
Genetic Variations and Antibiotic-Related Adverse Events



Abstract
1. Introduction
2. Antibiotics for Which the Role of Genetics in Conditioning Development of Adverse Events Is Definitively Demonstrated
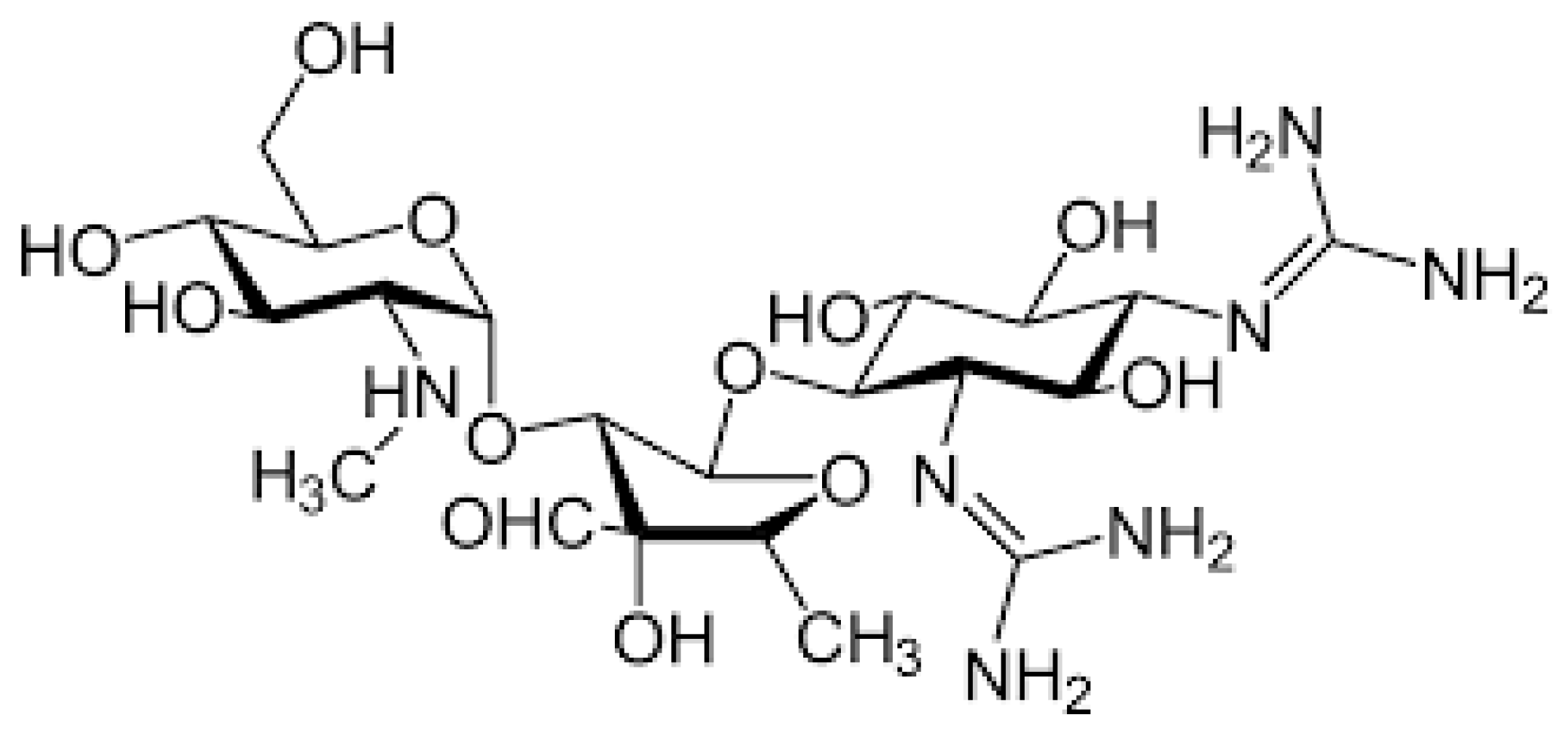
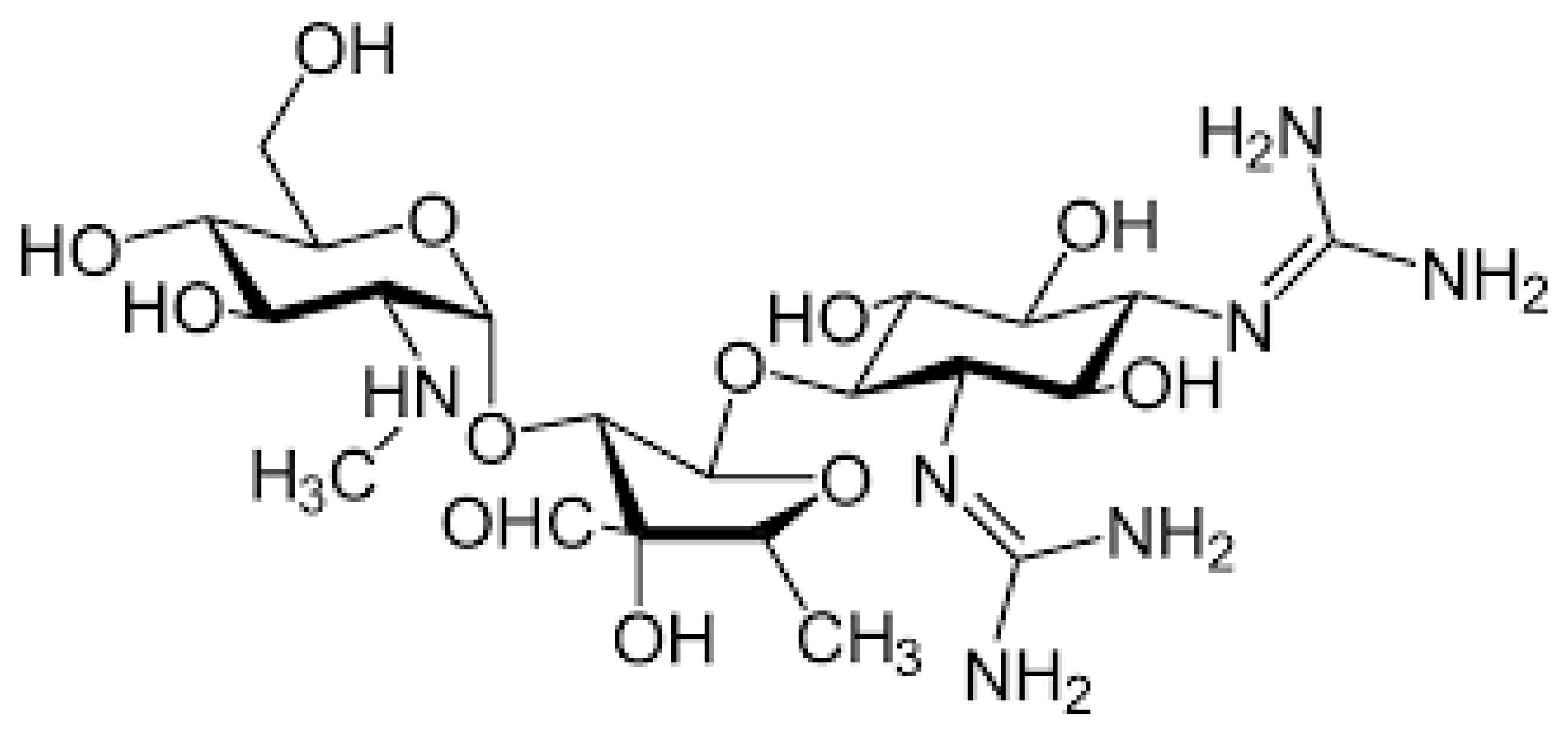
2.1. Aminoglycosides
2.2. Beta-Lactams
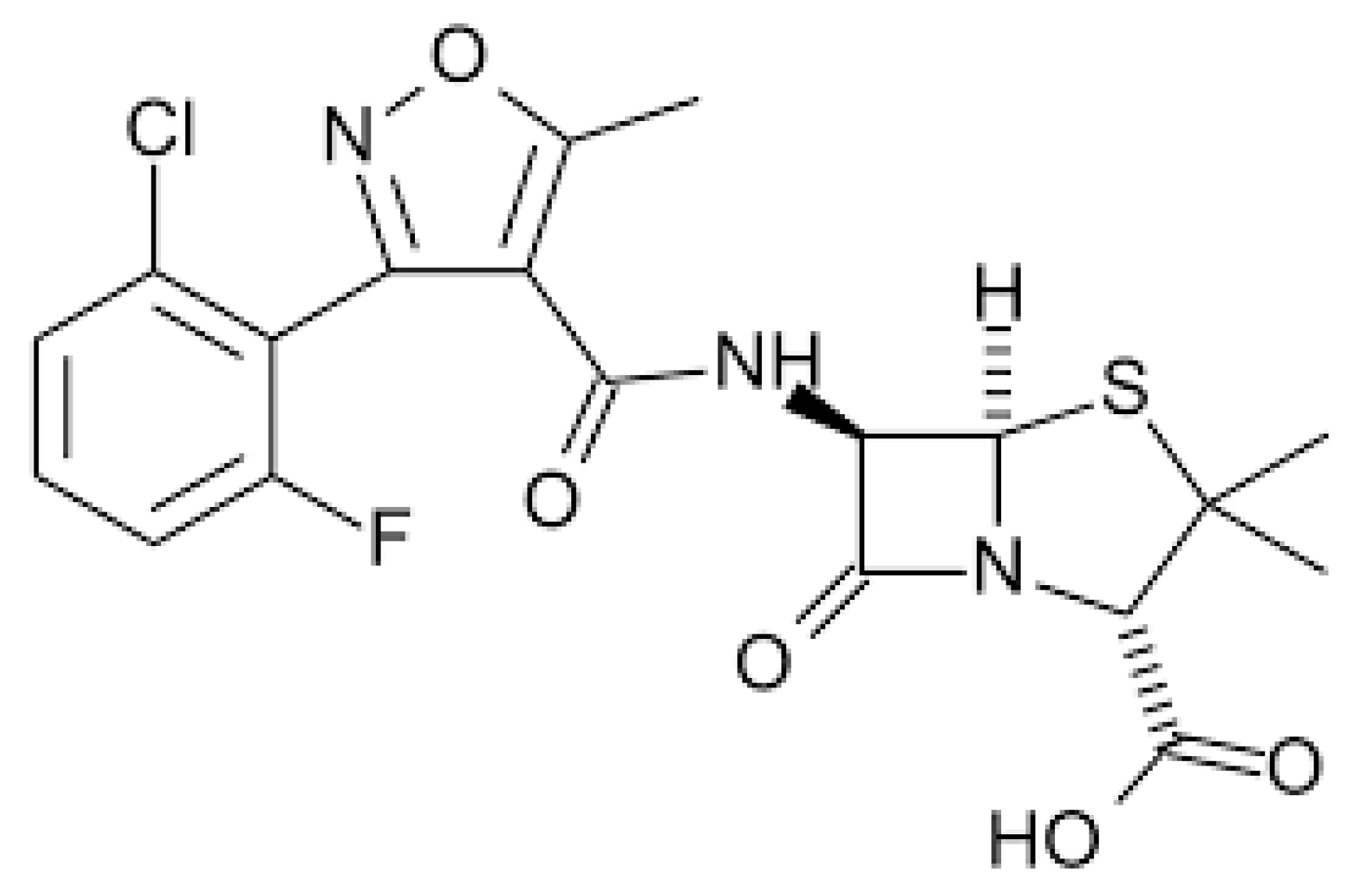
