
Activation of AMPK/mTOR-Driven Autophagy and Suppression of the HMGB1/TLR4 Pathway with Pentoxifylline Attenuates Doxorubicin-Induced Hepatic Injury in Rats

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
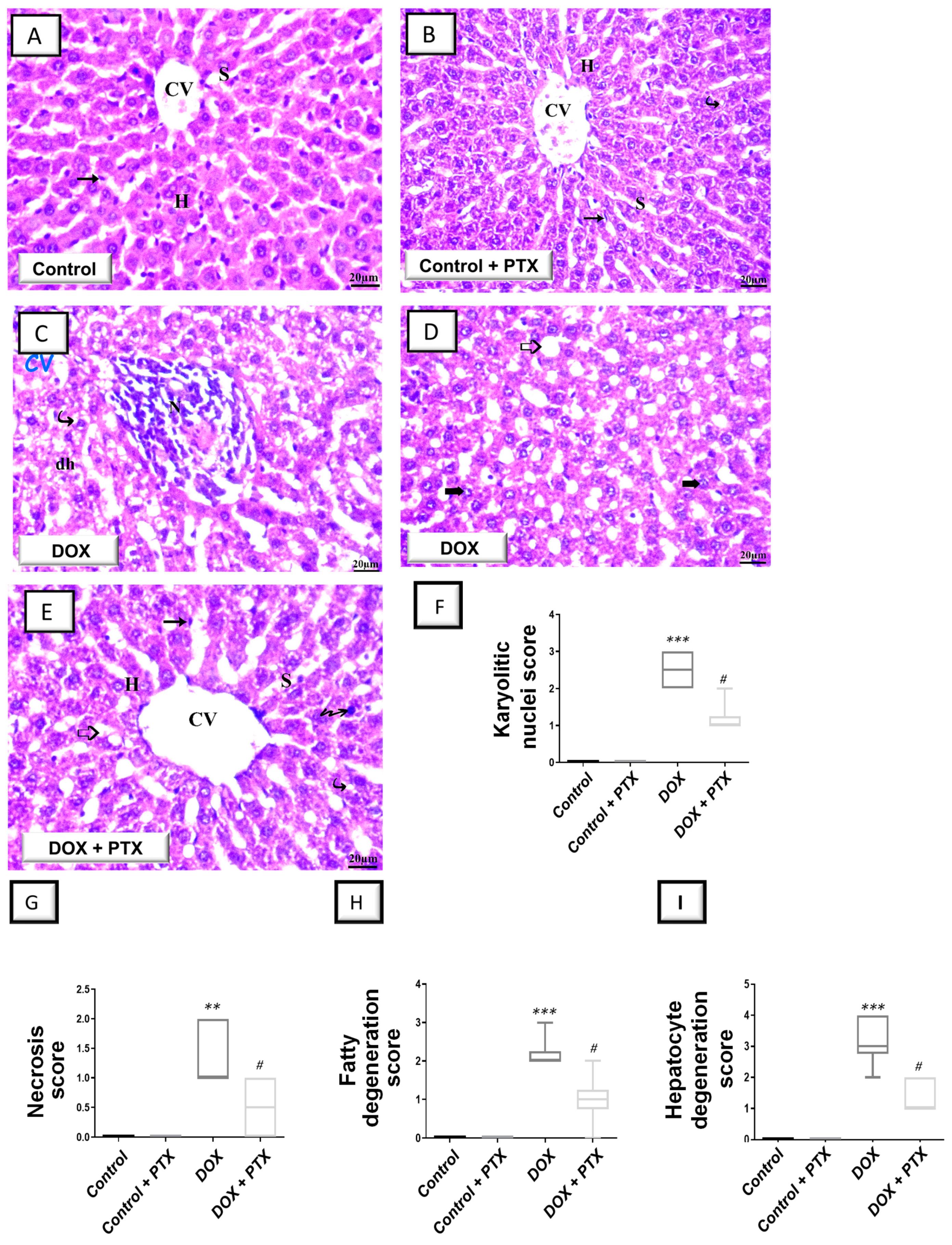
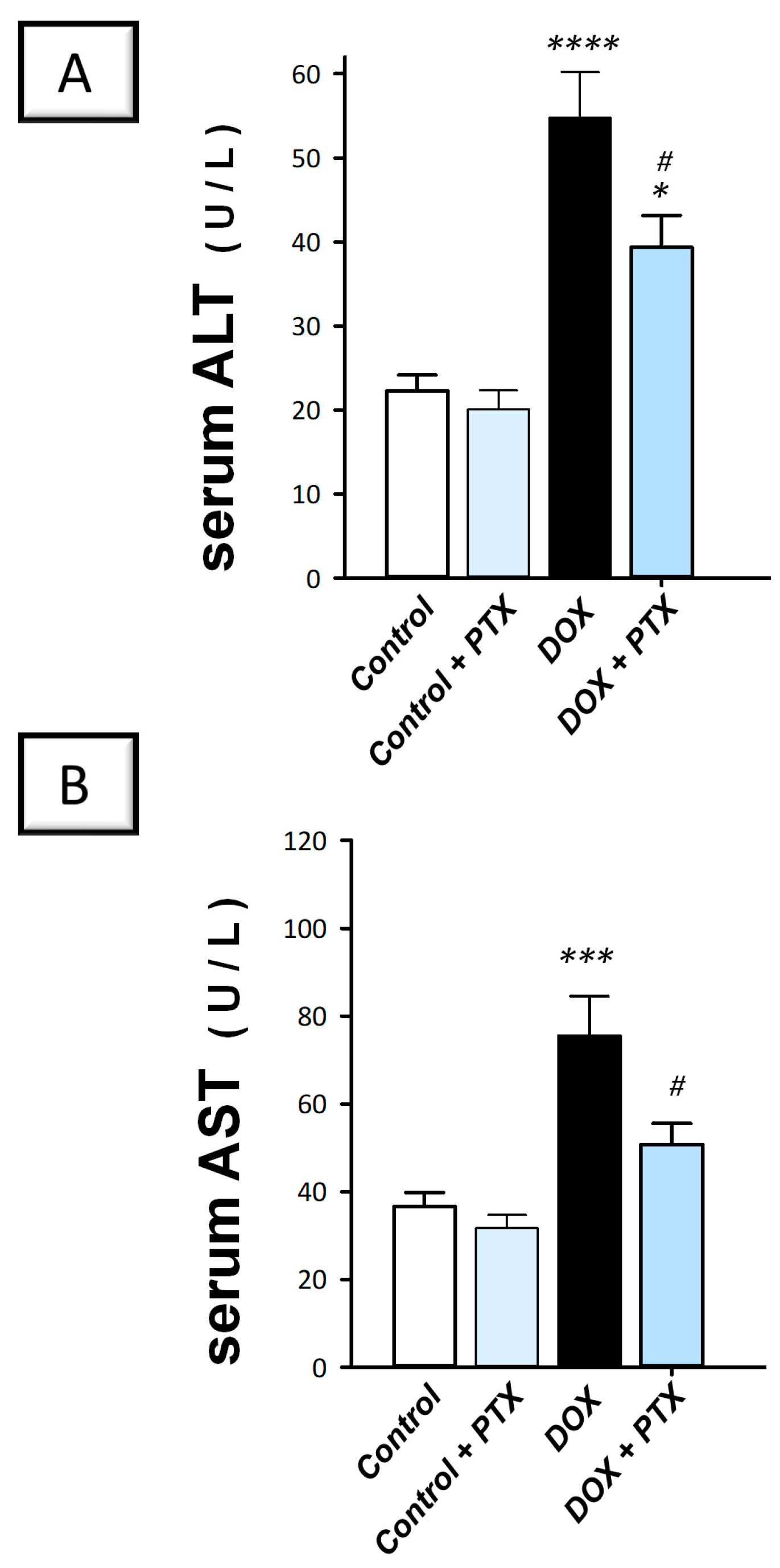
2.1. PTX Improved Hepatic Pathological Signs and Hepatic Cellular Integrity Markers in DOX-Intoxicated Rats
2.2. PTX Suppressed the Pro-Inflammatory Response in the Hepatic Tissue of DOX-Intoxicated Animals
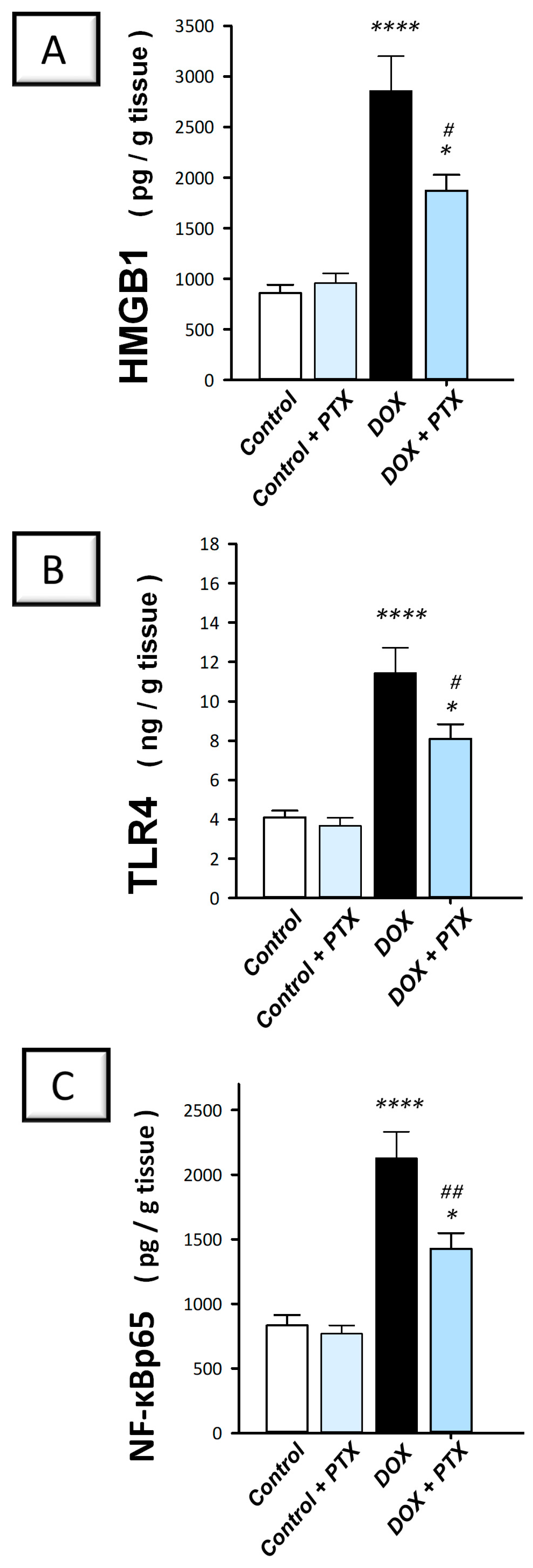
2.3. PTX Inhibited the Inflammation-Linked HMGB1/TLR4/NF-κB Axis in the Hepatic Tissue of DOX-Intoxicated Rats
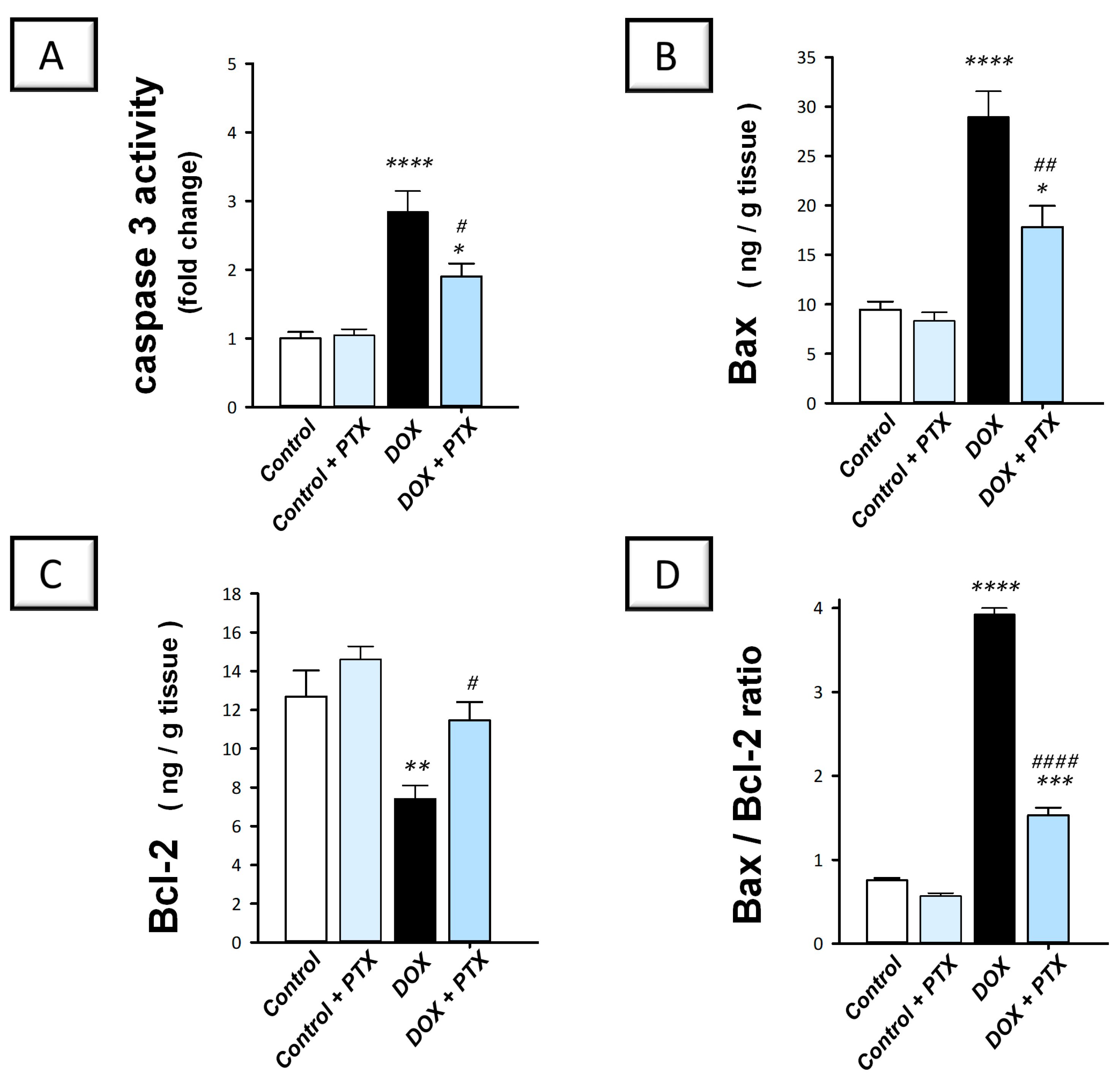
2.4. PTX Dampened the Pro-Apoptotic Machinery in the Hepatic Tissue of DOX-Intoxicated Rats
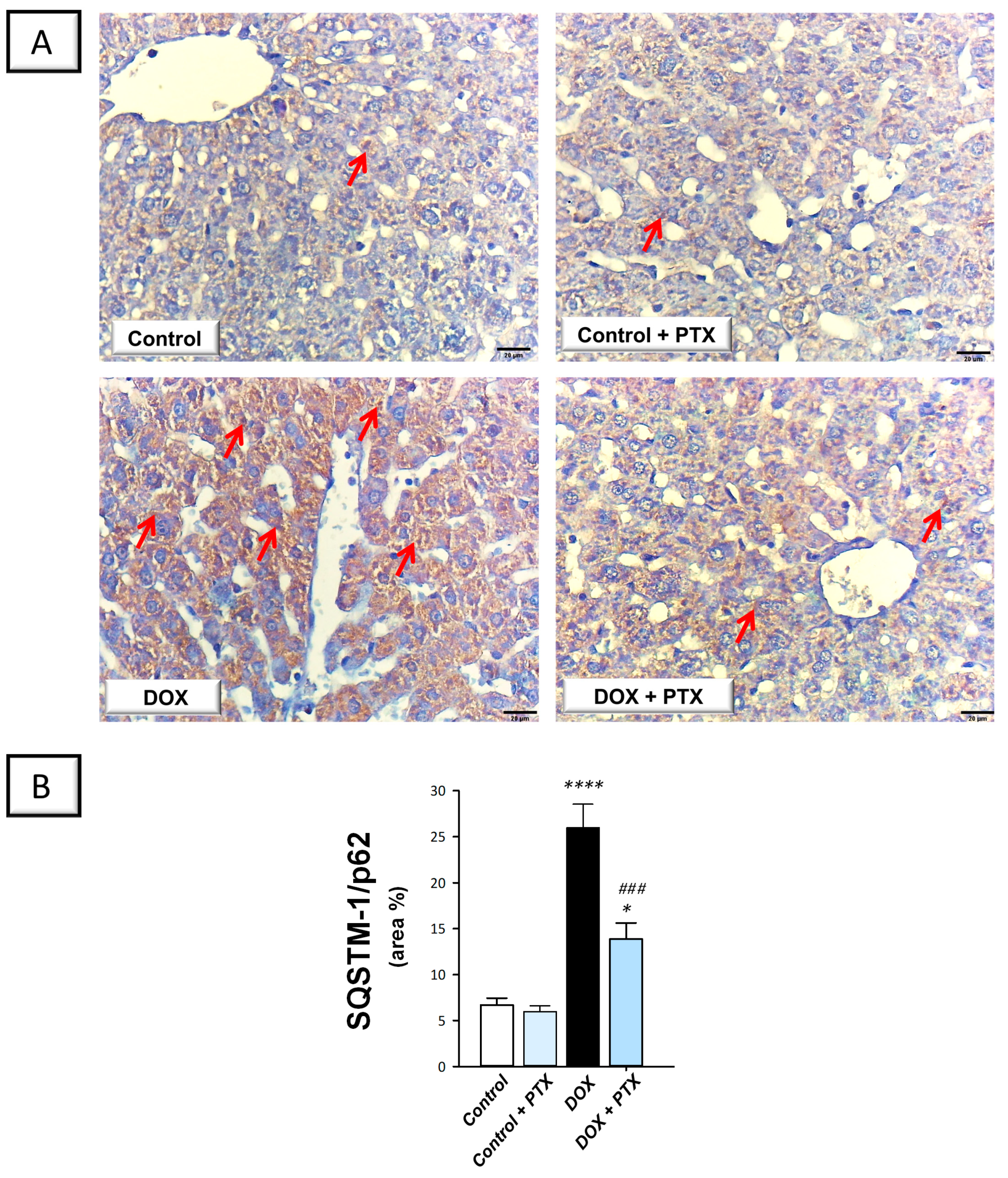
2.5. PTX Rescued the Impaired Autophagy Response in the Hepatic Tissue of DOX-Intoxicated Rats
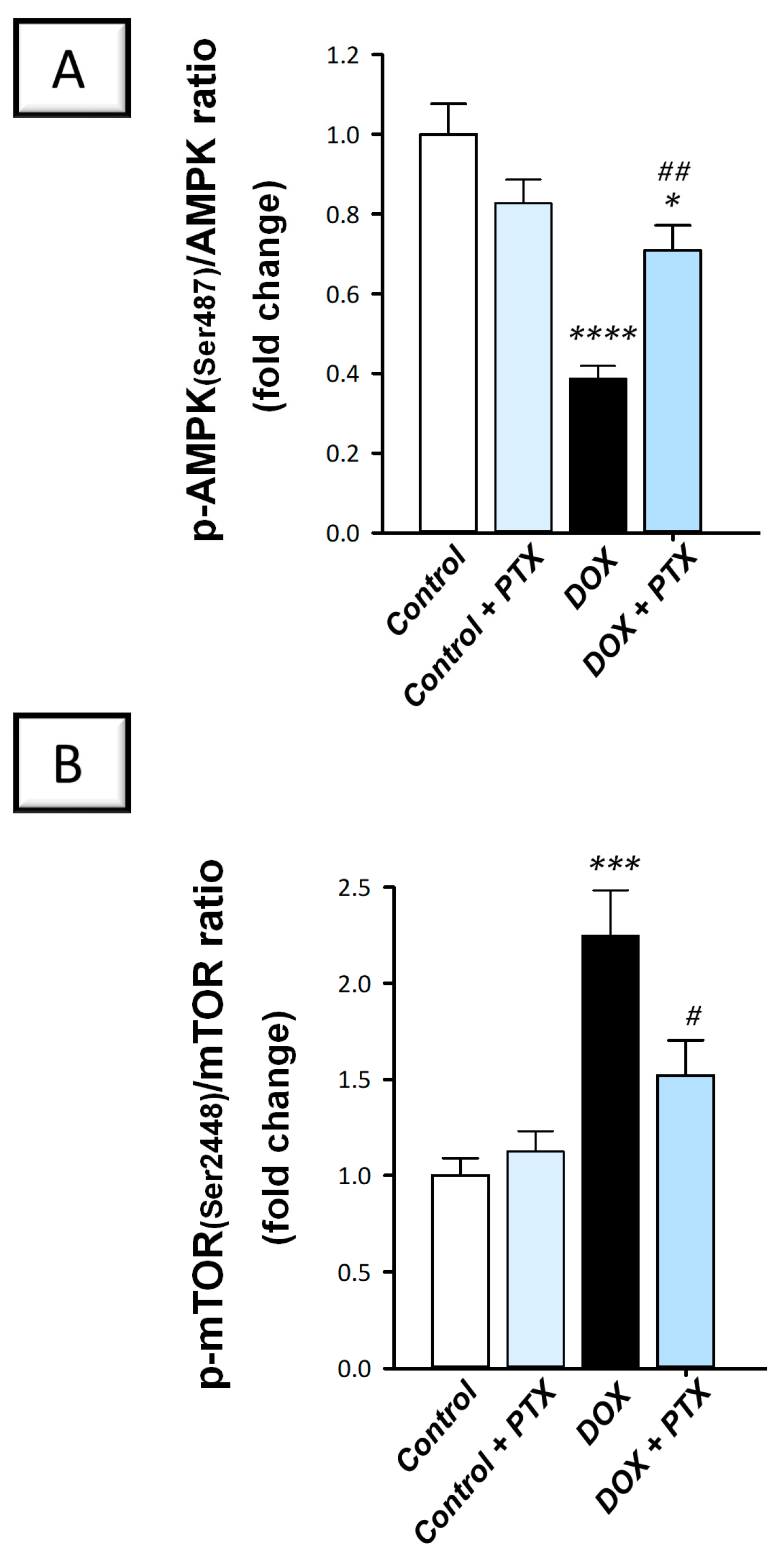
2.6. Hepatic AMPK/mTOR Pathway Was Activated by PTX in DOX-Intoxicated Rats
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Chemicals
4.3. Experimental Protocol
4.4. Tissue and Blood Collection
4.5. Liver Function Tests and Hepatic Inflammatory Cytokines
4.6. Histopathology
4.7. Hepatic HMGB1/TLR4/NF-κB Axis
4.8. Measurement of Apoptotic Events
4.9. Immunohistochemistry
4.10. Determination of the AMPK/mTOR Pathway
4.11. Statistics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Varela-Lopez, A.; Battino, M.; Navarro-Hortal, M.D.; Giampieri, F.; Forbes-Hernandez, T.Y.; Romero-Marquez, J.M.; Collado, R.; Quiles, J.L. An update on the mechanisms related to cell death and toxicity of doxorubicin and the protective role of nutrients. Food Chem. Toxicol. 2019, 134, 110834. [Google Scholar] [CrossRef] [PubMed]
- Bagdasaryan, A.A.; Chubarev, V.N.; Smolyarchuk, E.A.; Drozdov, V.N.; Krasnyuk, I.I.; Liu, J.; Fan, R.; Tse, E.; Shikh, E.V.; Sukocheva, O.A. Pharmacogenetics of Drug Metabolism: The Role of Gene Polymorphism in the Regulation of Doxorubicin Safety and Efficacy. Cancers 2022, 14, 5436. [Google Scholar] [CrossRef] [PubMed]
