


Isolation of Alpha-Glucosidase Inhibitors from the Panamanian Mangrove Plant Mora oleifera (Triana ex Hemsl.) Ducke



Abstract

1. Introduction
2. Results and Discussion
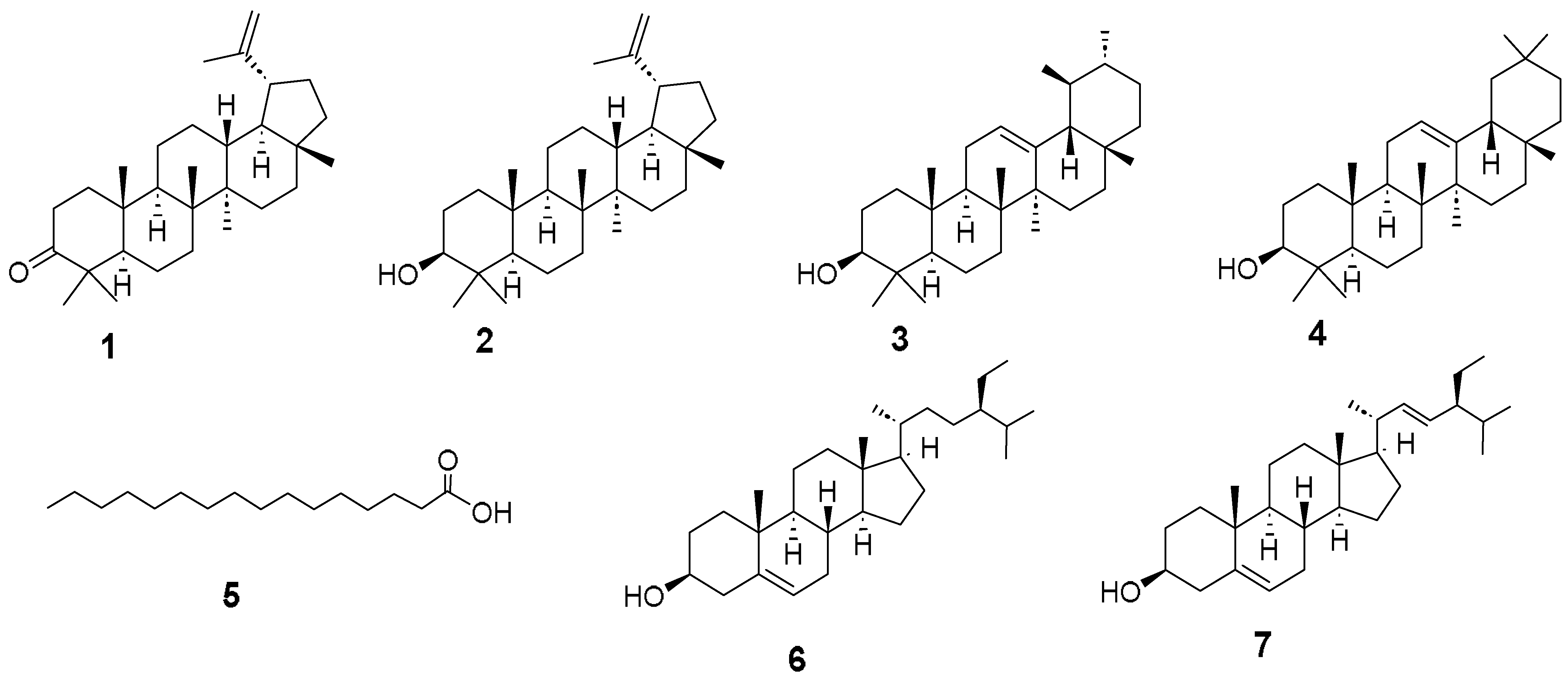
2.1. Activity-Guided Isolation and Identification
Spectral Compounds Data
2.2. Characterization of Alpha-Glucosidase Enzyme Inhibition by Isolated Compounds
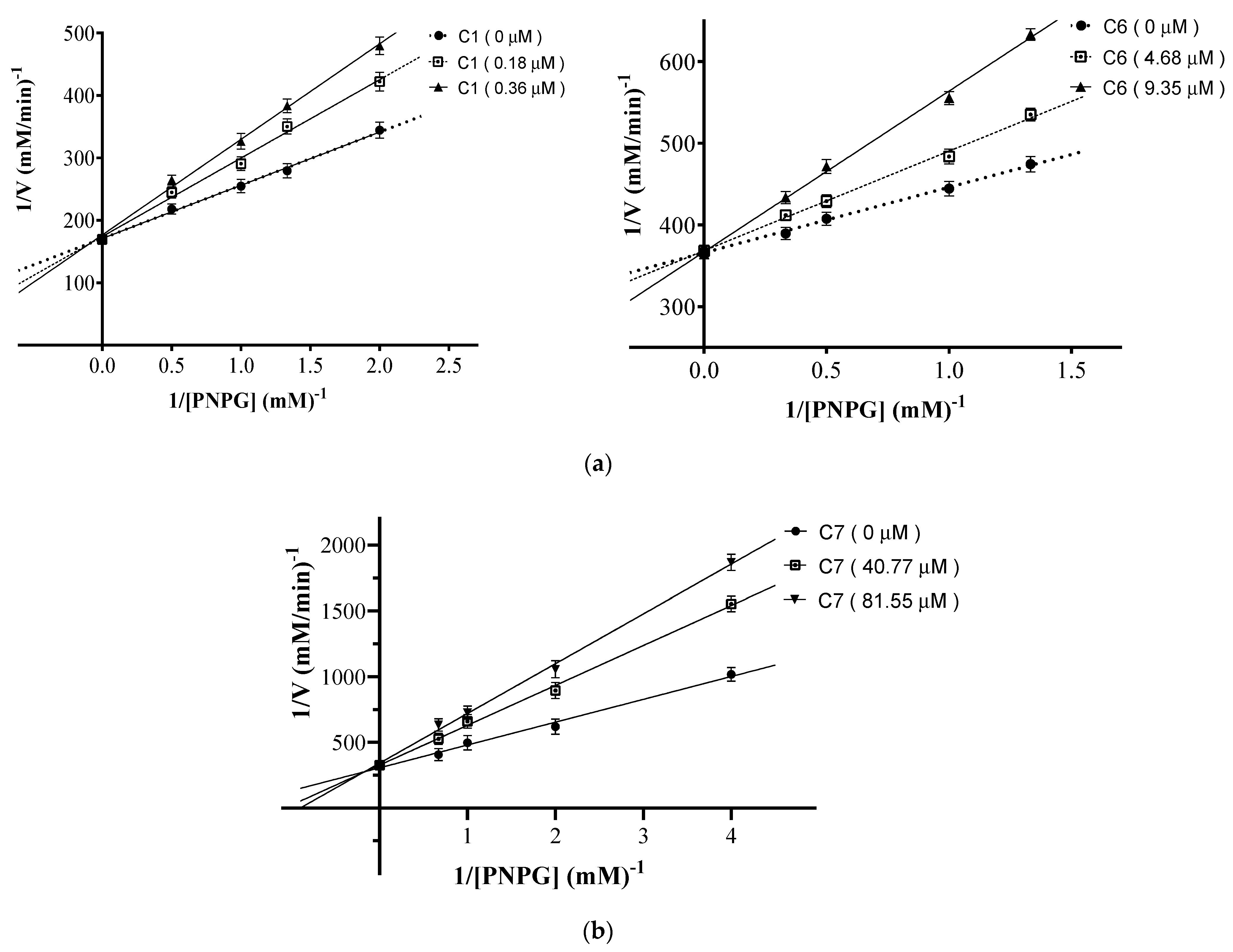
2.3. Mode of Inhibition of α-Glucosidase for Compounds 1, 6, and 7
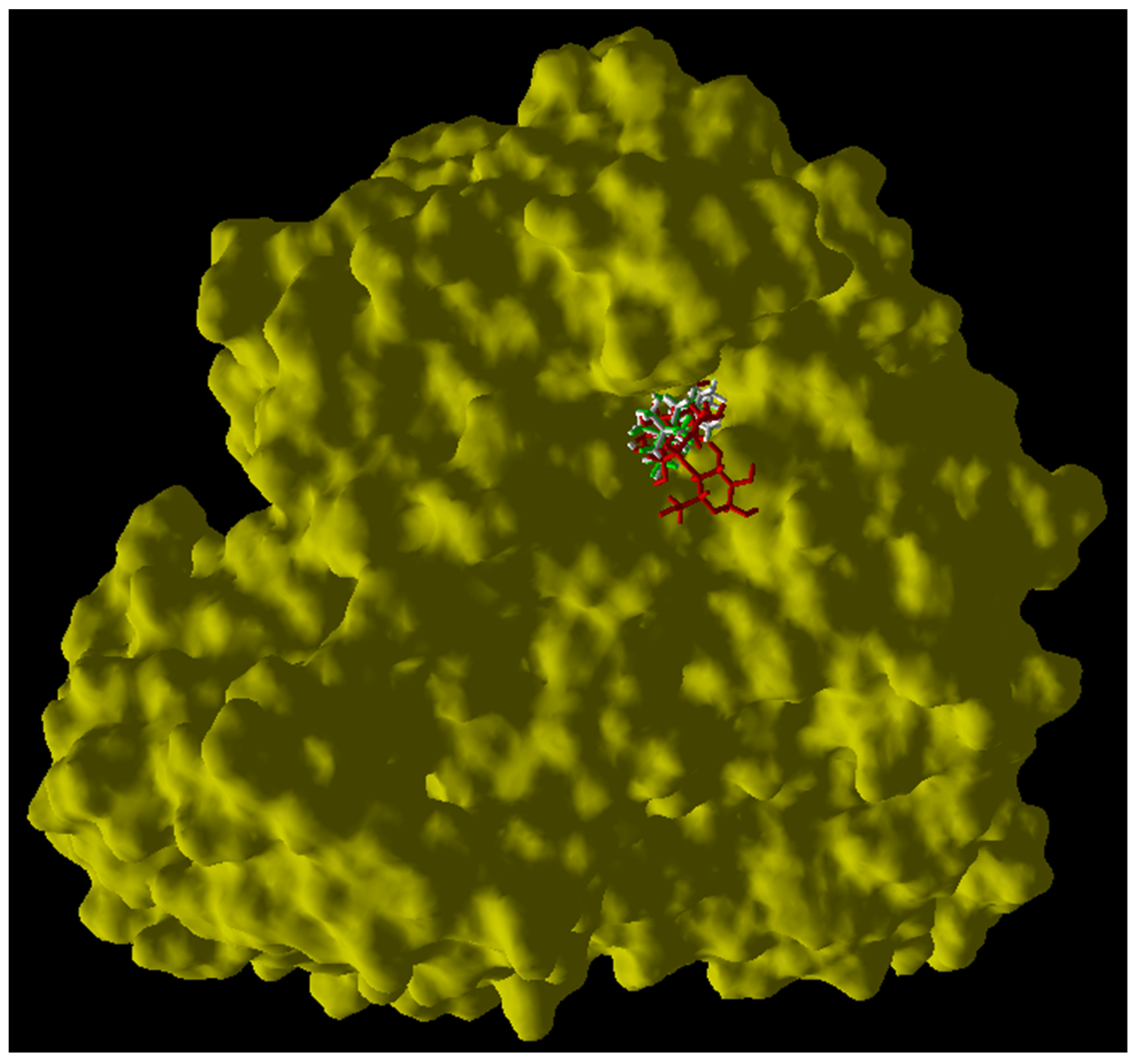
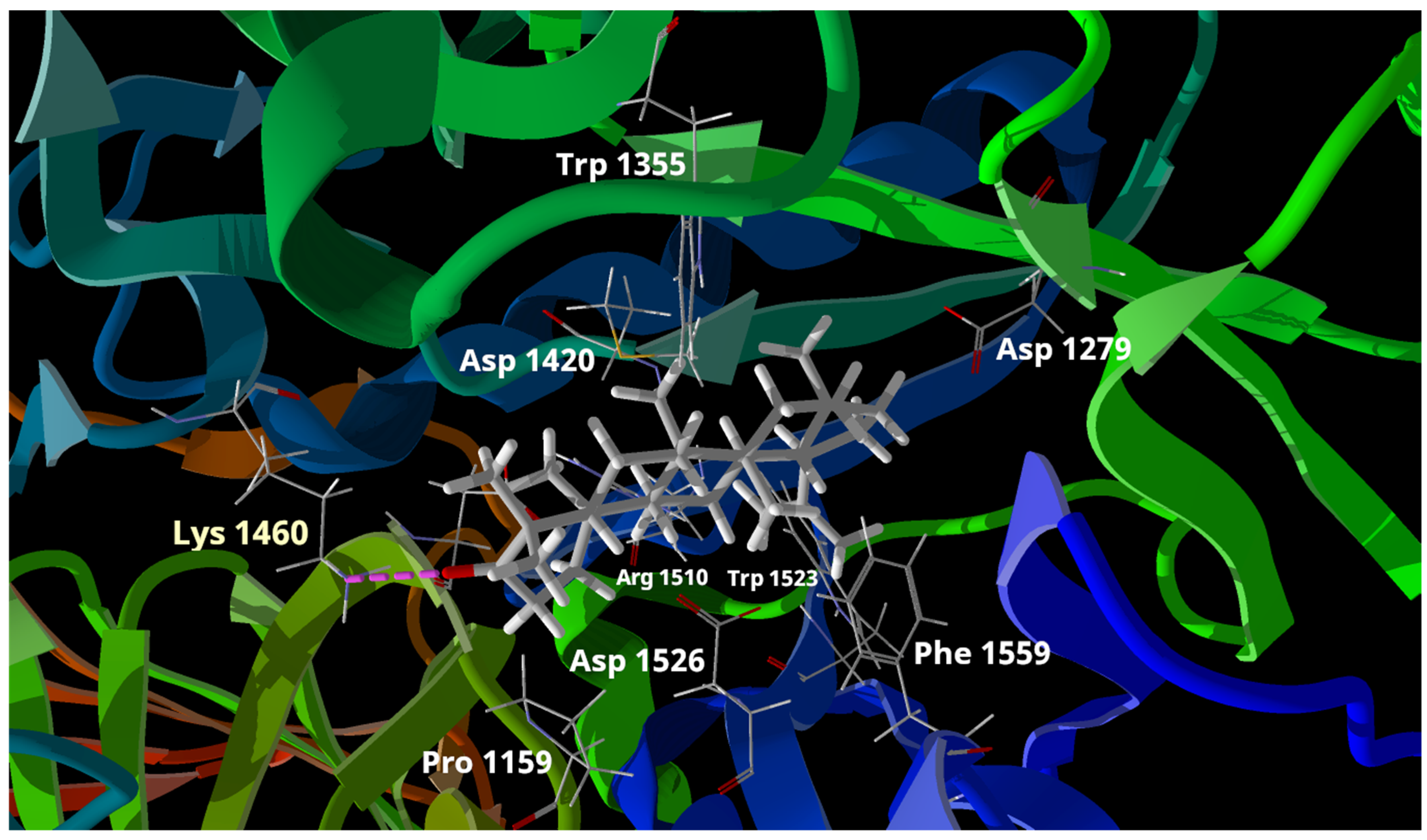
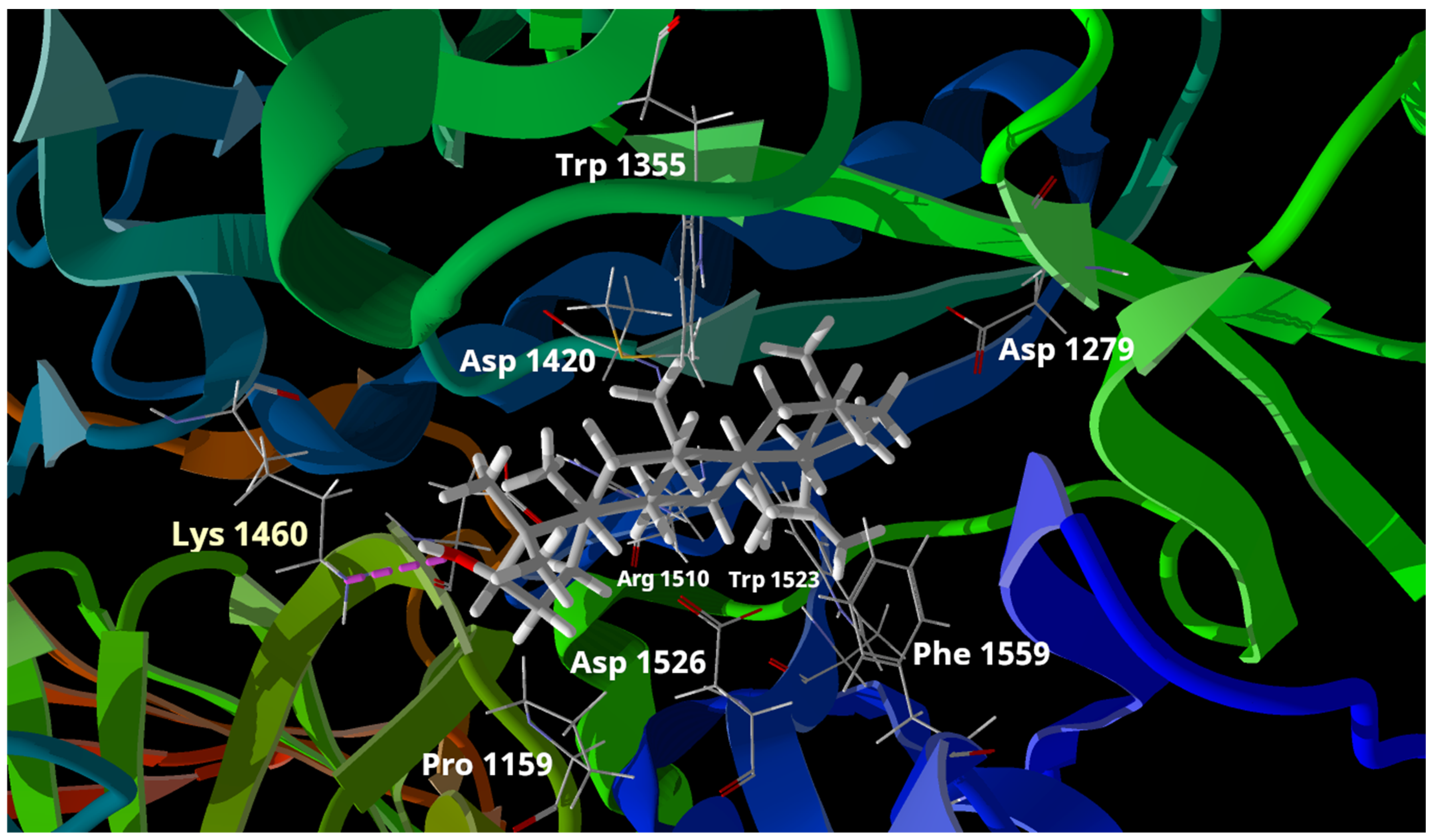
2.4. Docking Study for Compounds 1 and 2
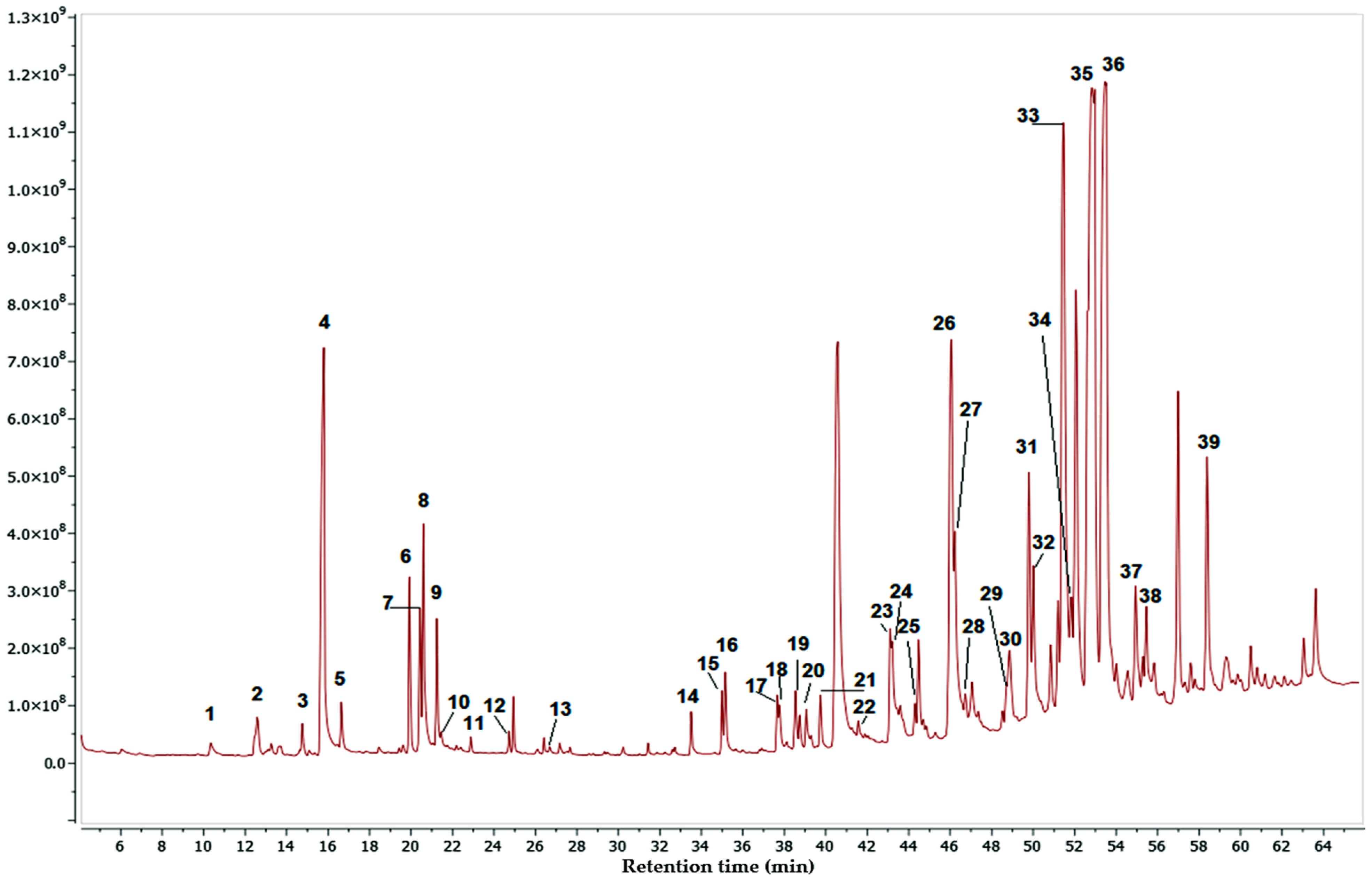
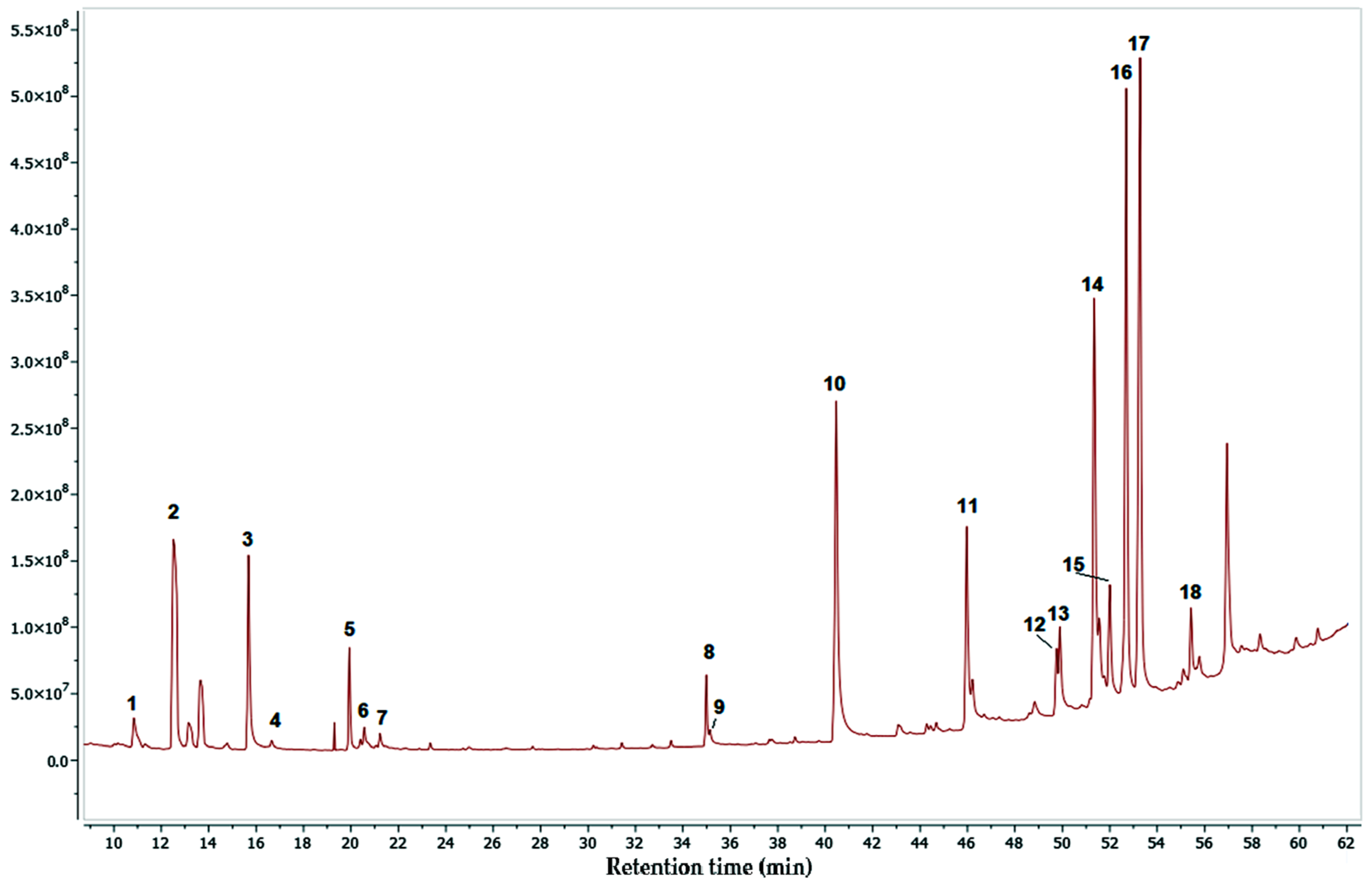
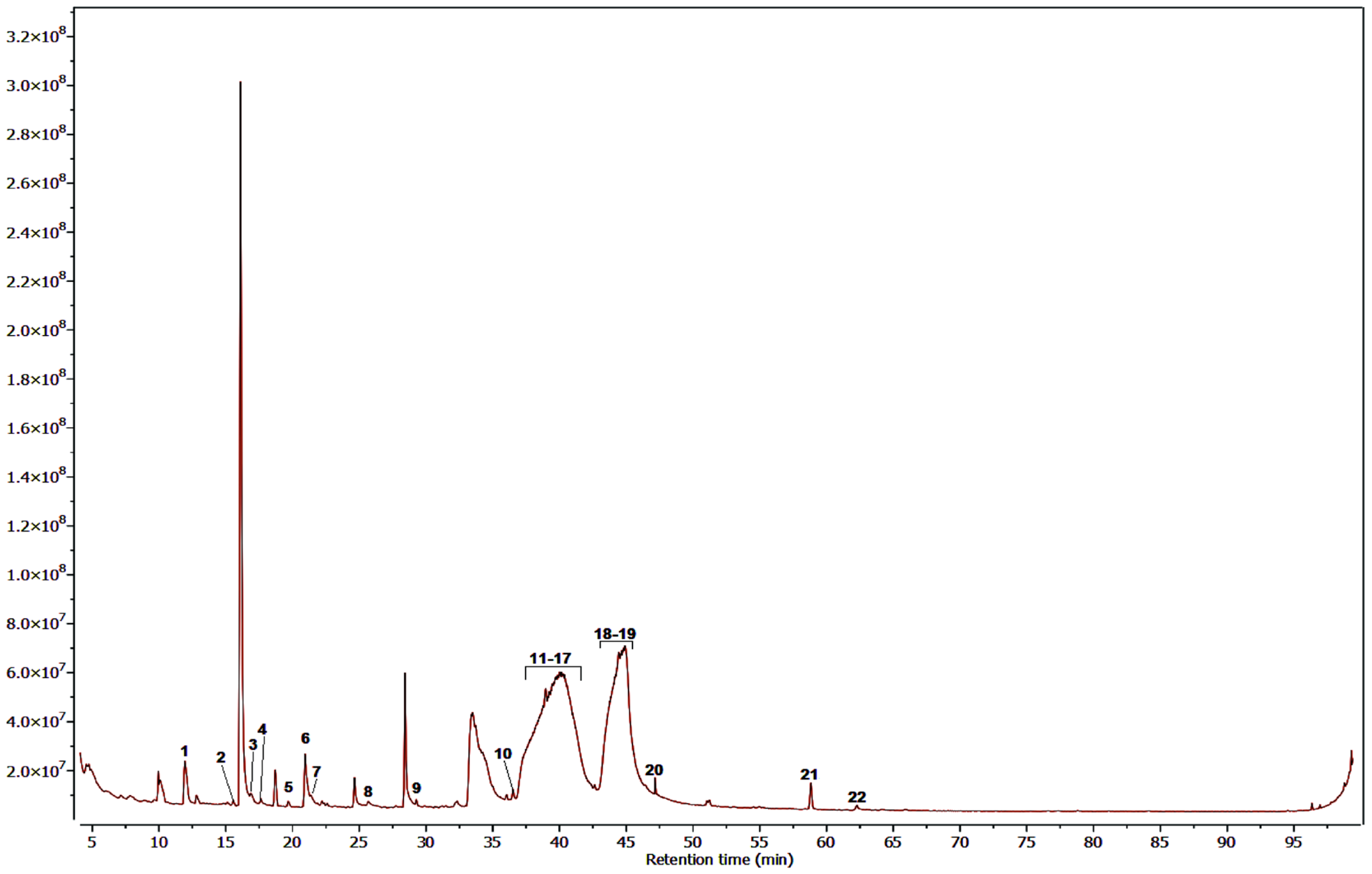
2.5. GC-MS-Based Metabolomic Analysis
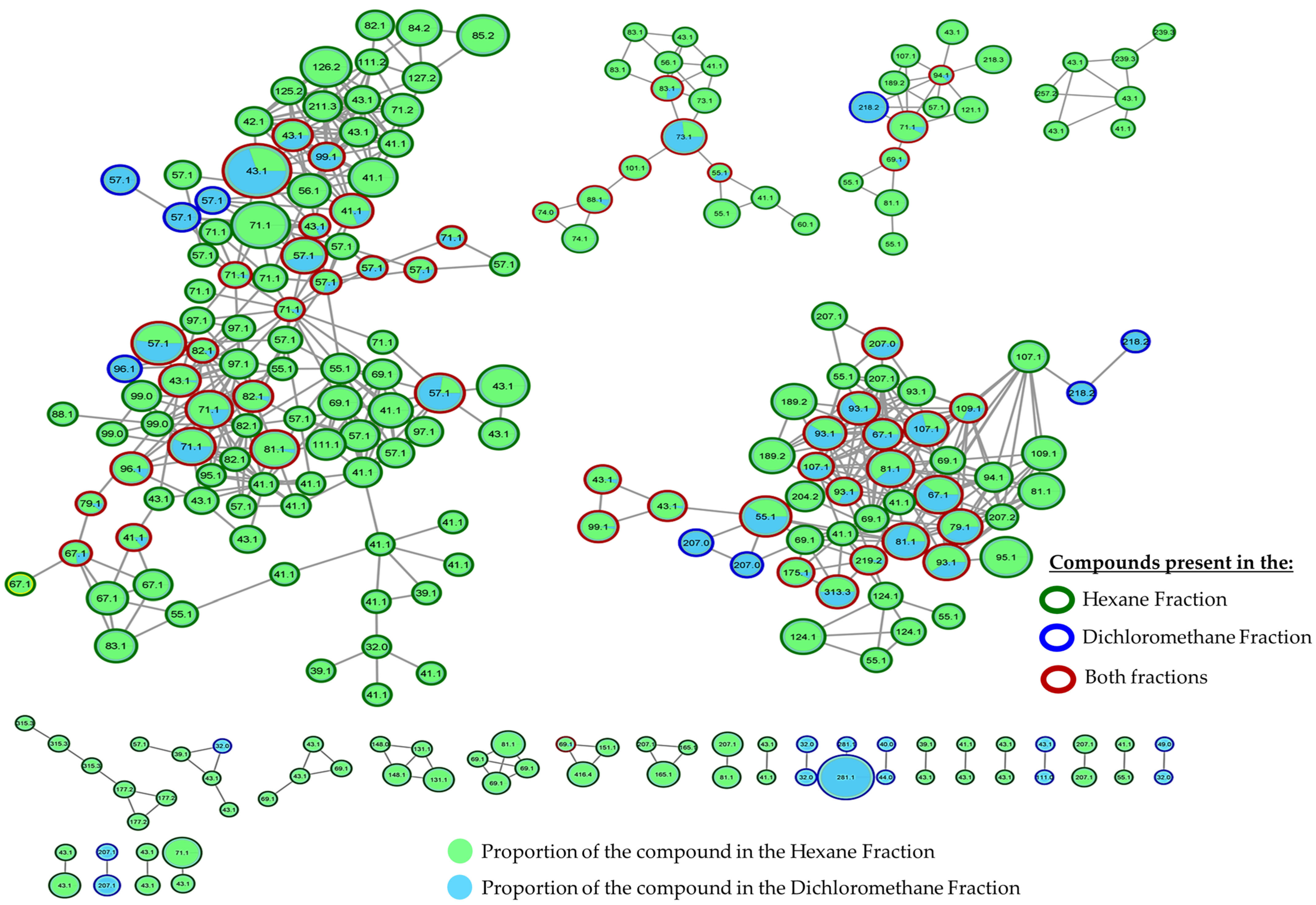
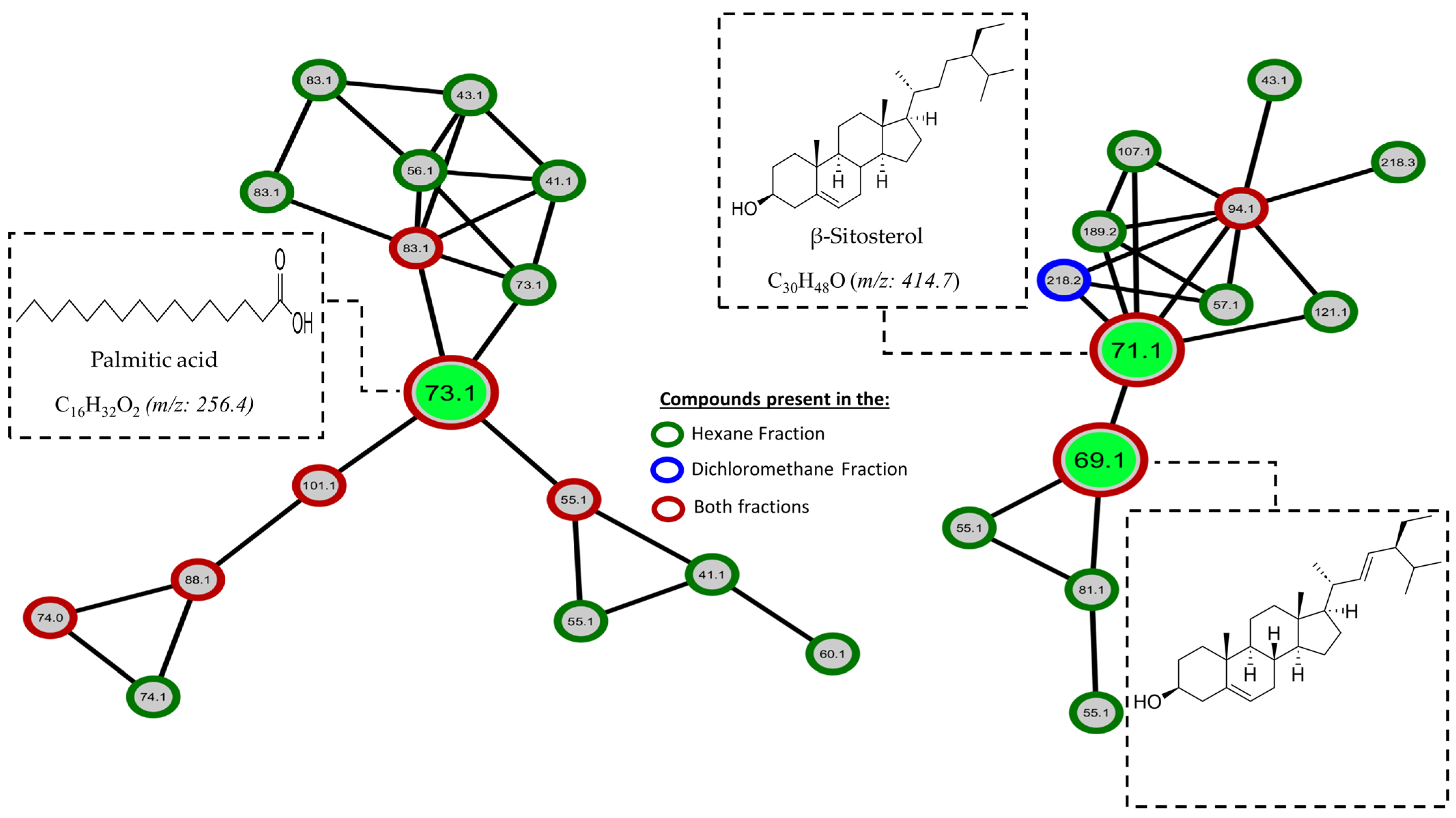
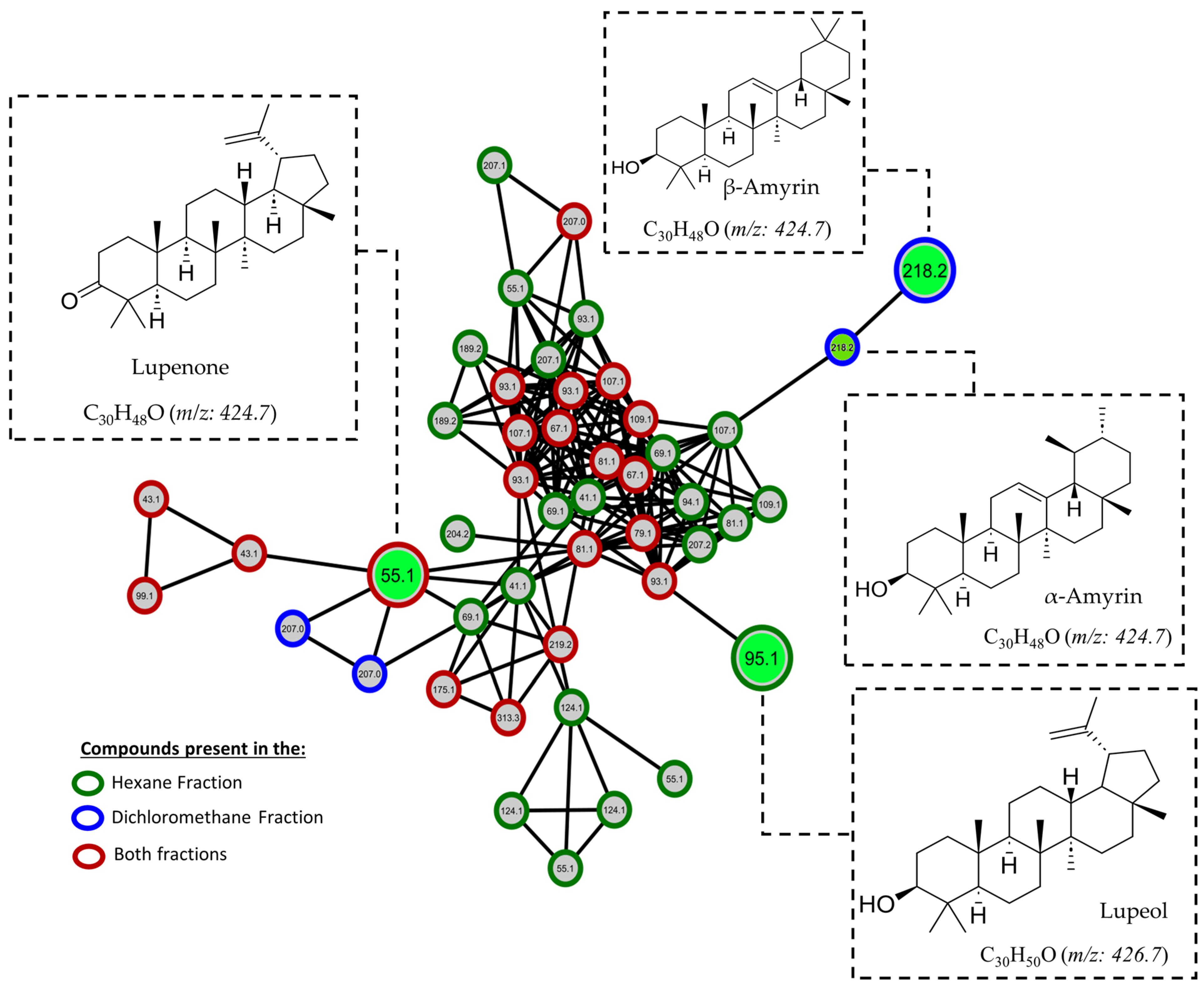
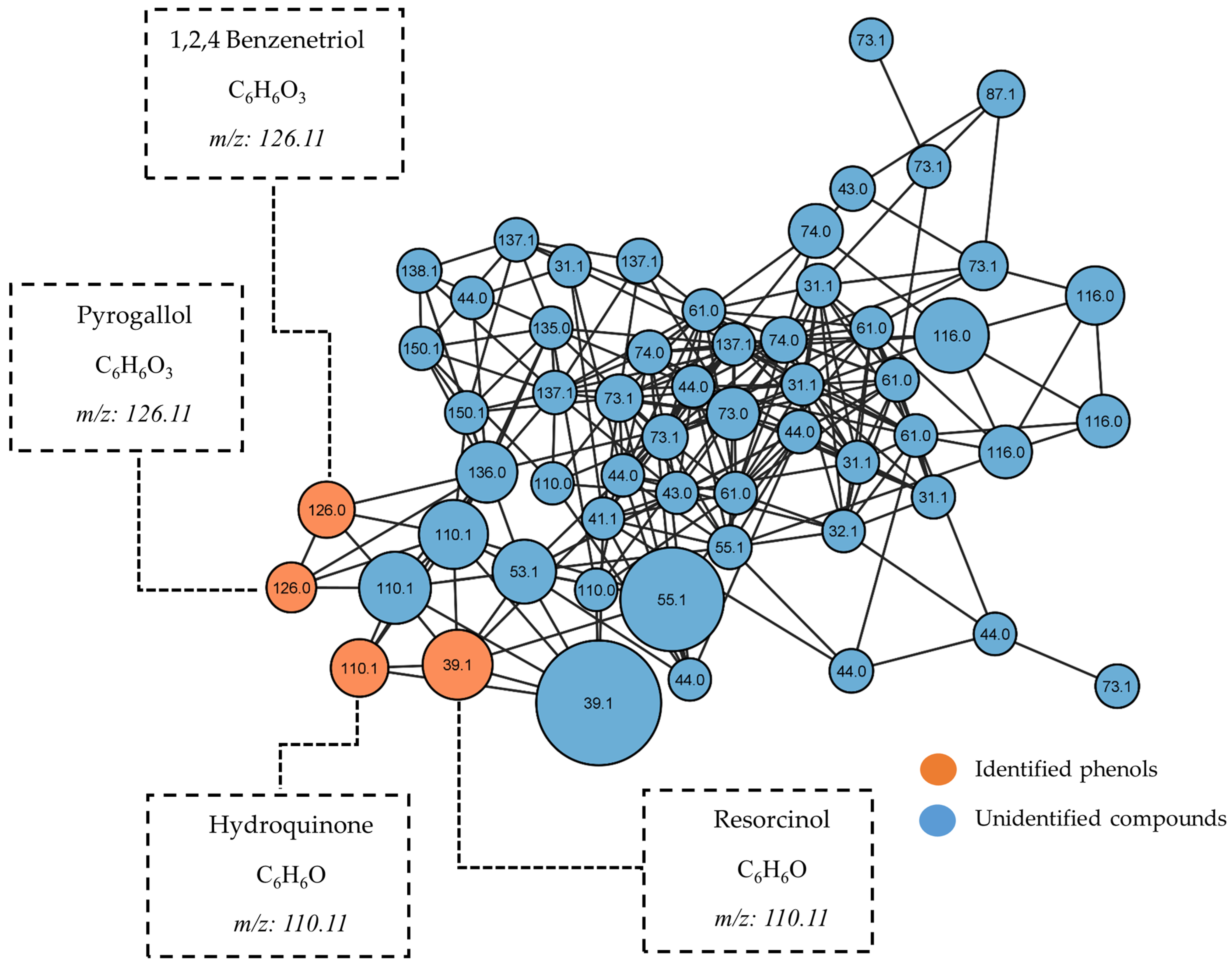
2.6. Molecular Networking
3. Experimental
3.1. Plant Material and Extract Preparation
3.2. Isolation of Compounds
3.3. Inhibition of Alpha-Glucosidase Assay
3.4. Kinetics of Alpha-Glucosidase Inhibition
3.5. Docking Study
3.6. GC-MS-Based Metabolomic Analysis
3.7. Molecular Networking
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mitra, S.; Naskar, N.; Chaudhuri, P. A Review on Potential Bioactive Phytochemicals for Novel Therapeutic Applications with Special Emphasis on Mangrove Species. Phytomed. Plus 2021, 1, 100107. [Google Scholar] [CrossRef]
- Dirir, A.M.; Daou, M.; Yousef, A.F.; Yousef, L.F. A Review of Alpha-Glucosidase Inhibitors from Plants as Potential Candidates for the Treatment of Type-2 Diabetes. Phytochem. Rev. 2022, 21, 1049–1079. [Google Scholar] [CrossRef] [PubMed]
