
Identification of Dihydropyrazolo[1,5-a]pyrazin-4(5H)-ones as Cyclic Products of β-Amidomethyl Vinyl Sulfone Alphavirus Cysteine Protease Inhibitors

 , , and
, , and
Abstract

1. Introduction
2. Results and Discussion
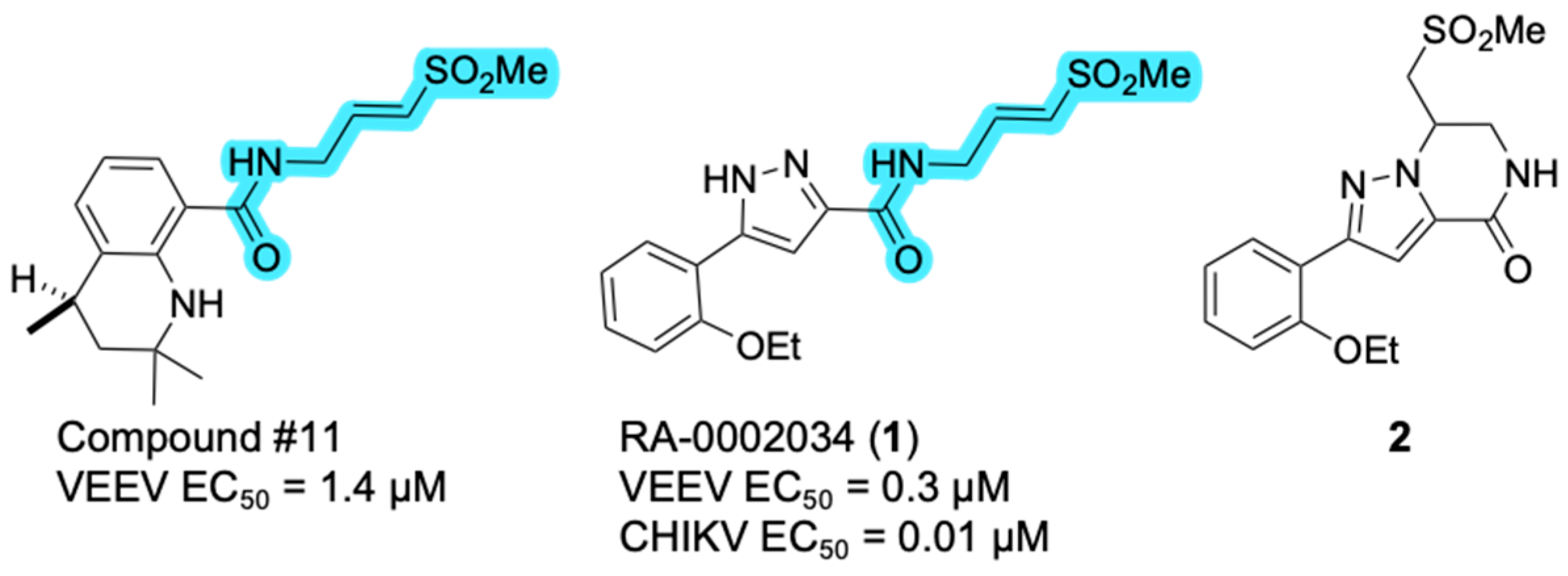
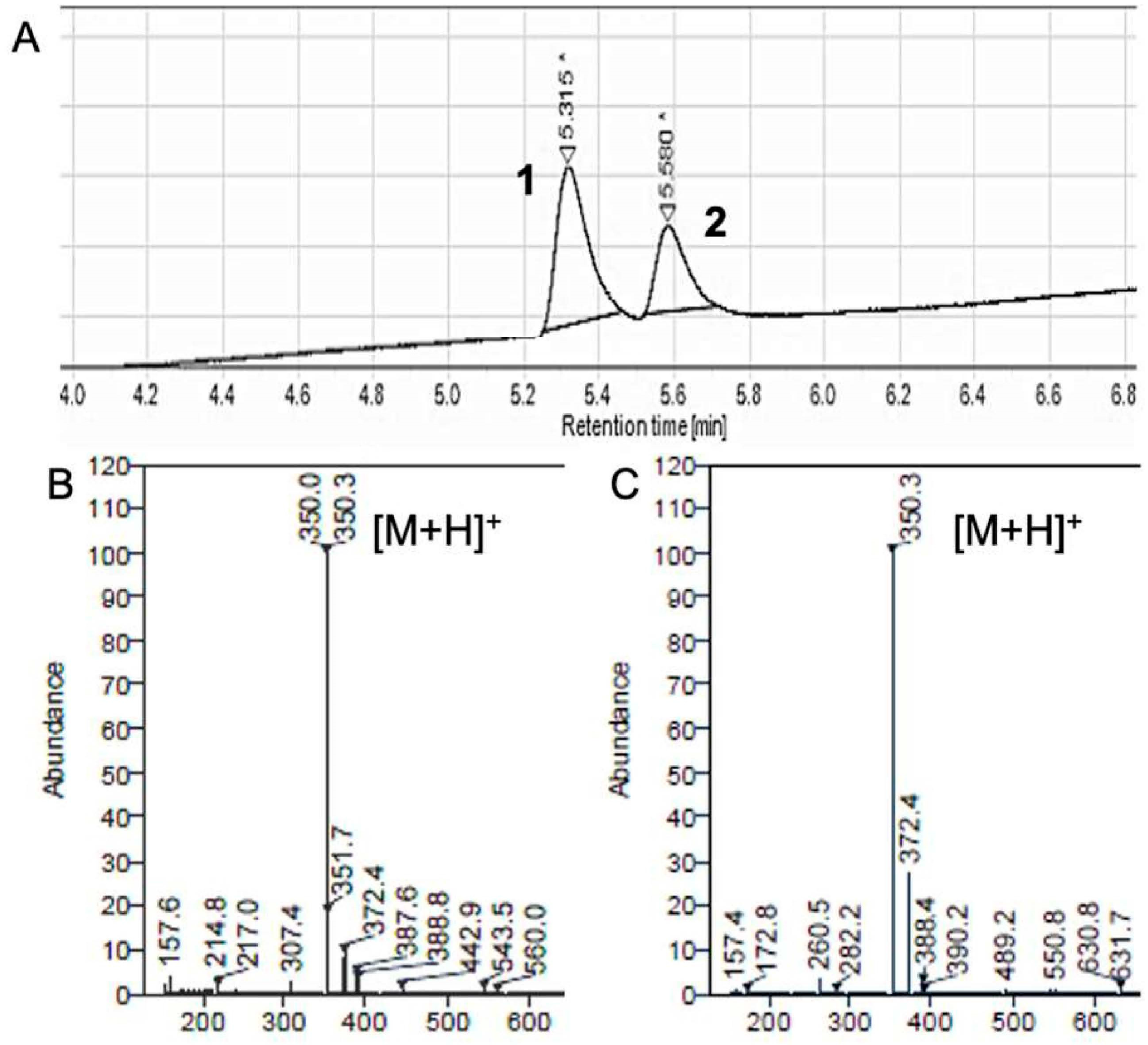
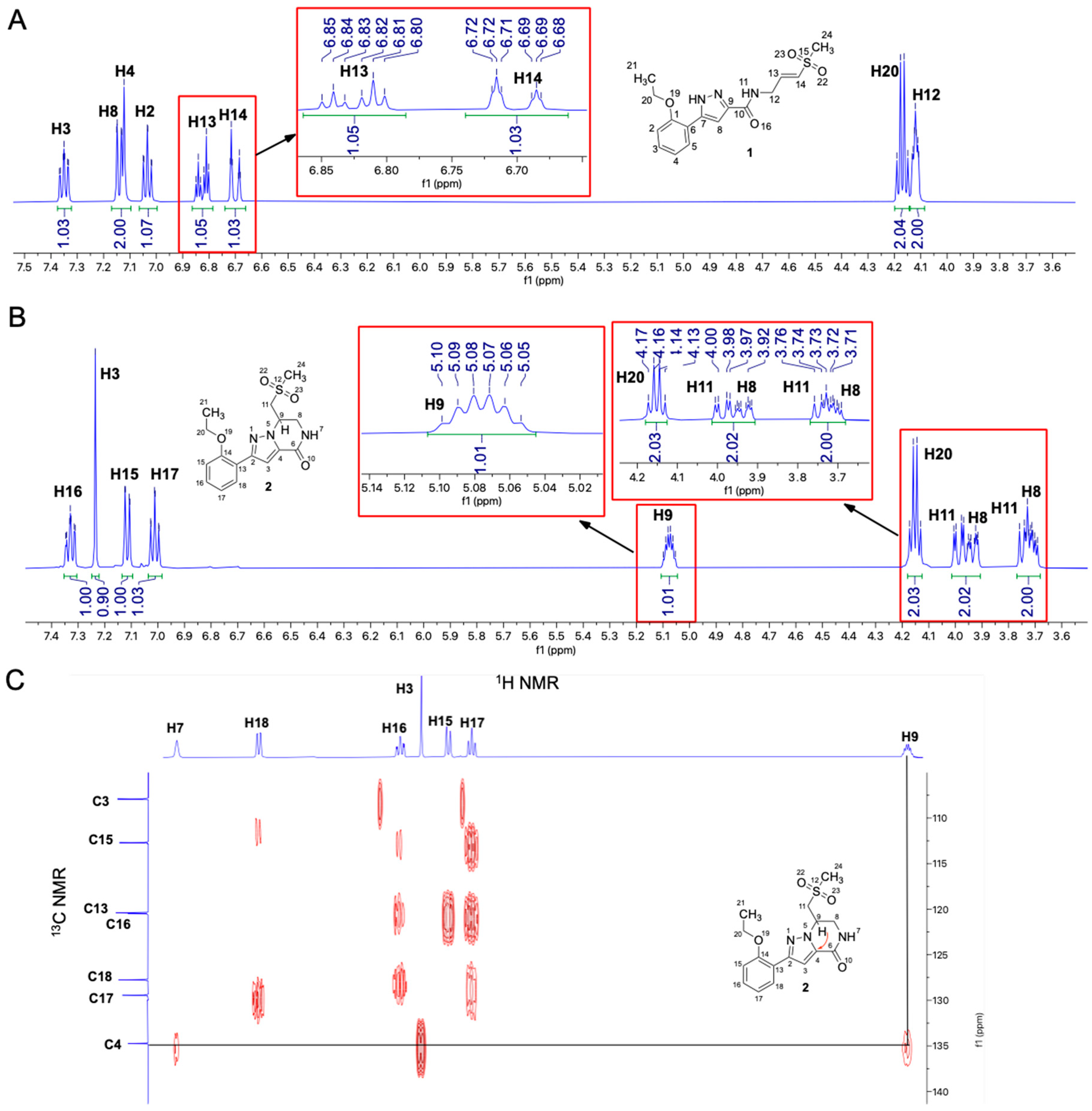
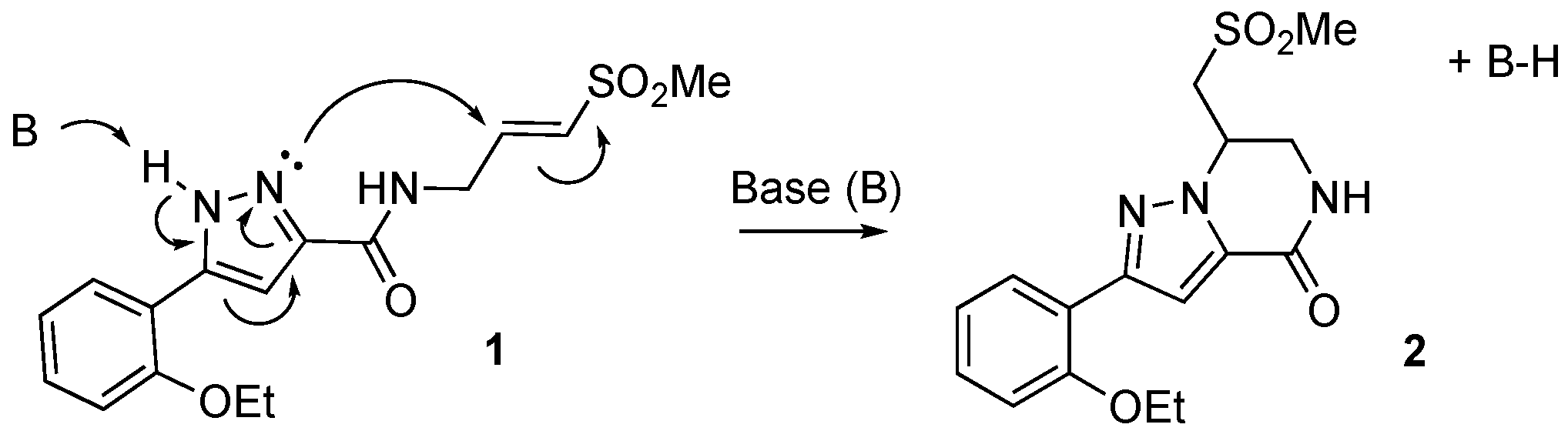
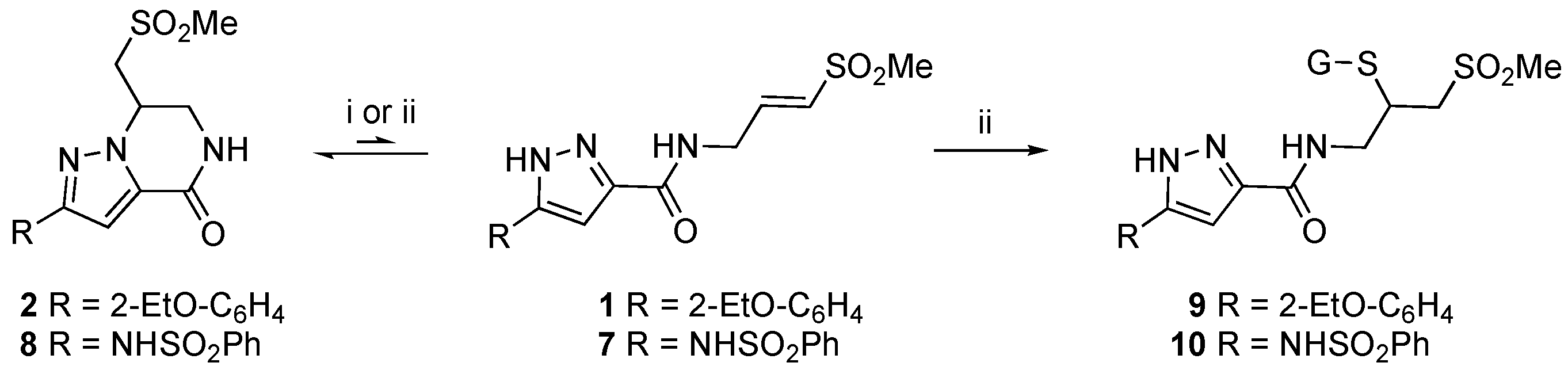
2.1. Identification of a Cyclic Product of Pyrazole-Substituted β-Amidomethyl Vinyl Sulfone 1
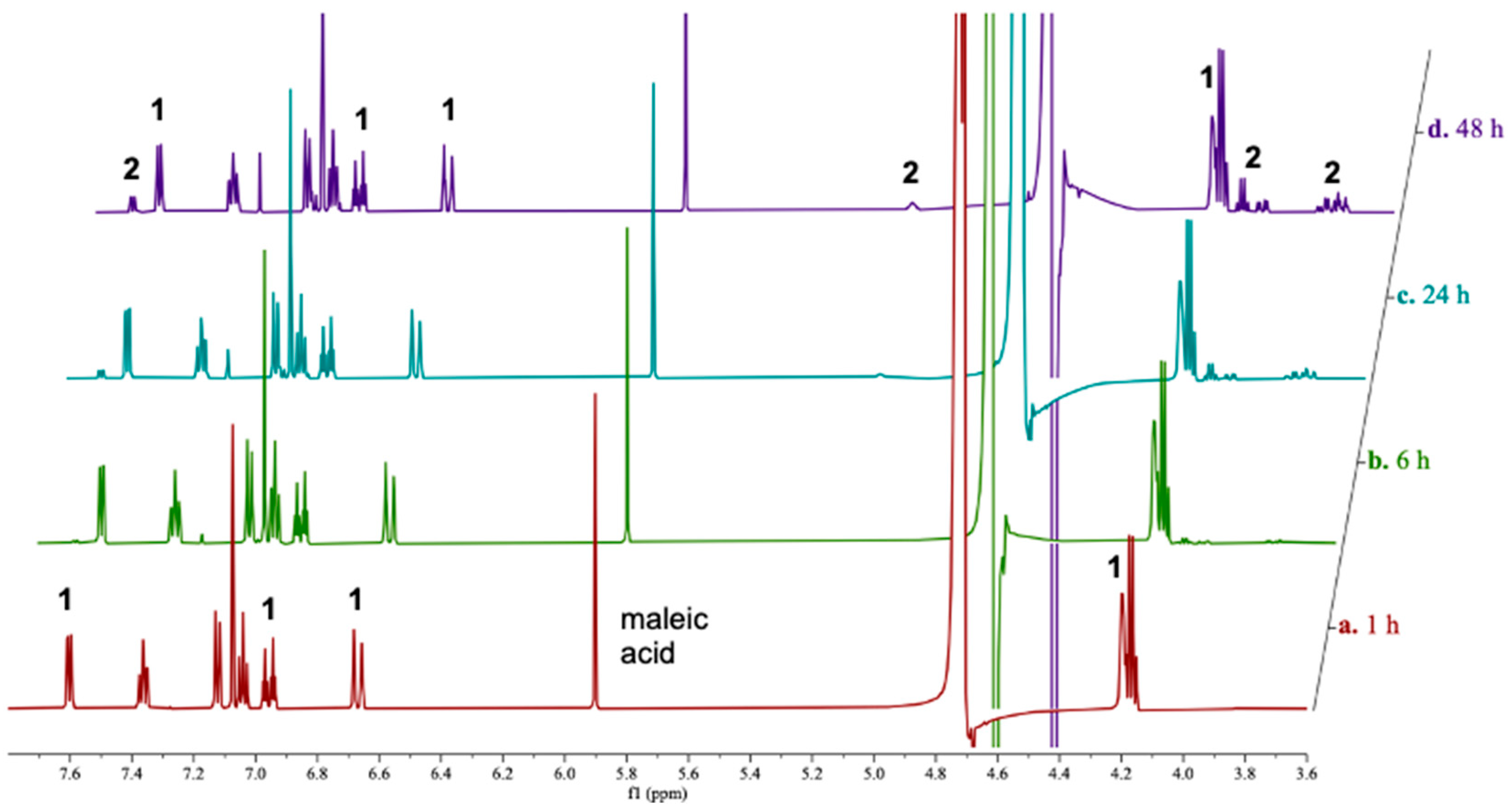
2.2. Stability of 1 in Neutral Phosphate Buffer
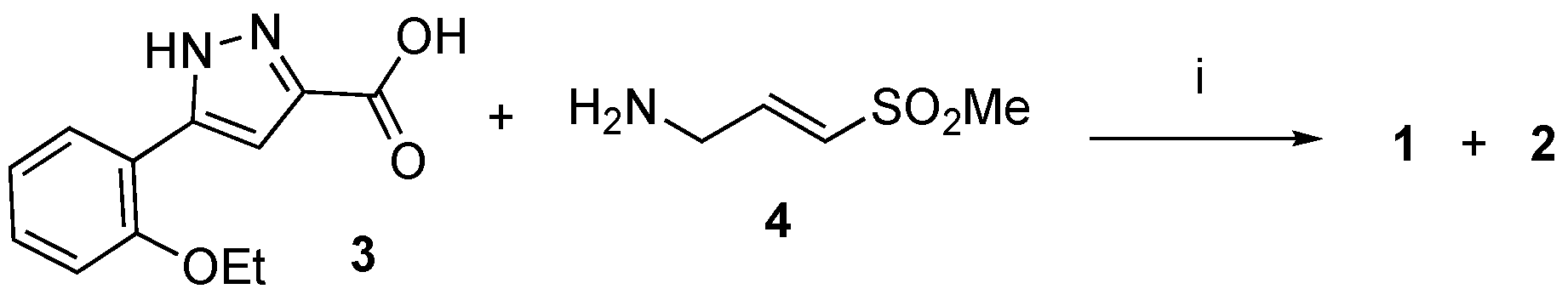
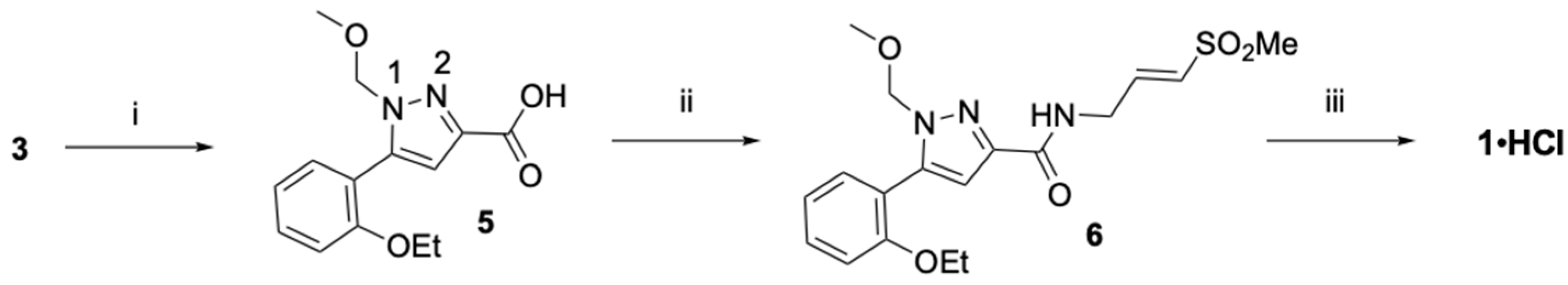
2.3. Optimized Synthesis of β-Amidomethyl Vinyl Sulfone 1
2.4. Optimized Synthesis of Dihydropyrazolo[1,5-a]pyrazin-4(5H)-one 2
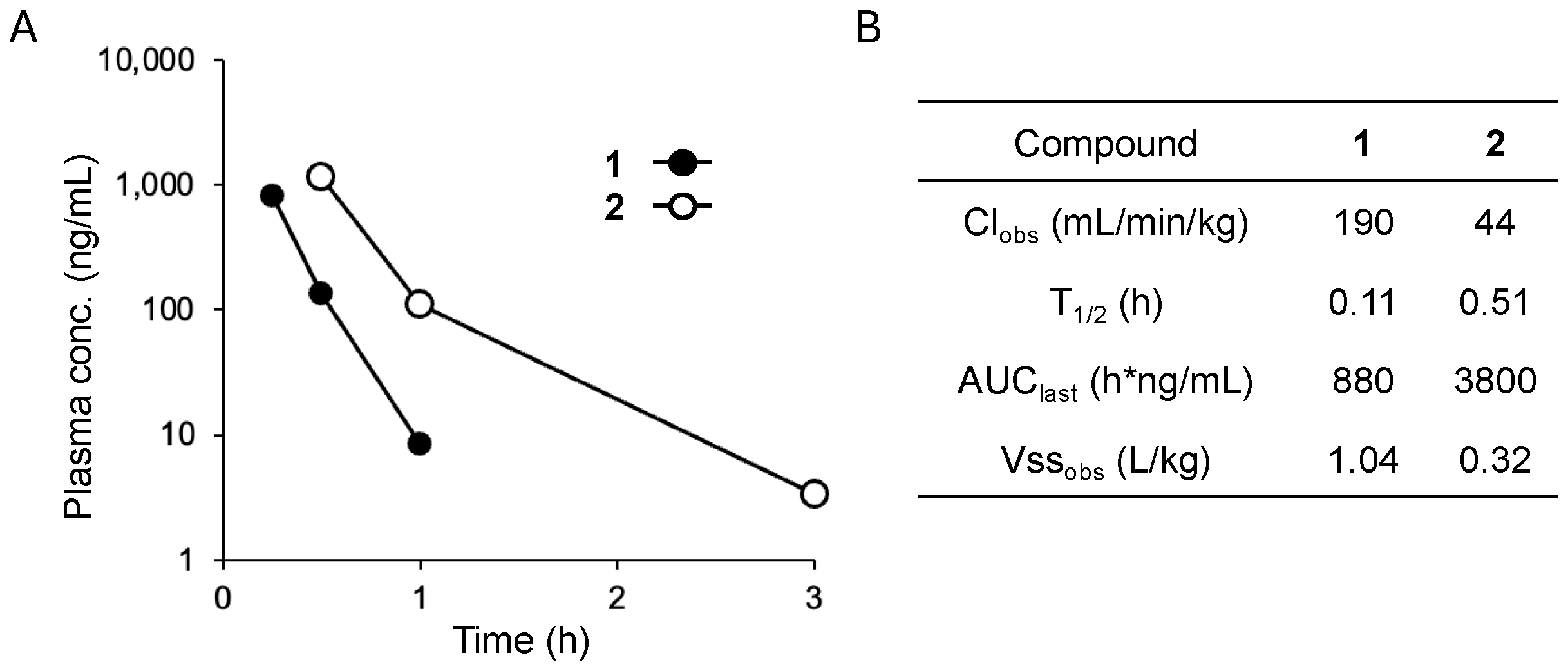
2.5. Pharmacokinetic Properties of 1 and 2
