
Impact of Pharmacogenomics in Clinical Practice



Abstract
:1. Introduction
2. Genetic Variations and Impact on Drug Transportation and Metabolism
2.1. Normal Mechanisms of Drug Transportation and Metabolism
2.2. Impact of Genetic Variants on Pharmacokinetics
2.2.1. Polymorphisms of the Most Important Phase I Metabolism Enzymes
2.2.2. Polymorphisms of the Most Important Phase II Metabolism Enzymes
2.2.3. Polymorphisms of the Most Important Transporters
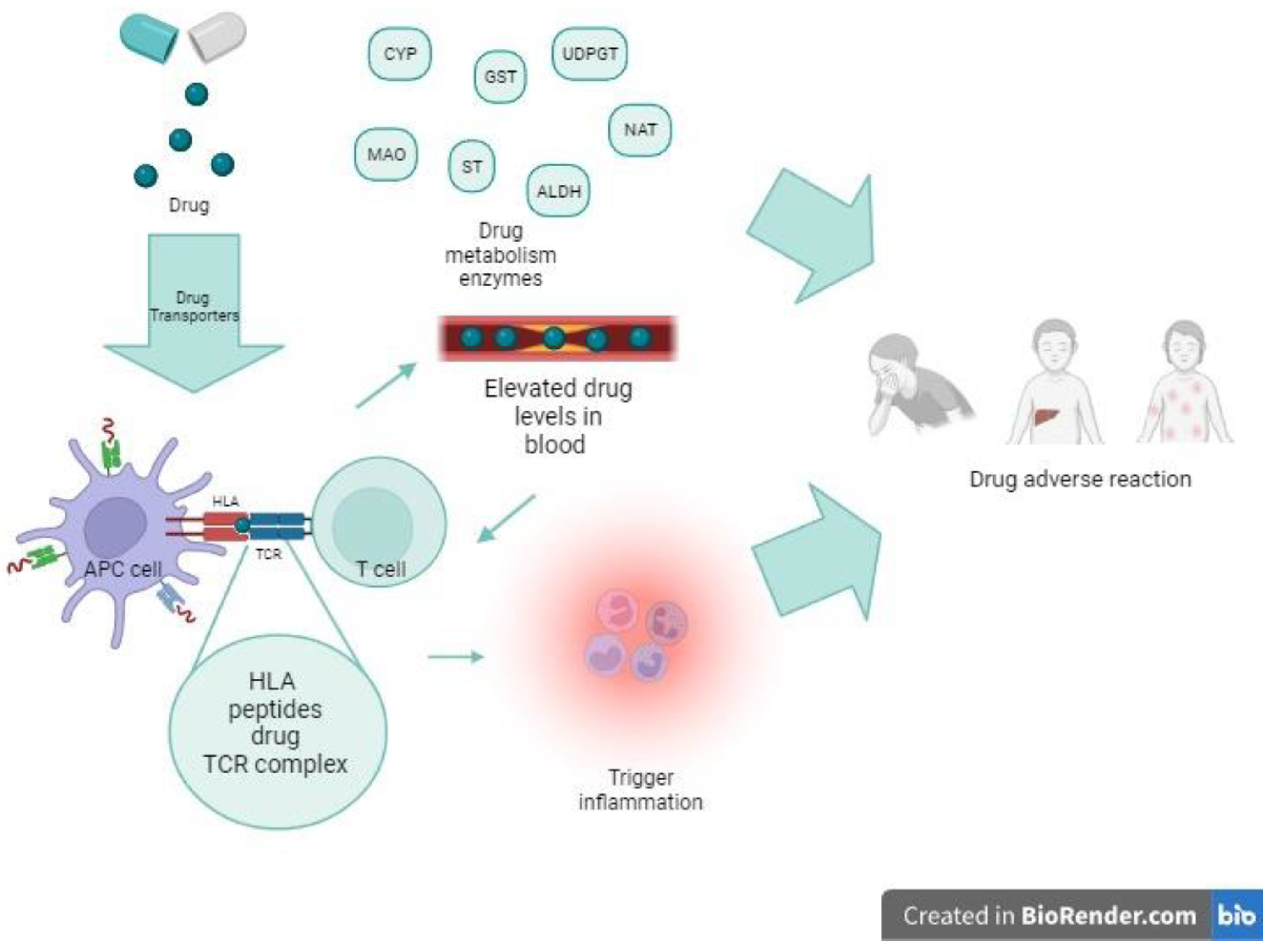
3. Genetic Variants That Affect Immune Response to Drugs
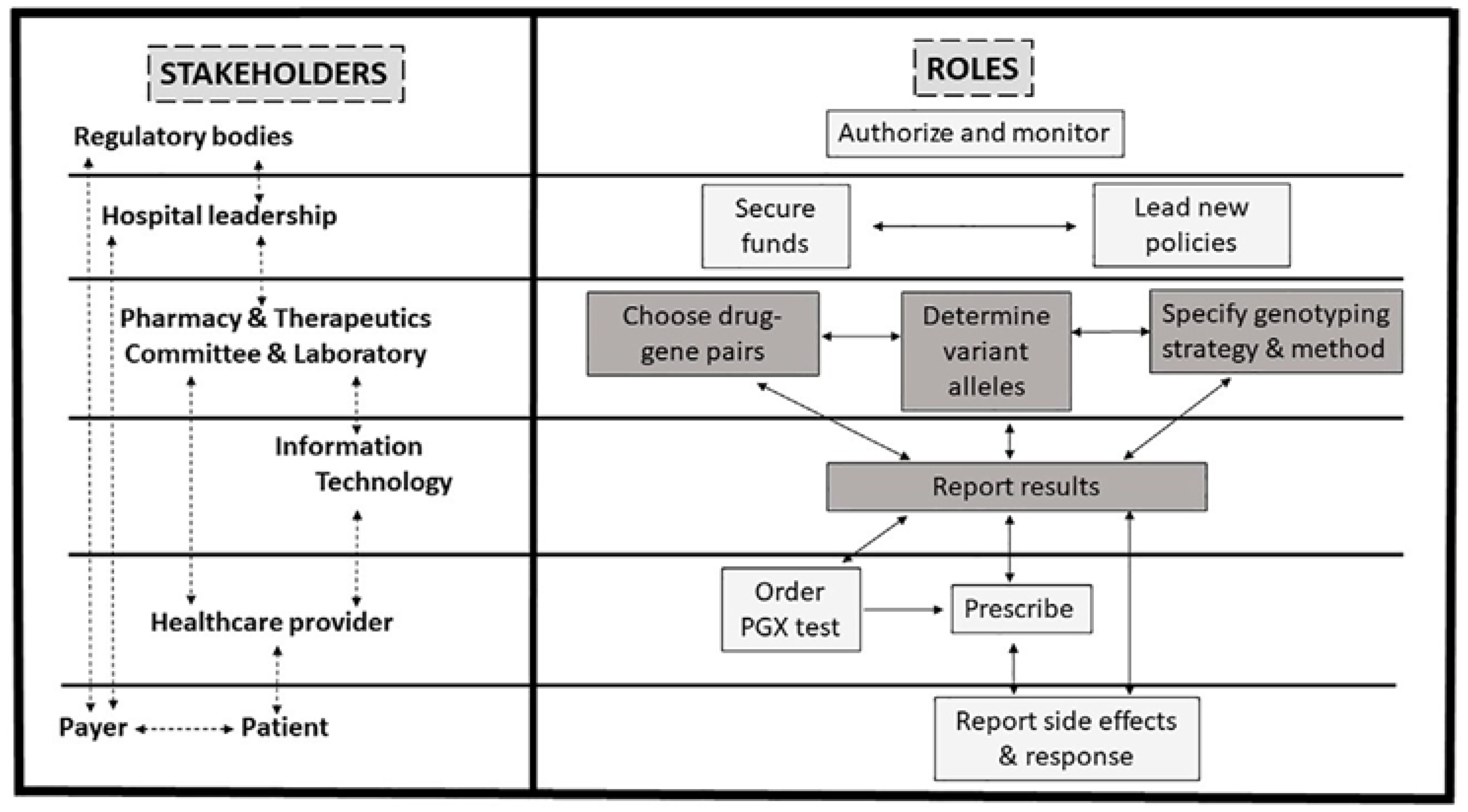
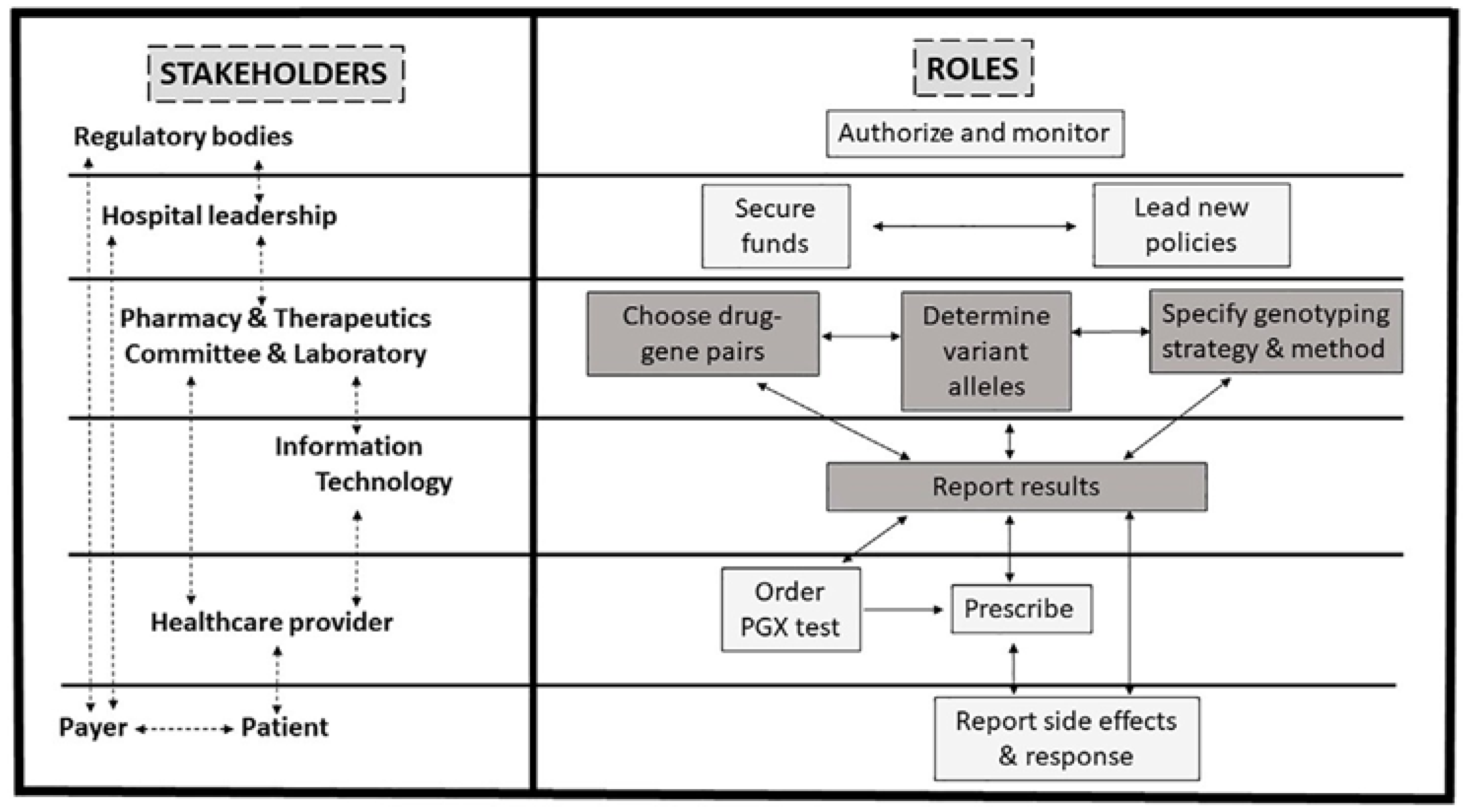
4. Implementation of Pharmacogenomics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Thummel, K.E.; Lin, Y.S. Sources of interindividual variability. In Enzyme Kinetics in Drug Metabolism; Humana Press: Totowa, NJ, USA, 2014; Volume 1113, pp. 363–415. [Google Scholar]
