
Discovery of Bacterial Key Genes from 16S rRNA-Seq Profiles That Are Associated with the Complications of SARS-CoV-2 Infections and Provide Therapeutic Indications
 ,
,  , , ,
, , ,
Abstract
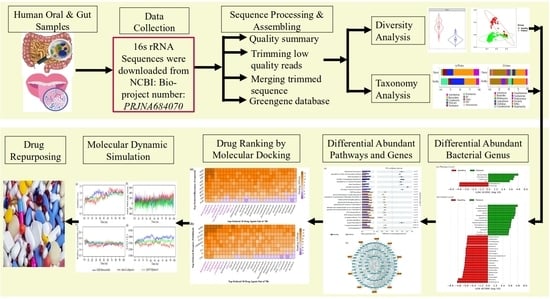
1. Introduction
2. Results
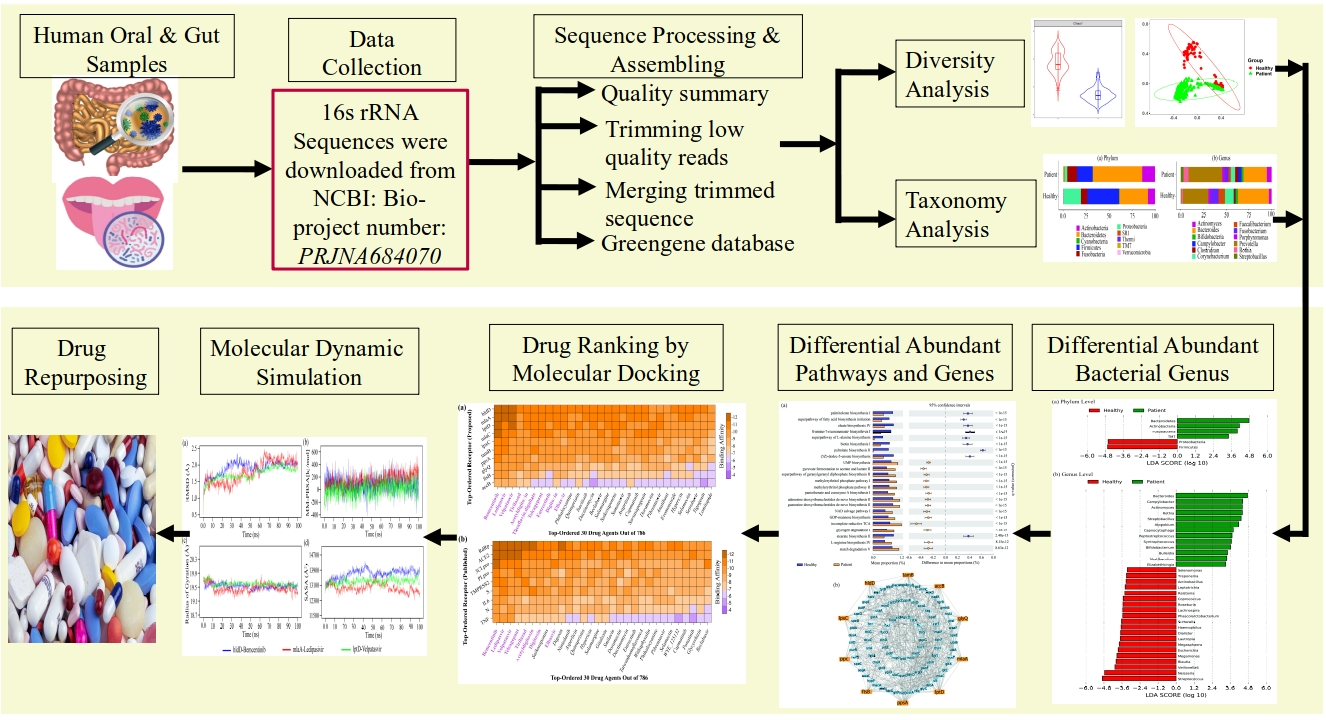
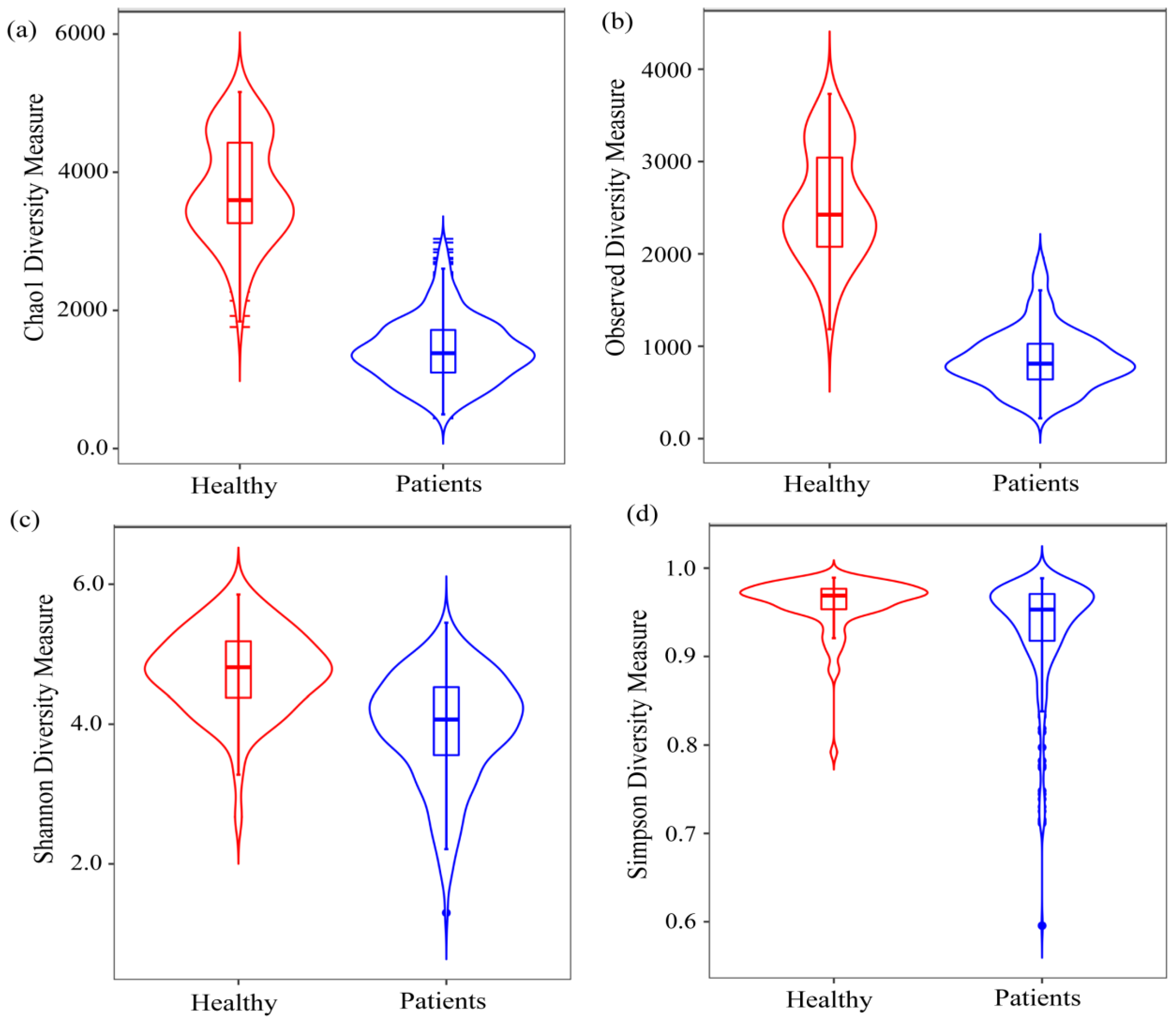
2.1. Diversity Analysis
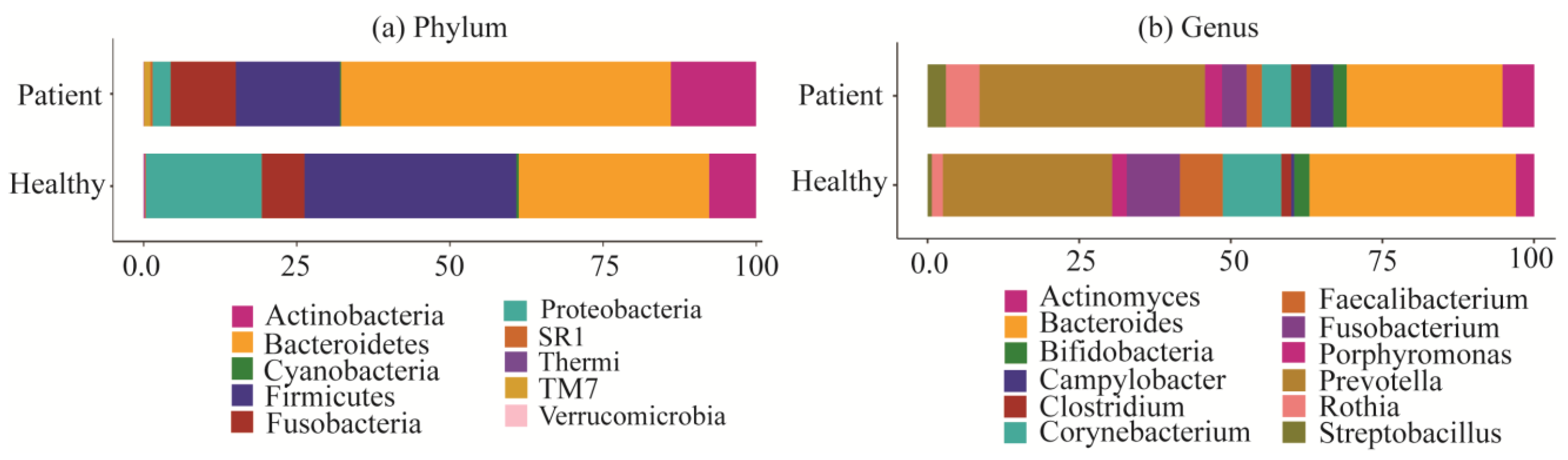
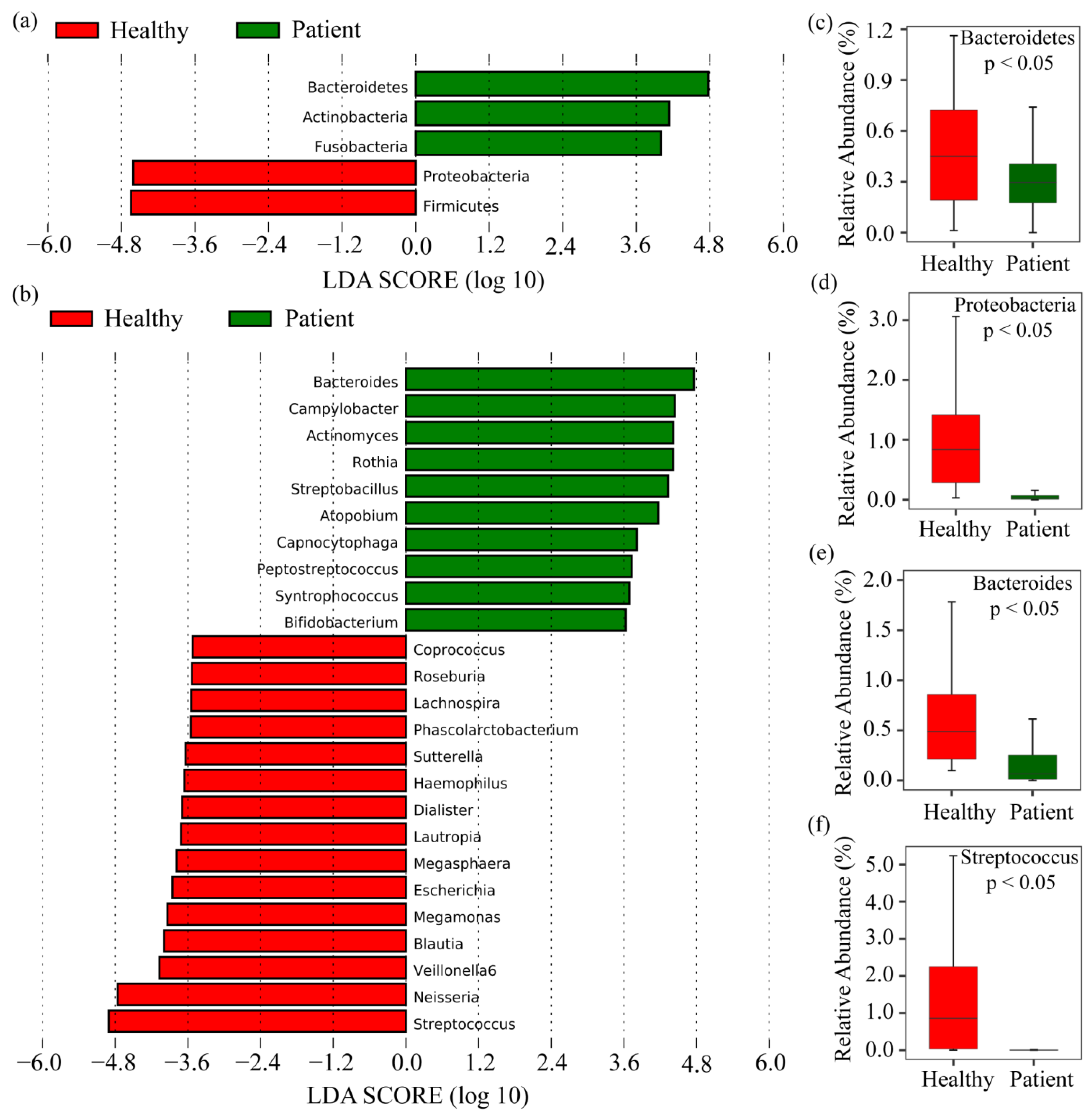
2.2. Taxonomy Analysis and Identification of Differentially Abundant Bacterial Compositions (DABCs)
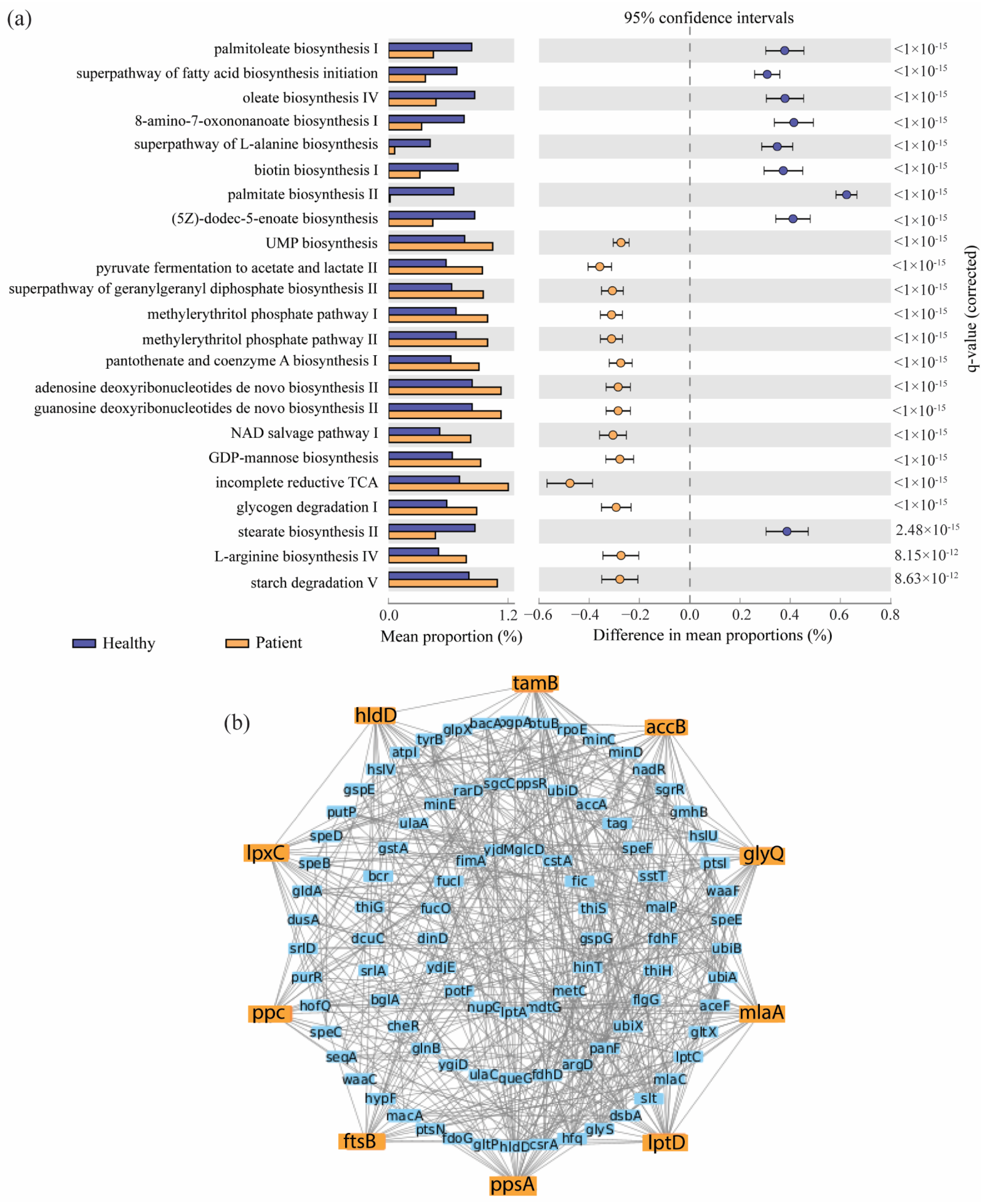
2.3. Identification of Pathway-Based Bacterial Key Genes (bKGs) from DABCs
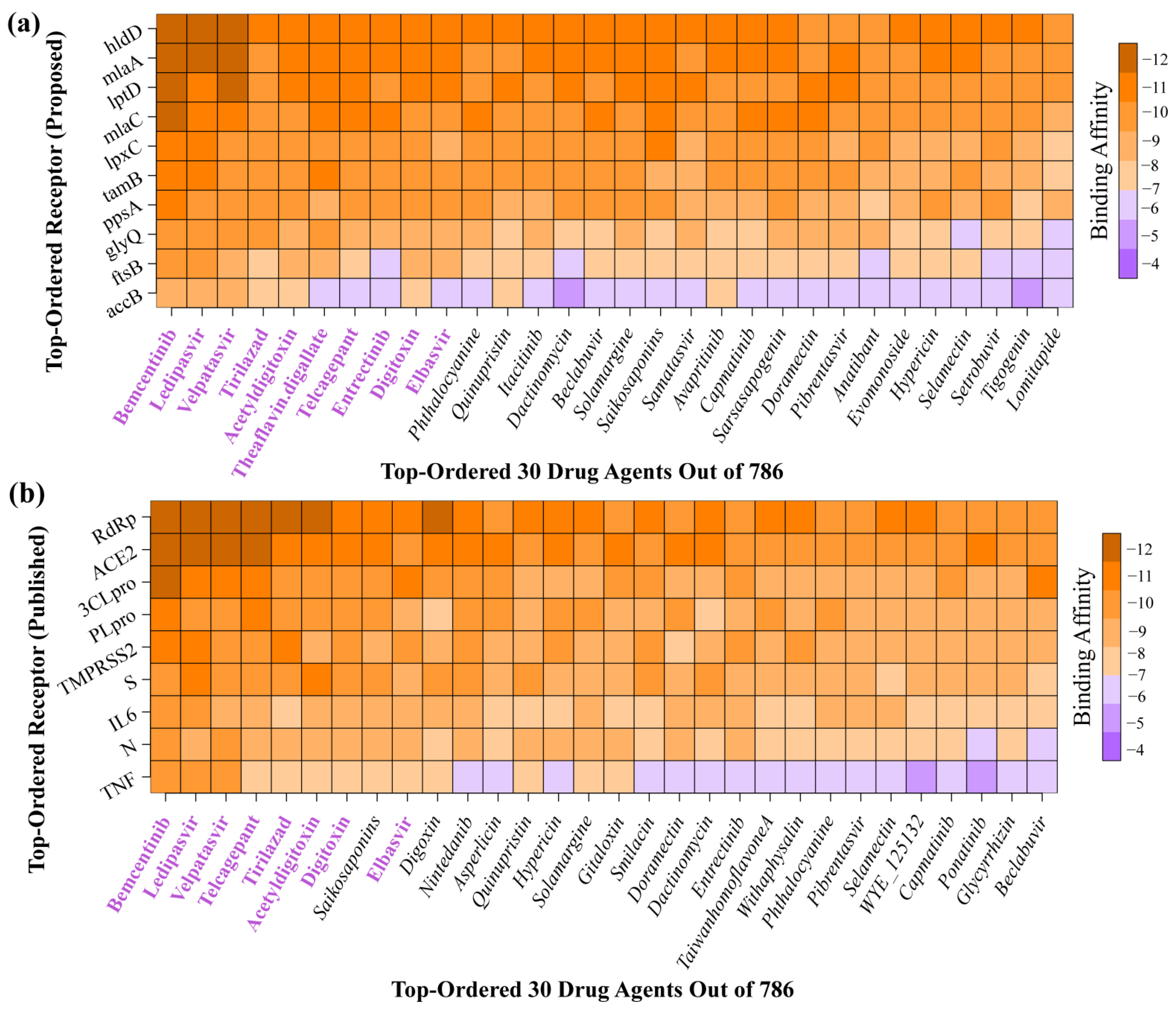
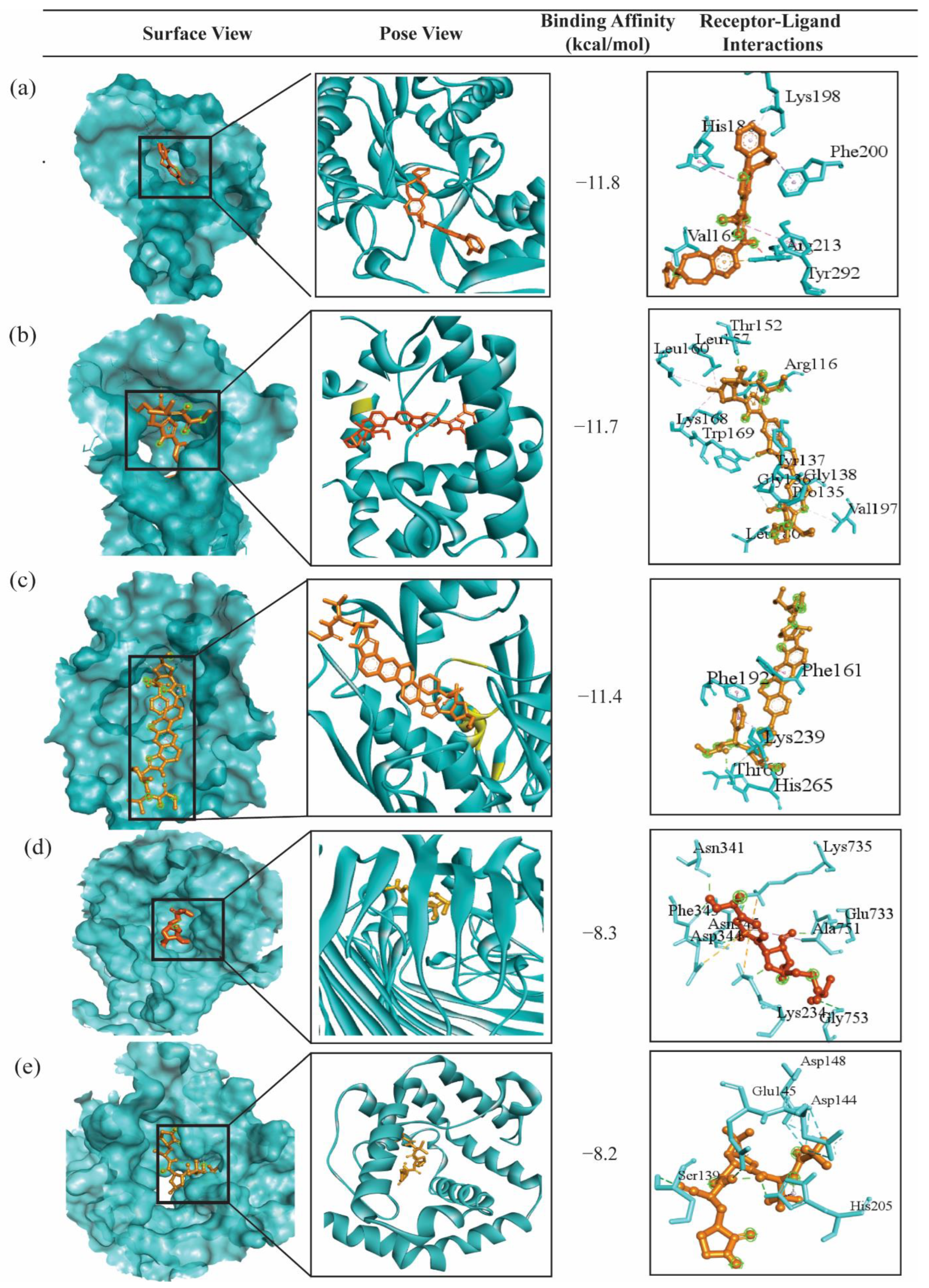
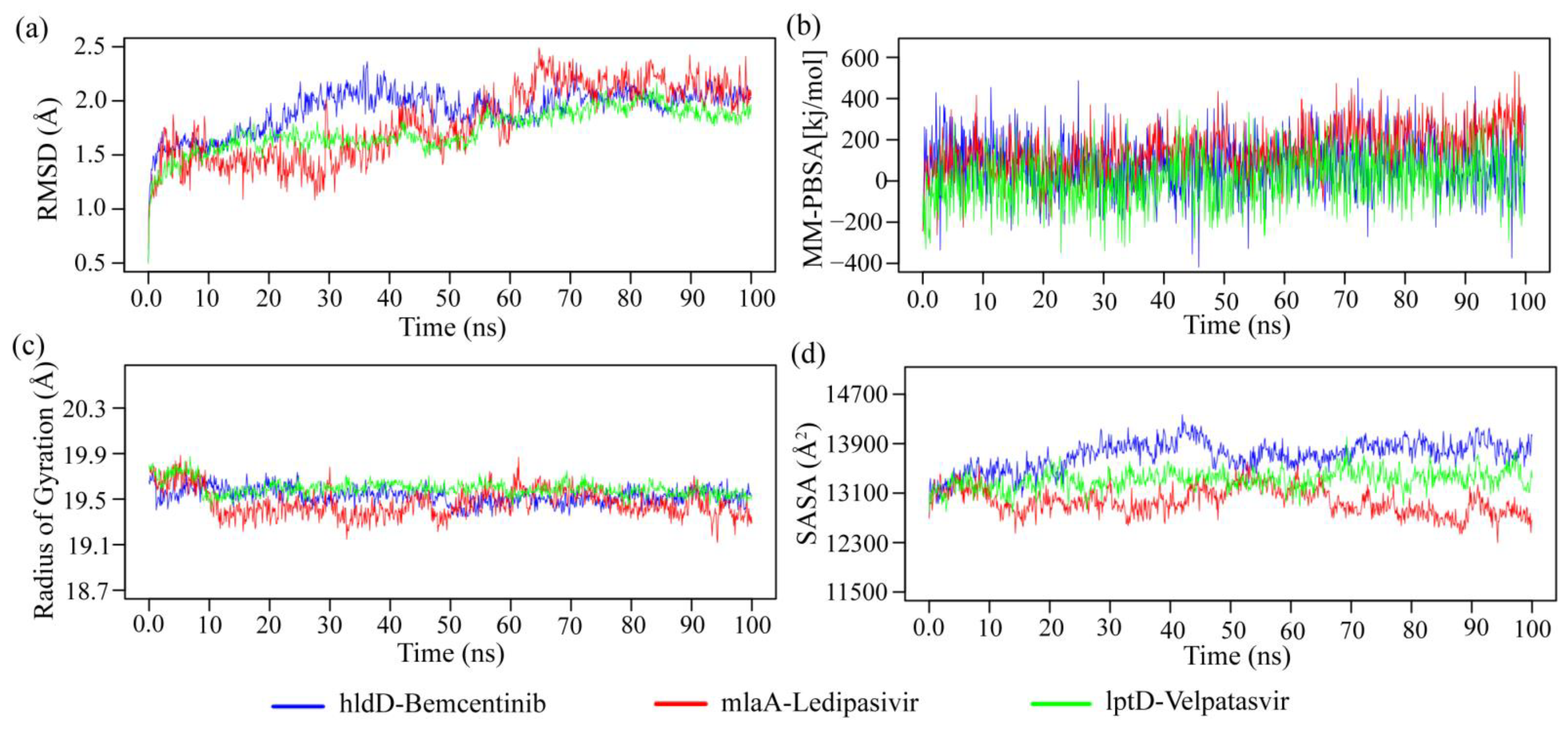
2.4. bKGs-Guided Drug Repurposing by Molecular Docking
3. Discussion
4. Limitations and Commercial Applications
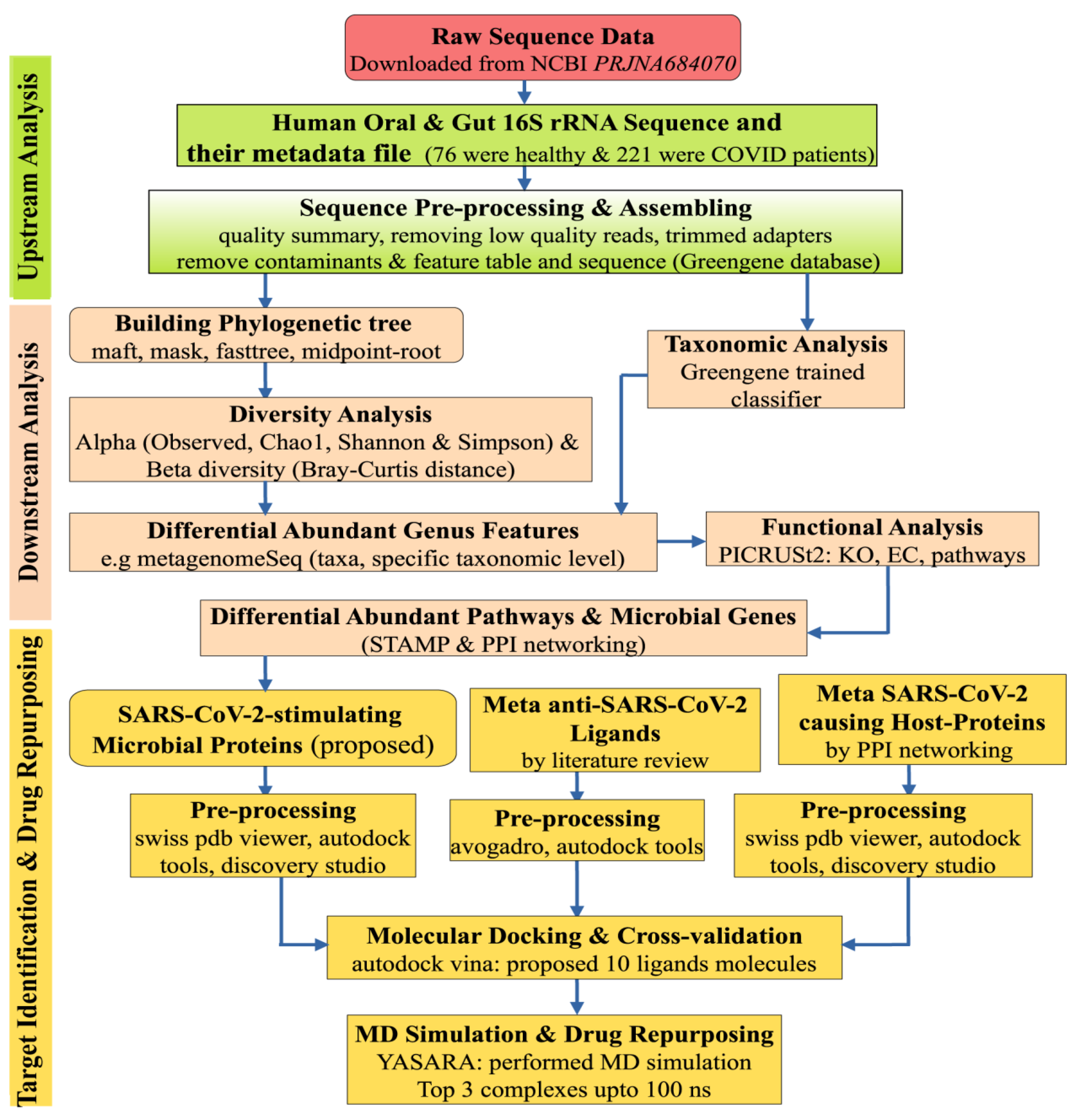
5. Materials and Methods
5.1. Data Source and Description
5.1.1. Collection of 16S rRNA Sequence Data
5.1.2. Collection of Metadata on Drug Molecules
5.2. Statistics and Bioinformatics Analysis
5.2.1. Preprocessing of 16S rRNA Sequence Profiles
5.2.2. Diversity Analysis
5.2.3. Taxonomy Analysis and Identification of Differentially Abundant Bacterial Compositions (DABCs)
5.2.4. Identification of Pathway-Based Bacterial Key Genes (bKGs) from DABCs
5.2.5. Bacterial Key Genes Guided Drug Repurposing by Molecular Docking
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. COVID-19 Epidemiological Update—15 March 2024; Epidemiology; WHO: Geneva, Switzerland, 2024; pp. 6–8. [Google Scholar]
- Markov, P.V.; Ghafari, M.; Beer, M.; Lythgoe, K.; Simmonds, P.; Stilianakis, N.I.; Katzourakis, A. The Evolution of SARS-CoV-2. Nat. Rev. Microbiol. 2023, 21, 361–379. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Abbasi, H.W.; Shahid, S.; Gul, S.; Abbasi, S.W. Molecular Docking, Simulation and MM-PBSA Studies of Nigella Sativa Compounds: A Computational Quest to Identify Potential Natural Antiviral for COVID-19 Treatment. J. Biomol. Struct. Dyn. 2021, 39, 4225–4233. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, Q.; Guo, D. Emerging Coronaviruses: Genome Structure, Replication, and Pathogenesis. J. Med. Virol. 2020, 92, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Mengist, H.M.; Dilnessa, T.; Jin, T. Structural Basis of Potential Inhibitors Targeting SARS-CoV-2 Main Protease. Front. Chem. 2021, 9, 622898. [Google Scholar] [CrossRef] [PubMed]
- Huff, S.; Kummetha, I.R.; Tiwari, S.K.; Huante, M.B.; Clark, A.E.; Wang, S.; Bray, W.; Smith, D.; Carlin, A.F.; Endsley, M.; et al. Discovery and Mechanism of SARS-CoV-2 Main Protease Inhibitors. J. Med. Chem. 2022, 65, 2866–2879. [Google Scholar] [CrossRef] [PubMed]
- Vicenti, I.; Zazzi, M.; Saladini, F. SARS-CoV-2 RNA-Dependent RNA Polymerase as a Therapeutic Target for COVID-19. Expert Opin. Ther. Pat. 2021, 31, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Rathore, A.S. RNA Dependent RNA Polymerase (RdRp) as a Drug Target for SARS-CoV-2. J. Biomol. Struct. Dyn. 2022, 40, 6039–6051. [Google Scholar] [CrossRef] [PubMed]
- Beck, B.R.; Shin, B.; Choi, Y.; Park, S.; Kang, K. Predicting Commercially Available Antiviral Drugs That May Act on the Novel Coronavirus (SARS-CoV-2) through a Drug-Target Interaction Deep Learning Model. Comput. Struct. Biotechnol. J. 2020, 18, 784–790. [Google Scholar] [CrossRef] [PubMed]
- Pałka, A.; Kujawska, A.; Hareza, D.; Gajda, M.; Wordliczek, J.; Jachowicz-Matczak, E.; Owsianka, I.; Żółtowska, B.; Chmielarczyk, A.; Romaniszyn, D.; et al. Secondary Bacterial Infections & Extensively Drug-Resistant Bacteria among COVID-19 Hospitalized Patients at the University Hospital in Kraków. Ann. Clin. Microbiol. Antimicrob. 2023, 22, 77. [Google Scholar] [CrossRef]
- Bedenić, B.; Bratić, V.; Mihaljević, S.; Lukić, A.; Vidović, K.; Reiner, K.; Schöenthaler, S.; Barišić, I.; Zarfel, G.; Grisold, A. Multidrug-Resistant Bacteria in a COVID-19 Hospital in Zagreb. Pathogens 2023, 12, 117. [Google Scholar] [CrossRef]
- Wu, H.Y.; Chang, P.H.; Chen, K.Y.; Lin, I.F.; Hsih, W.H.; Tsai, W.L.; Chen, J.A.; Lee, S.S.J. Coronavirus Disease 2019 (COVID-19) Associated Bacterial Coinfection: Incidence, Diagnosis and Treatment. J. Microbiol. Immunol. Infect. 2022, 55, 985–992. [Google Scholar] [CrossRef] [PubMed]
