
Towards Effective Targeted Alpha Therapy for Neuroendocrine Tumours: A Review


Abstract
1. Introduction
1.1. Molecular Radiotherapy
1.2. Neuroendocrine Tumours
1.3. PRRT for NETs—Targeting the Somatostatin Subtype 2 Receptor
1.4. Evolving Standard of Care in PRRT
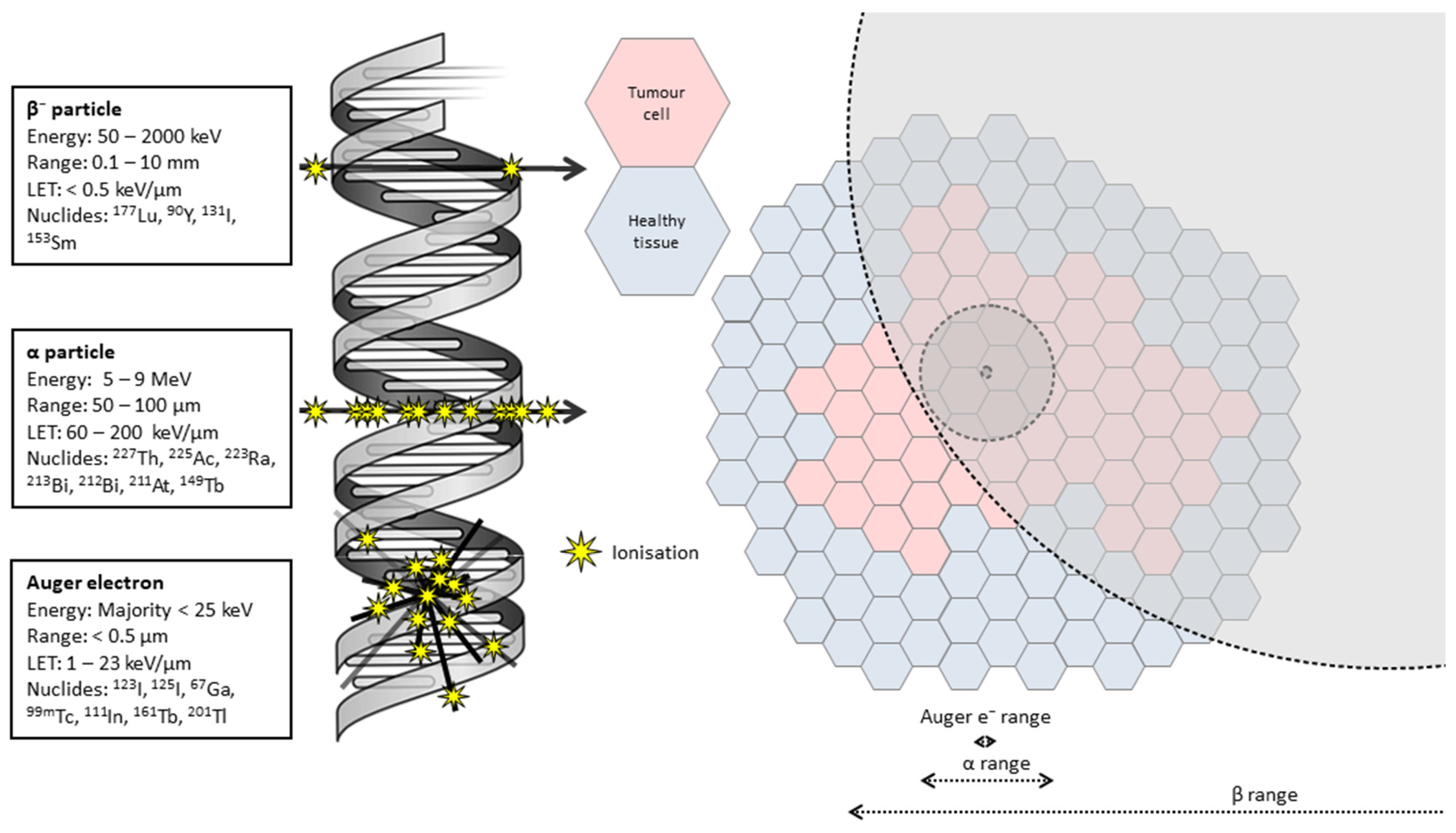
2. Targeted Alpha Therapy (TAT)
2.1. Radiobiological Basis for TAT
2.2. Evaluation of Candidate Radionuclides for TAT
3. Literature Review
3.1. Overview
3.2. Preclinical Studies
3.2.1. In Vitro RBE
3.2.2. In Vivo Efficacy
3.2.3. In Vivo Healthy Tissue Toxicity
3.2.4. In Vivo Dosimetry
3.3. Clinical Applications
3.3.1. Clinical Administration Regimen
3.3.2. Clinical Efficacy
3.3.3. Clinical Toxicity
3.4. Ongoing Clinical Trials
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hope, T.A.; Pavel, M.; Bergsland, E.K. Neuroendocrine Tumors and Peptide Receptor Radionuclide Therapy: When Is the Right Time? J. Clin. Oncol. 2022, 40, 2818. [Google Scholar] [CrossRef]
- Rindi, G.; Klimstra, D.S.; Abedi-Ardekani, B.; Asa, S.L.; Bosman, F.T.; Brambilla, E.; Busam, K.J.; Krijger, R.R.d.; Dietel, M.; El-Naggar, A.K.; et al. A common classification framework for neuroendocrine neoplasms: An International Agency for Research on Cancer (IARC) and World Health Organization (WHO) expert consensus proposal. Mod. Pathol. 2018, 31, 1770–1786. [Google Scholar] [CrossRef]
- Modlin, I.M.; Oberg, K.; Chung, D.C.; Jensen, R.T.; de Herder, W.W.; Thakker, R.V.; Caplin, M.; Delle Fave, G.; Kaltsas, G.A.; Krenning, E.P.; et al. Gastroenteropancreatic neuroendocrine tumours. Lancet Oncol. 2008, 9, 61–72. [Google Scholar] [CrossRef]
- Metz, D.C.; Choi, J.; Strosberg, J.; Heaney, A.P.; Howden, C.W.; Klimstra, D.; Yao, J.C. A rationale for multidisciplinary care in treating neuroendocrine tumours. Curr. Opin. Endocrinol. Diabetes Obes. 2012, 19, 306. [Google Scholar] [CrossRef] [PubMed]
- Sorbye, H.; Kong, G.; Grozinsky-Glasberg, S. PRRT in high-grade gastroenteropancreatic neuroendocrine neoplasms (WHO G3). Endocr.-Relat. Cancer 2020, 27, R67–R77. [Google Scholar] [CrossRef] [PubMed]
- Hicks, R.J.; Kwekkeboom, D.J.; Krenning, E.; Bodei, L.; Grozinsky-Glasberg, S.; Arnold, R.; Borbath, I.; Cwikla, J.; Toumpanakis, C.; Kaltsas, G.; et al. ENETS Consensus Guidelines for the Standards of Care in Neuroendocrine Neoplasia: Peptide Receptor Radionuclide Therapy with Radiolabeled Somatostatin Analogues. Neuroendocrinology 2017, 105, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Zaknun, J.J.; Bodei, L.; Mueller-Brand, J.; Pavel, M.E.; Baum, R.P.; Hörsch, D.; O’Dorisio, M.S.; O’Dorisiol, T.M.; Howe, J.R.; Cremonesi, M.; et al. The joint IAEA, EANM, and SNMMI practical guidance on peptide receptor radionuclide therapy (PRRNT) in neuroendocrine tumours. Eur. J. Nucl. Med. Mol. Imaging 2013, 40, 800–816. [Google Scholar] [CrossRef]
- Theodoropoulou, M.; Stalla, G.K. Somatostatin receptors: From signaling to clinical practice. Front. Neuroendocrinol. 2013, 34, 228–252. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Özgüven, S.; Filizoğlu, N.; Kesim, S.; Öksüzoğlu, K.; Şen, F.; Öneş, T.; İnanır, S.; Turoğlu, H.T.; Erdil, T.Y. Physiological Biodistribution of 68Ga-DOTA-TATE in Normal Subjects. Mol. Imaging Radionucl. Ther. 2021, 30, 39–46. [Google Scholar] [CrossRef]
- Reubi, J.C.; Laissue, J.; Krenning, E.; Lamberts, S.W.J. Somatostatin receptors in human cancer: Incidence, characteristics, functional correlates and clinical implications. J. Steroid Biochem. Mol. Biol. 1992, 43, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Bousquet, C.; Puente, E.; Buscail, L.; Vaysse, N.; Susini, C. Antiproliferative effect of somatostatin and analogs. Chemotherapy 2001, 47 (Suppl. 2), 30–39. [Google Scholar] [CrossRef]
- Krenning, E.P.; Bakker, W.H.; Kooij, P.P.M.; Breeman, W.a.P.; Oei, H.Y.; Jong, M.d.; Reubi, J.C.; Visser, T.J.; Bruns, C.; Kwekkeboom, D.J.; et al. Somatostatin Receptor Scintigraphy with Indium-111-DTPA-D-Phe-1-Octreotide in Man: Metabolism, Dosimetry and Comparison with Iodine-123-Tyr-3-Octreotide. J. Nucl. Med. 1992, 33, 652–658. [Google Scholar]
- Krenning, E.P.; Kwekkeboom, D.J.; Bakker, W.H.; Breeman, W.A.; Kooij, P.P.; Oei, H.Y.; van Hagen, M.; Postema, P.T.; de Jong, M.; Reubi, J.C. Somatostatin receptor scintigraphy with [111In-DTPA-D-Phe1]- and [123I-Tyr3]-octreotide: The Rotterdam experience with more than 1000 patients. Eur. J. Nucl. Med. 1993, 20, 716–731. [Google Scholar] [CrossRef]
- Krenning, E.P.; Kooij, P.P.; Bakker, W.H.; Breeman, W.A.; Postema, P.T.; Kwekkeboom, D.J.; Oei, H.Y.; de Jong, M.; Visser, T.J.; Reijs, A.E. Radiotherapy with a radiolabeled somatostatin analogue, [111In-DTPA-D-Phe1]-octreotide. A case history. Ann. N. Y. Acad. Sci. 1994, 733, 496–506. [Google Scholar] [CrossRef] [PubMed]
- Valkema, R.; de Jong, M.; Bakker, W.H.; Breeman, W.A.P.; Kooij, P.P.M.; Lugtenburg, P.J.; de Jong, F.H.; Christiansen, A.; Kam, B.L.R.; de Herder, W.W.; et al. Phase I study of peptide receptor radionuclide therapy with [111In-DTPA0]octreotide: The rotterdam experience. Semin. Nucl. Med. 2002, 32, 110–122. [Google Scholar] [CrossRef]