- Dahibhate, N.L.; Saddhe, A.A.; Kumar, K. Mangrove Plants as a Source of Bioactive Compounds: A Review. Nat. Prod. J. 2019, 9, 86–97. [Google Scholar] [CrossRef]
- Vinoth, R.; Kumaravel, S.; Ranganathan, R. Therapeutic and Traditional Uses of Mangrove Plants. J. Drug Deliv. Ther. 2019, 9, 849–854. [Google Scholar] [CrossRef]
- Sadeer, N.B.; Mahomoodally, M.F. Biopharmaceutical Applications of Mangrove Plants: Opening a New Door to Disease Management and Prevention. In Mangroves with Therapeutic Potential for Human Health; Elsevier: Amsterdam, The Netherlands, 2022; pp. 63–96. [Google Scholar] [CrossRef]
- Autoridad Nacional del Ambiente [ANAM] y Autoridad de los Recursos Acuáticos de Panamá [ARAP]. Manglares de Panamá: Importancia, Mejores Prácticas y Regulaciones Vigentes; Tarté, A., Ed.; Editora Novo Art: Panama City, Panama, 2013. [Google Scholar]
- Spalding, M.; Kainuma, M.; Collins, L. World Atlas of Mangroves; Earthscan: London, UK, 2010. [Google Scholar] [CrossRef]
- Cherigo, L.; López, D.; Spadafora, C.; Mejia, L.C.; Martínez-Luis, S. Exploring the Biomedical Potential of Endophytic Fungi Isolated from Panamanian Mangroves. Nat. Prod. Commun. 2024, 19, 1934578X241228152. [Google Scholar] [CrossRef]
- Duke, N. Mora oleifera. IUCN Red List. Threat. Species 2010, 2010, e.T178858A7629292. [Google Scholar] [CrossRef]
- Correa, A.; Mireya, D.; Galdames, C.; Stapt, M. Catálogo de Las Plantas Vasculares de Panamá, 1st ed.; Novo Art; Instituto Smithsonian de Investigaciones Tropicales: Ciudad de Panama, Panama, 2004. [Google Scholar]
- Lopez, D.; Cherigo, L.; de Sedas, A.; Spadafora, C.; Martinez-Luis, S. Evaluation of Antiparasitic, Anticancer, Antimicrobial and Hypoglycemic Properties of Organic Extracts from Panamanian Mangrove Plants. Asian Pac. J. Trop. Med. 2018, 11, 32. [Google Scholar] [CrossRef]