- Ritter, L.A.; Britton, N.; Heil, E.L.; Teeter, W.A.; Murthi, S.B.; Chow, J.H.; Ricotta, E.; Chertow, D.S.; Grazioli, A.; Levine, A.R. The Impact of Corticosteroids on Secondary Infection and Mortality in Critically Ill COVID-19 Patients. J. Intensive Care Med. 2021, 36, 1201–1208. [Google Scholar] [CrossRef]
- Langford, B.J.; So, M.; Raybardhan, S.; Leung, V.; Westwood, D.; MacFadden, D.R.; Soucy, J.P.R.; Daneman, N. Bacterial Co-Infection and Secondary Infection in Patients with COVID-19: A Living Rapid Review and Meta-Analysis. Clin. Microbiol. Infect. 2020, 26, 1622–1629. [Google Scholar] [CrossRef] [PubMed]
- Gomez, A.C.; Ortiz, T.; Valenzuela, A.; Egoávil-Espejo, R.; Huerto-Huanuco, R.; Pinto, J.A.; Lagos, J.; Ruiz, J. Super-Infection by Multiple Microorganisms in COVID-19 Patients. Front. Mol. Biosci. 2023, 10, 1113969. [Google Scholar] [CrossRef] [PubMed]
- Cataño-Correa, J.C.; Cardona-Arias, J.A.; Mancilla, J.P.P.; García, M.T. Bacterial Superinfection in Adults with COVID-19 Hospitalized in Two Clinics in Medellín-Colombia, 2020. PLoS ONE 2021, 16, e0254671. [Google Scholar] [CrossRef]
- Feldman, C.; Anderson, R. The Role of Co-Infections and Secondary Infections in Patients with COVID-19. Pneumonia 2021, 13, 5. [Google Scholar] [CrossRef]
- Chong, W.H.; Saha, B.K.; Ramani, A.; Chopra, A. State-of-the-Art Review of Secondary Pulmonary Infections in Patients with COVID-19 Pneumonia. Infection 2021, 49, 591–605. [Google Scholar] [CrossRef] [PubMed]
- Ding, F.M.; Feng, Y.; Han, L.; Zhou, Y.; Ji, Y.; Hao, H.J.; Xue, Y.S.; Yin, D.N.; Xu, Z.C.; Luo, S.; et al. Early Fever Is Associated with Clinical Outcomes in Patients with Coronavirus Disease. Front. Public Health 2021, 9, 712190. [Google Scholar] [CrossRef] [PubMed]
- Song, W.J.; Hui, C.K.; Hull, J.H.; Birring, S.S.; McGarvey, L.; Mazzone, S.B.; Chung, K.F. Confronting COVID-19-Associated Cough and the Post-COVID Syndrome: Role of Viral Neurotropism, Neuroinflammation, and Neuroimmune Responses. Lancet Respir. Med. 2021, 9, 533–544. [Google Scholar] [CrossRef]
- Mahase, E. COVID-19: Sore Throat, Fatigue, and Myalgia Are More Common with New UK Variant. BMJ 2021, 372, n288. [Google Scholar] [CrossRef]
- Zhang, Z. Diagnosis and Treatment Protocol for COVID-19 Patients (Trial Version 9). Health Care Sci. 2022, 1, 14–28. [Google Scholar] [CrossRef]
- Rain, M.; Puri, G.D.; Bhalla, A.; Avti, P.; Subramaniam, B.; Kaushal, V.; Srivastava, V.; Mahajan, P.; Singh, M.; Pandey, N.; et al. Effect of Breathing Intervention in Patients with COVID and Healthcare Workers. Front. Public Health 2022, 10, 945988. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, L.F.; Gonzáles, A.I.; Paiva, K.M.; Patatt, F.S.A.; Stolz, J.V.; Haas, P. Smell and Taste Alterations in COVID-19 Patients: A Systematic Review. Rev. Assoc. Med. Bras. 2021, 66, 1602–1608. [Google Scholar] [CrossRef] [PubMed]
- Widyadharma, I.P.E.; Sari, N.N.S.P.; Pradnyaswari, K.E.; Yuwana, K.T.; Adikarya, I.P.G.D.; Tertia, C.; Wijayanti, I.A.S.; Indrayani, I.A.S.; Utami, D.K.I. Pain as Clinical Manifestations of COVID-19 Infection and Its Management in the Pandemic Era: A Literature Review. Egypt. J. Neurol. Psychiatry Neurosurg. 2020, 56, 121. [Google Scholar] [CrossRef] [PubMed]
- Daria, S.; Katarzyna, S.A.; Jerzy, F.; Ivan, K. Muscle Pain and Muscle Weakness in COVID19 Patients: Cross-Talk with Statins—Preliminary Results. Biomed. Pharmacother. 2022, 148, 112757. [Google Scholar] [CrossRef] [PubMed]
- Mutiawati, E.; Kusuma, H.I.; Fahriani, M.; Harapan, H.; Syahrul, S.; Musadir, N. Headache in Post-COVID-19 Patients: Its Characteristics and Relationship with the Quality of Life. Medicina 2022, 58, 1500. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, F.; Baumgart, D.C.; Danese, S.; Peyrin-Biroulet, L. Diarrhea during COVID-19 Infection: Pathogenesis, Epidemiology, Prevention, and Management. Clin. Gastroenterol. Hepatol. 2020, 18, 1663–1672. [Google Scholar] [CrossRef]
- Hurn, E.; Dickinson, L.; Abraham, J.A. Bacterial Meningitis and COVID-19: A Complex Patient Journey. BMJ Case Rep. 2021, 14, e239533. [Google Scholar] [CrossRef]