- Bodei, L.; Ćwikla, J.B.; Kidd, M.; Modlin, I.M. The role of peptide receptor radionuclide therapy in advanced/metastatic thoracic neuroendocrine tumors. J. Thorac Dis. 2017, 9, S1511–S1523. [Google Scholar] [CrossRef]
- Otte, A.; Jermann, E.; Behe, M.; Goetze, M.; Bucher, H.C.; Roser, H.W.; Heppeler, A.; Mueller-Brand, J.; Maecke, H.R. DOTATOC: A powerful new tool for receptor-mediated radionuclide therapy. Eur. J. Nucl. Med. 1997, 24, 792–795. [Google Scholar] [CrossRef][Green Version]
- Otte, A.; Mueller-Brand, J.; Dellas, S.; Nitzsche, E.U.; Herrmann, R.; Maecke, H.R. Yttrium-90-labelled somatostatin-analogue for cancer treatment. Lancet 1998, 351, 417–418. [Google Scholar] [CrossRef]
- de Jong, M.; Breeman, W.A.P.; Bakker, W.H.; Kooij, P.P.M.; Bernard, B.F.; Hofland, L.J.; Visser, T.J.; Srinivasan, A.; Schmidt, M.A.; Erion, J.L.; et al. Comparison of 111In-labeled Somatostatin Analogues for Tumor Scintigraphy and Radionuclide Therapy1. Cancer Res. 1998, 58, 437–441. [Google Scholar] [PubMed]
- Strosberg, J.R.; Caplin, M.E.; Kunz, P.L.; Ruszniewski, P.B.; Bodei, L.; Hendifar, A.; Mittra, E.; Wolin, E.M.; Yao, J.C.; Pavel, M.E.; et al. 177Lu-Dotatate plus long-acting octreotide versus high-dose long-acting octreotide in patients with midgut neuroendocrine tumours (NETTER-1): Final overall survival and long-term safety results from an open-label, randomised, controlled, phase 3 trial. Lancet Oncol. 2021, 22, 1752–1763. [Google Scholar] [CrossRef]
- Hennrich, U.; Kopka, K. Lutathera®: The First FDA- and EMA-Approved Radiopharmaceutical for Peptide Receptor Radionuclide Therapy. Pharmaceuticals 2019, 12, 114. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. Lutathera Package Insert. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/208700s000lbl.pdf (accessed on 22 February 2024).
- Poty, S.; Francesconi, L.C.; McDevitt, M.R.; Morris, M.J.; Lewis, J.S. α-Emitters for Radiotherapy: From Basic Radiochemistry to Clinical Studies—Part 1. J. Nucl. Med. 2018, 59, 878–884. [Google Scholar] [CrossRef]
- Sgouros, G.; Roeske, J.C.; McDevitt, M.R.; Palm, S.; Allen, B.J.; Fisher, D.R.; Brill, A.B.; Song, H.; Howell, R.W.; Akabani, G. MIRD Pamphlet No. 22 (Abridged): Radiobiology and Dosimetry of α-Particle Emitters for Targeted Radionuclide Therapy. J. Nucl. Med. 2010, 51, 311–328. [Google Scholar] [CrossRef] [PubMed]
- Kassis, A.I. Molecular and cellular radiobiological effects of Auger emitting radionuclides. Radiat. Prot. Dosim. 2011, 143, 241–247. [Google Scholar] [CrossRef]
- Amin, I.K.; Adelstein, S.J. Radiobiologic Principles in Radionuclide Therapy. J. Nucl. Med. 2005, 46, 4S–12S. [Google Scholar]
- Lee, H.; Riad, A.; Martorano, P.; Mansfield, A.; Samanta, M.; Batra, V.; Mach, R.H.; Maris, J.M.; Pryma, D.A.; Makvandi, M. PARP-1-Targeted Auger Emitters Display High-LET Cytotoxic Properties In Vitro but Show Limited Therapeutic Utility in Solid Tumor Models of Human Neuroblastoma. J. Nucl. Med. 2020, 61, 850–856. [Google Scholar] [CrossRef] [PubMed]
- Munro, T.R. The Site of the Target Region for Radiation-Induced Mitotic Delay in Cultured Mammalian Cells. Radiat. Res. 1970, 44, 748–757. [Google Scholar] [CrossRef]
- Liu, B.-C.; Tang, T.-T.; Lv, L.-L.; Lan, H.-Y. Renal tubule injury: A driving force toward chronic kidney disease. Kidney Int. 2018, 93, 568–579. [Google Scholar] [CrossRef]
- Wulbrand, C.; Seidl, C.; Gaertner, F.C.; Bruchertseifer, F.; Morgenstern, A.; Essler, M.; Senekowitsch-Schmidtke, R. Alpha-Particle Emitting 213Bi-Anti-EGFR Immunoconjugates Eradicate Tumor Cells Independent of Oxygenation. PLoS ONE 2013, 8, e64730. [Google Scholar] [CrossRef]
- Barendsen, G.W. Modification of Radiation Damage by Fractionation of the Dose, Anoxia, and Chemical Protectors in Relation to Let. Ann. N. Y. Acad. Sci. 1964, 114, 96–114. [Google Scholar] [CrossRef]
- Feinendegen, L.E.; McClure, J.J. Alpha-Emitters for Medical Therapy: Workshop of the United States Department of Energy: Denver, Colorado, May 30–31, 1996. Radiat. Res. 1997, 148, 195–201. [Google Scholar] [CrossRef]
- Sundlöv, A.; Sjögreen-Gleisner, K. Peptide Receptor Radionuclide Therapy—Prospects for Personalised Treatment. Clin. Oncol. 2021, 33, 92–97. [Google Scholar] [CrossRef]
- Kręcisz, P.; Czarnecka, K.; Królicki, L.; Mikiciuk-Olasik, E.; Szymański, P. Radiolabeled Peptides and Antibodies in Medicine. Bioconjugate Chem. 2021, 32, 25–42. [Google Scholar] [CrossRef]
- Eychenne, R.; Bouvry, C.; Bourgeois, M.; Loyer, P.; Benoist, E.; Lepareur, N. Overview of Radiolabeled Somatostatin Analogs for Cancer Imaging and Therapy. Molecules 2020, 25, 4012. [Google Scholar] [CrossRef]
- McDevitt, M.R.; Ma, D.; Lai, L.T.; Simon, J.; Borchardt, P.; Frank, R.K.; Wu, K.; Pellegrini, V.; Curcio, M.J.; Miederer, M.; et al. Tumor Therapy with Targeted Atomic Nanogenerators. Science 2001, 294, 1537–1540. [Google Scholar] [CrossRef]
- Parker, C.; Nilsson, S.; Heinrich, D.; Helle, S.I.; O’Sullivan, J.M.; Fosså, S.D.; Chodacki, A.; Wiechno, P.; Logue, J.; Seke, M.; et al. Alpha Emitter Radium-223 and Survival in Metastatic Prostate Cancer. N. Engl. J. Med. 2013, 369, 213–223. [Google Scholar] [CrossRef]