2.6. Reversibility of the Aza-Michael Reaction
3. Materials and Methods
3.1. General Methods
3.2. Optimized Synthesis Pyrazole Vinyl Sulfones (1, 7) and Dihydropyrazolo[1,5-a]pyrazin-4(5H)-ones (2, 8)
3.2.1. (E)-5-(2-Ethoxyphenyl)-N-(3-(methylsulfonyl)allyl)-1H-pyrazole-3-carboxamide (1)
3.2.2. 2-(2-Ethoxyphenyl)-7-((methylsulfonyl)methyl)-6,7-dihydropyrazolo[1,5-a]pyrazin-4(5H)-one (2)
3.2.3. (E)-N-(3-(Methylsulfonyl)allyl)-5-(phenylsulfonamido)-1H-pyrazole-3-carboxamide (7)
3.2.4. N-(7-((Methylsulfonyl)methyl)-4-oxo-4,5,6,7-tetrahydropyrazolo[1,5-a]pyrazin-2-yl)benzenesulfonamide (8)
3.3. 1H NMR Stability Assay
3.4. Pharmacokinetic Methods
3.5. GSH Capture Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Skidmore, A.M.; Bradfute, S.B. The life cycle of the alphaviruses: From an antiviral perspective. Antivir. Res. 2023, 209, 105476. [Google Scholar] [CrossRef] [PubMed]