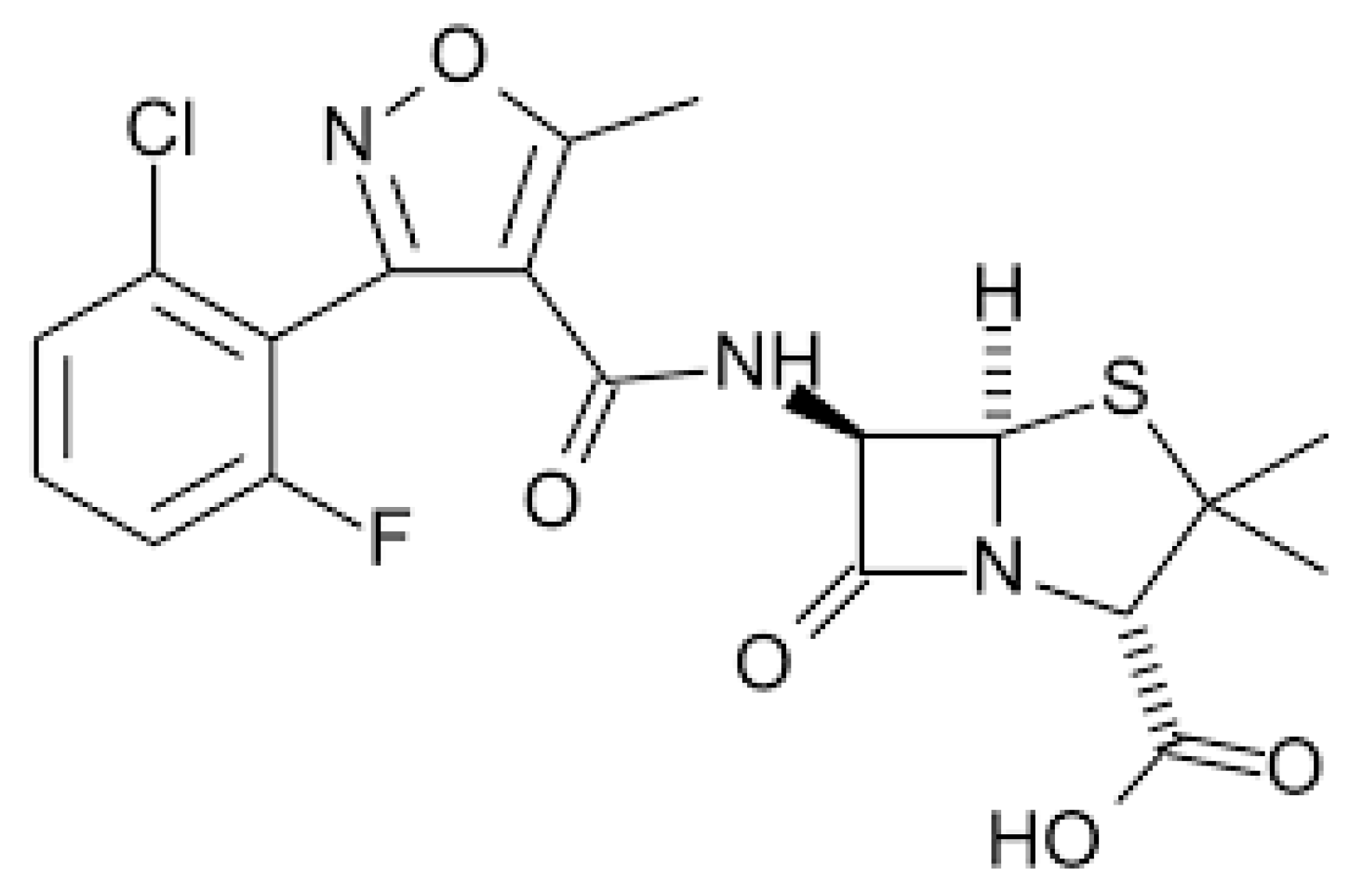
2.2.1. Amoxicillin–Clavulanic Acid
2.2.2. Flucloxacillin
2.3. Antituberculous Drugs
Isoniazid
2.4. Sulfonamides
Sulfamethoxazole
3. Antibiotics for Which the Role of Genetics in Conditioning the Development of Adverse Events Is not Definitively Demonstrated
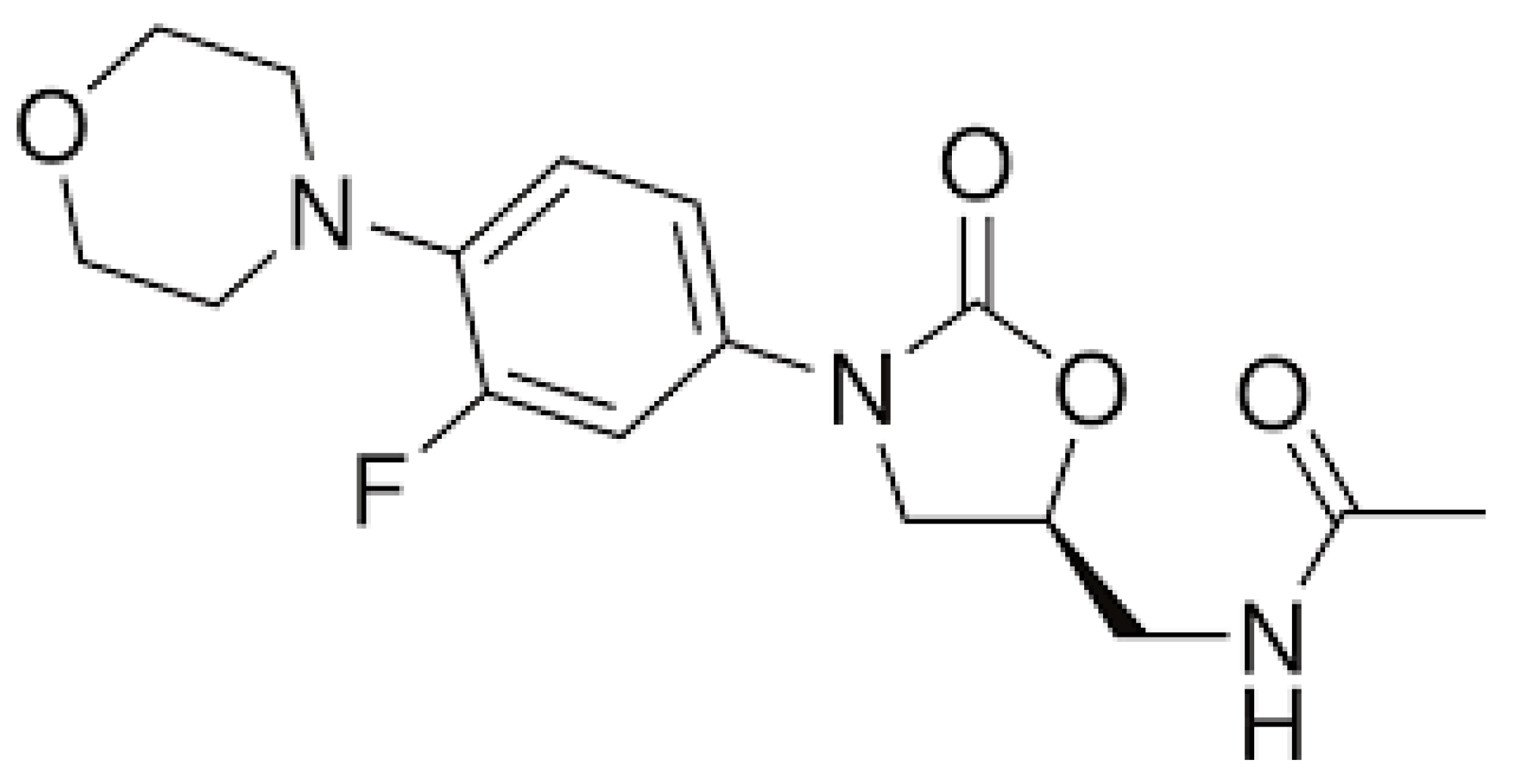
3.1. Linezolid