- Abou, D.S.; Thiele, N.A.; Gutsche, N.T.; Villmer, A.; Zhang, H.; Woods, J.J.; Baidoo, K.E.; Escorcia, F.E.; Wilson, J.J.; Thorek, D.L.J. Towards the stable chelation of radium for biomedical applications with an 18-membered macrocyclic ligand. Chem. Sci. 2021, 12, 3733–3742. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Jakobsson, V.; Greifenstein, L.; Khong, P.-L.; Chen, X.; Baum, R.P.; Zhang, J. Alpha-peptide receptor radionuclide therapy using actinium-225 labeled somatostatin receptor agonists and antagonists. Front. Med. 2022, 9, 1034315. [Google Scholar] [CrossRef]
- Khabibullin, A.R.; Karolak, A.; Budzevich, M.M.; McLaughlin, M.L.; Morse, D.L.; Woods, L.M. Structure and properties of DOTA-chelated radiopharmaceuticals within the (225)Ac decay pathway. Medchemcomm 2018, 9, 1155–1163. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, J.; Jaggi, J.S.; O’Donoghue, J.A.; Ruan, S.; McDevitt, M.; Larson, S.M.; Scheinberg, D.A.; Humm, J.L. Renal uptake of bismuth-213 and its contribution to kidney radiation dose following administration of actinium-225-labeled antibody. Phys. Med. Biol. 2011, 56, 721. [Google Scholar] [CrossRef]
- de Kruijff, R.M.; Wolterbeek, H.T.; Denkova, A.G. A Critical Review of Alpha Radionuclide Therapy—How to Deal with Recoiling Daughters? Pharmaceuticals 2015, 8, 321–336. [Google Scholar] [CrossRef]
- Mirzadeh, S.; Kumar, K.; Gansow, O.A. The Chemical Fate of 212Bi-DOTA Formed by β- Decay of 212Pb(DOTA)2. Radiochim. Acta 1993, 60, 1–10. [Google Scholar] [CrossRef]
- Su, F.M.; Beaumier, P.; Axworthy, D.; Atcher, R.; Fritzberg, A. Pretargeted radioimmunotherapy in tumored mice using an in vivo 212Pb/212Bi generator. Nucl. Med. Biol. 2005, 32, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Li, M.; Liu, D.; Baumhover, N.J.; Sagastume, E.A.; Marks, B.M.; Rastogi, P.; Pigge, F.C.; Menda, Y.; Johnson, F.L.; et al. Structural modifications toward improved lead-203/lead-212 peptide-based image-guided alpha-particle radiopharmaceutical therapies for neuroendocrine tumors. Eur. J. Nucl. Med. Mol. Imaging 2023, 51, 1147–1162. [Google Scholar] [CrossRef]
- Li, M.; Baumhover, N.J.; Liu, D.; Cagle, B.S.; Boschetti, F.; Paulin, G.; Lee, D.; Dai, Z.; Obot, E.R.; Marks, B.M.; et al. Preclinical Evaluation of a Lead Specific Chelator (PSC) Conjugated to Radiopeptides for 203Pb and 212Pb-Based Theranostics. Pharmaceutics 2023, 15, 414. [Google Scholar] [CrossRef] [PubMed]
- Nelson, B.J.B.; Wilson, J.; Schultz, M.K.; Andersson, J.D.; Wuest, F. High-yield cyclotron production of (203)Pb using a sealed (205)Tl solid target. Nucl. Med. Biol. 2023, 116–117, 108314. [Google Scholar] [CrossRef]
- Pretze, M.; Michler, E.; Runge, R.; Wetzig, K.; Tietze, K.; Brandt, F.; Schultz, M.K.; Kotzerke, J. Influence of the Molar Activity of (203/212)Pb-PSC-PEG(2)-TOC on Somatostatin Receptor Type 2-Binding and Cell Uptake. Pharmaceuticals 2023, 16, 1605. [Google Scholar] [CrossRef]
- Gustafsson, J.; Rodeno, E.; Minguez, P. Feasibility and limitations of quantitative SPECT for (223)Ra. Phys. Med. Biol. 2020, 65, 085012. [Google Scholar] [CrossRef]
- Muller, D.; Herrmann, H.; Schultz, M.K.; Solbach, C.; Ettrich, T.; Prasad, V. 203 Pb-VMT-alpha-NET Scintigraphy of a Patient With Neuroendocrine Tumor. Clin. Nucl. Med. 2023, 48, 54–55. [Google Scholar] [CrossRef]
- Michler, E.; Kastner, D.; Brogsitter, C.; Pretze, M.; Hartmann, H.; Freudenberg, R.; Schultz, M.K.; Kotzerke, J. First-in-human SPECT/CT imaging of [(212)Pb]Pb-VMT-alpha-NET in a patient with metastatic neuroendocrine tumor. Eur. J. Nucl. Med. Mol. Imaging 2023. [Google Scholar] [CrossRef] [PubMed]
- Stokke, C.; Gabiña, P.M.; Solný, P.; Cicone, F.; Sandström, M.; Gleisner, K.S.; Chiesa, C.; Spezi, E.; Paphiti, M.; Konijnenberg, M.; et al. Dosimetry-based treatment planning for molecular radiotherapy: A summary of the 2017 report from the Internal Dosimetry Task Force. EJNMMI Phys. 2017, 4, 27. [Google Scholar] [CrossRef] [PubMed]
- McNeil, B.L.; Robertson, A.K.H.; Fu, W.; Yang, H.; Hoehr, C.; Ramogida, C.F.; Schaffer, P. Production, purification, and radiolabeling of the 203Pb/212Pb theranostic pair. EJNMMI Radiopharm. Chem. 2021, 6, 6. [Google Scholar] [CrossRef]
- Kokov, K.V.; Egorova, B.V.; German, M.N.; Klabukov, I.D.; Krasheninnikov, M.E.; Larkin-Kondrov, A.A.; Makoveeva, K.A.; Ovchinnikov, M.V.; Sidorova, M.V.; Chuvilin, D.Y. (212)Pb: Production Approaches and Targeted Therapy Applications. Pharmaceutics 2022, 14, 189. [Google Scholar] [CrossRef] [PubMed]
- Radchenko, V.; Morgenstern, A.; Jalilian, A.R.; Ramogida, C.F.; Cutler, C.; Duchemin, C.; Hoehr, C.; Haddad, F.; Bruchertseifer, F.; Gausemel, H.; et al. Production and Supply of alpha-Particle-Emitting Radionuclides for Targeted alpha-Therapy. J. Nucl. Med. 2021, 62, 1495–1503. [Google Scholar] [CrossRef] [PubMed]
- U.S. Department of Energy Isotope Program 2022 Actinium-225 User Group Meeting. Available online: https://www.isotopes.gov/22UserGroupMeetings (accessed on 22 February 2024).
- World Nuclear News. CNL Eyes Dramatic Increase in Ac-225 Production. World Nuclear News. 2023. Available online: https://world-nuclear-news.org/Articles/CNL-eyes-dramatic-increase-in-Ac-225-production (accessed on 22 February 2024).