- Richards, T.; Stephen, J.; Lui, C.L. Severe Disseminated Veillonella parvula Infection Including Endocarditis, Bilateral Psoas Abscess, Discitis, and Osteomyelitis but Sparing Spinal and Hip Prostheses: A Case Report. J. Med. Case Rep. 2022, 16, 157. [Google Scholar] [CrossRef]
- Attaway, A.H.; Scheraga, R.G.; Bhimraj, A.; Biehl, M.; Hatipoğlu, U. Severe COVID-19 Pneumonia: Pathogenesis and Clinical Management. BMJ 2021, 372, n436. [Google Scholar] [CrossRef]
- Seong, Y.J.; Lee, S.H.; Kim, E.J.; Choi, Y.H.; Kim, T.J.; Lee, W.G.; Heo, J.Y. Campylobacter fetus Subspecies venerealis Meningitis Associated with a Companion Dog in a Young Adult: A Case Report. BMC Infect. Dis. 2021, 21, 1280. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, Q.; Yang, C.; Guo, M.; Cui, X.; Jing, Z.; Liu, Y.; Qiao, W.; Qi, H.; Zhang, H.; et al. Bacteroides Acidifaciens in the Gut Plays a Protective Role against CD95-Mediated Liver Injury. Gut Microbes 2022, 14, 2027853. [Google Scholar] [CrossRef] [PubMed]
- Alshaikh, F.S.; Baker, A.I. Prevalence of Bacterial Coinfection and Patterns of Antibiotics Prescribing in Patients with COVID-19: A Systematic Review and Meta-Analysis. Int. J. Pharm. Pract. 2022, 30, i21. [Google Scholar] [CrossRef]
- Mosharaf, M.P.; Reza, M.S.; Kibria, M.K.; Ahmed, F.F.; Kabir, M.H.; Hasan, S.; Mollah, M.N.H. Computational Identification of Host Genomic Biomarkers Highlighting Their Functions, Pathways and Regulators That Influence SARS-CoV-2 Infections and Drug Repurposing. Sci. Rep. 2022, 12, 4279. [Google Scholar] [CrossRef]
- Mosharaf, M.P.; Kibria, M.K.; Hossen, M.B.; Islam, M.A.; Reza, M.S.; Mahumud, R.A.; Alam, K.; Gow, J.; Mollah, M.N.H. Meta-Data Analysis to Explore the Hub of the Hub-Genes That Influence SARS-CoV-2 Infections Highlighting Their Pathogenetic Processes and Drugs Repurposing. Vaccines 2022, 10, 1248. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, F.F.; Reza, M.S.; Sarker, M.S.; Islam, M.S.; Mosharaf, M.P.; Hasan, S.; Mollah, M.N.H. Identification of Host Transcriptome-Guided Repurposable Drugs for SARS-CoV-1 Infections and Their Validation with SARS-CoV-2 Infections by Using the Integrated Bioinformatics Approaches. PLoS ONE 2022, 17, e0266124. [Google Scholar] [CrossRef]
- Sarker, B.; Rahaman, M.M.; Islam, M.A.; Alamin, M.H.; Husain, M.M.; Ferdousi, F.; Ahsan, M.A.; Mollah, M.N.H. Identification of Host Genomic Biomarkers from Multiple Transcriptomics Datasets for Diagnosis and Therapies of SARS-CoV-2 Infections. PLoS ONE 2023, 18, e0281981. [Google Scholar] [CrossRef]
- Weigert Muñoz, A.; Zhao, W.; Sieber, S.A. Monitoring Host-Pathogen Interactions Using Chemical Proteomics. RSC Chem. Biol. 2023, 5, 73–89. [Google Scholar] [CrossRef]
- Mahmud, S.; Hasan, M.R.; Biswas, S.; Paul, G.K.; Afrose, S.; Mita, M.A.; Sultana Shimu, M.S.; Promi, M.M.; Hani, U.; Rahamathulla, M.; et al. Screening of Potent Phytochemical Inhibitors Against SARS-CoV-2 Main Protease: An Integrative Computational Approach. Front. Bioinforma. 2021, 1, 717141. [Google Scholar] [CrossRef]
- Crua Asensio, N.; Munõz Giner, E.; De Groot, N.S.; Torrent Burgas, M. Centrality in the Host-Pathogen Interactome Is Associated with Pathogen Fitness during Infection. Nat. Commun. 2017, 8, 14092. [Google Scholar] [CrossRef]
- Balint, D.; Brito, I.L. Human–Gut Bacterial Protein–Protein Interactions: Understudied but Impactful to Human Health. Trends Microbiol. 2023. [Google Scholar] [CrossRef]
- Xu, R.; Lu, R.; Zhang, T.; Wu, Q.; Cai, W.; Han, X.; Wan, Z.; Jin, X.; Zhang, Z.; Zhang, C. Temporal Association between Human Upper Respiratory and Gut Bacterial Microbiomes during the Course of COVID-19 in Adults. Commun. Biol. 2021, 4, 240. [Google Scholar] [CrossRef]
- Rafiqul Islam, S.M.; Foysal, M.J.; Hoque, M.N.; Mehedi, H.M.H.; Rob, M.A.; Salauddin, A.; Tanzina, A.Y.; Biswas, S.; Noyon, S.H.; Siddiki, A.M.A.M.Z.; et al. Dysbiosis of Oral and Gut Microbiomes in SARS-CoV-2 Infected Patients in Bangladesh: Elucidating the Role of Opportunistic Gut Microbes. Front. Med. 2022, 9, 821777. [Google Scholar] [CrossRef] [PubMed]
- Moni, M.A.; Quinn, J.M.W.; Sinmaz, N.; Summers, M.A. Gene Expression Profiling of SARS-CoV-2 Infections Reveal Distinct Primary Lung Cell and Systemic Immune Infection Responses That Identify Pathways Relevant in COVID-19 Disease. Brief. Bioinform. 2021, 22, 1324–1337. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhang, J.; Zhang, D.; Ma, W.L.; Wang, X. Linking the Gut Microbiota to Persistent Symptoms in Survivors of COVID-19 after Discharge. J. Microbiol. 2021, 59, 941–948. [Google Scholar] [CrossRef]