- Puapairoj, P.; Naengchomnong, W.; Kijjoa, A.; Pinto, M.M.; Pedro, M.; Nascimento, M.S.; Silva, A.M.; Herz, W. Cytotoxic Activity of Lupane-Type Triterpenes from Glochidion Sphaerogynum and Glochidion Eriocarpum Two of Which Induce Apoptosis. Planta Med. 2005, 71, 208–213. [Google Scholar] [CrossRef]
- Yuca, H.; Özbek, H.; Demirezer, L.Ö.; Güvenalp, Z. Assessment of the α-Glucosidase and α-Amylase Inhibitory Potential of Paliurus Spina-Christi Mill. and Its Terpenic Compounds. Med. Chem. Res. 2022, 31, 1393–1399. [Google Scholar] [CrossRef]
- Alexandre, A.d.S.; Fachin-Espinar, M.T.; Nunez, C.V. Triterpenes, Steroids and Phenolic Isolated from Minquartia Guianensis Aubl. (Coulaceae) and Antibacterial Activity. Concilium 2023, 23, 883–895. [Google Scholar] [CrossRef]
- Cherigo, L.; Martínez-Luis, S. α-Glucosidase Inhibitor Isolated from Blechum Pyramidatum. Nat. Prod. Commun. 2018, 13, 461–464. [Google Scholar] [CrossRef]
- López, D.; Cherigo, L.; Spadafora, C.; Loza-Mejía, M.A.; Martínez-Luis, S. Phytochemical Composition, Antiparasitic and α-Glucosidase Inhibition Activities from Pelliciera Rhizophorae. Chem. Cent. J. 2015, 9, 53. [Google Scholar] [CrossRef] [PubMed]
- Cherigo, L.; Martínez-Luis, S. Identification of Major α-Glucosidase Inhibitors from Stem Bark of Panamanian Mangrove Plant Pelliciera Rhizophorae. Nat. Product. Commun. 2019, 14, 15–18. [Google Scholar] [CrossRef]
- Pinzi, L.; Rastelli, G. Molecular Docking: Shifting Paradigms in Drug Discovery. Int. J. Mol. Sci. 2019, 20, 4331. [Google Scholar] [CrossRef]
- Zhao, K.; Sun, S.; Wang, H.; Wang, L.; Qin, G.; Fan, J.; Guo, M.; Wang, W. α-Glucosidase Inhibitory Triterpenoids from Euonymus fortunei. Bioorg. Chem. 2021, 111, 104980. [Google Scholar] [CrossRef]