- Morsy, M.A.; El-Daly, M.; Kamel, B.A.; Rifaai, R.A.; Abdel-Gaber, S.A. Pregnenolone protects the liver against doxorubicin-induced cellular injury by anti-inflammatory, antioxidant, and antiapoptotic mechanisms: Role of Keap1/Nrf2/HO-1 and P-glycoprotein. Eur. Rev. Med. Pharmacol. Sci. 2023, 27, 4718–4734. [Google Scholar] [CrossRef] [PubMed]
- Al-Qahtani, W.H.; Alshammari, G.M.; Ajarem, J.S.; Al-Zahrani, A.Y.; Alzuwaydi, A.; Eid, R.; Yahya, M.A. Isoliquiritigenin prevents Doxorubicin-induced hepatic damage in rats by upregulating and activating SIRT1. Biomed. Pharmacother. 2022, 146, 112594. [Google Scholar] [CrossRef]
- Prasanna, P.L.; Renu, K.; Valsala Gopalakrishnan, A. New molecular and biochemical insights of doxorubicin-induced hepatotoxicity. Life Sci. 2020, 250, 117599. [Google Scholar] [CrossRef] [PubMed]
- El-Moselhy, M.A.; El-Sheikh, A.A. Protective mechanisms of atorvastatin against doxorubicin-induced hepato-renal toxicity. Biomed. Pharmacother. 2014, 68, 101–110. [Google Scholar] [CrossRef]
- Rawat, P.S.; Jaiswal, A.; Khurana, A.; Bhatti, J.S.; Navik, U. Doxorubicin-induced cardiotoxicity: An update on the molecular mechanism and novel therapeutic strategies for effective management. Biomed. Pharmacother. 2021, 139, 111708. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Shi, X.; Su, Z.; Hu, C.; Mu, X.; Pan, J.; Li, M.; Teng, F.; Ling, T.; Zhao, T.; et al. CD36 deficiency ameliorates drug-induced acute liver injury in mice. Mol. Med. 2021, 27, 57. [Google Scholar] [CrossRef] [PubMed]
- Mao, B.; Yuan, W.; Wu, F.; Yan, Y.; Wang, B. Autophagy in hepatic ischemia-reperfusion injury. Cell Death Discov. 2023, 9, 115. [Google Scholar] [CrossRef]
- Lian, N.; Tong, J.; Zhu, W.; Meng, Q.; Jiang, M.; Bian, M.; Li, Y. Ligustrazine and liguzinediol protect against doxorubicin-induced cardiomyocytes injury by inhibiting mitochondrial apoptosis and autophagy. Clin. Exp. Pharmacol. Physiol. 2023, 50, 867–877. [Google Scholar] [CrossRef]