- Zalutsky, M.R.; Pruszynski, M. Astatine-211: Production and Availability. Curr. Radiopharm. 2011, 4, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Handula, M.; Beekman, S.; Konijnenberg, M.; Stuurman, D.; de Ridder, C.; Bruchertseifer, F.; Morgenstern, A.; Denkova, A.; de Blois, E.; Seimbille, Y. First preclinical evaluation of [(225)Ac]Ac-DOTA-JR11 and comparison with [(177)Lu]Lu-DOTA-JR11, alpha versus beta radionuclide therapy of NETs. EJNMMI Radiopharm. Chem. 2023, 8, 13. [Google Scholar] [CrossRef]
- Chan, H.S.; Konijnenberg, M.W.; de Blois, E.; Koelewijn, S.; Baum, R.P.; Morgenstern, A.; Bruchertseifer, F.; Breeman, W.A.; de Jong, M. Influence of tumour size on the efficacy of targeted alpha therapy with 213Bi-[DOTA0,Tyr3]-octreotate. EJNMMI Res. 2016, 6, 6. [Google Scholar] [CrossRef]
- Chan, H.S.; de Blois, E.; Konijnenberg, M.W.; Morgenstern, A.; Bruchertseifer, F.; Norenberg, J.P.; Verzijlbergen, F.J.; de Jong, M.; Breeman, W.A.P. Optimizing labelling conditions of 213Bi-DOTATATE for preclinical applications of peptide receptor targeted alpha therapy. EJNMMI Radiopharm. Chem. 2016, 1, 9. [Google Scholar] [CrossRef][Green Version]
- Chan, H.S.; Konijnenberg, M.W.; Daniels, T.; Nysus, M.; Makvandi, M.; de Blois, E.; Breeman, W.A.; Atcher, R.W.; de Jong, M.; Norenberg, J.P. Improved safety and efficacy of 213Bi-DOTATATE-targeted alpha therapy of somatostatin receptor-expressing neuroendocrine tumors in mice pre-treated with l-lysine. EJNMMI Res. 2016, 6, 83. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.S.; de Blois, E.; Morgenstern, A.; Bruchertseifer, F.; de Jong, M.; Breeman, W.; Konijnenberg, M. In Vitro comparison of 213Bi- and 177Lu-radiation for peptide receptor radionuclide therapy. PLoS ONE 2017, 12, e0181473. [Google Scholar] [CrossRef]
- Chapeau, D.; Koustoulidou, S.; Handula, M.; Beekman, S.; de Ridder, C.; Stuurman, D.; de Blois, E.; Buchatskaya, Y.; van der Schilden, K.; de Jong, M.; et al. [(212)Pb]Pb-eSOMA-01: A Promising Radioligand for Targeted Alpha Therapy of Neuroendocrine Tumors. Pharmaceuticals 2023, 16, 985. [Google Scholar] [CrossRef]
- Cieslik, P.; Kubeil, M.; Zarschler, K.; Ullrich, M.; Brandt, F.; Anger, K.; Wadepohl, H.; Kopka, K.; Bachmann, M.; Pietzsch, J.; et al. Toward Personalized Medicine: One Chelator for Imaging and Therapy with Lutetium-177 and Actinium-225. J. Am. Chem. Soc. 2022, 144, 21555–21567. [Google Scholar] [CrossRef] [PubMed]
- Graf, F.; Fahrer, J.; Maus, S.; Morgenstern, A.; Bruchertseifer, F.; Venkatachalam, S.; Fottner, C.; Weber, M.M.; Huelsenbeck, J.; Schreckenberger, M.; et al. DNA Double Strand Breaks as Predictor of Efficacy of the Alpha-Particle Emitter Ac-225 and the Electron Emitter Lu-177 for Somatostatin Receptor Targeted Radiotherapy. PLoS ONE 2014, 9, e88239. [Google Scholar] [CrossRef]
- King, A.P.; Gutsche, N.T.; Raju, N.; Fayn, S.; Baidoo, K.E.; Bell, M.M.; Olkowski, C.S.; Swenson, R.E.; Lin, F.I.; Sadowski, S.M.; et al. 225Ac-Macropatate: A Novel Alpha Particle Peptide Receptor Radionuclide Therapy for Neuroendocrine Tumors. J. Nucl. Med. 2022, 64, 549–554. [Google Scholar] [CrossRef]
- Miederer, M.; Henriksen, G.; Alke, A.; Mossbrugger, I.; Quintanilla-Martinez, L.; Senekowitsch-Schmidtke, R.; Essler, M. Preclinical Evaluation of the α-Particle Generator Nuclide 225Ac for Somatostatin Receptor Radiotherapy of Neuroendocrine Tumors. Clin. Cancer Res. 2008, 14, 3555–3561. [Google Scholar] [CrossRef] [PubMed]
- Müller, C.; Vermeulen, C.; Köster, U.; Johnston, K.; Türler, A.; Schibli, R.; van der Meulen, N.P. Alpha-PET with terbium-149: Evidence and perspectives for radiotheragnostics. EJNMMI Radiopharm. Chem. 2016, 1, 5. [Google Scholar] [CrossRef]
- Nayak, T.K.; Norenberg, J.P.; Anderson, T.L.; Prossnitz, E.R.; Stabin, M.G.; Atcher, R.W. Somatostatin-receptor-targeted α-emitting 213Bi is therapeutically more effective than β−-emitting 177Lu in human pancreatic adenocarcinoma cells. Nucl. Med. Biol. 2007, 34, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Norenberg, J.P.; Krenning, B.J.; Konings, I.R.H.M.; Kusewitt, D.F.; Nayak, T.K.; Anderson, T.L.; de Jong, M.; Garmestani, K.; Brechbiel, M.W.; Kvols, L.K. 213Bi-[DOTA0, Tyr3]Octreotide Peptide Receptor Radionuclide Therapy of Pancreatic Tumors in a Preclinical Animal Model. Clin. Cancer Res. 2006, 12, 897–903. [Google Scholar] [CrossRef]
- Qin, S.; Yang, Y.; Zhang, J.; Yin, Y.; Liu, W.; Zhang, H.; Fan, X.; Yang, M.; Yu, F. Effective Treatment of SSTR2-Positive Small Cell Lung Cancer Using (211)At-Containing Targeted alpha-Particle Therapy Agent Which Promotes Endogenous Antitumor Immune Response. Mol. Pharm. 2023, 20, 5543–5553. [Google Scholar] [CrossRef] [PubMed]
- Stallons, T.A.R.; Saidi, A.; Tworowska, I.; Delpassand, E.S.; Torgue, J.J. Preclinical Investigation of 212Pb-DOTAMTATE for Peptide Receptor Radionuclide Therapy in a Neuroendocrine Tumor Model. Mol. Cancer Ther. 2019, 18, 1012–1021. [Google Scholar] [CrossRef] [PubMed]
- Tafreshi, N.K.; Pandya, D.N.; Tichacek, C.J.; Budzevich, M.M.; Wang, Z.; Reff, J.N.; Engelman, R.W.; Boulware, D.C.; Chiappori, A.A.; Strosberg, J.R.; et al. Preclinical evaluation of [225Ac]Ac-DOTA-TATE for treatment of lung neuroendocrine neoplasms. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 3408–3421. [Google Scholar] [CrossRef] [PubMed]
- Vaidyanathan, G.; Boskovitz, A.; Shankar, S.; Zalutsky, M.R. Radioiodine and 211At-labeled guanidinomethyl halobenzoyl octreotate conjugates: Potential peptide radiotherapeutics for somatostatin receptor-positive cancers. Peptides 2004, 25, 2087–2097. [Google Scholar] [CrossRef]
- Wharton, L.; Yang, H.; Jaraquemada-Pelaez, M.G.; Merkens, H.; Engudar, G.; Ingham, A.; Koniar, H.; Radchenko, V.; Kunz, P.; Schaffer, P.; et al. Rearmed Bifunctional Chelating Ligand for (225)Ac/(155)Tb Precision-Guided Theranostic Radiopharmaceuticals horizontal line H(4)noneunpaX. J. Med. Chem. 2023, 66, 13705–13730. [Google Scholar] [CrossRef]
- Zhao, B.; Qin, S.; Chai, L.; Lu, G.; Yang, Y.; Cai, H.; Yuan, X.; Fan, S.; Huang, Q.; Yu, F. Evaluation of astatine-211-labeled octreotide as a potential radiotherapeutic agent for NSCLC treatment. Bioorganic Med. Chem. 2018, 26, 1086–1091. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, R.F.; Howell, R.W.; Song, H.; Baechler, S.; Sgouros, G. Redefining Relative Biological Effectiveness in the Context of the EQDX Formalism: Implications for Alpha-Particle Emitter Therapy. Radiat. Res. 2014, 181, 90–98. [Google Scholar] [CrossRef]
- Vaziri, B.; Wu, H.; Dhawan, A.P.; Du, P.; Howell, R.W.; SNMMI MIRD Committee. MIRD Pamphlet No. 25: MIRDcell V2.0 Software Tool for Dosimetric Analysis of Biologic Response of Multicellular Populations. J. Nucl. Med. 2014, 55, 1557–1564. [Google Scholar] [CrossRef]
