
Synthesis and Biological Evaluation of a Series of New Hybrid Amide Derivatives of Triazole and Thiazolidine-2,4-dione

 , , ,
, , ,
Abstract
1. Introduction
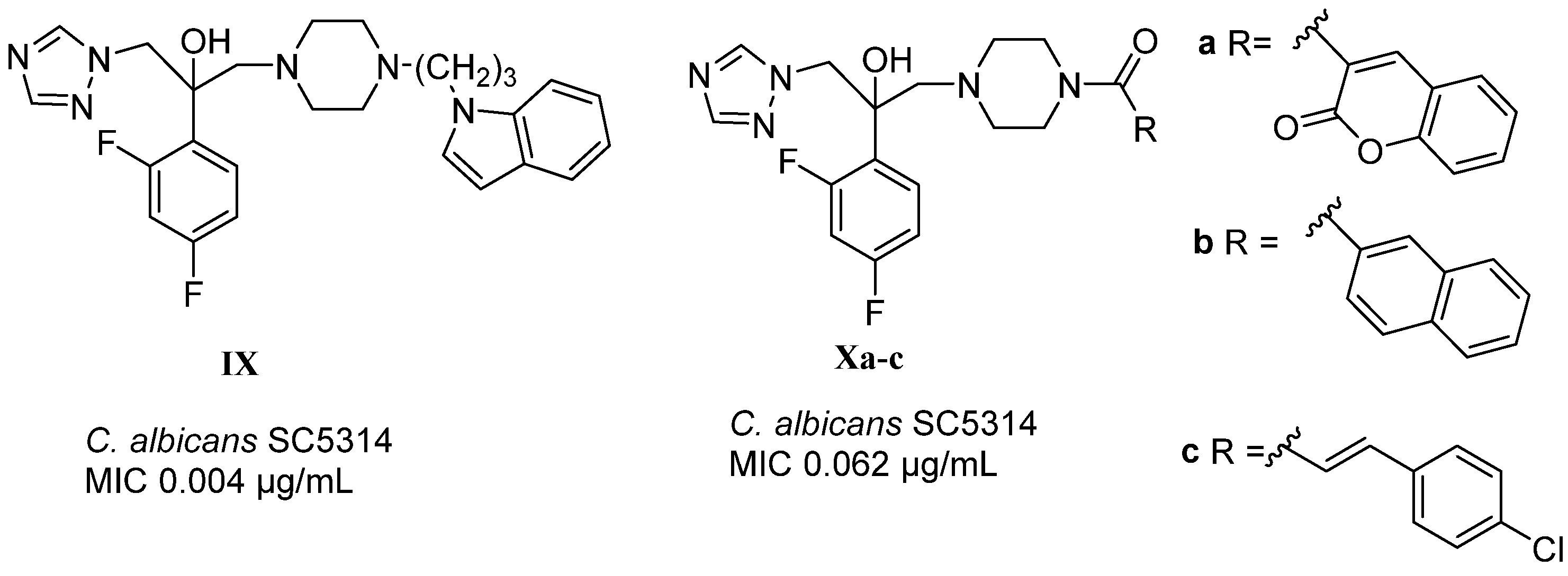
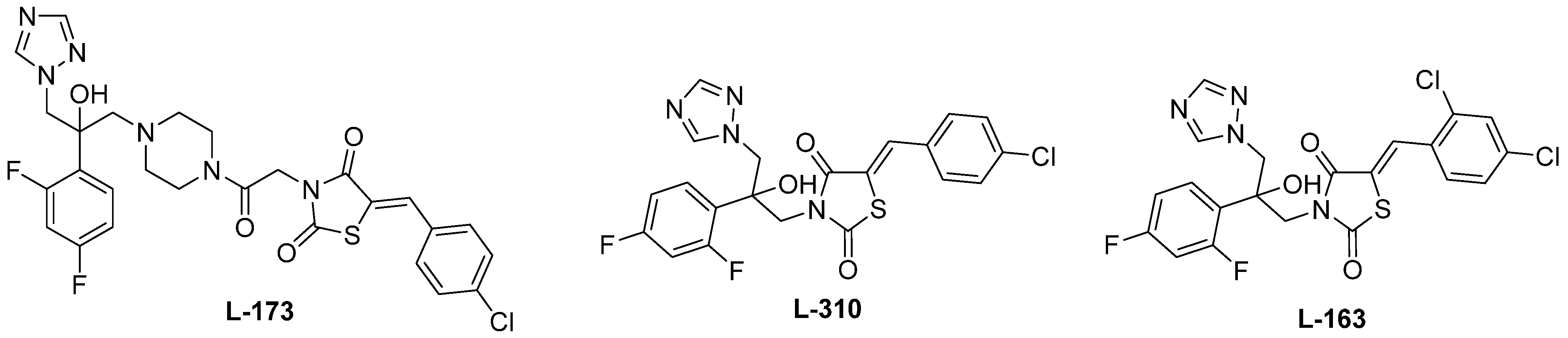
2. Results and Discussion
3. Microbiology
3.1. Characterization of the Compounds’ Biological Activity and SAR
3.2. Tests In Vivo
4. Molecular Modeling
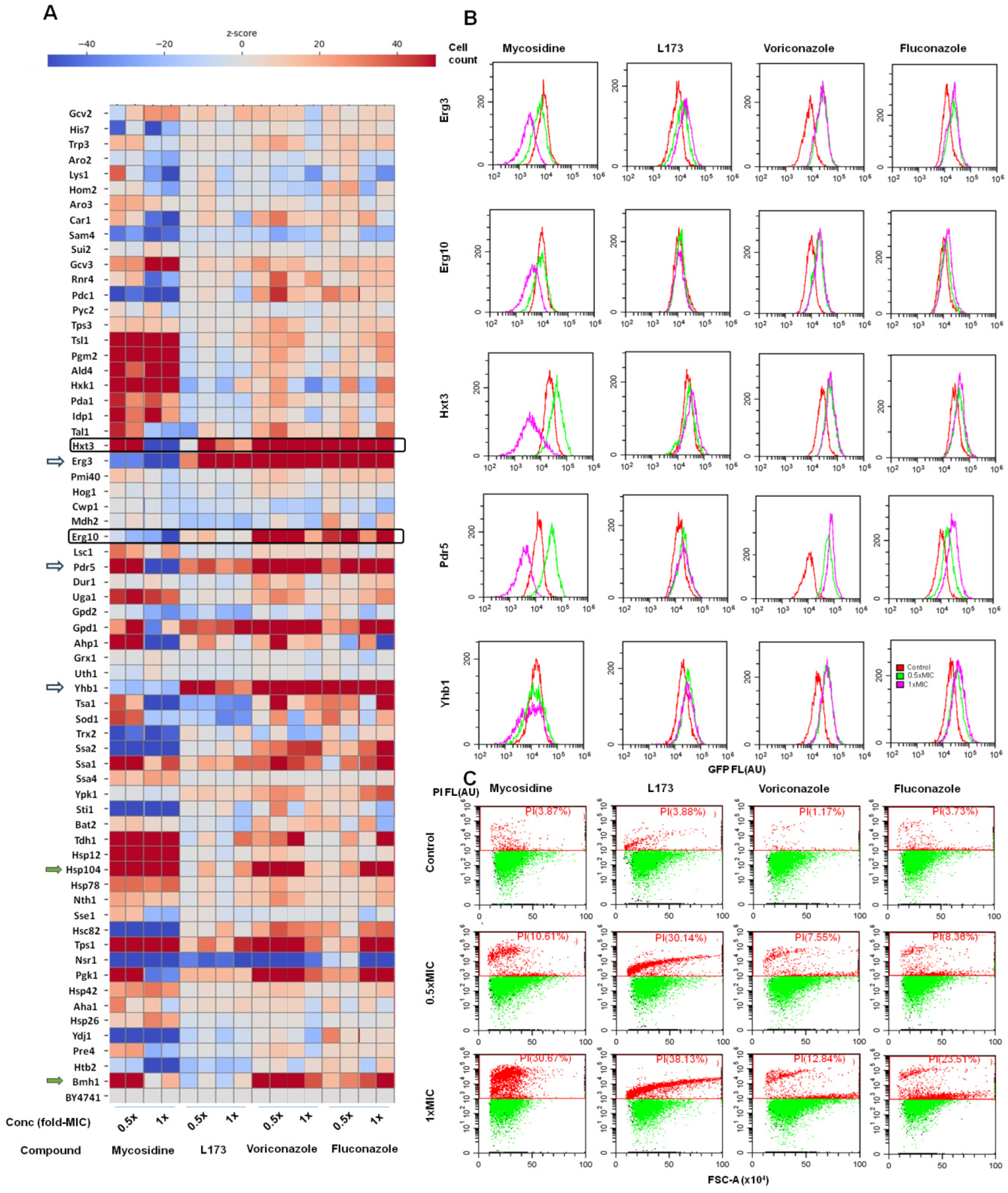