- Gaibani, P.; D’Amico, F.; Bartoletti, M.; Lombardo, D.; Rampelli, S.; Fornaro, G.; Coladonato, S.; Siniscalchi, A.; Re, M.C.; Viale, P.; et al. The Gut Microbiota of Critically Ill Patients with COVID-19. Front. Cell. Infect. Microbiol. 2021, 11, 670424. [Google Scholar] [CrossRef]
- Hua, Q.; Zhang, B.; Xu, G.; Wang, L.; Wang, H.; Lin, Z.; Yu, D.; Ren, J.; Zhang, D.; Zhao, L.; et al. CEMIP, a Novel Adaptor Protein of OGT, Promotes Colorectal Cancer Metastasis through Glutamine Metabolic Reprogramming via Reciprocal Regulation of β-Catenin. Oncogene 2021, 40, 6443–6455. [Google Scholar] [CrossRef]
- Uehara, O.; Abiko, Y.; Nagasawa, T.; Morikawa, T.; Hiraki, D.; Harada, F.; Kawano, Y.; Toraya, S.; Matsuoka, H.; Paudel, D.; et al. Alterations in the Oral Microbiome of Individuals with a Healthy Oral Environment Following COVID-19 Vaccination. BMC Oral Health 2022, 22, 50. [Google Scholar] [CrossRef] [PubMed]
- Perrone, M.R.; Romano, S.; De Maria, G.; Tundo, P.; Bruno, A.R.; Tagliaferro, L.; Maffia, M.; Fragola, M. Compositional Data Analysis of 16S RRNA Gene Sequencing Results from Hospital Airborne Microbiome Samples. Int. J. Environ. Res. Public Health 2022, 19, 10107. [Google Scholar] [CrossRef]
- Shi, Y.L.; He, M.Z.; Han, M.Z.; Gui, H.Y.; Wang, P.; Yu, J.L.; Ge, Y.L.; Sun, Y.; Huang, S.H. Characterization of Altered Oropharyngeal Microbiota in Hospitalized Patients with Mild SARS-CoV-2 Infection. Front. Cell. Infect. Microbiol. 2022, 12, 824578. [Google Scholar] [CrossRef]
- Mazzarelli, A.; Giancola, M.L.; Farina, A.; Marchioni, L.; Rueca, M.; Gruber, C.E.M.; Bartolini, B.; Bartoli, T.A.; Maffongelli, G.; Capobianchi, M.R.; et al. 16S RRNA Gene Sequencing of Rectal Swab in Patients Affected by COVID-19. PLoS ONE 2021, 16, e0247041. [Google Scholar] [CrossRef]
- Zuo, T.; Zhang, F.; Lui, G.C.Y.; Yeoh, Y.K.; Li, A.Y.L.; Zhan, H.; Wan, Y.; Chung, A.C.K.; Cheung, C.P.; Chen, N.; et al. Alterations in Gut Microbiota of Patients with COVID-19 during Time of Hospitalization. Gastroenterology 2020, 159, 944–955.e948. [Google Scholar] [CrossRef] [PubMed]
- Al Bataineh, M.T.; Henschel, A.; Mousa, M.; Daou, M.; Waasia, F.; Kannout, H.; Khalili, M.; Kayasseh, M.A.; Alkhajeh, A.; Uddin, M.; et al. Gut Microbiota Interplay with COVID-19 Reveals Links to Host Lipid Metabolism among Middle Eastern Populations. Front. Microbiol. 2021, 12, 761067. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Cheng, X.; Jiang, G.; Tang, H.; Ming, S.; Tang, L.; Lu, J.; Guo, C.; Shan, H.; Huang, X. Altered Oral and Gut Microbiota and Its Association with SARS-CoV-2 Viral Load in COVID-19 Patients during Hospitalization. npj Biofilms Microbiomes 2021, 7, 61. [Google Scholar] [CrossRef] [PubMed]
- Vila Domínguez, A.; Enrique Jiménez Mejías, M.; Smani, Y. Drugs Repurposing for Multi-Drug Resistant Bacterial Infections. Drug Repurposing—Hypothesis Mol. Asp. Ther. Appl. 2020, 6, 97–118. [Google Scholar] [CrossRef]
- Abubakar, U.; Al-Anazi, M.; Alanazi, Z.; Rodríguez-Baño, J. Impact of COVID-19 Pandemic on Multidrug Resistant Gram Positive and Gram Negative Pathogens: A Systematic Review. J. Infect. Public Health 2023, 16, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Clarridge, J.E., 3rd. Impact of 16S RRNA Gene Sequence Analysis for Identification of Bacteria on Clinical Microbiology and Infectious Diseases. Clin. Microbiol. Rev. 2004, 17, 840–862. [Google Scholar] [CrossRef] [PubMed]
- Sharma Manjul, A.; Shirkot, P.; Anshul Sharma Manjul, C. 6S RRNA Gene Sequencing for Bacterial Identification of Pullulanase Synthesizing Thermophilic Bacteria Contributing to Big Data. Int. J. Chem. Stud. 2018, 6, 2769–2773. [Google Scholar]
- Syed, Y.Y. Molnupiravir: First Approval. Drugs 2022, 82, 455–460. [Google Scholar] [CrossRef]
- Lamb, Y.N. Nirmatrelvir Plus Ritonavir: First Approval. Drugs 2022, 82, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Magne, F.; Gotteland, M.; Gauthier, L.; Zazueta, A.; Pesoa, S.; Navarrete, P.; Balamurugan, R. The Firmicutes/Bacteroidetes Ratio: A Relevant Marker of Gut Dysbiosis in Obese Patients? Nutrients 2020, 12, 1474. [Google Scholar] [CrossRef]