- Wu, J.; Li, K.; Liu, Y.; Feng, A.; Liu, C.; Adu-Amankwaah, J.; Ji, M.; Ma, Y.; Hao, Y.; Bu, H.; et al. Daidzein ameliorates doxorubicin-induced cardiac injury by inhibiting autophagy and apoptosis in rats. Food Funct. 2023, 14, 934–945. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, J.J.; Trivedi, P.C.; Yeung, P.; Kienesberger, P.C.; Pulinilkunnil, T. Doxorubicin impairs cardiomyocyte viability by suppressing transcription factor EB expression and disrupting autophagy. Biochem. J. 2016, 473, 3769–3789. [Google Scholar] [CrossRef]
- Kawaguchi, T.; Takemura, G.; Kanamori, H.; Takeyama, T.; Watanabe, T.; Morishita, K.; Ogino, A.; Tsujimoto, A.; Goto, K.; Maruyama, R. Prior starvation mitigates acute doxorubicin cardiotoxicity through restoration of autophagy in affected cardiomyocytes. Cardiovas. Res. 2012, 96, 456–465. [Google Scholar] [CrossRef]
- Lv, X.; Zhu, Y.; Deng, Y.; Zhang, S.; Zhang, Q.; Zhao, B.; Li, G. Glycyrrhizin improved autophagy flux via HMGB1-dependent Akt/mTOR signaling pathway to prevent Doxorubicin-induced cardiotoxicity. Toxicology 2020, 441, 152508. [Google Scholar] [CrossRef] [PubMed]
- Petrović, A.; Bogojević, D.; Korać, A.; Golić, I.; Jovanović-Stojanov, S.; Martinović, V.; Ivanović-Matić, S.; Stevanović, J.; Poznanović, G.; Grigorov, I. Oxidative stress-dependent contribution of HMGB1 to the interplay between apoptosis and autophagy in diabetic rat liver. J. Physiol. Biochem. 2017, 73, 511–521. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Livesey, K.M.; Zeh, H.J., III; Lotze, M.T. High mobility group box 1 (HMGB1) activates an autophagic response to oxidative stress. Antioxid. Redox Signal. 2011, 15, 2185–2195. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.; Clissold, S.P. Pentoxifylline. A review of its pharmacodynamic and pharmacokinetic properties, and its therapeutic efficacy. Drugs 1987, 34, 50–97. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.M.; Liu, J.Z.; Zhao, C.Q.; Guo, P.; Wang, Z.; Wu, H.; Yu, W.; Liu, R.; Hai, C.X.; Zhang, X.D. Protective effects of pentoxifylline against chlorine-induced acute lung injury in rats. BMC Pharmacol. Toxicol. 2023, 24, 12. [Google Scholar] [CrossRef]