5. Biochemistry
6. Materials and Methods
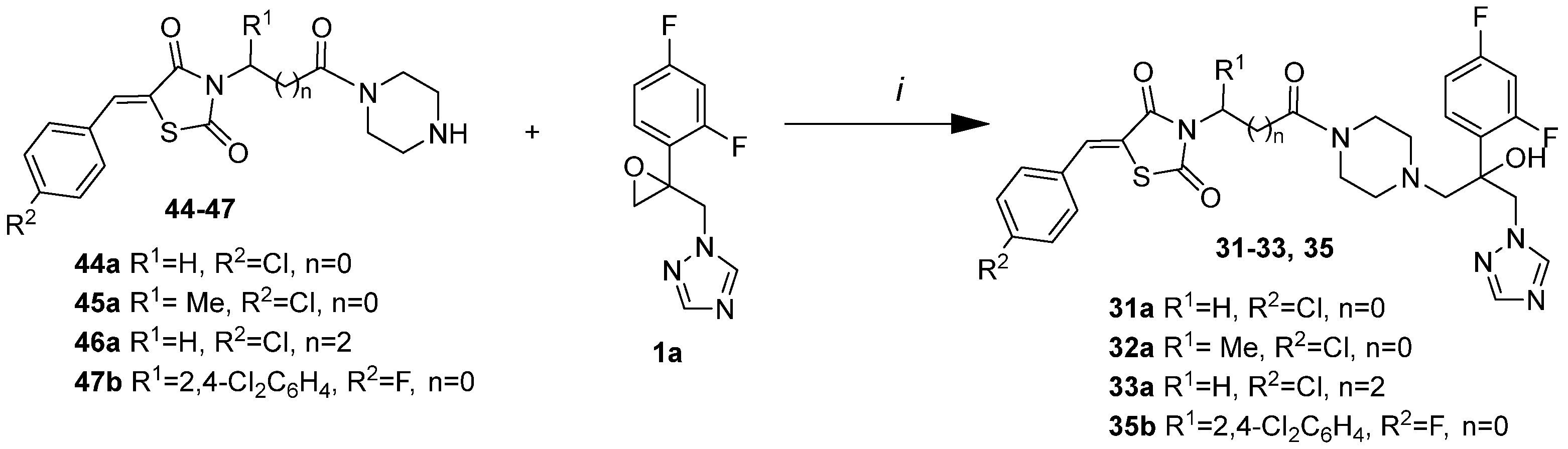
6.1. Chemistry
6.2. Biochemistry and Microbiology
6.3. Molecular Modeling
6.4. Analysis of Homology
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. Fungal Priority Pathogens List to Guide Research, Development and Public Health Action. Available online: www.who.int/publications/i/item/9789240060241 (accessed on 29 May 2024).