- Suhrbier, A.; Jaffar-Bandjee, M.C.; Gasque, P. Arthritogenic alphaviruses—An overview. Nat. Rev. Rheumatol. 2012, 8, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Morens, D.M.; Folkers, G.K.; Fauci, A.S. Eastern Equine Encephalitis Virus—Another Emergent Arbovirus in the United States. N. Engl. J. Med. 2019, 381, 1989–1992. [Google Scholar] [CrossRef] [PubMed]
- Abu Bakar, F.; Ng, L.F.P. Nonstructural Proteins of Alphavirus-Potential Targets for Drug Development. Viruses 2018, 10, 71. [Google Scholar] [CrossRef]
- Zhang, H.; Harmon, M.; Radoshitzky, S.R.; Soloveva, V.; Kane, C.D.; Duplantier, A.J.; Ogungbe, I.V. Vinyl Sulfone-Based Inhibitors of Nonstructural Protein 2 Block the Replication of Venezuelan Equine Encephalitis Virus. ACS Med. Chem. Lett. 2020, 11, 2139–2145. [Google Scholar] [CrossRef]
- Merten, E.M.; Sears, J.D.; Leisner, T.M.; Hardy, P.B.; Ghoshal, A.; Hossain, M.A.; Asressu, K.H.; Brown, P.J.; Stashko, M.A.; Herring, L.E.; et al. Discovery of a cell-active chikungunya virus nsP2 protease inhibitor using a covalent fragment-based screening approach. bioRxiv 2024. [Google Scholar] [CrossRef]
- Ahmadi, R.; Emami, S. Recent applications of vinyl sulfone motif in drug design and discovery. Eur. J. Med. Chem. 2022, 234, 114255. [Google Scholar] [CrossRef] [PubMed]
- Hillebrand, L.; Liang, X.J.; Serafim, R.A.M.; Gehringer, M. Emerging and Re-emerging Warheads for Targeted Covalent Inhibitors: An Update. J. Med. Chem. 2024, 67, 7668–7758. [Google Scholar] [CrossRef] [PubMed]
- McDowell, S.T.; Stirling, C.J.M. Elimination–addition. Part X. Rates of addition of amines to p-tolyl vinyl sulphone. J. Chem. Soc. B 1967, 343–348. [Google Scholar] [CrossRef]
- McDowell, S.T.; Stirling, C.J.M. Elimination–addition. Part XI. Electronic effects upon the reactivity of aryl vinyl sulphones towards amines. J. Chem. Soc. B 1967, 348–350. [Google Scholar] [CrossRef]