- Gołuński, G.; Borowik, A.; Derewońko, N.; Kawiak, A.; Rychłowski, M.; Woziwodzka, A.; Piosik, J. Pentoxifylline as a modulator of anticancer drug doxorubicin. Part II: Reduction of doxorubicin DNA binding and alleviation of its biological effects. Biochimie 2016, 123, 95–102. [Google Scholar] [CrossRef]
- Al-Kharashi, L.; Attia, H.; Alsaffi, A.; Almasri, T.; Arafa, M.; Hasan, I.; Alajami, H.; Ali, R.; Badr, A. Pentoxifylline and thiamine ameliorate rhabdomyolysis-induced acute kidney injury in rats via suppressing TLR4/NF-kappaB and NLRP-3/caspase-1/gasdermin mediated-pyroptosis. Toxicol. Appl. Pharmacol. 2023, 461, 116387. [Google Scholar] [CrossRef]
- Hendawy, N. Pentoxifylline attenuates cytokine stress and Fas system in syngeneic liver proteins induced experimental autoimmune hepatitis. Biomed. Pharmacother. 2017, 92, 316–323. [Google Scholar] [CrossRef]
- Taye, A.; El-Moselhy, M.A.; Hassan, M.K.; Ibrahim, H.M.; Mohammed, A.F. Hepatoprotective effect of pentoxifylline against D-galactosamine-induced hepatotoxicity in rats. Ann. Hepatol. 2009, 8, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Acedo, S.C.; Caria, C.R.; Gotardo, E.M.; Pereira, J.A.; Pedrazzoli, J.; Ribeiro, M.L.; Gambero, A. Role of pentoxifylline in non-alcoholic fatty liver disease in high-fat diet-induced obesity in mice. World J. Hepatol. 2015, 7, 2551–2558. [Google Scholar] [CrossRef]
- Massart, J.; Robin, M.A.; Noury, F.; Fautrel, A.; Letteron, P.; Bado, A.; Eliat, P.A.; Fromenty, B. Pentoxifylline aggravates fatty liver in obese and diabetic ob/ob mice by increasing intestinal glucose absorption and activating hepatic lipogenesis. Br. J. Pharmacol. 2012, 165, 1361–1374. [Google Scholar] [CrossRef] [PubMed]
- Satapathy, S.K.; Sakhuja, P.; Malhotra, V.; Sharma, B.C.; Sarin, S.K. Beneficial effects of pentoxifylline on hepatic steatosis, fibrosis and necroinflammation in patients with non-alcoholic steatohepatitis. J. Gastroenterol. Hepatol. 2007, 22, 634–638. [Google Scholar] [CrossRef]