- Sears, C.L.; Islam, S.; Saha, A.; Arjumand, M.; Alam, N.H.; Faruque, A.S.G.; Salam, M.A.; Shin, J.; Hecht, D.; Weintraub, A.; et al. Association of Enterotoxigenic Bacteroides Fragilis Infection with Inflammatory Diarrhea. Clin. Infect. Dis. 2008, 47, 797–803. [Google Scholar] [CrossRef]
- Kaur, U.S.; Shet, A.; Rajnala, N.; Gopalan, B.P.; Moar, P.; Himanshu, D.; Singh, B.P.; Chaturvedi, R.; Tandon, R. High Abundance of Genus Prevotella in the Gut of Perinatally HIV-Infected Children Is Associated with IP-10 Levels despite Therapy. Sci. Rep. 2018, 8, 17679. [Google Scholar] [CrossRef] [PubMed]
- Ramsheh, M.Y.; Haldar, K.; Esteve-Codina, A.; Purser, L.F.; Richardson, M.; Müller-Quernheim, J.; Greulich, T.; Nowinski, A.; Barta, I.; Stendardo, M.; et al. Lung Microbiome Composition and Bronchial Epithelial Gene Expression in Patients with COPD versus Healthy Individuals: A Bacterial 16S RRNA Gene Sequencing and Host Transcriptomic Analysis. Lancet Microbe 2021, 2, e300–e310. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.; Hirata, M.; Hossain, N.; Kunz, A. Prevotella Melaninogenica Meningitis and Abscess: Normal Flora in an Abnormal Location. Pediatrics 2018, 142, 602. [Google Scholar] [CrossRef]
- Lertpiriyapong, K.; Whary, M.T.; Muthupalani, S.; Lofgren, J.L.; Gamazon, E.R.; Feng, Y.; Ge, Z.; Wang, T.C.; Fox, J.G. Gastric Colonisation with a Restricted Commensal Microbiota Replicates the Promotion of Neoplastic Lesions by Diverse Intestinal Microbiota in the Helicobacter Pylori INS-GAS Mouse Model of Gastric Carcinogenesis. Gut 2014, 63, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Dias-Jácome, E.; Libânio, D.; Borges-Canha, M.; Galaghar, A.; Pimentel-Nunes, P. Gastric Microbiota and Carcinogenesis: The Role of Non-Helicobacter Pylori Bacteria—A Systematic Review. Rev. Esp. Enfermedades Dig. 2016, 108, 530–540. [Google Scholar] [CrossRef] [PubMed]
- Dahmus, J.D.; Kotler, D.L.; Kastenberg, D.M.; Kistler, C.A. The Gut Microbiome and Colorectal Cancer: A Review of Bacterial Pathogenesis. J. Gastrointest. Oncol. 2018, 9, 769–777. [Google Scholar] [CrossRef] [PubMed]
- Newsome, R.C.; Gauthier, J.; Hernandez, M.C.; Abraham, G.E.; Robinson, T.O.; Williams, H.B.; Sloan, M.; Owings, A.; Laird, H.; Christian, T.; et al. The Gut Microbiome of COVID-19 Recovered Patients Returns to Uninfected Status in a Minority-Dominated United States Cohort. Gut Microbes 2021, 13, 1926840. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.; Lambert, J.R. Campylobacter Jejuni Causing Flare-Up in Inflammatory Bowel Disease. Lancet 1980, 316, 919. [Google Scholar] [CrossRef] [PubMed]
- Starr, J. Clostridium Difficile Associated Diarrhoea: Diagnosis and Treatment. Bmj 2005, 331, 498. [Google Scholar] [CrossRef][Green Version]
- Kahler, C.M. Neisseria Species and Their Complicated Relationships with Human Health. Microbiol. Aust. 2021, 42, 79–83. [Google Scholar] [CrossRef]
- Dewhirst, F.E.; Chen, T.; Izard, J.; Paster, B.J.; Tanner, A.C.R.; Yu, W.H.; Lakshmanan, A.; Wade, W.G. The Human Oral Microbiome. J. Bacteriol. 2010, 192, 5002–5017. [Google Scholar] [CrossRef] [PubMed]
- Signat, B.; Roques, C.; Poulet, P.; Duffaut, D. Role of Fusobacterium Nucleatum in Periodontal Health and Disease. Curr. Issues Mol. Biol. 2011, 13, 25–36. [Google Scholar]
- Knoop, F.C. Haemophilus Infections. In xPharm: The Comprehensive Pharmacology Reference; Elsevier Inc.: Oxford, UK, 2011; pp. 1–4. [Google Scholar] [CrossRef]
- Quévrain, E.; Maubert, M.A.; Michon, C.; Chain, F.; Marquant, R.; Tailhades, J.; Miquel, S.; Carlier, L.; Bermúdez-Humarán, L.G.; Pigneur, B.; et al. Identification of an Anti-Inflammatory Protein from Faecalibacterium Prausnitzii, a Commensal Bacterium Deficient in Crohn’s Disease. Gut 2016, 65, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Jorth, P.; Turner, K.H.; Gumus, P.; Nizam, N.; Buduneli, N.; Whiteley, M. Metatranscriptomics of the Human Oral Microbiome during Health and Disease. MBio 2014, 5, 10–128. [Google Scholar] [CrossRef] [PubMed]