- Bolch, W.E.; Eckerman, K.F.; Sgouros, G.; Thomas, S.R. MIRD Pamphlet No. 21: A Generalized Schema for Radiopharmaceutical Dosimetry—Standardization of Nomenclature. J. Nucl. Med. 2009, 50, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Li, M.; Bednarz, B.; Schultz, M.K. Modeling Cell and Tumor-Metastasis Dosimetry with the Particle and Heavy Ion Transport Code System (PHITS) Software for Targeted Alpha-Particle Radionuclide Therapy. Radiat. Res. 2018, 190, 236–247. [Google Scholar] [CrossRef]
- Ladjohounlou, R.; Lozza, C.; Pichard, A.; Constanzo, J.; Karam, J.; Le Fur, P.; Deshayes, E.; Boudousq, V.; Paillas, S.; Busson, M.; et al. Drugs That Modify Cholesterol Metabolism Alter the p38/JNK-Mediated Targeted and Nontargeted Response to Alpha and Auger Radioimmunotherapy. Clin. Cancer Res. 2019, 25, 4775–4790. [Google Scholar] [CrossRef] [PubMed]
- de Jong, M.; Breeman, W.A.P.; Bernard, B.F.; van Gameren, A.; de Bruin, E.; Bakker, W.H.; van der Pluijm, M.E.; Visser, T.J.; Mäcke, H.R.; Krenning, E.P. Tumour uptake of the radiolabelled somatostatin analogue [DOTA0,TYR3]octreotide is dependent on the peptide amount. Eur. J. Nucl. Med. 1999, 26, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Beauregard, J.-M.; Hofman, M.S.; Kong, G.; Hicks, R.J. The tumour sink effect on the biodistribution of 68Ga-DOTA-octreotate: Implications for peptide receptor radionuclide therapy. Eur. J. Nucl. Med. Mol. Imaging 2012, 39, 50–56. [Google Scholar] [CrossRef]
- Li, M.; Robles-Planells, C.; Liu, D.; Graves, S.A.; Vasquez-Martinez, G.; Mayoral-Andrade, G.; Lee, D.; Rastogi, P.; Marks, B.M.; Sagastume, E.A.; et al. Pre-clinical evaluation of biomarkers for the early detection of nephrotoxicity following alpha-particle radioligand therapy. Eur. J. Nucl. Med. Mol. Imaging 2023, 1–14. [Google Scholar] [CrossRef]
- de Jong, M.; Krenning, E. New Advances in Peptide Receptor Radionuclide Therapy. J. Nucl. Med. 2002, 43, 617–620. [Google Scholar] [PubMed]
- Strigari, L.; Konijnenberg, M.; Chiesa, C.; Bardies, M.; Du, Y.; Gleisner, K.S.; Lassmann, M.; Flux, G. The evidence base for the use of internal dosimetry in the clinical practice of molecular radiotherapy. Eur. J. Nucl. Med. Mol. Imaging 2014, 41, 1976–1988. [Google Scholar] [CrossRef]
- Bodei, L.; Cremonesi, M.; Ferrari, M.; Pacifici, M.; Grana, C.M.; Bartolomei, M.; Baio, S.M.; Sansovini, M.; Paganelli, G. Long-term evaluation of renal toxicity after peptide receptor radionuclide therapy with 90Y-DOTATOC and 177Lu-DOTATATE: The role of associated risk factors. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 1847–1856. [Google Scholar] [CrossRef]
- Ballal, S.; Yadav, M.P.; Bal, C.; Sahoo, R.K.; Tripathi, M. Broadening horizons with 225Ac-DOTATATE targeted alpha therapy for gastroenteropancreatic neuroendocrine tumour patients stable or refractory to 177Lu-DOTATATE PRRT: First clinical experience on the efficacy and safety. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 934–946. [Google Scholar] [CrossRef] [PubMed]
- Ballal, S.; Yadav, M.P.; Tripathi, M.; Sahoo, R.K.; Bal, C. Survival Outcomes in Metastatic Gastroenteropancreatic Neuroendocrine Tumor Patients receiving Concomitant 225Ac-DOTATATE Targeted Alpha Therapy and Capecitabine: A Real-world Scenario Management Based Long-term Outcome Study. J. Nucl. Med. 2022, 64, 211–218. [Google Scholar] [CrossRef]
- Delpassand, E.S.; Tworowska, I.; Esfandiari, R.; Torgue, J.; Hurt, J.; Shafie, A.; Núñez, R. Targeted Alpha-Emitter Therapy With 212Pb-DOTAMTATE for the Treatment of Metastatic SSTR-Expressing Neuroendocrine Tumors: First-in-Human, Dose-Escalation Clinical Trial. J. Nucl. Med. 2022, 63, 1326–1333. [Google Scholar] [CrossRef] [PubMed]
- Demirci, E.; Alan Selcuk, N.; Beydagi, G.; Ocak, M.; Toklu, T.; Akcay, K.; Kabasakal, L. Initial Findings on the Use of [(225)Ac]Ac-DOTATATE Therapy as a Theranostic Application in Patients with Neuroendocrine Tumors. Mol. Imaging Radionucl. Ther. 2023, 32, 226–232. [Google Scholar] [CrossRef]
- Giesel, F.; Wulfert, S.; Zechmann, C.; Kuder, T.; Kauczor, H.-U.; Haberkorn, U.; Kratochwil, C. Monitoring of perfusion changes after systemic versus selective arterial 177Lu/90Y-DOTATOC and 213Bi-DOTATOC radiopeptide therapy using contrast-enhanced ultrasound in liver metastatic neuroendocrine cancer. J. Nucl. Med. 2011, 52, 1727. [Google Scholar]
- Kratochwil, C.; Giesel, F.L.; Bruchertseifer, F.; Mier, W.; Apostolidis, C.; Boll, R.; Murphy, K.; Haberkorn, U.; Morgenstern, A. 213Bi-DOTATOC receptor-targeted alpha-radionuclide therapy induces remission in neuroendocrine tumours refractory to beta radiation: A first-in-human experience. Eur. J. Nucl. Med. Mol. Imaging 2014, 41, 2106–2119. [Google Scholar] [CrossRef] [PubMed]
- Kratochwil, C.; Apostolidis, L.; Rathke, H.; Apostolidis, C.; Bicu, F.; Bruchertseifer, F.; Choyke, P.L.; Haberkorn, U.; Giesel, F.L.; Morgenstern, A. Dosing 225Ac-DOTATOC in patients with somatostatin-receptor-positive solid tumors: 5-year follow-up of hematological and renal toxicity. Eur. J. Nucl. Med. Mol. Imaging 2021, 49, 54–63. [Google Scholar] [CrossRef]
- Yadav, M.P.; Ballal, S.; Sahoo, R.K.; Bal, C. Efficacy and safety of 225Ac-DOTATATE targeted alpha therapy in metastatic paragangliomas: A pilot study. Eur. J. Nucl. Med. Mol. Imaging 2022, 49, 1595–1606. [Google Scholar] [CrossRef]
- Zhang, J.; Singh, A.; Kulkarni, H.R.; Schuchardt, C.; Müller, D.; Wester, H.-J.; Maina, T.; Rösch, F.; van der Meulen, N.P.; Müller, C.; et al. From Bench to Bedside—The Bad Berka Experience With First-in-Human Studies. Semin. Nucl. Med. 2019, 49, 422–437. [Google Scholar] [CrossRef] [PubMed]
- Imhof, A.; Brunner, P.; Marincek, N.; Briel, M.; Schindler, C.; Rasch, H.; Mäcke, H.R.; Rochlitz, C.; Müller-Brand, J.; Walter, M.A. Response, Survival, and Long-Term Toxicity After Therapy With the Radiolabeled Somatostatin Analogue [90Y-DOTA]-TOC in Metastasized Neuroendocrine Cancers. J. Clin. Oncol. 2011, 29, 2416–2423. [Google Scholar] [CrossRef] [PubMed]
- Bodei, L.; Kidd, M.; Paganelli, G.; Grana, C.M.; Drozdov, I.; Cremonesi, M.; Lepensky, C.; Kwekkeboom, D.J.; Baum, R.P.; Krenning, E.P.; et al. Long-term tolerability of PRRT in 807 patients with neuroendocrine tumours: The value and limitations of clinical factors. Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 5–19. [Google Scholar] [CrossRef]
- Sharma, M.; McCarthy, E.T.; Sharma, R.; Fish, B.L.; Savin, V.J.; Cohen, E.P.; Moulder, J.E. Arachidonic Acid Metabolites Mediate the Radiation-Induced Increase in Glomerular Albumin Permeability. Exp Biol. Med. 2006, 231, 99–106. [Google Scholar] [CrossRef]
- U.S. Department of Health and Human Services. Common Terminology Criteria for Adverse Events (CTCAE) Version 4.0; National Institutes of Health: New York, NY, USA, 2009; pp. 1–9.