- Nogueira-Machado, J.A.; Volpe, C.M.d.O.; Veloso, C.A.; Chaves, M.M. HMGB1, TLR and RAGE: A functional tripod that leads to diabetic inflammation. Expert Opin. Ther. Targets 2011, 15, 1023–1035. [Google Scholar] [CrossRef]
- Nagai, K.; Oda, A.; Konishi, H. Theanine prevents doxorubicin-induced acute hepatotoxicity by reducing intrinsic apoptotic response. Food Chem. Toxicol. 2015, 78, 147–152. [Google Scholar] [CrossRef]
- Bektas, S.; Karakaya, K.; Can, M.; Bahadir, B.; Guven, B.; Erdogan, N.; Ozdamar, S.O. The effects of tadalafil and pentoxifylline on apoptosis and nitric oxide synthase in liver ischemia/reperfusion injury. Kaohsiung J. Med. Sci. 2016, 32, 339–347. [Google Scholar] [CrossRef]
- Kara, E.; Coskun, T.; Kaya, Y.; Yumus, O.; Vatansever, S.; Var, A. Effects of silymarin and pentoxifylline on matrix metalloproteinase-1 and -2 expression and apoptosis in experimental hepatic fibrosis. Curr. Ther. Res. Clin. Exp. 2008, 69, 488–502. [Google Scholar] [CrossRef]
- Abdel Salam, O.M.; Baiuomy, A.R.; El-Shenawy, S.M.; Hassan, N.S. Effect of pentoxifylline on hepatic injury caused in the rat by the administration of carbon tetrachloride or acetaminophen. Pharmacol. Rep. 2005, 57, 596–603. [Google Scholar]
- Abdelmageed, M.E.; Abdelrahman, R.S. Canagliflozin attenuates thioacetamide-induced liver injury through modulation of HMGB1/RAGE/TLR4 signaling pathways. Life Sci. 2023, 322, 121654. [Google Scholar] [CrossRef]
- Du, H.; Tong, S.; Kuang, G.; Gong, X.; Jiang, N.; Yang, X.; Liu, H.; Li, N.; Xie, Y.; Xiang, Y.; et al. Sesamin Protects against APAP-Induced Acute Liver Injury by Inhibiting Oxidative Stress and Inflammatory Response via Deactivation of HMGB1/TLR4/NFkappaB Signal in Mice. J. Immunol. Res. 2023, 2023, 1116841. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Guo, E.; Yang, J.; Yang, Y.; Liu, S.; Hu, J.; Jiang, X.; Dirsch, O.; Dahmen, U.; Dong, W. Carbon monoxide ameliorates hepatic ischemia/reperfusion injury via sirtuin 1-mediated deacetylation of high-mobility group box 1 in rats. Liver Transpl. 2017, 23, 510–526. [Google Scholar] [CrossRef]