3.2. Fluoroquinolones
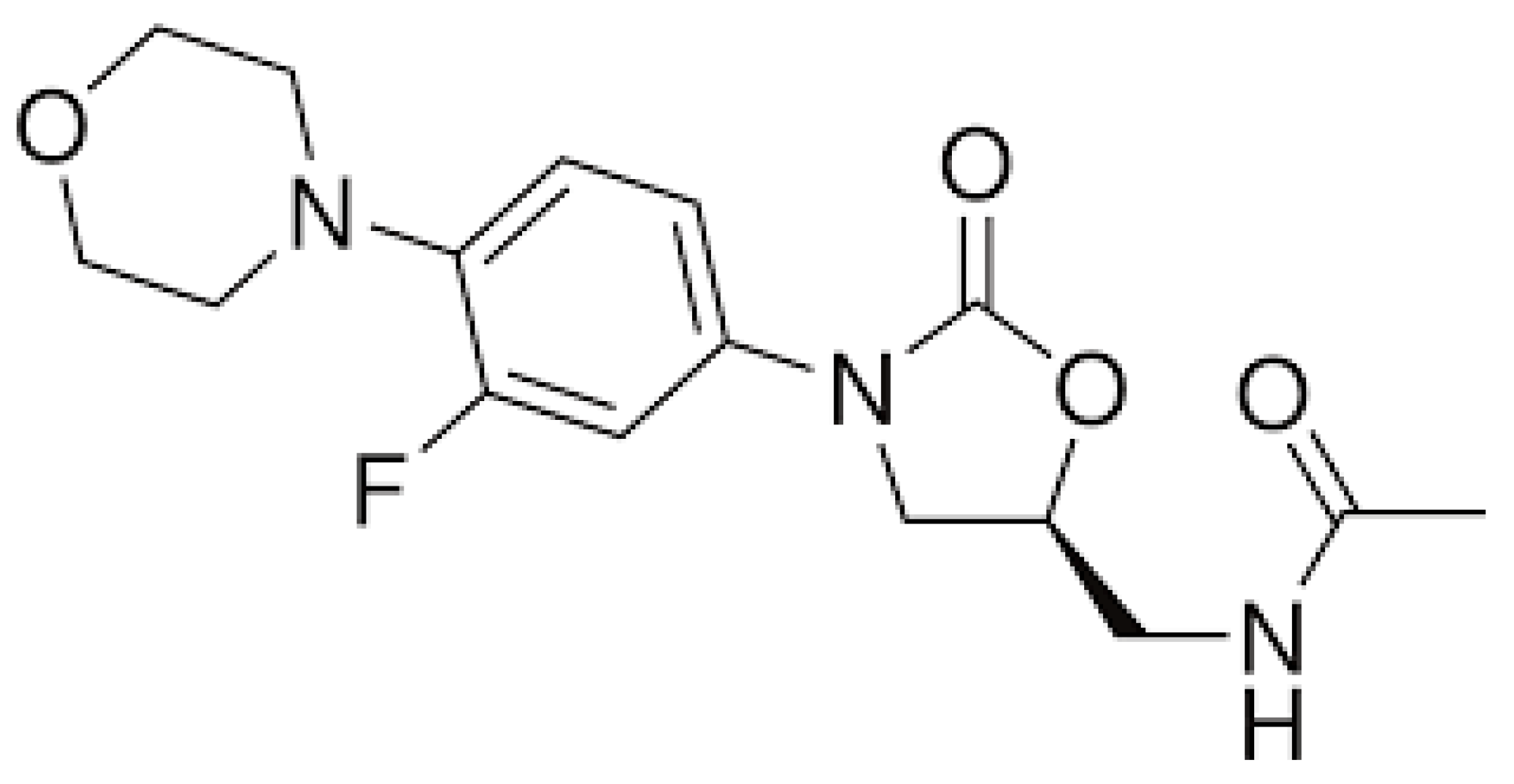
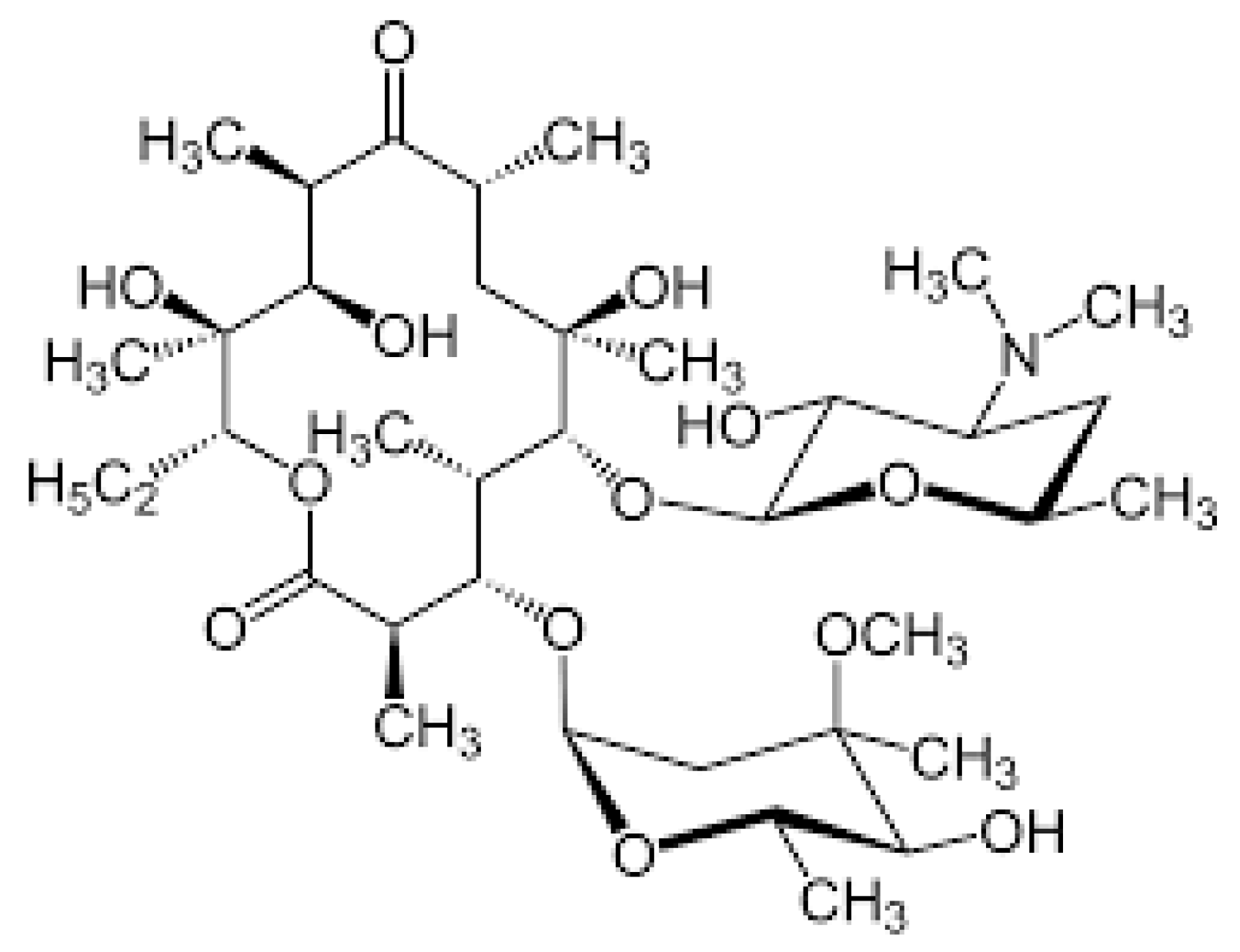
3.3. Macrolides
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Child Mortality and Causes of Death. Available online: https://www.who.int/data/gho/data/themes/topics/topic-details/GHO/child-mortality-and-causes-of-death (accessed on 13 December 2023).