- McCarty, T.P.; Pappas, P.G. Antifungal Pipeline. Front. Cell. Infect. Microbiol. 2021, 11, 732223. [Google Scholar] [CrossRef]
- Shafiei, M.; Peyton, L.; Hashemzadeh, M.; Foroumadi, A. History of the development of antifungal azoles: A review on structures, SAR, and mechanism of action. Bioorg. Chem. 2020, 104, 104240. [Google Scholar] [CrossRef]
- Hu, Y.; Liu, Z.; Zha, G.; Long, S.; Sridhara, M.B.; Kumar, K.S.S.; Rakesh, K.P. Triazole derivatives as potential antifungal agents: A structure-activity relationship (SAR) studies. Process Biochem. 2023, 135, 102–118. [Google Scholar] [CrossRef]
- Emami, S.; Ghobadi, E.; Saednia, S.; Hashemi, S.M. Current advances of triazole alcohols derived from fluconazole: Design, in vitro and in silico studies. Eur. J. Med. Chem. 2019, 170, 173–194. [Google Scholar] [CrossRef]
- Zirngibl, L. Antifungal Azoles: A Comprehensive Survey of Their Structures and Properties; Wiley-VCH: Hoboken, NJ, USA, 1998. [Google Scholar]
- Ghobadi, E.; Saednia, S.; Emami, S. Synthetic approaches and structural diversity of triazolylbutanols derived from voriconazole in the antifungal drug development. Eur. J. Med. Chem. 2022, 231, 114161. [Google Scholar] [CrossRef]
- Teixeira, M.M.; Carvalho, D.T.; Sousa, E.; Pinto, E. New Antifungal Agents with Azole Moieties. Pharmaceuticals 2022, 15, 1427. [Google Scholar] [CrossRef]
- Jović, Z.; Janković, S.M.; Zečević, D.R.; Milovanović, D.; Stefanović, S.; Folić, M.; Milovanović, J.; Kostić, M. Clinical Pharmacokinetics of Second Generation Triazoles for the Treatment of Invasive Aspergillosis and Candidiasis. Eur. J. Drug Metab. Pharmacokinet. 2019, 44, 139–157. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.F.; Rose, M.E.; Ellis, S.W.; England, H.; Kelly, S.L. Defective sterol C5-6 desaturation and azole resistance: A new hypothesis for the mode of action of azole antifungals. Biochem. Biophys. Res. Commun. 1989, 164, 1170–1175. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, R.S.; Robbins, N.; Cowen, L.E. Regulatory Circuitry Governing Fungal Development, Drug Resistance, and Disease. Microbiol. Mol. Biol. Rev. 2011, 75, 213–267. [Google Scholar] [CrossRef]
- Chen, X.; Xue, W.; Zhou, J.; Zhang, Z.; Wei, S.; Liu, X.; Sun, X.; Wang, W.; Li, S. De-repression of CSP-1 activatesadaptive responses to antifungalazoles. Sci. Rep. 2015, 6, 19447. [Google Scholar] [CrossRef]
- Levshin, I.B.; Simonov, A.Y.; Lavrenov, S.N.; Panov, A.A.; Grammatikova, N.A.; Alexandrov, A.A.; Ghazy, E.S.M.O.; Savin, N.A.; Gorelkin, P.V.; Erofeev, A.S.; et al. Antifungal Thiazolidines: Synthesis and Biological Evaluation of Mycosidine Congeners. Pharmaceuticals 2022, 15, 563. [Google Scholar] [CrossRef] [PubMed]
- Borate, H.B.; Sawargave, S.P.; Chavan, S.P.; Chandavarkar, M.A.; Iyer, R.; Tawte, A.; Kangire, G.S. Novel hybrids of fluconazole and furanones: Design, synthesis and antifungal activity. Bioorg. Med. Chem. Lett. 2011, 21, 4873–4878. [Google Scholar] [CrossRef]
- Wu, S.; Zhang, Y.; He, X.; Che, X.; Wang, S.; Liu, Y.; Sheng, C. From Antidiabetic to Antifungal: Discovery of Highly Potent Triazole-Thiazolidinedione Hybrids as Novel Antifungal Agents. ChemMedChem 2014, 9, 2639–2646. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Chen, H.; Chen, X.; Wang, X.; Kong, L.; Yang, M. Novel Triazoles with potent and broad-spectrum Antifiungal actovity in vitro and in vivo. J. Med. Chem. 2023, 66, 7497. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.G. New Triadimenol Derivative with Antifungal Activity and Its Preparation Process and Medicinal Use. CN17068334A, 14 December 2005. [Google Scholar]
- Tay, M.Z.; Poh, C.M.; Renia, L.; Macary, P.A.; Ng, L.F.P. The trinity of COVID-19: Immunity, inflammation and intervention. Nat. Rev. Immunol. 2020, 20, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Levshin, I.B.; Sandulenko, Y.B.; Polyakova, M.V.; Grammatikova, N.E.; Vasileva, N.V.; Bogomolova, T.S. Hybrid Amides Based on Triazole and Thiazolidine Having Antimicrobial Activity. Patent RU 2703997C1 (WO2020122766A1), 13 December 2018. [Google Scholar]
