.7 | −7.3 |
| (-)-Cannabidivarin (CBDV) | Cannabidiol | −7.6 | −7.6 |
| Cannabidiol monomethylether (CBDM) | Cannabidiol | −7.3 | −7.4 |
| Cannabidiol-C4 (CBDB) | Cannabidiol | −6.9 | −7.8 |
| Cannabidiolic acid (CBDA) | Cannabidiol | −5.7 | −8.3 |
| Cannabinerolic acid | Cannabigerol | −6.7 | −7.7 |
| Camagerol | Cannabigerol | −6.6 | −6.0 |
| (-)-6,7-cis-Epoxycannabigerolic acid | Cannabigerol | −6.3 | −6.9 |
| Cannabigerol (CBG) | Cannabigerol | −6.1 | −6.9 |
| Cannabigerovarinic acid (CBGVA) | Cannabigerol | −5.8 | −6.3 |
| (-)-6,7-trans-Epoxycannabigerolic acid | Cannabigerol | −5.4 | −6.9 |
| 5-Acetyl-4-hydroxy-cannabigerol | Cannabigerol | −5.1 | −7.0 |
| Cannabielsoin acid A | Cannabielsoin | −5.9 | −5.2 |
| Cannabielsoin acid B | Cannabielsoin | −5.1 | −6.0 |
| Cannabicyclol | Cannabicyclol | −9.8 | −8.9 |
| Cannabicyclovarin | Cannabicyclol | −9.3 | −7.8 |
| Cannabicyclolic acid | Cannabicyclol | −5.5 | −8.7 |
| Cannabichromevarin | Cannabichromene | −7.1 | −7.6 |
| Cannabichromenic acid (CBCA) | Cannabichromene | −6.4 | −7.7 |
| Cannabichromene (CBC) | Cannabichromene | −6.0 | −7.0 |
| (-)-7-Hydroxycannabichromene | Cannabichromene | −5.5 | −8.5 |
| Cannabichromevarinic acid | Cannabichromene | −5.2 | −7.6 |
| 8-Hydroxycannabinol | Cannabinol | −7.9 | −6.0 |
| Cannabinol (CBN) | Cannabinol | −5.9 | −8.9 |
| 8-Hydroxycannabinolic acid A | Cannabinol | −4.8 | −5.4 |
| (-)-trans-Cannabitriol-OEt-C3 * | Cannabitriol | −3.5 | −7.5 |
| (-)-Δ7-trans-(1R,3R,6R)-isotetrahydrocannabinol-C5 | Miscellaneous | −9.8 | −6.6 |
| Cannabimovone | Miscellaneous | −8.4 | −7.2 |
| Cannabichromanone C | Miscellaneous | −7.8 | −6.7 |
| Dehydrocannabifuran | Miscellaneous | −7.3 | −8.5 |
| Cannabichromanone B | Miscellaneous | −7.0 | −6.4 |
| (-)-(7R)-Cannabicoumarononic acid | Miscellaneous | −6.5 | −6.4 |
| Cannabicoumaronome-C5 | Miscellaneous | −6.0 | −7.2 |
| Cannabichromanone-C3 | Miscellaneous | −5.4 | −7.5 |
| Cannabinoid | Aromatase Inhibition (%) ± SEM | Binding Affinity—Aromatase (kcal/mol) |
|---|---|---|
| CBC | 8.9 ± 1.7 | −6.0 |
| CBCA | 3.0 ± 0.9 | −6.4 |
| CBD | 83.2 ± 0.6 | −8.7 |
| CBDA | 3.4 ± 1.3 | −5.7 |
| CBDB | 6.4 ± 3.6 | −6.9 |
| CBDM | 4.5 ± 2.8 | −7.3 |
| CBDV | 12.0 ± 2.5 | −7.6 |
| CBG | 3.6 ± 2.3 | −6.1 |
| CBGVA | 7.8 ± 0.9 | −5.8 |
| CBN | 3.8 ± 0.5 | −5.9 |
| Ana | 98.8 ± 0.2 | −8.2 |
| Let | 99.4 ± 0.2 | −7.3 |
| Exe | 98.4 ± 0.3 | −8.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almeida, C.F.; Palmeira, A.; Valente, M.J.; Correia-da-Silva, G.; Vinggaard, A.M.; Sousa, M.E.; Teixeira, N.; Amaral, C. Molecular Targets of Minor Cannabinoids in Breast Cancer: In Silico and In Vitro Studies. Pharmaceuticals 2024, 17, 1245. https://doi.org/10.3390/ph17091245
Almeida CF, Palmeira A, Valente MJ, Correia-da-Silva G, Vinggaard AM, Sousa ME, Teixeira N, Amaral C. Molecular Targets of Minor Cannabinoids in Breast Cancer: In Silico and In Vitro Studies. Pharmaceuticals. 2024; 17(9):1245. https://doi.org/10.3390/ph17091245
Chicago/Turabian StyleAlmeida, Cristina Ferreira, Andreia Palmeira, Maria João Valente, Georgina Correia-da-Silva, Anne Marie Vinggaard, Maria Emília Sousa, Natércia Teixeira, and Cristina Amaral. 2024. "Molecular Targets of Minor Cannabinoids in Breast Cancer: In Silico and In Vitro Studies" Pharmaceuticals 17, no. 9: 1245. https://doi.org/10.3390/ph17091245
APA StyleAlmeida, C. F., Palmeira, A., Valente, M. J., Correia-da-Silva, G., Vinggaard, A. M., Sousa, M. E., Teixeira, N., & Amaral, C. (2024). Molecular Targets of Minor Cannabinoids in Breast Cancer: In Silico and In Vitro Studies. Pharmaceuticals, 17(9), 1245. https://doi.org/10.3390/ph17091245