- World Health Organization. GHE: Life Expectancy and Healthy Life Expectancy. Available online: https://www.who.int/data/gho/data/themes/mortality-and-global-health-estimates/ghe-life-expectancy-and-healthy-life-expectancy (accessed on 13 December 2023).
- World Health Organization. Antimicrobial Resistance. Available online: https://www.who.int/news-room/fact-sheets/detail/antimicrobial-resistance (accessed on 10 December 2023).
- Agostoni, C.; Esposito, S.; Nobili, A. Dietary Supplements in Infants and Children: Only Beneficial? J Pediatr Gastroenterol Nutr. 2016, 63, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Principi, N.; Esposito, S. Antibiotic-related adverse events in paediatrics: Unique characteristics. Expert Opin. Drug Saf. 2019, 18, 795–802. [Google Scholar] [CrossRef] [PubMed]
- Shehab, N.; Lovegrove, M.C.; Geller, A.I.; Rose, K.O.; Weidle, N.J.; Budnitz, D.S. US Emergency Department Visits for Outpatient Adverse Drug Events, 2013–2014. JAMA 2016, 316, 2115–2125. [Google Scholar] [CrossRef] [PubMed]
- Shehab, N.; Patel, P.R.; Srinivasan, A.; Budnitz, D.S. Emergency department visits for antibiotic-associated adverse events. Clin. Infect. Dis. 2008, 47, 735–743. [Google Scholar] [CrossRef]
- Lovegrove, M.C.; Geller, A.I.; Fleming-Dutra, K.E.; Shehab, N.; Sapiano, M.R.P.; Budnitz, D.S. US Emergency Department Visits for Adverse Drug Events From Antibiotics in Children, 2011–2015. J. Pediatr. Infect. Dis. Soc. 2019, 8, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Principi, N.; Esposito, S. Antimicrobial stewardship in paediatrics. BMC Infect. Dis. 2016, 16, 424. [Google Scholar] [CrossRef] [PubMed]
- Conroy, S.; Choonara, I.; Impicciatore, P.; Mohn, A.; Arnell, H.; Rane, A.; Knoeppel, C.; Seyberth, H.; Pandolfini, C.; Raffaelli, M.P.; et al. Survey of unlicensed and off label drug use in paediatric wards in European countries. European network for drug investigation in children. BMJ 2000, 320, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Porta, A.; Esposito, S.; Menson, E.; Spyridis, N.; Tsolia, M.; Sharland, M.; Principi, N. Off-label antibiotic use in children in three European countries. Eur. J. Clin. Pharmacol. 2010, 66, 919–927. [Google Scholar] [CrossRef]
- British Society for Antimicrobial Chemotherapy. Antimicrobial Stewardship. From Principles to Practice. Available online: https://www.bsac.org.uk/antimicrobialstewardshipebook/BSAC-AntimicrobialStewardship-FromPrinciplestoPractice-eBook.pdf (accessed on 10 December 2023).
- Downes, K.J.; Hahn, A.; Wiles, J.; Courter, J.D.; Vinks, A.A. Dose optimisation of antibiotics in children: Application of pharmacokinetics/pharmacodynamics in paediatrics. Int. J. Antimicrob. Agents 2014, 43, 223–230. [Google Scholar] [CrossRef]
- Matalová, P.; Urbánek, K.; Anzenbacher, P. Specific features of pharmacokinetics in children. Drug Metab. Rev. 2016, 48, 70–79. [Google Scholar] [CrossRef]
- Principi, N.; Esposito, S. Appropriate use of fluoroquinolones in children. Int. J. Antimicrob. Agents 2015, 45, 341–346. [Google Scholar] [CrossRef]
- Wang, J.; Zou, D.; Li, Y.; Liu, P.; Guo, C. Drug-induced tooth discoloration: An analysis of the US food and drug administration adverse event reporting system. Front. Pharmacol. 2023, 14, 1161728. [Google Scholar] [CrossRef]
- Daly, A.K. Relevance of Pharmacogenomics to the Safe Use of Antimicrobials. Antibiotics 2023, 12, 425. [Google Scholar] [CrossRef]
- Principi, N.; Petropulacos, K.; Esposito, S. Impact of Pharmacogenomics in Clinical Practice. Pharmaceuticals 2023, 16, 1596. [Google Scholar] [CrossRef]
- Relling, M.V.; Klein, T.E. CPIC: Clinical Pharmacogenetics Implementation Consortium of the Pharmacogenomics ResearchNetwork. Clin. Pharmacol. Ther. 2011, 89, 464–467. [Google Scholar] [CrossRef]
- Relling, M.V.; Klein, T.E.; Gammal, R.S.; Whirl-Carrillo, M.; Hoffman, J.M.; Caudle, K.E. The Clinical Pharmacogenetics Implementation Consortium: 10 years later. Clin. Pharmacol. Ther. 2020, 107, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Swen, J.J.; Wilting, I.; Goede, A.L. Pharmacogenetics: From benchto byte. Clin. Pharmacol Ther. 2008, 83, 781–787. [Google Scholar] [CrossRef] [PubMed]
- Thorn, C.F.; Klein, T.E.; Altman, R.B. PharmGKB: The pharmacogenomics knowledge base. In Pharmacogenomics: Methods and Protocols; Humana Press: Totowa, NJ, USA, 2013; Volume 1015, pp. 311–320. [Google Scholar]
- Mingeot-Leclercq, M.-P.; Glupczynski, Y.; Tulkens, P.M. Aminoglycosides: Activity and resistance. Antimicrob. Agents Chemother. 1999, 43, 727–737. [Google Scholar] [CrossRef] [PubMed]
- Krause, K.M.; Serio, A.W.; Kane, T.R.; Connolly, L.E. Aminoglycosides: An Overview. Cold Spring Harb. Perspect. Med. 2016, 6, a027029. [Google Scholar] [CrossRef] [PubMed]
- Avent, M.L.; Rogers, B.A.; Cheng, A.C.; Paterson, D.L. Current use of aminoglycosides: Indications, pharmacokinetics and monitoring for toxicity. Intern. Med. J. 2011, 41, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Talaska, A.E.; Schacht, J. New developments in aminoglycoside therapy and ototoxicity. Hear. Res. 2011, 281, 28–37. [Google Scholar] [CrossRef]
- Rivetti, S.; Romano, A.; Mastrangelo, S.; Attinà, G.; Maurizi, P.; Ruggiero, A. Aminoglycosides-Related Ototoxicity: Mechanisms, Risk Factors, and Prevention in Pediatric Patients. Pharmaceuticals 2023, 16, 1353. [Google Scholar] [CrossRef]
- Yoshinaga-Itano, C.; Sedey, A.L.; Coulter, D.K.; Mehl, A.L. Language of early- and later-identified children with hearing loss. Pediatrics 1998, 102, 1161–1171. [Google Scholar] [CrossRef] [PubMed]
- Diepstraten, F.A.; Hoetink, A.E.; van Grotel, M.; Huitema, A.D.R.; Stokroos, R.J.; van den Heuvel-Eibrink, M.M.; Meijer, A.J.M. Aminoglycoside- and glycopeptide-induced ototoxicity in children: A systematic review. JAC-Antimicrob. Resist. 2021, 3, dlab184. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, N.; Lewis, J.S., 2nd. Aminoglycoside Therapeutic Drug Monitoring: On Paper vs in Practice. Clin. Infect. Dis. 2023, 77, 1737–1738. [Google Scholar] [CrossRef]
- Jiang, M.; Li, H.; Johnson, A.; Karasawa, T.; Zhang, Y.; Meier, W.B.; Taghizadeh, F.; Kachelmeier, A.; Steyger, P.S. Inflammation Up-Regulates Cochlear Expression of TRPV1 to Potentiate Drug-Induced Hearing Loss. Sci. Adv. 2019, 5, aaw1836. [Google Scholar] [CrossRef]
- Chai, Y.; He, W.; Yang, W.; Hetrick, A.P.; Gonzalez, J.G.; Sargsyan, L.; Wu, H.; Jung, T.T.K.; Li, H. Intratympanic Lipopolysaccharide Elevates Systemic Fluorescent Gentamicin Uptake in the Cochlea. Laryngoscope 2021, 131, E2573–E2582. [Google Scholar] [CrossRef] [PubMed]
- Guan, M.-X. Mitochondrial 12S rRNA mutations associated with aminoglycoside ototoxicity. Mitochondrion 2011, 11, 237–245. [Google Scholar] [CrossRef]
- Bitner-Glindzicz, M.; Pembrey, M.; Duncan, A.; Heron, J.; Ring, S.M.; Hall, A.; Rahman, S. Prevalence of mitochondrial 1555A→G mutation in European children. N. Engl. J. Med. 2009, 360, 640–642. [Google Scholar] [CrossRef] [PubMed]
- Vandebona, H.; Mitchell, P.; Manwaring, N.; Griffiths, K.; Gopinath, B.; Wang, J.J.; Sue, C.M. Prevalence of mitochondrial 1555A→G mutation in adults of European descent. N. Engl. J. Med. 2009, 360, 642–644. [Google Scholar] [CrossRef]
- Estivill, X.; Govea, N.; Barceló, A.; Perelló, E.; Badenas, C.; Romero, E.; Moral, L.; Scozzari, R.; D’Urbano, L.; Zeviani, M.; et al. Familial progressive sensorineural deafness is mainly due to the mtDNA A1555G mutation and is enhanced by treatment of aminoglycosides. Am. J. Hum. Genet. 1998, 62, 27–35. [Google Scholar] [CrossRef]
- Lu, J.; Li, Z.; Zhu, Y.; Yang, A.; Li, R.; Zheng, J.; Cai, Q.; Peng, G.; Zheng, W.; Tang, X.; et al. Mitochondrial 12S rRNA variants in 1642 Han Chinese pediatric subjects with aminoglycoside-induced and nonsyndromic hearing loss. Mitochondrion 2010, 10, 380–390. [Google Scholar] [CrossRef] [PubMed]
- Rigobello, R.; Shaw, J.; Ilg, D.; Zimmerman, R.; Edelmann, L.; Kornreich, R.; Scott, S.A.; Cody, N. Clinical Pharmacogenomic MT-RNR1 Screening for Aminoglycoside-Induced Ototoxicity and the Post-Test Counseling Conundrum. Clin Pharmacol Ther. 2023, 11, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Medicines and Healthcare Products Regulatory Agency. Drug Safety Update. 2021, Volume 14. Available online: https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/950307/Jan-2021-DSU-PDF-pub.pdf (accessed on 10 December 2023).