- Pujadas, G.; Vaque, M.; Ardevol, A.; Blade, C.; Salvado, M.; Blay, M.; Fernandez-Larrea, J.; Arola, L. Protein-Ligand Docking: A Review of Recent Advances and Future Perspectives. Curr. Pharm. Anal. 2008, 4, 1–19. [Google Scholar] [CrossRef]
- Madhavilatha, K.N.; Babu, G.R.M. Systematic Approach for Enrichment of Docking Outcome Using Consensus Scoring Functions. J. Phys. Conf. Ser. 2019, 1228, 012019. [Google Scholar] [CrossRef]
- Torres, P.H.M.; Sodero, A.C.R.; Jofily, P.; Silva, F.P., Jr. Key Topics in Molecular Docking for Drug Design. Int. J. Mol. Sci. 2019, 20, 4574. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and Community Curation of Mass Spectrometry Data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef]
- Chan, H.H.; Sun, H.D.; Reddy, M.V.B.; Wu, T.S. Potent α-Glucosidase Inhibitors from the Roots of Panax japonicus C. A. Meyer Var. Major. Phytochemistry 2010, 71, 1360–1364. [Google Scholar] [CrossRef]
- Copeland, R.A. Enzymes; Wiley: Hoboken, NJ, USA, 2023. [Google Scholar] [CrossRef]
- Spartan 10 for Windows; Wavefunction Inc.: Irvine, CA, USA, 2011.
- Ren, L.; Qin, X.; Cao, X.; Wang, L.; Bai, F.; Bai, G.; Shen, Y. Structural Insight into Substrate Specificity of Human Intestinal Maltase-Glucoamylase. Protein Cell 2011, 2, 827–836. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, R.; Christensen, M.H. MolDock: A New Technique for High-Accuracy Molecular Docking. J. Med. Chem. 2006, 49, 3315–3321. [Google Scholar] [CrossRef] [PubMed]
- Dubey, K.; Dubey, R. Computation Screening of Narcissoside a Glycosyloxyflavone for Potential Novel Coronavirus 2019 (COVID-19) Inhibitor. Biomed J. 2020, 43, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Pilon, A.C.; Del Grande, M.; Silvério, M.R.S.; Silva, R.R.; Albernaz, L.C.; Vieira, P.C.; Lopes, J.L.C.; Espindola, L.S.; Lopes, N.P. Combination of GC-MS Molecular Networking and Larvicidal Effect against Aedes Aegypti for the Discovery of Bioactive Substances in Commercial Essential Oils. Molecules 2022, 27, 1588. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- de Oliveira, G.; Carnevale Neto, F.; Demarque, D.; de Sousa Pereira-Junior, J.; Sampaio Peixoto Filho, R.; de Melo, S.; da Silva Almeida, J.; Lopes, J.; Lopes, N. Dereplication of Flavonoid Glycoconjugates from Adenocalymma Imperatoris-Maximilianii by Untargeted Tandem Mass Spectrometry-Based Molecular Networking. Planta Med. 2016, 83, 636–646. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (μM) | MolDock Score | Rerank Score | Interaction with Lys 1420 |
|---|---|---|---|---|
| 1 | 0.72 | −111.929 | 44.4241 | Hydrogen bond |
| 2 | 1.05 | −111.304 | 50.206 | Hydrogen bond |
| No. | RT (min) | Compound | P% | MW | MF | L |
|---|---|---|---|---|---|---|
| 1 | 10.36 | Tetradecanoic acid | 98 | 228.37 | C14H28O2 | Nist20 |
| 2 | 12.58 | Neophytadiene | 96 | 278.5 | C20H38 | Nist20 |
| 3 | 14.77 | Methyl Palmitate | 98 | 270.5 | C17H34O2 | Nist20 |
| 4 | 15.79 | Palmitic acid | 99 | 256.42 | C16H32O2 | Nist20 |
| 5 | 16.66 | Ethyl palmitate | 98 | 284.5 | C18H36O2 | Nist20 |
| 6 | 19.92 | Phytol | 91 | 296.5 | C20H40O | Nist20 |
| 7 | 20.44 | Isolinoleic acid | 91 | 280.4 | C18H32O2 | Nist20 |
| 8 | 20.60 | 6-octadecanoic acid | 94 | 282.5 | C18H34O2 | Nist20 |
| 9 | 21.24 | Stearic acid | 95 | 284.5 | C18H36O2 | Nist20 |
| 10 | 21.46 | 1-hexacosanol | 90 | 382.7 | C26H54O | Nist20 |
| 11 | 22.89 | 1-docosane | 92 | 310.61 | C22H46 | Nist20 |
| 12 | 24.74 | Dotriacontane | 99 | 450.9 | C32H66 | Nist20 |
| 13 | 26.68 | Hexyl palmitate | 96 | 340.6 | C22H44O2 | Nist20 |
| 14 | 33.51 | 1,21 docosadiene | 99 | 306.6 | C22H42 | Nist20 |
| 15 | 34.99 | Octacosyl acetate | 99 | 452.8 | C30H60O2 | Nist20 |