- Cremonesi, M.; Ferrari, M.E.; Bodei, L.; Chiesa, C.; Sarnelli, A.; Garibaldi, C.; Pacilio, M.; Strigari, L.; Summers, P.E.; Orecchia, R.; et al. Correlation of dose with toxicity and tumour response to 90Y- and 177Lu-PRRT provides the basis for optimization through individualized treatment planning. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 2426–2441. [Google Scholar] [CrossRef]
- Sundlöv, A.; Gleisner, K.S.; Tennvall, J.; Ljungberg, M.; Warfvinge, C.F.; Holgersson, K.; Hallqvist, A.; Bernhardt, P.; Svensson, J. Phase II trial demonstrates the efficacy and safety of individualized, dosimetry-based 177Lu-DOTATATE treatment of NET patients. Eur. J. Nucl. Med. Mol. Imaging 2022, 49, 3830–3840. [Google Scholar] [CrossRef]
- Strosberg, J.; Hofman, M.S.; Al-Toubah, T.; Hope, T.A. Rethinking Dosimetry: The Perils of Extrapolated External-Beam Radiotherapy Constraints to Radionuclide Therapy. J. Nucl. Med. 2024. [Google Scholar] [CrossRef]
- Gear, J.I.; Cox, M.G.; Gustafsson, J.; Gleisner, K.S.; Murray, I.; Glatting, G.; Konijnenberg, M.; Flux, G.D. EANM practical guidance on uncertainty analysis for molecular radiotherapy absorbed dose calculations. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 2456–2474. [Google Scholar] [CrossRef]
- Geenen, L.; Nonnekens, J.; Konijnenberg, M.; Baatout, S.; De Jong, M.; Aerts, A. Overcoming nephrotoxicity in peptide receptor radionuclide therapy using [(177)Lu]Lu-DOTA-TATE for the treatment of neuroendocrine tumours. Nucl. Med. Biol. 2021, 102–103, 1–11. [Google Scholar] [CrossRef]
- Hofmann, W.; Li, W.B.; Friedland, W.; Miller, B.W.; Madas, B.; Bardies, M.; Balashazy, I. Internal microdosimetry of alpha-emitting radionuclides. Radiat. Env. BioPhys. 2020, 59, 29–62. [Google Scholar] [CrossRef]
- Al-Toubah, T.; Montilla-Soler, J.; El-Haddad, G.; Haider, M.; Strosberg, J. Somatostatin Receptor Expression in Lung Neuroendocrine Tumors: An Analysis of dotatate pet Scans. J. Nucl. Med. 2023, 64, 1895–1898. [Google Scholar] [CrossRef]
- Reubi, J.C.; Maecke, H.R. Approaches to Multireceptor Targeting: Hybrid Radioligands, Radioligand Cocktails, and Sequential Radioligand Applications. J. Nucl. Med. 2017, 58, 10S–16S. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parent | Daughters | T1/2 | Decay Type | Energy (MeV) | Yield | Imaging |
|---|---|---|---|---|---|---|
| 227Th | 18.7 d | α | 5.76, 5.98, 6.04 | 0.20, 0.23, 0.24 | γ: 236 keV (0.13) | |
| 223Ra | 11.4 d | α | 5.60, 5.72 | 0.25, 0.51 | γ: 269 keV (0.13) | |
| 219Rn | 3.96 s | α | 6.55, 6.82 | 0.13, 0.79 | γ: 271 keV (0.11) | |
| 215Po | 1.78 ms | α | 7.39 | 1.00 | - | |
| 211Pb | 36.1 min | β- | 0.16, 0.47 | 0.06. 0.91 | γ: 405 keV (0.04) | |
| 211Bi | 2.14 min | α | 6.28, 6.62 | 0.16, 0.84 | γ: 351 keV (0.13) | |
| 207Tl | 4.77 min | β- | 0.493 | 1.00 | - | |
| 207Pb | Stable | |||||
| 225Ac | 10.0 d | α | 5.79, 5.83 | 0.18, 0.51 | - | |
| 221Fr | 4.80 min | α | 6.13, 6.24 6.34 | 0.15, 0.01, 0.83 | γ: 218 keV (0.13) | |
| 217At | 32.6 ms | α | 7.07 | 1.00 | - | |
| 213Bi | 45.6 min | α (0.02) | 5.86 | 0.02 | γ: 440 keV (0.26) | |
| β- (0.98) | 0.32, 0.49 | 0.30, 0.67 | ||||
| 213Po | 3.72 µs | α | 8.38 | 1.00 | - | |
| 209Tl | 2.16 min | β- | 0.660 | 0.97 | γ: 117 keV (0.76) | |
| 209Pb | 3.23 h | β- | 0.198 | 1.00 | - | |
| 209Bi | 2.0 × 1019 y | α | 2.88, 3.08 | 0.01, 0.99 | - | |
| 205Tl | Stable | |||||
| 224Ra | 3.66 d | α | 5.45, 5.69 | 0.05, 0.95 | γ: 241 keV (0.04) | |
| 220Rn | 55.6 s | α | 6.29 | 0.99 | - | |
| 216Po | 144 ms | α | 6.78 | 1.00 | - | |
| 212Pb | 10.6 h | β- | 0.41, 0.93, 0.17 | 0.05, 0.81, 0.14 | - | |
| 212Bi | 60.6 min | α (0.36) | 6.05, 6.09 | 0.25, 0.10 | γ: 727 keV (0.07) | |
| β- (0.64) | 0.53, 0.83 | 0.04, 0.55 | ||||
| 212Po | 17.1 ns | α | 10.2 | 0.42 | - | |
| 208Tl | 3.05 min | β- | 0.44, 0.54, 0.65 | 0.24, 0.22, 0.49 | γ: 277 keV (0.07) | |
| 208Pb | Stable | |||||
| 211At | 7.21 h | α (0.42) | 5.87 | 0.42 | X: 77–92 keV | |
| ε (0.58) | - | - | - | |||
| 211Po | 0.52 s | α | 7.45 | 0.99 | - | |
| 207Bi | 31.6 y | ε | - | - | γ: 570 keV (0.98) | |
| 207Pb | Stable | |||||
| 149Tb | 4.12 h | α (0.17) | 3.97 | 0.17 | β+: 639 keV (0.04) | |
| ε (0.83) | γ: 165 keV (0.27) | |||||
| 149Gd | 9.28 d | ε | - | - | γ: 150 keV (0.48) | |
| 149Eu | 93.1 d | ε | - | - | - | |
| 149Sm | Stable | |||||
| 145Eu | 5.93 d | ε | - | - | β+: 740 keV (0.02) | |
| 145Sm | 340 d | ε | - | - | - | |
| 145Pm | 17.7 y | α (2.8 × 10−7) | 2.24 | 2.80 × 10−7 | - | |
| ε (1.00) | - | - | - | |||
| 145Nd | Stable | |||||
| 141Pr | Stable |
| Author | Radiopharmaceutical | Aim | Findings |
|---|---|---|---|
| Chan [61] | [213Bi]Bi-DOTA-TATE | Determine whether TAT efficacy in vivo is related to tumour size in two SSTR2 +ve cell lines. | Improved OS, increased tumour doubling time vs. control in small (50 mm3) and large (200 mm3) CA20948 and H69 tumours. Several cures in small tumour cohort. No toxicity. |
| Chan [62] | [213Bi]Bi-DOTA-TATE | Investigate optimal radiolabelling conditions (peptide amount, quencher, pH) for [213Bi]Bi-DOTATATE. | >3.5 nmol DOTATATE required for >99% incorporation with 100 MBq 213Bi. Optimised conditions: pH = 8.3, TRIS = 0.15 mol/L in 800 µL. Ascorbic acid (0.9 mmol/L) required to avoid radiolysis. |
| Chan [63] | [213Bi]Bi-DOTA-TATE | Evaluate the therapeutic effect of TAT with and without renal protection using L lysine in vivo. | MTA in healthy mice = 13, 21.7 MBq with/without renal protection. In tumour-bearing, median OS > 30 d at 17 MBq, severe weight loss and mortality at 33 MBq. Renal protection improved OS. |
| Chan [64] | [213Bi]Bi-DOTA-TATE | Develop methods to determine relationship between absorbed dose and cell killing in vitro. Compare cytotoxicity across radiations in various cell lines. | In CA20948, D10 = 3 Gy, 18 Gy and 5 Gy for [213Bi]Bi-DOTATATE, [177Lu]Lu-DOTATATE and 137Cs. In BON, [177Lu]Lu-DOTATATE had no effect, D10 for [213Bi]Bi-DOTATATE, 137Cs = 2.5 Gy, 4.5 Gy. |