- Clinical Pharmacogenetics Implementation Consortium. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for the Use of Aminoglycosides Based on MT-RNR1 Genotype. Available online: https://files.cpicpgx.org/data/guideline/publication/aminoglycosides/2021/34032273.pdf (accessed on 10 December 2023).
- McDermott, J.H.; Mahaveer, A.; James, R.A.; Booth, N.; Turner, M.; Harvey, K.E.; Miele, G.; Beaman, G.M.; Stoddard, D.C.; Tricker, K.; et al. Rapid Point-of-Care Genotyping to Avoid Aminoglycoside-Induced Ototoxicity in Neonatal Intensive Care. JAMA Pediatr. 2022, 176, 486–492. [Google Scholar] [CrossRef]
- Evans, J.; Hanoodi, M.; Wittler, M. Amoxicillin Clavulanate. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK538164/ (accessed on 10 December 2023).
- Buccellato, E.; Melis, M.; Biagi, C.; Donati, M.; Motola, D.; Vaccheri, A. Use of Antibiotics in Pediatrics: 8-Years Survey in Italian Hospitals. PLoS ONE 2015, 10, e0139097. [Google Scholar] [CrossRef]
- Leitner, J.M.; Graninger, W.; Thalhammer, F. Hepatotoxicity of antibacterials: Pathomechanisms and clinical. Infection 2010, 38, 3–11. [Google Scholar] [CrossRef]
- Björnsson, E.S.; Bergmann, O.M.; Björnsson, H.K.; Kvaran, R.B.; Olafsson, S. Incidence, presentation, and outcomes in patients with drug-induced liver injury in the general population of Iceland. Gastroenterology 2013, 144, 1419–1425.e3. [Google Scholar] [CrossRef] [PubMed]
- Hita, E.O.; García, J.A.M.; Gonzalez, J.C.F.; Molina, A.A.; Cordero, M.A.; Escobar, J.S.; Ruiz-Extremera, A. Amoxicillin-clavulanic acid hepatotoxicity in children. J. Pediatr. Gastroenterol. Nutr. 2012, 55, 663–667. [Google Scholar] [CrossRef] [PubMed]
- Robles, M.; Andrade, R.J. Hepatotoxicity by antibiotics: Update in 2008. Rev. Esp. Quimioter. 2008, 21, 224–233. [Google Scholar]
- Robles, M.; Toscano, E.; Cotta, J.; Lucena, M.I.; Andrade, R.J. Antibiotic-induced liver toxicity: Mechanisms, clinical features and causality assessment. Curr. Drug Saf. 2010, 5, 212–222. [Google Scholar] [CrossRef]
- Fontana, R.J.; Shakil, A.O.; Greenson, J.K.; Boyd, I.; Lee, W.M. Acute liver failure due to amoxicillin and amoxicillin/clavulanate. Dig. Dis. Sci. 2005, 50, 1785–1790. [Google Scholar] [CrossRef] [PubMed]
- Andrade, R.J.; Lucena, M.I.; Kaplowitz, N.; García-Muņoz, B.; Borraz, Y.; Pachkoria, K.; García-Cortés, M.; Fernández, M.C.; Pelaez, G.; Rodrigo, L.; et al. Outcome of acute idiosyncratic drug-induced liver injury: Long-term follow-up in a hepatotoxicity registry. Hepatology 2006, 44, 1581–1588. [Google Scholar] [CrossRef] [PubMed]
- Lucena, M.I.; Molokhia, M.; Shen, Y.; Urban, T.J.; Aithal, G.P.; Andrade, R.J.; Day, C.P.; Ruiz–Cabello, F.; Donaldson, P.T.; Stephens, C.; et al. Susceptibility to Amoxicillin-Clavulanate-Induced Liver Injury Is Influenced by Multiple HLA Class I and II Alleles. Gastroenterology 2011, 141, 338–347. [Google Scholar] [CrossRef]
- Nicoletti, P.; Dellinger, A.; Li, Y.J.; Barnhart, H.X.; Chalasani, N.; Fontana, R.J.; Odin, J.A.; Serrano, J.; Stolz, A.; Etheridge, A.S.; et al. Identification of Reduced ERAP2 Expression and a Novel HLA Allele as Components of a Risk Score for Susceptibility to Liver Injury Due to Amoxicillin-Clavulanate. Gastroenterology 2023, 164, 454–466. [Google Scholar] [CrossRef]
- Urban, T.J.; Nicoletti, P.; Chalasani, N.; Serrano, J.; Stolz, A.; Daly, A.K.; Aithal, G.P.; Dillon, J.; Navarro, V.; Odin, J.; et al. Minocycline hepatotoxicity: Clinical characterization and identification of HLA-B∗35:02 as a risk factor. J. Hepatol. 2017, 67, 137–144. [Google Scholar] [CrossRef]
- Cirulli, E.T.; Nicoletti, P.; Abramson, K.; Andrade, R.J.; Bjornsson, E.S.; Chalasani, N.; Fontana, R.J.; Li, Y.J.; Lucena, M.I.; Long, N.; et al. A Missense Variant in PTPN22 is a Risk Factor for Drug-induced Liver Injury. Gastroenterology 2019, 156, 1707–1716.e2. [Google Scholar] [CrossRef] [PubMed]
- de Menezes, M.N.; de Marco, B.A.; Fiorentino, F.A.M.; Zimmermann, A.; Kogawa, A.C.; Salgado, H.R.N. Flucloxacillin: A Review of Characteristics, Properties and Analytical Methods. Crit. Rev. Anal. Chem. 2019, 49, 67–77. [Google Scholar] [CrossRef]
- Barker, C.I.S.; Kipper, K.; Lonsdale, D.O.; Wright, K.; Thompson, G.; Kim, M.; Turner, M.A.; Johnston, A.; Sharland, M.; Standing, J.F. The Neonatal and Paediatric Pharmacokinetics of Antimicrobials study (NAPPA): Investigating amoxicillin, benzylpenicillin, flucloxacillin and piperacillin pharmacokinetics from birth to adolescence. J. Antimicrob. Chemother. 2023, 78, 2148–2161. [Google Scholar] [CrossRef]
- Wing, K.; Bhaskaran, K.; Pealing, L.; Root, A.; Smeeth, L.; van Staa, T.P.; Klungel, O.H.; Reynolds, R.F.; Douglas, I. Quantification of the risk of liver injury associated with flucloxacillin: A UK population-based cohort study. J. Antimicrob. Chemother. 2017, 72, 2636–2646. [Google Scholar] [CrossRef]
- Russmann, S.; Kaye, J.A.; Jick, S.S.; Jick, H. Risk of cholestatic liver disease associated with flucloxacillin and flucloxacillin prescribing habits in the UK: Cohort study using data from the UK General Practice Research Database. Br. J. Clin. Pharmacol. 2005, 60, 76–82. [Google Scholar] [CrossRef]
- Miros, M.; Kerlin, P.; Walker, N.; Harris, O. Flucloxacillin induced delayed cholestatic hepatitis. Aust. N. Z. J. Med. 1990, 20, 251–253. [Google Scholar] [CrossRef]
- Koek, G.H.; Stricker, B.H.C.; Blok, A.P.R.; Schalm, S.W.; Desmet, V.J. Flucloxacillin-associated hepatic injury. Liver 1994, 14, 225–229. [Google Scholar] [CrossRef]
- Daly, A.K.; Donaldson, P.T.; Bhatnagar, P.; Shen, Y.; Pe’er, I.; Floratos, A.; Daly, M.J.; Goldstein, D.B.; John, S.; Nelson, M.R.; et al. HLA-B*5701 genotype is a major determinant of drug-induced liver injury due to flucloxacillin. Nat. Genet. 2009, 41, 816–819. [Google Scholar] [CrossRef]
- Kaplan, H.A.; Woloski, B.M.; Hellman, M.; Jamieson, J.C. Studies on the effect of inflammation on rat liver and serum sialyltransferase. Evidence that inflammation causes release of Gal beta 1 leads to 4GlcNAc alpha 2 leads to 6 sialyltransferase from liver. J. Biol. Chem. 1983, 258, 11505–11509. [Google Scholar] [CrossRef]
- Nicoletti, P.; Aithal, G.P.; Chamberlain, T.C.; Coulthard, S.; Alshabeeb, M.; Grove, J.I.; Andrade, R.J.; Bjornsson, E.; Dillon, J.F.; Hallberg, P.; et al. Drug-induced liver injury due to flucloxacillin: Relevance of multiple human leukocyte antigen alleles. Clin. Pharmacol. Ther. 2019, 106, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Karnes, J.H.; Miller, M.A.; White, K.D.; Konvinse, K.C.; Pavlos, R.K.; Redwood, A.J.; Peter, J.G.; Lehloenya, R.; Mallal, S.A.; Phillips, E.J. Applications of Immunopharmacogenomics: Predicting, Preventing, and Understanding Immune-Mediated Adverse Drug Reactions. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 463–486. [Google Scholar] [CrossRef] [PubMed]
- Management of drug-resistant TB in children. In Guidance for National Tuberculosis Programmes on the Management of Tuberculosis in Children, 2nd ed.; World Health Organization: Geneva, Switzerland, 2014. Available online: https://www.ncbi.nlm.nih.gov/books/NBK214453/ (accessed on 10 December 2023).