- Anand, S.; Kaur, H.; Mande, S.S. Comparative in Silico Analysis of Butyrate Production Pathways in Gut Commensals and Pathogens. Front. Microbiol. 2016, 7, 1945. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Yang, Z.; Rey, F.E.; Ridaura, V.K.; Davidson, N.O.; Gordon, J.I.; Semenkovich, C.F. Fatty Acid Synthase Modulates Intestinal Barrier Function through Palmitoylation of Mucin 2. Cell Host Microbe 2012, 11, 140–152. [Google Scholar] [CrossRef] [PubMed]
- Benoit, B.; Bruno, J.; Kayal, F.; Estienne, M.; Debard, C.; Ducroc, R.; Plaisancié, P. Saturated and Unsaturated Fatty Acids Differently Modulate Colonic Goblet Cells In Vitro and in Rat Pups. J. Nutr. 2015, 145, 1754–1762. [Google Scholar] [CrossRef] [PubMed]
- Zain, M.; Awan, F.R.; Najam, S.S.; Islam, M.; Khan, A.R.; Bilal, A.; Bellili, N.; Marre, M.; Roussel, R.; Fumeron, F. Association of ACACB Gene Polymorphism (Rs2268388, G > A) with Type 2 Diabetes and End Stage Renal Disease in Pakistani Punjabi Population. Meta Gene 2017, 12, 109–112. [Google Scholar] [CrossRef]
- Wang, Y.F.; Wang, G.; Li, J.L.; Qu, Y.X.; Liang, X.Y.; Chen, X.D.; Sima, Y.H.; Xu, S.Q. Influence of Hyperproteinemia on Insect Innate Immune Function of the Circulatory System in Bombyx mori. Biology 2021, 10, 112. [Google Scholar] [CrossRef]
- Ali, A.M.; Kunugi, H. Hypoproteinemia Predicts Disease Severity and Mortality in COVID-19: A Call for Action. Diagn. Pathol. 2021, 16, 31. [Google Scholar] [CrossRef]
- Pirzada, R.H.; Haseeb, M.; Batool, M.; Kim, M.S.; Choi, S. Remdesivir and Ledipasvir among the FDA-Approved Antiviral Drugs Have Potential to Inhibit SARS-CoV-2 Replication. Cells 2021, 10, 1052. [Google Scholar] [CrossRef] [PubMed]
- Gabra, H.; BerGenBio ASA. NCT04890509 A Study of Bemcentinib for the Treatment of COVID-19 in Hospitalised Patients. 2021. Available online: https://clinicaltrials.gov/show/NCT04890509 (accessed on 14 October 2021).
- Bonaventura, A.; Montecucco, F. Sofosbuvir/Velpatasvir: A Promising Combination. World J. Hepatol. 2016, 8, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Hill, R.L.; Singh, I.N.; Brelsfoard, J.; Hall, E.D. Pharmacological Inhibition of Lipid Peroxidative Damage by the 21-Aminosteroid U-74389G Improves Cortical Mitochondrial Function Following Traumatic Brain Injury in Young Adult Male Rats. Neuropharmacology 2020, 170, 108023. [Google Scholar] [CrossRef] [PubMed]
- Connor, K.M.; Shapiro, R.E.; Diener, H.C.; Lucas, S.; Kost, J.; Fan, X.; Fei, K.; Assaid, C.; Lines, C.; Ho, T.W. Randomized, Controlled Trial of Telcagepant for the Acute Treatment of Migraine. Neurology 2009, 73, 970–977. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.H.; Peng, C.Y.; Fang, Y.J.; Kao, W.Y.; Yang, S.S.; Lin, C.K.; Lai, H.C.; Su, W.P.; Fang, S.U.; Chang, C.C.; et al. Elbasvir/Grazoprevir for Hepatitis C Virus Genotype 1b East-Asian Patients Receiving Hemodialysis. Sci. Rep. 2020, 10, 9180. [Google Scholar] [CrossRef]
- Fang, C.; Mei, J.; Tian, H.; Liou, Y.L.; Rong, D.; Zhang, W.; Liao, Q.; Wu, N. CSF3 Is a Potential Drug Target for the Treatment of COVID-19. Front. Physiol. 2021, 11, 605792. [Google Scholar] [CrossRef] [PubMed]
- Tutusaus, A.; de Gregorio, E.; Cucarull, B.; Cristóbal, H.; Aresté, C.; Graupera, I.; Coll, M.; Colell, A.; Gausdal, G.; Lorens, J.B.; et al. A Functional Role of GAS6/TAM in Nonalcoholic Steatohepatitis Progression Implicates AXL as Therapeutic Target. Cmgh 2020, 9, 349–368. [Google Scholar] [CrossRef] [PubMed]
- Bohan, D.; Van Ert, H.; Ruggio, N.; Rogers, K.J.; Badreddine, M.; Briseno, J.A.A.; Elliff, J.M.; Chavez, R.A.R.; Gao, B.; Stokowy, T.; et al. Phosphatidylserine Receptors Enhance SARS-CoV-2 Infection. PLoS Pathog. 2021, 17, e1009743. [Google Scholar] [CrossRef]
- Bohan, D.; Van Ert, H.; Dhakal, S.; Ruggio, N.; Rogers, K.; Stokowy, T.; Gausdal, G.; Jackson, A.; Gabra, H.; Micklem, D.R.; et al. Targeting the Receptor Axl by Bemcentinib Prevents SARS-CoV-2 Infection. Top. Antivir. Med. 2021, 29, 137–138. [Google Scholar]
- Elgohary, M.A.S.; Hasan, E.M.; Ibrahim, A.A.; Abdelsalam, M.F.A.; Abdel-Rahman, R.Z.; Zaki, A.I.; Elaatar, M.B.; Elnagar, M.T.; Emam, M.E.; Hamada, M.M.; et al. Efficacy of Sofosbuvir plus Ledipasvir in Egyptian Patients with COVID-19 Compared to Standard Treatment: A Randomized Controlled Trial. J. Med. Life 2022, 15, 350–358. [Google Scholar] [CrossRef]