- Li, H.; Tan, G.; Tong, L.; Han, P.; Zhang, F.; Liu, B.; Sun, X. Pentoxifylline inhibits pulmonary inflammation induced by infrarenal aorticcross-clamping dependent of adenosine receptor A2A. Am. J. Transl. Res. 2016, 8, 2210–2221. [Google Scholar] [PubMed]
- Chi, M.; Gu, L.; Zhang, L.; Lin, J.; Xu, Q.; Jiang, N.; Wang, Y.; Qi, Y.; Diao, W.; Yi, W.; et al. Pentoxifylline treats Aspergillus fumigatus keratitis by reducing fungal burden and suppressing corneal inflammation. Eur. J. Pharmacol. 2023, 945, 175607. [Google Scholar] [CrossRef]
- Sishi, B.J.; Loos, B.; van Rooyen, J.; Engelbrecht, A.M. Autophagy upregulation promotes survival and attenuates doxorubicin-induced cardiotoxicity. Biochem. Pharmacol. 2013, 85, 124–134. [Google Scholar] [CrossRef]
- Gonzalez-Rodriguez, A.; Mayoral, R.; Agra, N.; Valdecantos, M.; Pardo, V.; Miquilena-Colina, M.; Vargas-Castrillón, J.; Iacono, O.L.; Corazzari, M.; Fimia, G. Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of NAFLD. Cell Death Dis. 2014, 5, e1179. [Google Scholar] [CrossRef]
- Arab, H.H.; Elhemiely, A.A.; El-Sheikh, A.A.K.; Khabbaz, H.J.A.; Arafa, E.A.; Ashour, A.M.; Kabel, A.M.; Eid, A.H. Repositioning Linagliptin for the Mitigation of Cadmium-Induced Testicular Dysfunction in Rats: Targeting HMGB1/TLR4/NLRP3 Axis and Autophagy. Pharmaceuticals 2022, 15, 852. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Loze, M.T.; Zeh, I.; Herbert, J.; Kang, R. The redox protein HMGB1 regulates cell death and survival in cancer treatment. Autophagy 2010, 6, 1181–1183. [Google Scholar] [CrossRef]
- Wang, Y.; Dong, L.; Li, J.; Luo, M.; Shang, B. Pentoxifylline induces apoptosis of HepG2 cells by reducing reactive oxygen species production and activating the MAPK signaling. Life Sci. 2017, 183, 60–68. [Google Scholar] [CrossRef]
- Kamran, M.Z.; Gude, R.P. Preclinical evaluation of the antimetastatic efficacy of Pentoxifylline on A375 human melanoma cell line. Biomed. Pharmacother. 2012, 66, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, L.A.; Taub, D.D.; Moors, M.A.; Blank, K.J. Methylxanthine-induced inhibition of the antigen- and superantigen-specific activation of T and B lymphocytes. Immunopharmacology 1992, 24, 203–217. [Google Scholar] [CrossRef] [PubMed]