- Volkova, T.V.; Levshin, I.B.; Perlovich, G.L. New antifungal compound: Solubility thermodynamics and partitioning processes in biologically relevant solvents. J. Mol. Liq. 2020, 310, 113148. [Google Scholar] [CrossRef]
- Volkova, T.V.; Perlovich, G.L. Comparative analysis of solubilization and complexation characteristics for new antifungal compound with cyclodextrins. Impact of cyclodextrins on distribution process. Eur. J. Pharm. Sci. 2020, 154, 105531. [Google Scholar] [CrossRef] [PubMed]
- Savin, N.; Erofeev, A.; Timoshenko, R.; Vaneev, A.; Rudenko, A.; Salikhov, S.; Grammatikova, N.; Levshin, I.; Korchev, Y.; Gorelkin, P. Investigation of the antifungal and anticancer effects of the novel synthesized thiazolidinedione by ion- conductance microscopy. Cells 2023, 12, 1666. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, K.; Bai, G.; Huang, L.; Wu, Q.; Pan, W.; Yu, S. Synthesis and antifungal activity of novel triazole compounds containing piperazine moiety. Molecules 2014, 19, 11333. [Google Scholar] [CrossRef]
- Lebouvier, N.; Giraud, F.; Corbin, T.; Min Na, Y.; Le Baut, G.; Marchand, P.; Le Borgne, M. Efficient microwave-assisted synthesis of 1-(1H-indol-1-yl)-2-phenyl-3-(1H-1,2,4-triazol-1-yl)propan-2-ols as antifungals. Tetrahedron Lett. 2006, 47, 6479–6483. [Google Scholar] [CrossRef]
- Xie, Y.; Liu, Y.; Gong, G.; Rinderspacher, A.; Deng, S.-X.; Smith, D.H.; Toebben, U.; Tzilianos, E.; Branden, L.; Vidović, D.; et al. Discovery of a novel submicromolar inhibitor of the lymphoid specific tyrosine phosphatase. Bioorg. Med. Chem. Lett. 2008, 18, 2840–2844. [Google Scholar] [CrossRef]
- Christoffa, R.M.; Soares da Costab, T.P.; Bayata, S.; Holienc, J.K.; Matthew, A.; Peruginib, M.A.; Abbotta, B.A. Synthesis and Structure-Activity Relationship Studies of 2,4-Thiazolidinediones and Analogous Heterocycles as Inhibitors of Dihydrodipicolinate Synthase. Bioorg. Med. Chem. 2021, 52, 116518. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Mao, Q.; Zhang, B.; Lv, J.; Ping, K.; Zhang, P.; Lin, F.; Zhao, J.; Feng, Y.; Yang, J.; et al. Thiazolidinedione-based structure modification of celastrol provides thiazolidinedione-conjugated derivatives as potent agents against non-small cell lung cancer cells through mitochondria-mediated apoptotic pathway. J. Nat. Prod. 2022, 85, 1147–1156. [Google Scholar] [CrossRef] [PubMed]
- Levshin, I.B.; Tsurkan, A.A.; V’yunov, K.A.; Ginak, A.I. ChemInform Abstract: Preparative method of obtaining 5-arylideneselena- and thiazolidine-2,4-diones. Chem. Informationsdienst 1983, 14. [Google Scholar] [CrossRef]
- Maccari, R.; Ottana, R.; Curinga, C.; Vigorita, M.G.; Rakowitz, D.; Steindl, T.; Langer, T. Structure–Activity Relationships and Molecular Modelling of 5-Arylidene-2,4-Thiazolidinediones Active as Aldose Reductase Inhibitors. Bioorg. Med. Chem. 2005, 13, 2809–2823. [Google Scholar] [CrossRef]
- Schaude, M.; Ackerbauer, H.; Mieth, H. Inhibitory Effect of anthifungal agents on gem tube formation in Candida albicans. Mykosen 1987, 6, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Ghazy, E.S.M.O.; Bidiuk, V.A.; Ryabov, F.; Riabova, O.; Mitkevich, O.V.; Stanishevskiy, Y.M.; Levshin, I.B.; Alexandrova, L.A.; Jasko, M.V.; Makarov, D.A.; et al. SCRAPPY—A single cell rapid assay of proteome perturbation in yeast uncovers a (joint) role of aromatic amino acids (and oxidative stress) in the toxicity of lipophilic nucleoside analogs. bioRxiv 2024. [Google Scholar] [CrossRef]
- Clinical and Laboratory Standards Institute. Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts and Fifilamentous Fungi; Approved Standard M27-A3 and M38-A2; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2008. [Google Scholar]
- ISO Standard No. 16256:2012; Clinical Laboratory Testing and In Vitro Diagnostic Test Systems—Reference Method for Testing the In Vitro Activity of Antimicrobial Agents against Yeast Fungi Involved in Infectious Diseases. International Organization for Standardization: Geneva, Switzerland, 2012. Available online: https://www.iso.org/ru/standard/56027.html (accessed on 14 May 2024).