- Zuccaro, V.; Lombardi, A.; Asperges, E.; Sacchi, P.; Bruno, R. PK/PD and Antiviral Activity of Anti-HCV Therapy: Is There Still a Role in the Choice of Treatment? Expert Opin. Drug Metab. Toxicol. 2020, 16, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and Chloroquine Effectively Inhibit the Recently Emerged Novel Coronavirus (2019-NCoV) In Vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Sayad, B.; Sobhani, M.; Khodarahmi, R. Sofosbuvir as Repurposed Antiviral Drug Against COVID-19: Why Were We Convinced to Evaluate the Drug in a Registered/Approved Clinical Trial? Arch. Med. Res. 2020, 51, 577–581. [Google Scholar] [CrossRef] [PubMed]
- Al-Karmalawy, A.A.; Alnajjar, R.; Dahabd, M.M.; Metwaly, A.M.; Eissa, I.H. Molecular Docking and Dynamics Simulations Reveal the Potential of Anti-HCV Drugs to Inhibit COVID-19 Main Protease. Pharm. Sci. 2021, 27, S109–S121. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, J.M. NGmerge: Merging Paired-End Reads via Novel Empirically-Derived Models of Sequencing Errors. BMC Bioinformatics 2018, 19, 536. [Google Scholar] [CrossRef] [PubMed]
- Ning, Y.; Yang, G.; Chen, Y.; Zhao, X.; Qian, H.; Liu, Y.; Chen, S.; Shi, G. Characteristics of the Urinary Microbiome from Patients with Gout: A Prospective Study. Front. Endocrinol. 2020, 11, 272. [Google Scholar] [CrossRef] [PubMed]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed]
- Roswell, M.; Dushoff, J.; Winfree, R. A Conceptual Guide to Measuring Species Diversity. Oikos 2021, 130, 321–338. [Google Scholar] [CrossRef]
- Cox, K.D.; Black, M.J.; Filip, N.; Miller, M.R.; Mohns, K.; Mortimor, J.; Freitas, T.R.; Greiter Loerzer, R.; Gerwing, T.G.; Juanes, F.; et al. Community Assessment Techniques and the Implications for Rarefaction and Extrapolation with Hill Numbers. Ecol. Evol. 2017, 7, 11213–11226. [Google Scholar] [CrossRef]
- Legendre, P.; Borcard, D.; Peres-Neto, P.R. Analyzing Beta Diversity: Partitioning the Spatial Variation of Community Composition Data. Ecol. Monogr. 2005, 75, 435–450. [Google Scholar] [CrossRef]
- McCafferty, J.; Mühlbauer, M.; Gharaibeh, R.Z.; Arthur, J.C.; Perez-Chanona, E.; Sha, W.; Jobin, C.; Fodor, A.A. Stochastic Changes over Time and Not Founder Effects Drive Cage Effects in Microbial Community Assembly in a Mouse Model. ISME J. 2013, 7, 2116–2125. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Bittinger, K.; Charlson, E.S.; Hoffmann, C.; Lewis, J.; Wu, G.D.; Collman, R.G.; Bushman, F.D.; Li, H. Associating Microbiome Composition with Environmental Covariates Using Generalized UniFrac Distances. Bioinformatics 2012, 28, 2106–2113. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Yang, W.; Chen, G.; Shafiq, M.; Javed, S.; Zaidi, S.S.A.; Shahid, R.; Liu, C.; Bokhari, H. Analysis of Gut Microbiota of Obese Individuals with Type 2 Diabetes and Healthy Individuals. PLoS ONE 2019, 14, e0226372. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, J.S.; Sikaroodi, M.; Gillevet, P.M.; Bajaj, J.S.; Thacker, L.R.; Fagan, A.; White, M.B.; Gavis, E.A.; Hylemon, P.B.; Brown, R.; et al. Gut Microbial RNA and DNA Analysis Predicts Hospitalizations in Cirrhosis. JCI Insight 2018, 3, e98019. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Guo, B.; Yi, N. Zero-Inflated Gaussian Mixed Models for Analyzing Longitudinal Microbiome Data. PLoS ONE 2020, 15, e0242073. [Google Scholar] [CrossRef] [PubMed]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G. PICRUSt2 for Prediction of Metagenome Functions. Nat. Biotechnol. 2020, 38, 669–673. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical Analysis of Taxonomic and Functional Profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING Database in 2017: Quality-Controlled Protein-Protein Association Networks, Made Broadly Accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef]
- Killcoyne, S.; Carter, G.W.; Smith, J.; Boyle, J. Cytoscape: A Community-Based Framework for Network Modeling. Methods Mol. Biol. 2009, 563, 219–239. [Google Scholar] [CrossRef]
- Di Muzio, E.; Toti, D.; Polticelli, F. DockingApp: A User Friendly Interface for Facilitated Docking Simulations with AutoDock Vina. J. Comput. Aided. Mol. Des. 2017, 31, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Studio, D. Dassault Systemes BIOVIA, Discovery Studio Modelling Environment, Release 4.5; Accelrys Software Inc.: San Diego, CA, USA, 2015.