| 16 | 35.16 | Heneicosane | 99 | 296.6 | C21H44 | Nist20 |
| 17 | 37.67 | 9-hexacosene | 99 | 364.7 | C26H52 | Nist20 |
| 18 | 37.78 | Tetracosane | 96 | 336.6 | C24H48 | Nist20 |
| 19 | 38.54 | Squalene | 95 | 410.7 | C30H50 | Nist20 |
| 20 | 39.07 | Alpha-tocospiro B | 91 | 448.7 | C28H48O4 | Nist20 |
| 21 | 39.75 | Alpha-tocospiro A | 93 | 462.7 | C29H50O4 | Nist20 |
| 22 | 41.58 | Delta-tocopherol | 96 | 402.65 | C27H46O2 | Nist20 |
| 23 | 43.10 | 1-nonadecene | 99 | 266.5 | C19H38 | Nist20 |
| 24 | 43.20 | Triacontane | 99 | 422.8 | C30H62 | Nist20 |
| 25 | 44.31 | Pentadecanal | 91 | 226.40 | C15H30O | Nist20 |
| 26 | 46.05 | Beta-amyrene (olean-12-ene) | 90 | 410.7 | C30H50 | Nist20 |
| 27 | 46.22 | D-Friedoolean-14-ene | 90 | 410.7 | C30H50 | Nist20 |
| 28 | 46.75 | Dl-alpha-tocopherol | 92 | 430.7 | C29H50O4 | Nist20 |
| 29 | 48.72 | Z-14-Nonacosene | 97 | 406.8 | C28H58 | Nist20 |
| 30 | 48.86 | Campesterol | 99 | 400.7 | C29H48O | Nist20 |
| 31 | 49.79 | Stigmasterol | 99 | 412.7 | C29H48O | Nist20 |
| 32 | 50.01 | Triacontanal | 90 | 436.8 | C30H60O | Nist20 |
| 33 | 51.46 | Sitosterol | 96 | 414.7 | C29H50O | Nist20 |
| 34 | 51.83 | alpha-amyrine | 90 | 426.7 | C30H50O | Nist20 |
| 35 | 52.97 | Lupenone | 99 | 424.7 | C30H48O | Nist20 |
| 36 | 53.46 | Lupeol | 91 | 426.7 | C30H50O | Nist20 |
| 37 | 54.94 | Sitostenone | 91 | 412.7 | C29H48O | Nist20 |
| 38 | 55.45 | Dotriacontanal | 95 | 464.8 | C32H64O | Nist20 |
| 39 | 58.38 | Phytyl stearate | 92 | 563.0 | C38H74O2 | Nist20 |
| No. | RT (min) | Compound | P% | MW | MF | L |
|---|---|---|---|---|---|---|
| 1 | 10.84 | Loliolide | 99 | 196.2 | C11H16O3 | Nist20 |
| 2 | 12.52 | Neophytadiene | 89 | 278.5 | C20H40O | Nist20 |
| 3 | 15.68 | Palmitic acid | 99 | 256.4 | C16H32 O2 | Nist20 |