- Allen, W.J.; Fochtman, B.C.; Balius, T.E.; Rizzo, R.C. Customizable de novo design strategies for DOCK: Application to HIVgp41 and other therapeutic targets. J. Comput. Chem. 2017, 38, 2641–2663. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Sagatova, A.; Keniya, M.V.; Wilson, R.K.; Monk, B.C.; Tyndall, J.D.A. Structural insights into binding of the antifungal drug fluconazole to Saccharomyces cerevisiae lanosterol 14α-demethylase. Antimicrob. Agents Chemother. 2015, 59, 4982–4989. [Google Scholar] [CrossRef]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef] [PubMed]
- Boratyn, G.M.; Camacho, C.; Cooper, P.S.; Coulouris, G.; Fong, A.; Ma, N.; Madden, T.L.; Matten, W.T.; McGinnis, S.D.; Merezhuk, Y.; et al. BLAST: A more efficient report with usability improvements. Nucleic Acids Res. 2013, 41, W29–W33. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Whiting, E.; Raje, M.R.; Chauhan, J.; Wilder, P.T.; Van Eker, D.; Hughes, S.J.; Bowen, N.G.; Vickers, G.E.A.; Fenimore, I.C.; Fletcher, S. Discovery of Mcl-1 inhibitors based on a thiazolidine-2,4-dione scaffold. Bioorg. Med. Chem. Lett. 2018, 28, 523–528. [Google Scholar] [CrossRef] [PubMed]
- Abd Alhameed, R.; Almarhoon, Z.; Bukhari, S.I.; El-Faham, A.; de la Torre, B.G.; Albericio, F. Synthesis and Antimicrobial Activity of a New Series of Thiazolidine-2,4-diones Carboxamide and Amino Acid Derivatives. Molecules 2020, 25, 105. [Google Scholar] [CrossRef]
- Council of Europe. European Convention for the Protection of Vertebrate Animals Used for Experimental and Other Purposes; Council of Europe: Strasbourg, France, 1986. [Google Scholar]
- National State Standard GOST P 53434-2009; The Russian Federation Standard. The Principles of Good Laboratory Practice. Standartinform: Moscow, Russia, 2010.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
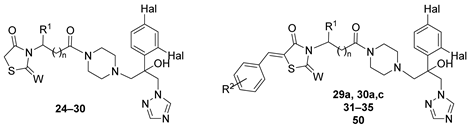 | |||||||
|---|---|---|---|---|---|---|---|
| № | W | n | R1 | R2 | Hal | Method | Yield, % |
| 24 | O | 0 | H | - | F | A | 52 |
| 25 | O | 0 | Me | - | F | A | 25 |
| 26 | O | 2 | H | - | F | A | 69 |
| 27 | O | 3 | H | - | F | A | 65 |
| 28 | O | 0 | 2,4-Cl2Ph | - | F | A | 34 |
| 29 | S | 0 | H | - | F | A | 20 |
| 30 | S | 0 | H | - | Cl | A | 28 |
| 31a | O | 0 | H | 4-Cl | F | A, B, K | 69, 51, 57 |
| 31b | O | 0 | H | 4-F | F | A | 72 |
| 31c | O | 0 | H | 4-OMe | F | A | 61 |
| 31d | O | 0 | H | 4-Et | F | A | 72 |
| 31e | O | 0 | H | 4-i-Pr | F | A | 65 |
| 31f | O | 0 | H | 4-t-Bu | F | A | 63 |
| 31g | O | 0 | H | 2,4-Cl2 | F | A, K | 58, 48 |
| 32a | O | 0 | Me | 4-Cl | F | A, B | 72, 46 |
| 33a | O | 2 | H | 4-Cl | F | A, B, K | 41, 32, 42 |
| 33b | O | 2 | H | 4-F | F | A | 37 |
| 33g | O | 2 | H | 2,4-Cl2 | F | A | 45 |
| 33h | O | 2 | H | 2-OH-3,5-Cl2 | F | K | 51 |
| 33i | O | 2 | H | 2-Cl-6-(4-MePip) | F | K | 46 |
| 34a | O | 3 | H | 4-Cl | F | K | 44 |
| 34b | O | 3 | H | 4-F | F | K | 48 |
| 34g | O | 3 | H | 2,4-Cl2 | F | K | 44 |