- DeLano, W.L. The PyMOL Molecular Graphics System, Version 2.3; Schrödinger LLC: New York, NY, USA, 2020.
- Kaplan, W.; Littlejohn, T.G. Swiss-PDB Viewer (Deep View). Brief. Bioinform. 2001, 2, 195–197. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeerschd, T.; Zurek, E.; Hutchison, G.R. Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Jaramillo-Botero, A.; Naserifar, S.; Goddard, W.A. General Multiobjective Force Field Optimization Framework, with Application to Reactive Force Fields for Silicon Carbide. J. Chem. Theory Comput. 2014, 10, 1426–1439. [Google Scholar] [CrossRef]
- Rostkowski, M.; Olsson, M.H.; Søndergaard, C.R.; Jensen, J.H. Graphical Analysis of PH-Dependent Properties of Proteins Predicted Using PROPKA. BMC Struct. Biol. 2011, 11, 6. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Li, W.; Zhou, Y.; Shen, J.; Wu, Z.; Liu, G.; Lee, P.W.; Tang, Y. AdmetSAR: A Comprehensive Source and Free Tool for Assessment of Chemical ADMET Properties. J. Chem. Inf. Model. 2012, 52, 3099–3105. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. PkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Land, H.; Humble, M.S. YASARA: A Tool to Obtain Structural Guidance in Biocatalytic Investigations. Methods Mol. Biol. 2018, 1685, 43–67. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber Biomolecular Simulation Programs. J. Comput. Chem. 2005, 26, 1668. [Google Scholar] [CrossRef]
- Dash, R.; Ali, M.C.; Dash, N.; Azad, M.A.K.; Zahid Hosen, S.M.; Hannan, M.A.; Moon, I.S. Structural and Dynamic Characterizations Highlight the Deleterious Role of SULT1A1 R213H Polymorphism in Substrate Binding. Int. J. Mol. Sci. 2019, 20, 6256. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, E.; Rajasekaran, R. Computational Investigation of Curcumin, a Natural Polyphenol That Inhibits the Destabilization and the Aggregation of Human SOD1 Mutant (Ala4Val). RSC Adv. 2016, 6, 102744–102753. [Google Scholar] [CrossRef]
- Swargiary, A.; Mahmud, S.; Saleh, M.A. Screening of Phytochemicals as Potent Inhibitor of 3-Chymotrypsin and Papain-like Proteases of SARS-CoV-2: An in Silico Approach to Combat COVID-19. J. Biomol. Struct. Dyn. 2020, 40, 2067–2081. [Google Scholar] [CrossRef] [PubMed]
- Mahmud, S.; Parves, M.R.; Riza, Y.M.; Sujon, K.M.; Ray, S.; Tithi, F.A.; Zaoti, Z.F.; Alam, S.; Absar, N. Exploring the Potent Inhibitors and Binding Modes of Phospholipase A2 through in Silico Investigation. J. Biomol. Struct. Dyn. 2020, 38, 4221–4231. [Google Scholar] [CrossRef] [PubMed]
- Bappy, S.S.; Sultana, S.; Adhikari, J.; Mahmud, S.; Khan, M.A.; Kibria, K.M.K.; Rahman, M.M.; Shibly, A.Z. Extensive Immunoinformatics Study for the Prediction of Novel Peptide-Based Epitope Vaccine with Docking Confirmation against Envelope Protein of Chikungunya Virus: A Computational Biology Approach. J. Biomol. Struct. Dyn. 2021, 39, 1139–1154. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, K.H.; Chowdhury, M.R.; Mahmud, S.; Tareq, A.M.; Hanif, N.B.; Banu, N.; Ali Reza, A.S.M.; Bin Emran, T.; Simal-Gandara, J. Drug Repurposing Approach against Novel Coronavirus Disease (COVID-19) through Virtual Screening Targeting SARS-CoV-2 Main Protease. Biology 2021, 10, 2. [Google Scholar] [CrossRef]
- Khan, M.A.; Mahmud, S.; Alam, A.S.M.R.U.; Rahman, M.E.; Ahmed, F.; Rahmatullah, M. Comparative Molecular Investigation of the Potential Inhibitors against SARS-CoV-2 Main Protease: A Molecular Docking Study. J. Biomol. Struct. Dyn. 2020, 39, 6317–6323. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phylum | Family | Genus | Species | log2FC | Adj. p-Value |
|---|---|---|---|---|---|
| Proteobacteria | Neisseriaceae | Neisseria | oralis | −4.42 | 0.000 |
| Firmicutes | Streptococcaceae | Streptococcus | infantis | −4.32 | 0.000 |
| Firmicutes | Gemellaceae | Unclassified | Unclassified | −4.29 | 0.000 |
| Firmicutes | Streptococcaceae | Streptococcus | Unclassified | −4.15 | 0.000 |
| Fusobacteria | Leptotrichiaceae | Leptotrichia | Unclassified | −4 | 0.000 |
| Proteobacteria | Oxalobacteraceae | Cupriavidus | Unclassified | −3.8 | 0.000 |
| Firmicutes | Veillonellaceae | Veillonella | parvula | −3.69 | 0.000 |
| Proteobacteria | Pasteurellaceae | Unclassified | Unclassified | −3.63 | 0.000 |
| Firmicutes | Unclassified | Unclassified | Unclassified | −3.52 | 0.000 |
| Proteobacteria | Burkholderiaceae | Lautropia | Unclassified | −3.49 | 0.000 |
| Proteobacteria | Pasteurellaceae | Haemophilus | influenzae | −3.01 | 0.000 |
| Firmicutes | Unclassified | Unclassified | Unclassified | 3.08 | 0.000 |
| Bacteroidetes | Porphyromonadaceae | Parabacteroides | gordonii | 3.18 | 0.000 |
| Actinobacteria | Actinomycetaceae | Actinomyces | hyovaginalis | 3.24 | 0.000 |
| Proteobacteria | Campylobacteraceae | Campylobacter | fetus | 3.26 | 0.000 |
| Firmicutes | Lachnospiraceae | Clostridium | difficile | 3.49 | 0.000 |
| Bacteroidetes | Prevotellaceae | Prevotella | melaninogenica | 3.62 | 0.000 |
| Actinobacteria | Coriobacteriaceae | Atopobium | Rimae | 3.87 | 0.000 |
| Firmicutes | Peptostreptococcaceae | Peptostreptococcus | Anaerobius | 3.95 | 0.000 |
| Bacteroidetes | Bacteroidaceae | Bacteroides | Acidifaciens | 5.1 | 0.000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kibria, M.K.; Ali, M.A.; Yaseen, M.; Khan, I.A.; Bhat, M.A.; Islam, M.A.; Mahumud, R.A.; Mollah, M.N.H. Discovery of Bacterial Key Genes from 16S rRNA-Seq Profiles That Are Associated with the Complications of SARS-CoV-2 Infections and Provide Therapeutic Indications. Pharmaceuticals 2024, 17, 432. https://doi.org/10.3390/ph17040432
Kibria MK, Ali MA, Yaseen M, Khan IA, Bhat MA, Islam MA, Mahumud RA, Mollah MNH. Discovery of Bacterial Key Genes from 16S rRNA-Seq Profiles That Are Associated with the Complications of SARS-CoV-2 Infections and Provide Therapeutic Indications. Pharmaceuticals. 2024; 17(4):432. https://doi.org/10.3390/ph17040432
Chicago/Turabian StyleKibria, Md. Kaderi, Md. Ahad Ali, Muhammad Yaseen, Imran Ahmad Khan, Mashooq Ahmad Bhat, Md. Ariful Islam, Rashidul Alam Mahumud, and Md. Nurul Haque Mollah. 2024. "Discovery of Bacterial Key Genes from 16S rRNA-Seq Profiles That Are Associated with the Complications of SARS-CoV-2 Infections and Provide Therapeutic Indications" Pharmaceuticals 17, no. 4: 432. https://doi.org/10.3390/ph17040432
APA StyleKibria, M. K., Ali, M. A., Yaseen, M., Khan, I. A., Bhat, M. A., Islam, M. A., Mahumud, R. A., & Mollah, M. N. H. (2024). Discovery of Bacterial Key Genes from 16S rRNA-Seq Profiles That Are Associated with the Complications of SARS-CoV-2 Infections and Provide Therapeutic Indications. Pharmaceuticals, 17(4), 432. https://doi.org/10.3390/ph17040432