- El-Faham, A.; Albericio, F. Peptide coupling reagents, more than a letter soup. Chem. Rev. 2011, 111, 6557–6602. [Google Scholar] [CrossRef] [PubMed]
- Almena, I.; Díz-Barra, E.; Hoz, A.D.L.; Ruiz, J.; Sínchez-Migallón, A.; Elguero, J. Alkylation and arylation of pyrazoles under solvent-free conditions: Conventional heating versus microwave irradiation. J. Heterocycl. Chem. 1998, 35, 1263–1268. [Google Scholar] [CrossRef]
- Huang, A.; Orcid, W.K.; Lee, S.Y.C.; Kneitschel, N.; Chang, J.; Zhu, K.; Mello, T.; Bancroft, L.; Norman, N.J.; Zheng, S.-L. Regioselective Synthesis, NMR, and Crystallographic Analysis of N1-Substituted Pyrazoles. J. Org. Chem. 2017, 82, 8864–8872. [Google Scholar] [CrossRef]
- Xiao, Y.C.; Chen, F.E. The vinyl sulfone motif as a structural unit for novel drug design and discovery. Expert Opin. Drug Discov. 2024, 19, 239–251. [Google Scholar] [CrossRef]
- Ghoshal, A.; Asressu, K.H.; Hossain, M.A.; Brown, P.J.; Merten, E.M.; Sears, J.D.; Perveen, S.; Pearce, K.H.; Popov, K.I.; Moorman, N.J.; et al. Structure Activity of β-Amidomethyl Vinyl Sulfones as Covalent Inhibitors of Chikungunya nsP2 Cysteine Protease with Anti-alphavirus Activity. bioRxiv 2024. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Coupling Agent | Base | pKa | Solvent | Temp (°C) | Time (h) | Ratio of 1:2 b |
|---|---|---|---|---|---|---|---|
| 1 | HBTU | DIPEA | 10.9 | DMF | 25 | 16 | 60:40 |
| 2 | HATU | DIPEA | 10.9 | DMF | 25 | 2 | 60:40 |
| 3 | PyBOP | DIPEA | 10.9 | DMF | 25 | 16 | 60:40 |
| 4 | T3P | TEA | 10.7 | DMF | 25 | 2 | 60:40 |
| 5 | DIC | DMAP | 9.7 | DMF | 25 | 16 | 60:40 |
| 6 | EDC | HOBt | 4.6 | MeCN | 25 | 2 | 70:30 |
| 7 | TBTU | Pyridine | 5.2 | Pyridine | 25 | 2 | 100:0 |
| Entry | Base | Eq. | Solvent | Ratio of 1:2 b |