- Fountain, F.F.; Tolley, E.; Chrisman, C.R. Isoniazid hepatotoxicity associated with treatment of latent tuberculosis infection: A 7-year evaluation from a public health tuberculosis clinic. Chest 2005, 128, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Tostmann, A.; Boeree, M.J.; Aarnoutse, R.E. Antituberculosis drug-induced hepatotoxicity: Concise up-to-date review. J. Gastroenterol. Hepatol. 2008, 23, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Donald, P.R. Antituberculosis drug-induced hepatotoxicity in children. Pediatr. Rep. 2011, 3, e16. [Google Scholar] [CrossRef]
- Principi, N.; Galli, L.; Lancella, L.; Tadolini, M.; Migliori, G.B.; Villani, A.; Esposito, S.; Italian Pediatric TB Study Group. TB Recommendations Concerning the First-Line Treatment of Children with Tuberculosis. Pediatr. Drugs 2016, 18, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Ostapowicz, G.; Fontana, R.J.; Schiødt, F.V.; Larson, A.; Davern, T.J.; Han, S.H.; McCashland, T.M.; Shakil, A.O.; Hay, J.E.; Hynan, L.; et al. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann. Intern. Med. 2002, 137, 947–954. [Google Scholar] [CrossRef]
- Kumar, R.; Shalimar; Bhatia, V.; Khanal, S.; Sreenivas, V.; Gupta, S.D.; Panda, S.K.; Acharya, S.K. Antituberculosis therapy-induced acute liver failure: Magnitude, profile, prognosis, and predictors of outcome. Hepatology 2010, 51, 1665–1674. [Google Scholar] [CrossRef] [PubMed]
- Garibaldi, R.A.; Drusin, R.E.; Ferebee, S.H.; Gregg, M.B. Isoniazid-associated hepatitis. Report of an outbreak. Am. Rev. Respir. Dis. 1972, 106, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Senousy, B.E.; Belal, S.I.; Draganov, P.V. Hepatotoxic effects of therapies for tuberculosis. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 543–556. [Google Scholar] [CrossRef] [PubMed]
- Ramappa, V.; Aithal, G.P. Hepatotoxicity Related to Anti-tuberculosis Drugs: Mechanisms and Management. J. Clin. Exp. Hepatol. 2013, 3, 37–49. [Google Scholar] [CrossRef]
- Snider, D.; Caras, G.J., Jr. Isoniazid-associated hepatitis deaths: A review of available information. Am. Rev. Respir. Dis. 1992, 145, 494–497. [Google Scholar] [CrossRef] [PubMed]
- Preziosi, P. Isoniazid: Metabolic aspects and toxicological correlates. Curr. Drug Metab. 2007, 8, 839–851. [Google Scholar] [CrossRef]
- Das Roy, P.; Majumder, M.; Roy, B. Pharmacogenomics of anti-TB drugs-related hepatotoxicity. Pharmacogenomics 2008, 9, 311–321. [Google Scholar]
- Gupta, V.H.; Amarapurkar, D.N.; Singh, M.; Sasi, P.; Joshi, J.M.; Baijal, R.; Ramegowda, P.H.; Amarapurkar, A.D.; Joshi, K.; Wangikar, P.P. Association of N-acetyltransferase 2 and cytochrome P450 2E1 gene polymorphisms with antituberculosis drug-induced hepatotoxicity in Western India. J. Gastroenterol. Hepatol. 2013, 28, 1368–1374. [Google Scholar] [CrossRef]
- Teixeira, R.L.d.F.; Morato, R.G.; Cabello, P.H.; Muniz, L.M.K.; Moreira, A.d.S.R.; Kritski, A.L.; Mello, F.C.Q.; Suffys, P.N.; de Miranda, A.B.; Santos, A.R. Genetic polymorphisms of NAT2, CYP2E1 and GST enzymes and the occurrence of antituberculosis drug-induced hepatitis in Brazilian TB patients. Mem. Inst. Oswaldo Cruz 2011, 106, 716–724. [Google Scholar] [CrossRef]
- Zhang, D.; Hao, J.; Hou, R.; Yu, Y.; Hu, B.; Wei, L. The role of NAT2 polymorphism and methylation in anti-tuberculosis drug-induced liver injury in Mongolian tuberculosis patients. J. Clin. Pharm. Ther. 2020, 45, 561–569. [Google Scholar] [CrossRef]
- Amorim, G.; Jaworski, J.; Cordeiro-Santos, M.; Kritski, A.L.; Figueiredo, M.C.; Turner, M.; Andrade, B.B.; Velez Edwards, D.R.; Santos, A.R.; Rolla, V.C.; et al. Pharmacogenetics of tuberculosis treatment toxicity and effectiveness in a large Brazilian cohort. medRxiv 2023, 2023.08.30.23294860. [Google Scholar]
- Azuma, J.; Ohno, M.; Kubota, R.; Yokota, S.; Nagai, T.; Tsuyuguchi, K.; Okuda, Y.; Takashima, T.; Kamimura, S.; Fujio, Y.; et al. Pharmacogenetics-based tuberculosis therapy research group. NAT2 genotype guided regimen reduces isoniazid-induced liver injury and early treatment failure in the 6-month four-drug standard treatment of tuberculosis: A randomized controlled trial for pharmacogenetics-based therapy. Eur. J. Clin. Pharmacol. 2013, 69, 1091–1101. [Google Scholar]
- Wang, P.; Pradhan, K.; Zhong, X.-B.; Ma, X. Isoniazid metabolism and hepatotoxicity. Acta Pharm. Sin. B 2016, 6, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-H.; Kim, S.-H.; Bahn, J.-W.; Kim, Y.-K.; Chang, Y.-S.; Shin, E.-S.; Kim, Y.-S.; Park, J.-S.; Kim, B.-H.; Jang, I.-J.; et al. Genetic polymorphisms of drug-metabolizing enzymes and anti-TB drug-induced hepatitis. Pharmacogenomics 2009, 10, 1767–1779. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.-J.; Koh, W.-J.; Ryu, Y.-J.; Ki, C.-S.; Nam, M.-H.; Kim, J.-W.; Lee, S.-Y. Genetic polymorphisms of NAT2 and CYP2E1 associated with antituberculosis drug-induced hepatotoxicity in Korean patients with pulmonary tuberculosis. Tuberculosis 2007, 87, 551–556. [Google Scholar] [CrossRef]
- Huang, Y.; Chern, H.; Su, W.; Wu, J.; Chang, S.; Chiang, C.; Chang, F.; Lee, S. Cytochrome P450 2E1 genotype and the susceptibility to antituberculosis drug-induced hepatitis. Hepatology 2003, 37, 924–930. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Gao, L.; Shi, Z.; Li, Y.; Zhu, L.; Li, S.; Zhang, P.; Zheng, G.; Ren, Q.; Li, Y.; et al. Involvement of cytochrome P450 1A1 and glutathione S-transferase P1 polymorphisms and promoter hypermethylation in the progression of anti-tuberculosis drug-induced liver injury: A case-control study. PLoS ONE 2015, 10, e0119481. [Google Scholar] [CrossRef] [PubMed]
- Kantemirova, B.I.; Bogorodskaya, E.M.; Poptsova, M.S.; Sychev, D.A.; Tsimbal, E.A.; Stepanova, N.A. Research of Russian physicians’ opinions on tuberculosis pharmacogenetics. Int. J. Risk Saf. Med. 2023, 35, 25–36. [Google Scholar] [CrossRef]
- Roy, B.; Ghosh, S.K.; Sutradhar, D.; Sikdar, N.; Mazumder, S.; Barman, S. Predisposition of antituberculosis drug induced hepatotoxicity by cytochrome P450 2E1 genotype and haplotype in pediatric patients. J. Gastroenterol. Hepatol. 2006, 21, 784–786. [Google Scholar] [CrossRef]
- Cai, Y.; Yi, J.; Zhou, C.; Shen, X. Pharmacogenetic study of drug-metabolising enzyme polymorphisms on the risk of anti-tuberculosis drug-induced liver injury: A meta-analysis. PLoS ONE 2012, 7, e47769. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. Table of Pharmacogenetic Associations. Available online: https://www.fda.gov/medical-devices/precision-medicine/table-pharmacogenetic-associations#section2 (accessed on 10 December 2023).