- Joy, M.S. Impact of glomerular kidney diseases on the clearance of drugs. J. Clin. Pharmacol. 2012, 52 (Suppl. S1), 23S–34S. [Google Scholar] [CrossRef] [PubMed]
- Weersink, R.A.; Bouma, M.; Burger, D.M.; Drenth, J.P.; Hunfeld, N.G.; Kranenborg, M.; Monster-Simons, M.H.; van Putten, S.A.; Metselaar, H.J.; Taxis, K.; et al. Evaluating the safety and dosing of drugs in patients with liver cirrhosis by literature review and expert opinion. BMJ Open 2016, 6, e012991. [Google Scholar] [CrossRef] [PubMed]
- Ruggiero, A.; Ariano, A.; Triarico, S.; Capozza, M.A.; Ferrara, P.; Attinà, G. Neonatal pharmacology and clinical implications. Drugs Context 2019, 8, 212608. [Google Scholar] [CrossRef] [PubMed]
- Porta, A.; Esposito, S.; Menson, E.; Spyridis, N.; Tsolia, M.; Sharland, M.; Principi, N. Off-label antibiotic use in children in three European countries. Eur. J. Clin. Pharmacol. 2010, 66, 919–927. [Google Scholar] [CrossRef]
- Kalow, W.; Tang, B.K.; Endrenyi, L. Hypothesis: Comparisons of inter- and intra-individual variations can substitute for twin studies in drug research. Pharmacogenetics 1998, 8, 283–289. [Google Scholar]
- Relling, M.; Evans, W. Pharmacogenomics in the clinic. Nature 2015, 526, 343–350. [Google Scholar] [CrossRef]
- Wang, C.-W.; Preclaro, I.A.C.; Lin, W.-H.; Chung, W.-H. An Updated Review of Genetic Associations with Severe Adverse Drug Reactions: Translation and Implementation of Pharmacogenomic Testing in Clinical Practice. Front. Pharmacol. 2022, 13, 886377. [Google Scholar] [CrossRef]
- European Medicines Agency. Use of Pharmacogenetic Methodologies in the Pharmacokinetic Evaluation of Medicinal Products—Scientific Guideline. Available online: https://www.ema.europa.eu/en/use-pharmacogenetic-methodologies-pharmacokinetic-evaluation-medicinal-products-scientific-guideline (accessed on 1 September 2023).
- U.S Food and Drug Administration. Administration Clinical Pharmacogenomics: Premarket Evaluation in Early-Phase Clinical Studies and Recommendations for Labeling. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/clinical-pharmacogenomics-premarket-evaluation-early-phase-clinical-studies-and-recommendations (accessed on 1 September 2023).
- U.S. Food and Drug Administration. Table of Pharmacogenetic Associations. Available online: www.fda.gov/medical-devices/precision-medicine/table-pharmacogenetic-associations (accessed on 1 September 2023).