|---|---|---|---|---|
| 1 | K2CO3 | 3.5 | EtOH | 0:100 |
| 2 | K2CO3 | 3.5 | H2O | 100:0 |
| 3 | Na2CO3 | 3.0 | Dioxane/H2O | 0:100 |
| 4 | NaHCO3 | 3.0 | MeOH | 12:88 |
| 5 | NaHCO3 | 3.0 | H2O | 47:53 |
| 6 | TEA | 3.0 | MeOH | 51:49 |
| 7 | DIPEA | 3.0 | DMF | 4:96 |
| 8 | DBU | 0.5 | ACN | 8:92 |
| Time (h) | GSH-Adduct Formation (%) a | |||
|---|---|---|---|---|
| Acyclic | Cyclic | |||
| 1 | 7 | 2 | 8 | |
| 8 | 100 | 93 | 0 | 3 |
| 24 | 100 | 100 | 0 | 6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghoshal, A.; Magalhães, Á.F.; Asressu, K.H.; Hossain, M.A.; Todd, M.H.; Willson, T.M. Identification of Dihydropyrazolo[1,5-a]pyrazin-4(5H)-ones as Cyclic Products of β-Amidomethyl Vinyl Sulfone Alphavirus Cysteine Protease Inhibitors. Pharmaceuticals 2024, 17, 836. https://doi.org/10.3390/ph17070836