- Viladkar, A.; Juvekar, A.; Chitnis, M.; Advani, S. Amelioration of doxorubicin resistance by pentoxifylline in human chronic myeloid leukemia cells in vitro. Sel. Cancer Ther. 1991, 7, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Goel, P.N.; Gude, R.P. Delineating the anti-metastatic potential of pentoxifylline in combination with liposomal doxorubicin against breast cancer cells. Biomed. Pharmacother. 2014, 68, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhou, W.; Mao, Q.; Gao, D.; Xiong, L.; Hu, X.; Zheng, Y.; Xu, X. HMGB1 Promotes Resistance to Doxorubicin in Human Hepatocellular Carcinoma Cells by Inducing Autophagy via the AMPK/mTOR Signaling Pathway. Front. Oncol. 2021, 11, 739145. [Google Scholar] [CrossRef] [PubMed]
- Karim, S.; Madani, B.; Burzangi, A.S.; Alsieni, M.; Bazuhair, M.A.; Jamal, M.; Daghistani, H.; Barasheed, M.O.; Alkreathy, H.; Khan, M.A. Urolithin A’s Antioxidative, Anti-Inflammatory, and Antiapoptotic Activities Mitigate Doxorubicin-Induced Liver Injury in Wistar Rats. Biomedicines 2023, 11, 1125. [Google Scholar] [CrossRef]
- Arab, H.H.; Eid, A.H.; El-Sheikh, A.A.K.; Arafa, E.A.; Ashour, A.M. Irbesartan reprofiling for the amelioration of ethanol-induced gastric mucosal injury in rats: Role of inflammation, apoptosis, and autophagy. Life Sci. 2022, 308, 120939. [Google Scholar] [CrossRef]
- Arab, H.H.; Eid, A.H.; Alsufyani, S.E.; Ashour, A.M.; El-Sheikh, A.A.K.; Darwish, H.W.; Georgy, G.S. Neuroprotective Impact of Linagliptin against Cadmium-Induced Cognitive Impairment and Neuropathological Aberrations: Targeting SIRT1/Nrf2 Axis, Apoptosis, and Autophagy. Pharmaceuticals 2023, 16, 1065. [Google Scholar] [CrossRef]
- Arab, H.H.; Ashour, A.M.; Eid, A.H.; Arafa, E.A.; Al Khabbaz, H.J.; Abd El-Aal, S.A. Targeting oxidative stress, apoptosis, and autophagy by galangin mitigates cadmium-induced renal damage: Role of SIRT1/Nrf2 and AMPK/mTOR pathways. Life Sci. 2022, 291, 120300. [Google Scholar] [CrossRef]