- Relling, M.V.; Klein, T.E. CPIC: Clinical Pharmacogenetics Implementation Consortium of the Pharmacogenomics Research Network. Clin. Pharmacol. Ther. 2011, 89, 464–467. [Google Scholar] [CrossRef]
- Relling, M.V.; Klein, T.E.; Gammal, R.S.; Whirl-Carrillo, M.; Hoffman, J.M.; Caudle, K.E. The Clinical Pharmacogenetics Implementation Consortium: 10 years later. Clin. Pharmacol. Ther. 2020, 107, 171–175. [Google Scholar] [CrossRef]
- Swen, J.J.; Wilting, I.; Goede, A.L. Pharmacogenetics: From benchto byte. Clin. Pharmacol. Ther. 2008, 83, 781–787. [Google Scholar] [CrossRef]
- Thorn, C.F.; Klein, T.E.; Altman, R.B. PharmGKB: The pharmacogenomics knowledge base. Pharmacogenomics Methods Protoc. 2013, 1015, 311–320. [Google Scholar]
- Borobia, A.M.; Dapia, I.; Tong, H.Y.; Arias, P.; Muñoz, M.; Tenorio, J.; Hernández, R.; García García, I.; Gordo, G.; Ramírez, E.; et al. Clinical Implementation of Pharmacogenetic Testing in a Hospital of the Spanish National Health System: Strategy and Experience over 3 Years. Clin. Transl. Sci. 2018, 11, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; O’Donnell, P.H.; Middlestadt, M.; Ruhnke, G.W.; Danahey, K.; van Wijk, X.M.; Choksi, A.; Knoebel, R.; Hartman, S.; Yeo, K.T.J.; et al. Implementation of pharmacogenomics into inpatient general medicine. Pharmacogenetics Genom. 2023, 33, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, L.B.; Ong, H.H.; Schildcrout, J.S.; Shi, Y.; Tang, L.A.; Hicks, J.K.; El Rouby, N.; Cavallari, L.H.; Tuteja, S.; Aquilante, C.L.; et al. Prescribing Prevalence of Medications with Potential Genotype-Guided Dosing in Pediatric Patients. JAMA Netw. Open 2020, 3, e2029411. [Google Scholar] [CrossRef]
- Nigam, S.K. What do drug transporters really do? Nat. Rev. Drug Discov. 2015, 14, 29–44. [Google Scholar] [CrossRef]
- Yee, S.W.; Brackman, D.J.; Ennis, E.A.; Sugiyama, Y.; Kamdem, L.K.; Blanchard, R.; Galetin, A.; Zhang, L.; Giacomini, K.M. Influence of Transporter Polymorphisms on Drug Disposition and Response: A Perspective from the International Transporter Consortium. Clin. Pharmacol. Ther. 2018, 104, 803–817. [Google Scholar] [CrossRef]
- Iversen, D.B.; Andersen, N.E.; Dalgård Dunvald, A.C.; Pottegård, A.; Stage, T.B. Drug metabolism and drug transport of the 100 most prescribed oral drugs. Basic Clin. Pharmacol. Toxicol. 2022, 131, 311–324. [Google Scholar] [CrossRef]
- Ingelman-Sundberg, M. Human drug metabolising cytochrome P450 enzymes: Properties and polymorphisms. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2004, 369, 89–104. [Google Scholar]
- Ahmed, S.; Zhou, Z.; Zhou, J.; Chen, S.Q. Pharmacogenomics of Drug Metabolizing Enzymes and Transporters: Relevance to Precision Medicine. Genom. Proteom. Bioinform. 2016, 14, 298–313. [Google Scholar] [CrossRef]
- Li, J.; Bluth, M.H. Pharmacogenomics of drug metabolizing enzymes and transporters: Implications for cancer therapy. Pharmacogenomics Pers. Med. 2011, 4, 11–33. [Google Scholar]
- Sim, S.C.; Kacevska, M.; Ingelman-Sundberg, M. Pharmacogenomics of drug-metabolizing enzymes: A recent update on clinical implications and endogenous effects. Pharmacogenom. J. 2013, 13, 1–11. [Google Scholar] [CrossRef]