| 34h | O | 3 | H | 2-OH-3,5-Cl2 | F | K | 41 |
| 34i | O | 3 | H | 2-Cl-6-(4-MePip) | F | K | 38 |
| 35b | O | 0 | 2,4-Cl2Ph | 4-F | F | K | 33 |
| 50a | O | 1 | H | 4-Cl | F | C | 18 |
| 50c | O | 1 | H | 4-OMe | F | C | 10 |
| 50g | O | 1 | H | 2,4-Cl2 | F | C | 20 |
| 29a | S | 0 | H | 4-Cl | F | K | 60 |
| 30a | S | 0 | H | 4-Cl | Cl | K | 58 |
| 30c | S | 0 | H | 4-OMe | Cl | K | 39 |
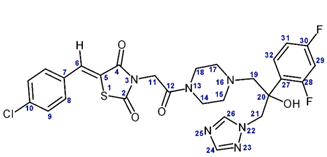  | |||||
|---|---|---|---|---|---|
| Position | L-173 | L-310 | |||
| δ(1H), ppm | δ(13C), ppm | δ(1H), ppm | δ(13C), ppm | ||
| 2 | - | 166.8 | - | 167.4 | - |
| 3 | - | - | - | - | 162.6 |
| 4 | - | 165.2 | - | 166.0 | - |
| 5 | - | 121.7 | - | 122.0 | - |
| 6 | 7.92 | 132.1 | 7.86 | 131.5 | - |
| 7 | - | 135.4 | - | 135.3 | - |
| 8 | 7.63 | 131.8 | 7.62 | 131.9 | - |
| 9 | 7.58 | 129.5 | 7.58 | 129.6 | - |
| 10 | - | 131.7 | - | 132.2 | - |
| 11 | 4.54 | 42.6 | 4.11; 4.13 | 48.1 | - |
| 12 | - | 162.9 | - | - | - |
| 13 | - | - | - | - | - |
| 14 | 3.32 | 41.8 | - | - | - |
| 15 | 2.38; 2.41 | 53.7 | - | - | - |
| 16 | - | - | - | - | - |
| 17 | 2.43; 2.48 | 54.0 | - | - | - |
| 18 | 3.37 | 44.1 | - | - | - |
| 19 | 2.71; 2.88 | 63.4 | - | - | - |
| 20 | 5.74 (OH) | 74.7 | 6.11 (OH) | 74.3 | - |
| 21 | 4.58 | 55.5 | 4.59; 4.81 | 55.0 | - |
| 22 | - | - | - | - | 211.6 |
| 23 | - | - | - | - | 298.4 |
| 24 | 7.75 | 150.5 | 7.69 | 150.6 | - |
| 25 | - | - | - | - | 252.3 |
| 26 | 8.29 | 144.9 | 8.27 | 145.0 | - |
| 27 | - | 126.0 | - | 123.9 | - |
| 28 | - | 158.9 | - | 159.5 | - |
| 29 | 7.14 | 103.8 | 7.17 | 104.0 | - |
| 30 | - | 161.7 | - | 162.1 | - |
| 31 | 6.95 | 110.8 | 6.87 | 110.9 | - |
| 32 | 7.41 | 129.8 | 7.25 | 129.7 | - |
| Strains |  |  |  |  | ||||
|---|---|---|---|---|---|---|---|---|
| 3a | 4a | L-31016 | L-16316 | |||||
| 24 h | 48 h | 24 h | 48 h | 24 h | 48 h | 24 h | 48 h | |
| C. parapsilosis ATCC 22019 | 1 | 2–4 | 32 | >32 | 0.5 | 1.0 | 0.5–1.0 | 2 |
| A. niger 37a | 8 | 16–32 | 4 | 16 | ||||
| M. canis B-200 | 16 | 32 | 0.5 | 0.5–1.0 | ||||
| T. rubrum 2002 | 16 | >32 | 4 | 4 | ||||
| № | C. parapsilosis ATCC 22019 | A. niger 37a | A. fumigatus 46645 | M. canis B-200 | T. rubrum 2002 | |
|---|---|---|---|---|---|---|
| 24 h | 48 h | |||||
| 38a | >32 | >32 | >32 | >32 | >32 | >32 |
| 39a | 4 | 8 | >32 | >32 | >32 | >32 |
| 39b | 4 | 8 | >32 | >32 | >32 | >32 |
| 44a | 16 | 32 | >32 | >32 | 32 | 32 |
| 45a | 32 | 32 | >32 | >32 | 32 | 32 |
| 46a | 32 | 32 | >32 | >32 | 32 | 32 |
| 47a | 0.25 | 0.5 | 32 | 32 | 8 | 8–16 |
| 47b | 0.25 | 0.5 | 32 | 32 | 8 | 8 |
| Strains | C. parapsilosis ATCC 22019 | C. albicans ATCC 24433 | C. parapsilosis 58L | C. tropicalis 3019 | C. glabrata 61L | Cryptococcus neoformans | C. albicans 8 (R) | C. krusei 432M | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 24 h | 48 h | 24 h | 48 h | 24 h | 48 h | 24 h | 48 h | 24 h | 48 h | 24 h | 48 h | 24 h | 48 h | 24 h | 48 h | |
| 47a | 0.5 | 2 | 16 | 32 | 0.5 | 2 | 0.5 | 2 | 4 | 32 | 16 | 16 | 2 | 32 | 8 | 32 |