- Whirl-Carrillo, M.; McDonagh, E.M.; Hebert, J.M.; Gong, L.; Sangkuhl, K.; Thorn, C.F.; Altman, R.B.; Klein, T.E. Pharmacogenomics Knowledge for Personalized Medicine. Clin. Pharmacol. Ther. 2012, 92, 414–417. [Google Scholar] [CrossRef] [PubMed]
- Ovung, A.; Bhattacharyya, J. Sulfonamide drugs: Structure, antibacterial property, toxicity, and biophysical interactions. Biophys. Rev. 2021, 13, 259–272. [Google Scholar] [CrossRef]
- Kemnic, T.R.; Coleman, M. Trimethoprim Sulfamethoxazole. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK513232/ (accessed on 10 December 2023).
- Khan, D.A.; Knowles, S.R.; Shear, N.H. Sulfonamide Hypersensitivity: Fact and Fiction. J. Allergy Clin. Immunol. Pract. 2021, 7, 2116–2123. [Google Scholar] [CrossRef]
- Asyraf, P.A.; Kusnadi, I.F.; Stefanus, J.; Khairinisa, M.A.; Abdulah, R. Clinical Manifestations and Genetic Influences in Sulfonamide-Induced Hypersensitivity. Drug Healthc. Patient Saf. 2022, 14, 113–124. [Google Scholar] [CrossRef]
- Pirmohamed, M.; Alfirevic, A.; Vilar, J.; Stalford, A.; Wilkins, E.G.L.; Sim, E.; Park, B.K. Association analysis of drug metabolizing enzyme gene polymorphisms in HIV-positive patients with co-trimoxazole hypersensitivity. Pharmacogenetics 2000, 10, 705–713. [Google Scholar] [CrossRef]
- Tabet Aoul, A.; Al-Nasseri, A.; Hall, C.; He, C.; Abernathy, J. Stevens-Johnson Syndrome in a Patient on Concomitant Treatment with Levetiracetam and Trimethoprim/Sulfamethoxazole. Am J Case Rep. 2024, 25, e942982. [Google Scholar] [CrossRef] [PubMed]
- Wung, C.-H.; Wang, C.-W.; Lai, K.-C.; Chen, C.-B.; Chen, W.-T.; Hung, S.-I.; Chung, W.-H.; Taiwan Severe Cutaneous Adverse Reaction Consortium. Current understanding of genetic associations with delayed hypersensitivity reactions induced by antibiotics and anti-osteoporotic drugs. Front. Pharmacol. 2023, 14, 1183491. [Google Scholar] [CrossRef]
- Kloypan, C.; Koomdee, N.; Satapornpong, P.; Tempark, T.; Biswas, M.; Sukasem, C. A Comprehensive Review of HLA and Severe Cutaneous Adverse Drug Reactions: Implication for Clinical Pharmacogenomics and Precision Medicine. Pharmaceuticals 2021, 14, 1077. [Google Scholar] [CrossRef]
- Serrano-Arias, B.; Araya-Zúñiga, A.; Waterhouse-Garbanzo, J.; Rojas-Barrantes, Z.; Arguedas-Chacón, S.; Zavaleta-Monestel, E. A Comprehensive Review of Sulfonamide Hypersensitivity: Implications for Clinical Practice. Clin. Rev. Allergy Immunol. 2023, 65, 433–442. [Google Scholar] [CrossRef]
- Garazzino, S.; Krzysztofiak, A.; Esposito, S.; Castagnola, E.; Plebani, A.; Galli, L.; Cellini, M.; Lipreri, R.; Scolfaro, C.; Bertaina, C.; et al. Use of linezolid in infants and children: A retrospective multicentre study of the Italian Society for Paediatric Infectious Diseases. J. Antimicrob. Chemother. 2011, 66, 2393–2397. [Google Scholar] [CrossRef]
- Zhang, X.; Falagas, M.E.; Vardakas, K.Z.; Wang, R.; Qin, R.; Wang, J.; Liu, Y. Systematic review and meta-analysis of the efficacy and safety of therapy with linezolid containing regimens in the treatment of multidrug-resistant and extensively drug-resistant tuberculosis. J. Thorac. Dis. 2015, 7, 603–615. [Google Scholar]
- Batts, D.H. Linezolid—A new option for treating Gram-positive infections. Oncology 2000, 14 (Suppl. 6), 23–29. [Google Scholar]
- Hashemian, S.M.; Farhadi, T.; Ganjparvar, M. Linezolid: A review of its properties, function, and use in critical care. Drug Des. Dev. Ther. 2018, 12, 1759–1767. [Google Scholar] [CrossRef] [PubMed]
- McKee, E.E.; Ferguson, M.; Bentley, A.T.; Marks, T.A. Inhibition of mammalian mitochondrial protein synthesis by oxazolidinones. Antimicrob. Agents Chemother. 2006, 50, 2042–2049. [Google Scholar] [CrossRef] [PubMed]
- Garrabou, G.; Soriano, A.; López, S.; Guallar, J.P.; Giralt, M.; Villarroya, F.; Martínez, J.A.; Casademont, J.; Cardellach, F.; Mensa, J.; et al. Reversible inhibition of mitochondrial protein synthesis during linezolid-related hyperlactatemia. Antimicrob. Agents Chemother. 2007, 51, 962–967. [Google Scholar] [CrossRef] [PubMed]
- Garrabou, G.; Soriano, À.; Pinós, T.; Casanova-Mollà, J.; Pacheu-Grau, D.; Morén, C.; García-Arumí, E.; Morales, M.; Ruiz-Pesini, E.; Catalán-Garcia, M.; et al. Influence of Mitochondrial Genetics on the Mitochondrial Toxicity of Linezolid in Blood Cells and Skin Nerve Fibers. Antimicrob. Agents Chemother. 2017, 61, e00542-17. [Google Scholar] [CrossRef] [PubMed]
- Allegra, S.; Di Paolo, A.; Cusato, J.; Fatiguso, G.; Arrigoni, E.; Danesi, R.; Corcione, S.; D’Avolio, A. A Common mdr1 Gene Polymorphism is Associated With Changes in Linezolid Clearance. Ther. Drug Monit. 2018, 40, 602–609. [Google Scholar] [CrossRef]
- Penman, S.L.; Carter, A.S.; Chadwick, A.E. Investigating the importance of individual mitochondrial genotype in susceptibility to drug-induced toxicity. Biochem. Soc. Trans. 2020, 48, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Esposito, S.; Tagliabue, C.; Bosis, S.; Principi, N. Levofloxacin for the treatment of Mycoplasma pneumoniae-associated meningoencephalitis in childhood. Int. J. Antimicrob. Agents 2011, 37, 472–475. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. Fluoroquinolone Antibiotics: Reminder of Measures to Reduce the Risk of Long-Lasting, Disabling and Potentially Irreversible Side Effects. Available online: https://www.ema.europa.eu/en/news/fluoroquinolone-antibiotics-reminder-measures-reduce-risk-long-lasting-disabling-and-potentially-irreversible-side-effects (accessed on 10 January 2024).
- U.S. Food and Drug Administration. FDA Drug Safety Communication: FDA Advises Restricting Fluoroquinolone Antibiotic Use for Certain Uncomplicated Infections; Warns about Disabling Side Effects that Can Occur Together. Available online: https://www.fda.gov/drugs/drug-safety-and-availability/fda-drug-safety-communication-fda-advises-restricting-fluoroquinolone-antibiotic-use-certain (accessed on 10 January 2024).