- Zhou, Y.; Nevosadová, L.; Eliasson, E.; Lauschke, V.M. Global distribution of functionally important CYP2C9 alleles and their inferred metabolic consequences. Hum. Genom. 2023, 17, 15. [Google Scholar] [CrossRef] [PubMed]
- Aithal, G.P.; Day, C.P.; Kesteven, P.J.; Daly, A.K. Association of polymorphisms in the cytochrome P450 CYP2C9 with warfarin dose requirement and risk of bleeding complications. Lancet 1999, 353, 717–719. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, A.L.; Prince, C.; Fitzgerald, G.; Hanson, A.; Downing, J.; Reynolds, J.; Zhang, J.E.; Alfirevic, A.; Pirmohamed, M. Implementation of genotype-guided dosing of warfarin with point-of-care genetic testing in three UK clinics: A matched cohort study. BMC Med. 2019, 17, 76. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.E.; Yee, J.; Lee, G.Y.; Chung, J.E.; Seong, J.M.; Chang, B.C.; Gwak, H.S. Genotype-guided warfarin dosing may benefit patients with mechanical aortic valve replacements: Randomized controlled study. Sci. Rep. 2020, 10, 6988. [Google Scholar] [CrossRef] [PubMed]
- Al-Metwali, B.Z.; Rivers, P.; Goodyer, L.; O’Hare, L.; Young, S.; Mulla, H. Personalised Warfarin Dosing in Children Post-cardiac Surgery. Pediatr. Cardiol. 2019, 40, 1735–1744. [Google Scholar] [CrossRef] [PubMed]
- Karnes, J.H.; Rettie, A.E.; Somogyi, A.A.; Huddart, R.; Fohner, A.E.; Formea, C.M.; Ta Michael Lee, M.; Llerena, A.; Whirl-Carrillo, M.; Klein, T.E.; et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for CYP2C9 and HLA-B Genotypes and Phenytoin Dosing: 2020 Update. Clin. Pharmacol. Ther. 2021, 109, 302–309. [Google Scholar] [CrossRef]
- Dean, L.; Kane, M. Phenytoin Therapy and HLA-B*15:02 and CYP2C9 Genotype. In Medical Genetics Summaries [Internet]; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2012. Available online: https://www.ncbi.nlm.nih.gov/books/NBK385287/ (accessed on 21 October 2023).
- Theken, K.N.; Lee, C.R.; Gong, L.; Caudle, K.E.; Formea, C.M.; Gaedigk, A.; Klein, T.E.; Agúndez, J.A.; Grosser, T. Clinical Pharmacogenetics Implementation Consortium Guideline (CPIC) for CYP2C9 and Nonsteroidal Anti-Inflammatory Drugs. Clin. Pharmacol. Ther. 2020, 108, 191–200. [Google Scholar] [CrossRef]
- Isoherranen, N.; Thummel, K.E. Drug metabolism and transport during pregnancy: How does drug disposition change during pregnancy and what are the mechanisms that cause such changes? Drug Metab. Dispos. 2013, 41, 256–262. [Google Scholar] [CrossRef]
- Durrmeyer, X.; Hovhannisyan, S.; Medard, Y.; Jacqz-Aigrain, E.; Decobert, F.; Barre, J.; Alberti, C.; Aujard, Y.; Danan, C.; Baud, O. Are Cytochrome P450 CYP2C8 and CYP2C9 Polymorphisms Associated with Ibuprofen Response in Very Preterm Infants? PLoS ONE 2010, 5, e12329. [Google Scholar] [CrossRef]
- Dehbozorgi, M.; Kamalidehghan, B.; Hosseini, I.; Dehghanfard, Z.; Sangtarash, M.H.; Firoozi, M.; Ahmadipour, F.; Meng, G.Y.; Houshmand, M. Prevalence of the CYP2C19*2 (681 G>A), *3 (636 G>A) and *17 (-806 C>T) alleles among an Iranian population of different ethnicities. Mol. Med. Rep. 2018, 17, 4195–4202. [Google Scholar] [CrossRef]