Ghoshal A, Magalhães ÁF, Asressu KH, Hossain MA, Todd MH, Willson TM. Identification of Dihydropyrazolo[1,5-a]pyrazin-4(5H)-ones as Cyclic Products of β-Amidomethyl Vinyl Sulfone Alphavirus Cysteine Protease Inhibitors. Pharmaceuticals. 2024; 17(7):836. https://doi.org/10.3390/ph17070836
Chicago/Turabian StyleGhoshal, Anirban, Álvaro F. Magalhães, Kesatebrhan Haile Asressu, Mohammad Anwar Hossain, Matthew H. Todd, and Timothy M. Willson. 2024. "Identification of Dihydropyrazolo[1,5-a]pyrazin-4(5H)-ones as Cyclic Products of β-Amidomethyl Vinyl Sulfone Alphavirus Cysteine Protease Inhibitors" Pharmaceuticals 17, no. 7: 836. https://doi.org/10.3390/ph17070836
APA StyleGhoshal, A., Magalhães, Á. F., Asressu, K. H., Hossain, M. A., Todd, M. H., & Willson, T. M. (2024). Identification of Dihydropyrazolo[1,5-a]pyrazin-4(5H)-ones as Cyclic Products of β-Amidomethyl Vinyl Sulfone Alphavirus Cysteine Protease Inhibitors. Pharmaceuticals, 17(7), 836. https://doi.org/10.3390/ph17070836