| 4 | 16.66 | Ethyl palmitate | 95 | 284.5 | C18H36O2 | Nist20 |
| 5 | 19.34 | Phytol | 97 | 296.5 | C20H40O | Nist20 |
| 6 | 20.56 | 6-Octadecenoic acid | 95 | 282.5 | C18H34O2 | Nist20 |
| 7 | 21.23 | Octadecanoic acid | 99 | 284.48 | C18H36O2 | Nist20 |
| 8 | 34.99 | Octacosyl acetate | 99 | 452.8 | C30H60O2 | Nist20 |
| 9 | 35.14 | Tricosane | 95 | 324.6 | C23H48 | Nist20 |
| 10 | 40.47 | 1-tetracosene | 99 | 334.6 | C24H48 | Nist20 |
| 11 | 45.97 | Eicosane | 92 | 282.5 | C20H42 | Nist20 |
| 12 | 49.76 | Stigmasterol | 91 | 412.7 | C29H48O | Nist20 |
| 13 | 49.90 | Triacontanal | 97 | 436.8 | C30H60O | Nist20 |
| 14 | 51.34 | Sitosterol | 99 | 414.7 | C29H50O | Nist20 |
| 15 | 52.00 | Beta-amyrin | 97 | 426.7 | C30H50O | Nist20 |
| 16 | 52.69 | Lupenone | 99 | 424.7 | C30H48O | Nist20 |
| 17 | 53.28 | Lupeol | 96 | 426.7 | C30H50O | Nist20 |
| 18 | 55.43 | Dotriacontanal | 93 | 464.8 | C32H64O | Nist20 |
| No. | RT (min) | Compound | P% | MW | MF | L |
|---|---|---|---|---|---|---|
| 1 | 11.93 | Catechol | 96 | 110.11 | C6H6O2 | Nist20 |
| 2 | 15.57 | Hydroquinone | 90 | 110.11 | C6H6O2 | Nist20 |
| 3 | 16.88 | Resorcinol | 93 | 110.11 | C6H6O2 | Nist20 |
| 4 | 17.62 | 2-methoxy-4-vinylphenol | 86 | 150.17 | C9H10O2 | Nist20 |
| 5 | 19.68 | Syringol | 89 | 154.16 | C6H6O3 | Nist20 |
| 6 | 20.94 | 1,2,4-Benzenetriol | 98 | 126.11 | C6H6O3 | Nist20 |
| 7 | 21.37 | Pyrogallol | 97 | 126.11 | C6H6O3 | Nist20 |
| 8 | 25.67 | 2-hydroxy-5-methylbenzaldehyde | 86 | 136.15 | C8H8O2 | Nist20 |
| 9 | 29.29 | Dihydroactinidiolide | 91 | 180.24 | C11H16O2 | Nist20 |
| 10 | 36.51 | Dihydroconiferyl alcohol | 94 | 182.22 | C10H14O3 | Nist20 |
| 11 | 37–42 | -Methyl-4-O-methyl-D-arabinopyranoside | 72 | 178.18 | C7H14O5 | Nist20 |
| 12 | -3,4, Di-O-methyl-L-arabinopyranose | 72 | 178.18 | C7H14O5 | Nist20 | |