- Abdel-Fattah, M.M.; Hassanein, E.H.M.; Sayed, A.M.; Alsufyani, S.E.; El-Sheikh, A.A.K.; Arab, H.H.; Mohamed, W.R. Targeting SIRT1/FoxO3a/Nrf2 and PI3K/AKT Pathways with Rebamipide Attenuates Acetic Acid-Induced Colitis in Rats. Pharmaceuticals 2023, 16, 533. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arab, H.H.; Eid, A.H.; Alsufyani, S.E.; Ashour, A.M.; Alnefaie, A.M.; Alsharif, N.M.; Alshehri, A.M.; Almalawi, A.A.; Alsowat, A.A.; Abd El Aal, H.A.; et al. Activation of AMPK/mTOR-Driven Autophagy and Suppression of the HMGB1/TLR4 Pathway with Pentoxifylline Attenuates Doxorubicin-Induced Hepatic Injury in Rats. Pharmaceuticals 2024, 17, 681. https://doi.org/10.3390/ph17060681
Arab HH, Eid AH, Alsufyani SE, Ashour AM, Alnefaie AM, Alsharif NM, Alshehri AM, Almalawi AA, Alsowat AA, Abd El Aal HA, et al. Activation of AMPK/mTOR-Driven Autophagy and Suppression of the HMGB1/TLR4 Pathway with Pentoxifylline Attenuates Doxorubicin-Induced Hepatic Injury in Rats. Pharmaceuticals. 2024; 17(6):681. https://doi.org/10.3390/ph17060681
Chicago/Turabian StyleArab, Hany H., Ahmed H. Eid, Shuruq E. Alsufyani, Ahmed M. Ashour, Alwaleed M. Alnefaie, Nasser M. Alsharif, Abdullah M. Alshehri, Abdulmajeed A. Almalawi, Abdulmajeed A. Alsowat, Hayat A. Abd El Aal, and et al. 2024. "Activation of AMPK/mTOR-Driven Autophagy and Suppression of the HMGB1/TLR4 Pathway with Pentoxifylline Attenuates Doxorubicin-Induced Hepatic Injury in Rats" Pharmaceuticals 17, no. 6: 681. https://doi.org/10.3390/ph17060681
APA StyleArab, H. H., Eid, A. H., Alsufyani, S. E., Ashour, A. M., Alnefaie, A. M., Alsharif, N. M., Alshehri, A. M., Almalawi, A. A., Alsowat, A. A., Abd El Aal, H. A., Hassan, E. S. G., Elesawy, W. H., & Elhemiely, A. A. (2024). Activation of AMPK/mTOR-Driven Autophagy and Suppression of the HMGB1/TLR4 Pathway with Pentoxifylline Attenuates Doxorubicin-Induced Hepatic Injury in Rats. Pharmaceuticals, 17(6), 681. https://doi.org/10.3390/ph17060681