| 47b | 0.5 | 1 | 16 | 32 | 0.5 | 1 | 0.5 | 1 | 4 | 16 | 16 | 16 | 2 | 32 | 8 | 16 |
| Flc * | 4 | 4 | >32 | >32 | 8 | 16 | 16 | 16 | 8 | 16 | 32 | 32 | >32 | >32 | 32 | >32 |
| № | C. parapsilosis ATCC 22019 | A. niger 37a | A. fumigatus 46645 | M. canis B-200 | T. rubrum 2002 | |
|---|---|---|---|---|---|---|
| 24 h | 48 h | |||||
| 24 | >32 | >32 | >32 | >32 | >32 | >32 |
| 25 | >32 | >32 | >32 | >32 | >32 | >32 |
| 26 | >32 | >32 | >32 | >32 | >32 | >32 |
| 27 | >32 | >32 | >32 | >32 | >32 | >32 |
| 28 | >32 | >32 | >32 | >32 | >32 | >32 |
| 29 | >32 | >32 | >32 | >32 | >32 | >32 |
| 29a | 0.03 | 0.25 | >32 | >32 | 0.5–1 | 0.5 |
| 30 | >32 | >32 | >32 | >32 | >32 | >32 |
| 30a | 0.5 | 0.5–1 | >32 | >32 | 0.5–1 | 0.5 |
| 30c | <0.015 | 0.06 | 8 | 16 | 0.125–0.015 | 1.0–0.015 |
| 31a (L-173) | 0.03 | 0.03 | 1 | 2 | 0.015 | 0.015 |
| 31b | 0.25 | 2 | 4 | 4 | 0.25 | 0.25 |
| 31c | 0.03 | 0.06 | >32 | >32 | 0.125–0.03 | 1.0–0.03 |
| 31d | 0.03 | 0.5 | 0.25–0.5 | 4 | 1 | 0.5 |
| 31e | 0.125 | 0.25 | 0.5 | 4 | 2 | 0.5–1 |
| 31f | 1 | 2 | 1 | 2–4 | 2 | 0.5 |
| 31g | 0.03 | 0.06 | 2 | 4 | 0.06 | 0.03 |
| 32a | 0.125 | 0.5 | 2 | 2 | 0.5 | 1 |
| 33a | 0.015 | 0.5–1 | 4 | 4 | 0.5–1 | 0.25 |
| 33b | 4 | 8–16 | 8 | 8 | 0.5 | 0.25 |
| 33h | 8 | 8 | >32 | >32 | 32 | 32 |
| 33i | 2 | 2–4 | >32 | >32 | 16 | 16 |
| 34a | 2 | 2–4 | 4 | 4 | 0.5 | 0.25 |
| 34b | 0.5 | 16 | 4 | 4 | 0.5 | 0.25 |
| 34h | 4 | 4 | >32 | >32 | 16 | 8 |
| 34i | 8 | 8 | >32 | >32 | 16 | 8 |
| 35b | >32 | >32 | >32 | >32 | >32 | >32 |
| 37g | 1 | 1–2 | 4 | 4–8 | 0.5–1 | 32 |
| 38g | 2 | >32 | 8 | 8 | 1 | 0.25 |
| 50a | 0.125 | 0.25 | 4 | 4 | 0.25 | 0.125 |
| 50c | 1 | 1 | 8 | 8–16 | 4 | 2 |
| 50g | 0.5 | 0.5–1 | 8 | 8 | 0.25 | 0.125 |
| Flu | 0.5 | 4 | >32 | 16 | 32 | >32 |
| Itra | 0.03 | 0.06 | 0.5 | 2 | 0.5 | 0.25 |
| AmB | 1 | 2 | 0.5 | 0.25–0.5 | 0.125 | 0.25 |
| Strain \ Compound | C. albicans 8P | C. albicans 8 R | C. albicans 604M | C. parapsilosis 58L | C. utilis 84 | C. tropicalis 3019 | C. glabrata 61L | C. krusei 432M | Cryptococcus neoformans | |
|---|---|---|---|---|---|---|---|---|---|---|
| Itraconazole | 24 h | 16 | 32 | 32 | 0.25 | 0.125 | 2 | 0.25 | 0.5 | 0.125 |
| 48 h | 16–32 | 32 | 32 | 0.5 | 0.5 | 4 | 1 | 0.5 | 0.125 | |
| Ketoconazole | 24 h | 8 | 8 | 8 | 0.06 | 0.03 | 0.25 | 0.25 | 0.25 | 0.03 |
| 48 h | 8 | 8 | 8 | 0.5 | 0.03 | 0.5 | 0.25 | 1 | 0.03 | |
| 29a | 24 h | 8 | 8 | 8 | 0.5 | 1 | 1 | 1 | 1 | 0.25 |
| 48 h | 8 | 16 | 32 | 2 | 2 | 1 | 16 | 2 | 0.25 | |
| 30a | 24 h | 16 | 32 | 8 | 8 | 8 | 32 | 1 | 16 | 0.5 |
| 48 h | 16 | 32 | 16 | 16 | 16 | 32 | 2 | 32 | 0.5 | |
| 31a | 24 h | 0.015 | 4 | 32 | 0.125 | 0.125 | 0.5 | 0.5 | 2 | 0.03 |
| 48 h | 4 | 32 | 32 | 0.125 | 0.25 | 0.5 | 2 | 4 | 0.03 | |
| 31d | 24 h | 32 | 32 | 16 | 0.25 | 2 | 0.25 | 0.25 | 1 | 0.25 |
| 48 h | >32 | 32 | >32 | 1 | 8 | 1 | 2 | 2 | 4 | |
| 31e | 24 h | 32 | 32 | 16 | 0.125 | 16 | 1 | 0.25 | 4 | 1 |