- UpToDate. Fluoroquinolones. Available online: https://www-uptodate-com.pros2.lib.unimi.it/contents/fluoroquinolones?search=fluoroquinolones&source=search_result&selectedTitle=2~143&usage_type=default&display_rank=1 (accessed on 10 January 2024).
- Pranger, A.D.; van der Werf, T.S.; Kosterink, J.G.W.; Alffenaar, J.W.C. The Role of Fluoroquinolones in the Treatment of Tuberculosis in 2019. Drugs 2019, 79, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, A.I.; Pérez, M.; Prieto, J.G.; Molina, A.J.; Real, R.; Merino, G. Fluoroquinolone efflux mediated by ABC transporters. J. Pharm. Sci. 2008, 97, 3483–3493. [Google Scholar] [CrossRef]
- de Lange, E.C.; Marchand, S.; van den Berg, D.; van der Sandt, I.C.; de Boer, A.G.; Delon, A.; Bouquet, S.; Couet, W. In vitro and in vivo investigations on fluoroquinolones; effects of the P-glycoprotein efflux transporter on brain distribution of sparfloxacin. Eur. J. Pharm. Sci. 2000, 12, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Gervasoni, C.; Cattaneo, D.; Falvella, F.S.; Vitiello, P.; Cheli, S.; Milazzo, L.; Clementi, E.; Riva, A. Levofloxacin-induced seizures in a patient without predisposing risk factors: The impact of pharmacogenetics. Eur. J. Clin. Pharmacol. 2013, 69, 1611–1613. [Google Scholar] [CrossRef]
- Naidoo, A.; Naidoo, K.; McIlleron, H.; Essack, S.; Padayatchi, N. A Review of Moxifloxacin for the Treatment of Drug-Susceptible Tuberculosis. J. Clin. Pharmacol. 2017, 57, 1369–1386. [Google Scholar] [CrossRef] [PubMed]
- Weiner, M.; Burman, W.; Luo, C.-C.; Peloquin, C.A.; Engle, M.; Goldberg, S.; Agarwal, V.; Vernon, A. Effects of rifampin and multidrug resistance gene polymorphism on concentrations of moxifloxacin. Antimicrob. Agents Chemother. 2007, 51, 2861–2866. [Google Scholar] [CrossRef] [PubMed]
- Principi, N.; Esposito, S. Comparative tolerability of erythromycin and newer macrolide antibacterials in paediatric patients. Drug Saf. 1999, 20, 25–41. [Google Scholar] [CrossRef] [PubMed]
- Tenson, T.; Lovmar, M.; Ehrenberg, M. The mechanism of action of macrolides, lincosamides and streptogramin B reveals the nascent peptide exit path in the ribosome. J. Mol. Biol. 2003, 330, 1005–1014. [Google Scholar] [CrossRef]
- Morozumi, M.; Iwata, S.; Hasegawa, K.; Chiba, N.; Takayanagi, R.; Matsubara, K.; Nakayama, E.; Sunakawa, K.; Ubukata, K. Increased macrolide resistance of Mycoplasma pneumoniae in pediatric patients with community-acquired pneumonia. Antimicrob. Agents Chemother. 2008, 52, 348–350. [Google Scholar] [CrossRef]
- Dinos, G.P. The macrolide antibiotic renaissance. Br. J. Pharmacol. 2017, 174, 2967–2983. [Google Scholar] [CrossRef]
- Albert, R.K.; Schuller, J.L.; COPD Clinical Research Network. Macrolide antibiotics and the risk of cardiac arrhythmias. Am. J. Respir. Crit. Care Med. 2014, 189, 1173–1180. [Google Scholar] [CrossRef] [PubMed]
- PHARMGKB. Macrolide Antibiotic Pathway, Pharmacokinetics/Pharmacodynamics. Available online: https://www.pharmgkb.org/pathway/PA166160731/overview (accessed on 10 January 2024).
- Tsai, D.; Jamal, J.-A.; Davis, J.S.; Lipman, J.; Roberts, J.A. Interethnic differences in pharmacokinetics of antibacterials. Clin. Pharmacokinet. 2014, 54, 243–260. [Google Scholar] [CrossRef]
- Franke, R.M.; Lancaster, C.S.; Peer, C.J.; Gibson, A.A.; Kosloske, A.M.; Orwick, S.J.; Mathijssen, R.H.; Figg, W.D.; Baker, S.D.; Sparreboom, A.; et al. Effect of ABCC2 (MRP2) transport function on erythromycin metabolism. Clin. Pharmacol. Ther. 2011, 89, 693–701. [Google Scholar] [CrossRef]
- Lancaster, C.S.; Bruun, G.H.; Peer, C.J.; Mikkelsen, T.S.; Corydon, T.J.; Gibson, A.A.; Hu, S.; Orwick, S.J.; Mathijssen, R.H.J.; Figg, W.D.; et al. OATP1B1 polymorphism as a determinant of erythromycin disposition. Clin. Pharmacol. Ther. 2012, 92, 642–650. [Google Scholar] [CrossRef] [PubMed]
- He, X.-J.; Zhao, L.-M.; Qiu, F.; Sun, Y.-X.; Li-Ling, J. Influence of ABCB1 gene polymorphisms on the pharmacokinetics of azithromycin among healthy Chinese Han ethnic subjects. Pharmacol. Rep. 2009, 61, 843–850. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation | Prevalence in Patients with AG-Related Deafness |
|---|---|
| m1555A>G | 5–33% |
| 1095 T>C | <5% |
| 1494 C>T | <5% |
| Variation | Increase in DILI Risk |
|---|---|
| DRB1*15:01-DQB1*06:02 | x3 |
| HLA-A*02:01 | x3 |
| HLA-B*15-18 | x3 |
| PTPN22 gene | x2 |
| ERAP2 gene | x2 |
| Variation | Increase in Risk of Cholestatic Hepatitis |
|---|---|
| HLA-B*57:01 | X80 |
| HLA-B*57:03 | x37 |
| Variation | Increase in Risk of Hepatotoxicity |
|---|---|
| NAT2*7, NAT2*6 andNAT2*5 | x3–7 |
| CYP2E1 c1/c1 genotype | x2.5 |
| CYP2E1*6 allele | x2 |
| CYP2E1*1A-*6-*1D haplotype | x2 |
| GSTM1 null genotype | x2 |
| Test | Pros | Cons |
|---|---|---|
| Pharmacogenomics | Personalized Medicine: Pharmacogenomics allows for the customization of antibiotic therapy based on an individual’s genetic makeup. This can lead to more effective and safer treatment by targeting the specific genetic factors affecting drug metabolism and response. Reduced Adverse Effects: By identifying genetic variations that affect drug metabolism and response, pharmacogenomics can help prevent adverse drug reactions and toxicity, leading to safer antibiotic use. Optimized Drug Selection: Pharmacogenomic testing can guide clinicians in selecting the most appropriate antibiotic for a particular patient, based on their genetic profile. This can enhance treatment efficacy and reduce the risk of treatment failure. Improved Antibiotic Stewardship: By tailoring antibiotic therapy to individual patients, pharmacogenomics can contribute to antibiotic stewardship efforts by minimizing the unnecessary use of broad-spectrum antibiotics and reducing the risk of antibiotic resistance. | Cost: Pharmacogenomic testing can be expensive, which may limit its widespread adoption, especially in resource-constrained healthcare settings. Complexity: Interpreting pharmacogenomic test results and integrating them into clinical decision making can be complex and time consuming for healthcare providers. Limited Evidence: While pharmacogenomics holds promise for optimizing antibiotic therapy, the evidence supporting its clinical utility in this context is still emerging, and more research is needed to fully understand its impact on patient outcomes. Ethical and Privacy Concerns: Pharmacogenomic testing raises ethical and privacy concerns related to the storage and use of genetic information, as well as potential implications for insurance coverage and employment discrimination. |
| Therapeutic drug monitoring |
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Principi, N.; Petropulacos, K.; Esposito, S. Genetic Variations and Antibiotic-Related Adverse Events. Pharmaceuticals 2024, 17, 331. https://doi.org/10.3390/ph17030331
Principi N, Petropulacos K, Esposito S. Genetic Variations and Antibiotic-Related Adverse Events. Pharmaceuticals. 2024; 17(3):331. https://doi.org/10.3390/ph17030331
Chicago/Turabian StylePrincipi, Nicola, Kyriakoula Petropulacos, and Susanna Esposito. 2024. "Genetic Variations and Antibiotic-Related Adverse Events" Pharmaceuticals 17, no. 3: 331. https://doi.org/10.3390/ph17030331
APA StylePrincipi, N., Petropulacos, K., & Esposito, S. (2024). Genetic Variations and Antibiotic-Related Adverse Events. Pharmaceuticals, 17(3), 331. https://doi.org/10.3390/ph17030331