| 13 | -2-O-Methyl-D-mannopyranosa | 77 | 194.18 | C7H14O6 | Nist20 | |
| 14 | -3-O-methyl-D-fructose | 77 | 194.18 | C7H14O6 | Nist20 | |
| 15 | -Alpha-methyl mannofuranoside | 73 | 194.18 | C7H14O6 | Nist20 | |
| 16 | -4-O-methyl-mannose | 70 | 194.18 | C7H14O6 | Nist20 | |
| 17 | -4,6-di-O-methyl-alpha-d-galactose | 75 | 208.21 | C8H16O6 | Nist20 | |
| 18 | 43–46 | 3,4,6-tri-O-methyl-D-glucose | 77 | 222.24 | C9H18O6 | Nist20 |
| 19 | Methyl(methyl-4-O-methyl-alpha-D-mannopyranoside)uronate | 73 | 236.22 | C9H16O7 | Nist20 | |
| 20 | Neophytadiene | 97 | 278.5 | C20H38 | Nist20 | |
| 21 | 58.83 | Methyl Palmitate | 99 | 270.5 | C17H34O2 | Nist20 |
| 22 | 62.31 | Palmitic acid | 98 | 256.42 | C16H32O2 | Nist20 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cherigo, L.; Liao-Luo, J.; Fernández, J.; Martínez-Luis, S. Isolation of Alpha-Glucosidase Inhibitors from the Panamanian Mangrove Plant Mora oleifera (Triana ex Hemsl.) Ducke. Pharmaceuticals 2024, 17, 890. https://doi.org/10.3390/ph17070890
Cherigo L, Liao-Luo J, Fernández J, Martínez-Luis S. Isolation of Alpha-Glucosidase Inhibitors from the Panamanian Mangrove Plant Mora oleifera (Triana ex Hemsl.) Ducke. Pharmaceuticals. 2024; 17(7):890. https://doi.org/10.3390/ph17070890
Chicago/Turabian StyleCherigo, Lilia, Javier Liao-Luo, Juan Fernández, and Sergio Martínez-Luis. 2024. "Isolation of Alpha-Glucosidase Inhibitors from the Panamanian Mangrove Plant Mora oleifera (Triana ex Hemsl.) Ducke" Pharmaceuticals 17, no. 7: 890. https://doi.org/10.3390/ph17070890
APA StyleCherigo, L., Liao-Luo, J., Fernández, J., & Martínez-Luis, S. (2024). Isolation of Alpha-Glucosidase Inhibitors from the Panamanian Mangrove Plant Mora oleifera (Triana ex Hemsl.) Ducke. Pharmaceuticals, 17(7), 890. https://doi.org/10.3390/ph17070890