| Chapeau [65] | [212Pb]Pb-eSOMA-01 | Develop new octreotate derivatives with non-DOTA chelators and assess their potential for TAT of NETs with Pb. | New SSTR2-targetting ligands labelled successfully with 212/203Pb, eSOMA-01 showed favourable biodistribution compared to DOTAM-TATE. |
| Cieslik [66] | [225Ac]Ac-L1-TATE | Assess feasibility of L1 as chelator with 177Lu, 211At, 225Ac in two SSTR2 +ve cell lines, evaluate biodistribution in MPC tumour bearing mice. | L1 can bind radionuclides for imaging and therapy. Preferable fast and mild labelling compared to DOTA. [225Ac]Ac-L1 produced with molar activity > 0.25 MBq/nmol. |
| Graf [67] | [225Ac]Ac-DOTA-TOC | Assess γH2AX foci formation as biomarker of cytotoxicity and response to [225Ac]Ac-DOTATOC and [177Lu]Lu-DOTATOC in vitro and in vivo. | High tumour control rate with single treatment of both agents. Number of γH2AX foci correlated with apoptosis (in vitro) and tumour growth, showing potential as biomarker. |
| Handula [60] | [225Ac]Ac-DOTA-JR11 | Investigate potential of [225Ac]Ac-DOTA-JR11 (antagonist) for therapy of NETs via mouse model. | Low tumour-to-kidney ratio of absorbed dose is limiting for therapeutic use of [225Ac]Ac-DOTA-JR11. |
| King [68] | [225Ac]Ac-MACROPA-TATE | Synthesise and characterise MACROPA TATE, compare performance with DOTA TATE in labelling efficiency, stability, binding, efficacy. | [225Ac]Ac-MACROPATATE showed higher renal and liver uptake and toxicity at lower activities, DOTATATE deemed superior. |
| Lee [46] | [212Pb]Pb-PSC-PEG2-TOC | Improve SSTR2 targeting over DOTA-based conjugates via click-chemistry-based cyclization, improved chelator design and insertion of PEG linkers. | Development of lead-specific chelator (PSC) and insertion of PEG linkers results in improved tumour uptake, retention and quicker renal clearance, and dose-dependent therapeutic effect with acceptable toxicity. |
| Li [47] | [212Pb]Pb-PSC-PEG-TOC | Characterise Pb-specific chelator for radiolabelling yield, stability and in vivo biodistribution. | 212Pb and 212Bi stably incorporated in PSC-PEG-TOC. Biodistribution of 212Pb/212Bi-PSC-PEG-TOC were comparable. 203/212Pb showed comparable biodistribution. |
| Miederer [69] | [225Ac]Ac-DOTA-TOC | Compare biodistribution, toxicity and anti-tumour effect of [225Ac]Ac-DOTATOC and [177Lu]Lu-DOTATOC. | Activities > 30 kBq of 225Ac-induced tubular necrosis, weight loss. 225Ac (20 kBq) showed improved tumour growth delay vs. 177Lu (0.45 MBq). |
| Müller [70] | [149Tb]Tb-DOTA-NOC | Letter to the editor to highlight the potential of 149Tb for ‘α PET’. | High quality PET image of mouse injected with 7 MBq [149Tb]Tb-DOTANOC showing high tumour uptake. |
| Nayak [71] | [213Bi]Bi-DOTA-TOC | Compare binding, cytotoxicity, induction of apoptosis between [213Bi/177Lu]Lu-DOTATOC in human pancreatic adenocarcinoma cells. | RBE of [213Bi]Bi-DOTATOC, [177Lu]Lu-DOTATOC relative to 137Cs = 3.4, 1.0. 213Bi induced greater release of apoptosis markers in Capan-2 cells. |
| Norenberg [72] | [213Bi]Bi-DOTA-TOC | Evaluate quantitative labelling methods, stability, biodistribution, safety, and efficacy in vivo. | Activity-related decrease in tumour growth rate observed (>11 MBq). Mild acute but no chronic nephrotoxicity. No haemato-toxicity. |
| Pretze [49] | [212Pb]Pb-PSC-PEG2-TOC | Investigate the influence of different molar activities of [203/212Pb]Pb-PSC2-TOC on cell uptake. | Uptake increased with molar activity, 15–40 MBq/nmol showed highest cell uptake. |
| Qin [73] | [211At]At-SAB-Oct | Develop octreotide SAB conjugate to be labelled with 211At and evaluate therapeutic efficacy against SCLC. | Anti-tumour response against SCLC model demonstrated, with acceptable toxicity profile. |
| Stallons [74] | [212Pb]Pb-DOTAM-TATE | Determine binding and cell kill in vitro. Assess biodistribution in vivo. Establish tolerable regimen and efficacy as mono and combination therapy. | Non-toxic at <45 µCi, toxicity overcome by fractionation into 3 cycles. 79% cure rate with 3 × 10 µCi in combination with 5FU. Benefits of ascorbic acid and nephro protection demonstrated. |
| Tafreshi [75] | [225Ac]Ac-DOTA-TATE | Assess toxicity, biodistribution, dosimetry and efficacy in lung neuroendocrine model (H727/H69) in vivo. | Chronic progressive nephropathy at >111 kBq. Single admin produced tumour growth delay and reduction in tumour volume vs. control. |
| Vaidyanathan [76] | [211At]At-GIMBO | Synthesise octreotate analogue with guanidine-containing template for 211At labelling, assess in comparison with Glu-TOCA in vitro and in vivo. | Single step process to synthesise radioiodinated and astatinated octreotide analogue with positive template reported. Affinity for SSTR2 demonstrated, but high uptake in normal tissue is limiting. |
| Wharton [77] | [225Ac]Ac-H4noneupaX-TATE | Develop novel bifunctional chelator capable of complexing 225Ac and 155Tb for theragnostics. | H4noneupaX was characterised, then labelling of 225Ac and 155Tb assessed. SPECT/CT imaging of 155Tb demonstrates potential as theragnostic pair isotope for 225Ac therapy. |
| Zhao [78] | [211At]At-SPC-TOC | Investigate possible use of 211Ac-labelled octreotide to treat NSCLC. | [211At]At-SPC-octreotide showed elevated and activity-dependent apoptosis induction compared to PBS, cold peptide and unlabelled 211At. |
| Author | Cell Line | Radiopharmaceutical | Reference Radiation | End Point | RBE |
|---|---|---|---|---|---|
| Chan [64] | CA20948 (rat pancreatic) | [213Bi]Bi-DTPA | 137Cs | D10 | 2.0 |
| [213Bi]Bi-DOTATATE | 137Cs | D10 | 1.5 | ||
| [213Bi]Bi-DOTATATE | [177Lu]Lu-DOTATATE | D10 | 5.4 | ||
| [213Bi]Bi-DOTATATE | [177Lu]Lu-DOTATATE | D10 | 5.7 | ||
| BON (human carcinoid) | [213Bi]Bi-DTPA | 137Cs | D10 | 1.8 | |
| [213Bi]Bi-DOTATATE | 137Cs | D10 | 1.7 | ||
| Graf [67] | AR42J (rat pancreatic) | [225Ac]Ac-DOTATOC | [177Lu]Lu-DOTATOC | ED50 (kBq/mL) | 5.5 |
| Nayak [71] | Capan-2 (human pancreatic) | [213Bi]Bi-DOTATOC | 137Cs | D20 | 3.4 |