| 48 h | >32 | 32 | >32 | 0.25 | 16 | 2 | >32 | 8 | 8 | |
| 31f | 24 h | 16 | >32 | 8 | 0.25 | 16 | 1 | 1 | 1 | 0.015 |
| 48 h | 16 | >32 | 16 | 0.5 | 16 | 2 | >32 | 2 | 0.06 | |
| 31g | 24 h | 8 | 8 | 8 | 0.25 | 0.25 | 2 | 2 | 1 | 0.06 |
| 48 h | 8 | 8 | 8 | 0.5 | 0.25 | 4 | 4 | 1 | 0.06 | |
| 32a | 24 h | 16 | 32 | 32 | 0.5 | 2 | 2 | 1 | 4 | <0.5 |
| 48 h | 32 | 32 | 32 | 2 | 4 | 8 | 4 | 8 | <0.5 | |
| Strain | Strain-N | HKI 12034018, L-173 | Voriconazole | Remark |
|---|---|---|---|---|
| Candida albicans | 2016-303 | <0.016 | <0.016/0.03 | Ecninocandin-R (Mut) |
| Candida auris | 2019-731 | 2 | 8 | Azol-R |
| Candida glabrata | 2018-606 | >8 | >8 | Ecninocandin-R (Mut) |
| Candida guilliermondii | 2020-194 | 0.25 | 0.125 | |
| Candida krusei | 2017-144 | 2 | 0.25 | |
| Candida parapsilosis | ATCC 22019 | 0.03 | 0.03 | |
| Aspergillus fumigatus | 2020-364 | >8 | >8 | Azol-R (Mut) |
| Aspergillus fumigatus | ATCC 204305 | 1 | 0.5 | Azol-S |
| Fusarium petrolifilum | 2020-183 | >8 | >8 | |
| Lomentospora prolificans | 2020-066 | >8 | >8 | |
| Rhizopus arrhizus | 2020-227 | 0.25 | 4 | |
| Scedosporium apiospermum | 2020-135 | >8 | 0.5 |
| Animal Group | Administered Compound | Dose, mg/kg | % Suppression ** | p *** |
|---|---|---|---|---|
| 1 | L-173 * | 5 | 22.33 | 0.00001 |
| 2 | L-173 * | 12 | 30.86 | 0.00001 |
| 3 | L-173 * | 25 | 41.04 | 0.00001 |
| 4 | Voriconazole * | 12 | 17.00 | 0.00001 |
| D (control) | - | 0 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Levshin, I.B.; Simonov, A.Y.; Panov, A.A.; Grammatikova, N.E.; Alexandrov, A.I.; Ghazy, E.S.M.O.; Ivlev, V.A.; Agaphonov, M.O.; Mantsyzov, A.B.; Polshakov, V.I. Synthesis and Biological Evaluation of a Series of New Hybrid Amide Derivatives of Triazole and Thiazolidine-2,4-dione. Pharmaceuticals 2024, 17, 723. https://doi.org/10.3390/ph17060723
Levshin IB, Simonov AY, Panov AA, Grammatikova NE, Alexandrov AI, Ghazy ESMO, Ivlev VA, Agaphonov MO, Mantsyzov AB, Polshakov VI. Synthesis and Biological Evaluation of a Series of New Hybrid Amide Derivatives of Triazole and Thiazolidine-2,4-dione. Pharmaceuticals. 2024; 17(6):723. https://doi.org/10.3390/ph17060723
Chicago/Turabian StyleLevshin, Igor B., Alexander Yu. Simonov, Alexey A. Panov, Natalia E. Grammatikova, Alexander I. Alexandrov, Eslam S. M. O. Ghazy, Vasiliy A. Ivlev, Michael O. Agaphonov, Alexey B. Mantsyzov, and Vladimir I. Polshakov. 2024. "Synthesis and Biological Evaluation of a Series of New Hybrid Amide Derivatives of Triazole and Thiazolidine-2,4-dione" Pharmaceuticals 17, no. 6: 723. https://doi.org/10.3390/ph17060723
APA StyleLevshin, I. B., Simonov, A. Y., Panov, A. A., Grammatikova, N. E., Alexandrov, A. I., Ghazy, E. S. M. O., Ivlev, V. A., Agaphonov, M. O., Mantsyzov, A. B., & Polshakov, V. I. (2024). Synthesis and Biological Evaluation of a Series of New Hybrid Amide Derivatives of Triazole and Thiazolidine-2,4-dione. Pharmaceuticals, 17(6), 723. https://doi.org/10.3390/ph17060723