| [177Lu]Lu-DOTATOC | 137Cs | D20 | 1.0 |
| Author | Radiopharmaceutical | Tumour Bearing | Cell Line | Nephro-Protection | ADC (Gy/MBq) | T:K | |
|---|---|---|---|---|---|---|---|
| Tumour | Kidneys | ||||||
| Chan [61] | [213Bi]Bi-DOTATATE | + | CA20948 | − | 0.8 | 1.6 | 0.49 |
| + | H69 | − | 0.5 | 2.0 | 0.23 | ||
| Chan [63] | [213Bi]Bi-DOTATATE | + | AR42J | + | 0.7 | 0.6 | 1.18 |
| + | AR42J | − | 0.7 | 1.1 | 0.64 | ||
| − | N/A | + | N/A | 0.5 | N/A | ||
| − | N/A | − | N/A | 1.0 | N/A | ||
| Chapeau [65] | [212Pb]Pb-DOTAM-TATE | + | H69 | − | 26.6 | 140.0 | 0.19 |
| [212Pb]Pb-eSOMA-01 | + | H69 | − | 35.5 | 121.7 | 0.29 | |
| [212Pb]Pb-eSOMA-02 | + | H69 | − | 14.7 | 147.4 | 0.10 | |
| Handula [60] | [225Ac]Ac-DOTA-JR11 | + | H69 | − | 328.5 | 952.6 | 0.34 |
| Lee [46] | [212Pb]Pb-DOTA-TOC | + | AR42J | + | 2.4 | 7.0 | 0.35 |
| [212Pb]Pb-PSC-TOC | + | AR42J | + | 9.2 | 5.4 | 1.70 | |
| [212Pb]Pb-PSC-PEG2-TOC | + | AR42J | + | 12.7 | 6.2 | 2.04 | |
| [212Pb]Pb-PSC-PEG2-TOC | + | AR42J | + | 8.7 | 3.2 | 2.69 | |
| Tafreshi [75] | [225Ac]Ac-DOTATATE | − | N/A | − | N/A | 6.8 | N/A |
| Author | Indication | Radiopharmaceutical | N | Aim | Findings |
|---|---|---|---|---|---|
| Ballal [90] | GEP-NETs | [225Ac]Ac-DOTA-TATE | 32 | Present early results on safety, efficacy, QoL following TAT in patients stable or refractory to [177Lu]Lu-DOTATATE | Morphological response assessed in 24/34 patients, n = 15 PR, n = 9 SD. No disease progression. Therapy was well tolerated in this population. |
| Ballal [91] | GEP-NETs | [225Ac]Ac-DOTA-TATE | 91 | Evaluate long-term outcome of TAT in GEP-NET patients in mixed population of PRRT naive and pre-treated. | TAT improved OS, even in patients refractory to prior 177Lu, with transient and acceptable toxicity. |
| Delpassand [92] | GEP-NETs | [212Pb]Pb-DOTAM-TATE | 20 | Establish safety of 212Pb-DOTAM-TATE in phase 1 dose-escalation study. | TAT well tolerated, no serious TEAEs related to the study drug. ORR of 80% at 2.50 MBq/kg/cycle, showing potential benefit over approved therapies. |
| Demirci [93] | NETs | [225Ac]Ac-DOTA-TATE | 11 | Retrospective study including 11 patients with NETs of different primary sites treated with [225Ac]Ac-DOTA-TATE. | Nine patients had PET/CT follow up. No grade III/IV toxicity, 4/9 partial response, 8/9 disease control. 225Ac is safe and effective in treatment of patients refractory to β-PRRT. |
| Giesel [94] | Hepatic NET mets | [213Bi]Bi-DOTA-TOC | 14 * | Investigate the role of contrast enhanced ultrasound in monitoring tumour response to α/β PRRT. | CE-US comparable to CE-CT and suitable for monitoring PRRT response. Decrease in perfusion indicative of tumour response. |
| Kratochwil [95] | NETs | [213Bi]Bi-DOTA-TOC | 8 | Report first in-human experience in PRRT pre-treated patients with [213Bi]Bi-DOTA-TOC. | Specific tumour uptake shown on imaging. TAT produced enduring response with moderate nephrotoxicity, is effective against β-refractory disease. |
| Kratochwil [96] | NETs | [225Ac]Ac-DOTA-TOC | 39 | Estimate optimal single cycle and cumulative activity for [225Ac]Ac-DOTA-TOC. | ~20 MBq/cycle (4-month interval) and cumulative activity ≤ 60–80 MBq avoided acute and chronic grade III/IV haemato-toxicity, some chronic renal toxicity. |
| Yadav [97] | Metastatic paraganglioma | [225Ac]Ac-DOTA-TATE | 9 | Evaluate the efficacy and safety of TAT in advanced stage paragangliomas. | 50% PR, 37.5% SD, 12.5% PD, with symptoms decreased. No grade III/IV renal or haematological toxicity. Benefit even in patients refractory to β-PRRT. |
| Zhang [98] | NETs | [225Ac]Ac-DOTA-TOC | 10 | Discuss experience with first-in-human use of novel radiopharmaceuticals, including [225Ac]Ac-DOTA-TOC, at Bad Berka. | α-PRRT was well tolerated and effective, including in one patient treated intra-arterially. |
| Author | Radiopharmaceutical | Activity/Cycle (MBq) | N Cycles | Interval (Weeks) | Cumulative Activity (MBq) | Co-Admin |
|---|---|---|---|---|---|---|
| Ballal [90] | [225Ac]Ac-DOTA-TATE | 0.1/kg (8/80 kg) | 1–4 | 8 | 23 (8–33) | Amino acid |
| Ballal [91] | [225Ac]Ac-DOTA-TATE | 0.1/kg (8/80 kg) | 1–10 (med = 4) | 8 | 36 (22–59) | Amino acid, radiosensitiser |
| Delpassand [92] | [212Pb]Pb-DOTAM-TATE | 1.13/kg (90/80 kg) | 1 | 8 | 84 | Amino acid |
| 1.48/kg (118/80 kg) | 1 | 8 | 112 | Amino acid | ||
| 1.92/kg (154/80 kg) | 3 | 8 | 406 | Amino acid | ||
| 2.50/kg (200/80 kg) | 4 | 8 | 791 | Amino acid | ||
| Demirci [93] | [225Ac]Ac-DOTA-TATE | 0.1–0.12/kg (8–9.6/80 kg) | 1–3 | 18 | N/A | Amino acid |
| Giesel [94] | [213Bi]Bi-DOTA-TOC | N/A | N/A | N/A | N/A | N/A |
| Kratochwil [95] | [213Bi]Bi-DOTA-TOC | 1000–10,500 | 1–5 (med = 4.5) | 8 | 45 | Amino acid |
| Kratochwil [96] | [225Ac]Ac-DOTA-TOC | 6–60 | 1–5 (med = 4.5) | 8–52 (med = 16) | 15,800 (3300–20,600) | Amino acid, diuretic |
| Yadav [97] | [225Ac]Ac-DOTA-TATE | 0.1/kg (8/80 kg) | 2–9 (med = 3) | 8 | 42.4 (15.5–86.6) | Amino acid, radiosensitiser |
| Zhang [98] | [225Ac]Ac-DOTA-TOC | N/A | N/A | N/A | N/A | N/A |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gape, P.M.D.; Schultz, M.K.; Stasiuk, G.J.; Terry, S.Y.A. Towards Effective Targeted Alpha Therapy for Neuroendocrine Tumours: A Review. Pharmaceuticals 2024, 17, 334. https://doi.org/10.3390/ph17030334
Gape PMD, Schultz MK, Stasiuk GJ, Terry SYA. Towards Effective Targeted Alpha Therapy for Neuroendocrine Tumours: A Review. Pharmaceuticals. 2024; 17(3):334. https://doi.org/10.3390/ph17030334
Chicago/Turabian StyleGape, Paul M. D., Michael K. Schultz, Graeme J. Stasiuk, and Samantha Y. A. Terry. 2024. "Towards Effective Targeted Alpha Therapy for Neuroendocrine Tumours: A Review" Pharmaceuticals 17, no. 3: 334. https://doi.org/10.3390/ph17030334
APA StyleGape, P. M. D., Schultz, M. K., Stasiuk, G. J., & Terry, S. Y. A. (2024). Towards Effective Targeted Alpha Therapy for Neuroendocrine Tumours: A Review. Pharmaceuticals, 17(3), 334. https://doi.org/10.3390/ph17030334