- Jung, F.; Richardson, T.H.; Raucy, J.L.; Johnson, E.F. Diazepam metabolism by cDNA-expressed human 2C P450s: Identification of P4502C18 and P4502C19 as low K(M) diazepam N-demethylases. Drug Metab. Dispos. 1997, 25, 133–139. [Google Scholar] [PubMed]
- Dean, L. Diazepam Therapy and CYP2C19 Genotype. In Medical Genetics Summaries [Internet]; Pratt, V.M., Scott, S.A., Pirmohamed, M., Esquivel, B., Kattman, B.L., Malheiro, A.J., Eds.; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2012. [Google Scholar]
- Baldwin, R.M.; Ohlsson, S.; Pedersen, R.S.; Mwinyi, J.; Ingelman-Sundberg, M.; Eliasson, E.; Bertilsson, L. Increased omeprazole metabolism in carriers of the CYP2C19*17 allele; a pharmacokinetic study in healthy volunteers. Br. J. Clin. Pharmacol. 2008, 65, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Schwab, M.; Schaeffeler, E.; Klotz, U.; Treiber, G. CYP2C19 polymorphism is a major predictor of treatment failure in white patients by use of lansoprazole-based quadruple therapy for eradication of Helicobacter pylori. Clin. Pharmacol. Ther. 2004, 76, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Sibbing, D.; Koch, W.; Gebhard, D.; Schuster, T.; Braun, S.; Stegherr, J. Cytochrome 2C19*17 allelic variant, platelet aggregation, bleeding events, and stent thrombosis in clopidogrel-treated patients with coronary stent placement. Circulation 2010, 121, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Kubica, A.; Kozinski, M.; Grzesk, G.; Fabiszak, T.; Navarese, E.P.; Goch, A. Genetic determinants of platelet response to clopidogrel. J. Thromb. Thrombolysis 2011, 32, 459–466. [Google Scholar] [CrossRef]
- Tilen, R.; Paioni, P.; Goetschi, A.N.; Goers, R.; Seibert, I.; Müller, D.; Bielicki, J.A.; Berger, C.; Krämer, S.D.; zu Schwabedissen, H.E.M. Pharmacogenetic Analysis of Voriconazole Treatment in Children. Pharmaceutics 2022, 14, 1289. [Google Scholar] [CrossRef]
- Ingelman-Sundberg, M. Genetic polymorphisms of cytochrome P450 2D6 (CYP2D6): Clinical consequences, evolutionary aspects and functional diversity. Pharmacogenom. J. 2005, 5, 6–13. [Google Scholar] [CrossRef]
- Zhou, S.-F.; Di, Y.M.; Chan, E.; Du, Y.-M.; Chow, V.D.-W.; Xue, C.C.; Lai, X.; Wang, J.-C.; Li, C.G.; Tian, M.; et al. Clinical pharmacogenetics and potential application in personalized medicine. Curr. Drug Metab. 2008, 9, 738–784. [Google Scholar] [CrossRef]
- Dean, L.; Kane, M. Codeine Therapy and CYP2D6 Genotype. In Medical Genetics Summaries [Internet]; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2012. Available online: https://www.ncbi.nlm.nih.gov/books/NBK100662/ (accessed on 1 September 2023).
- Molden, E.; Jukić, M.M. CYP2D6 Reduced Function Variants and Genotype/Phenotype Translations of CYP2D6 Intermediate Metabolizers: Implications for Personalized Drug Dosing in Psychiatry. Front. Pharmacol. 2021, 12, 650750. [Google Scholar] [CrossRef]
- Jukic, M.M.; Smith, R.L.; Haslemo, T.; Molden, E.; Ingelman-Sundberg, M. Effect of CYP2D6 genotype on exposure and efficacy of risperidone and aripiprazole: A retrospective, cohort study. Lancet Psychiatry 2019, 6, 418–426. [Google Scholar] [CrossRef]
- Sindrup, S.H.; Brøsen, K. The pharmacogenetics of codeine hypoalgesia. Pharmacogenetics 1995, 5, 246–335. [Google Scholar] [CrossRef] [PubMed]
- Hinderer, M.; Boeker, M.; Wagner, S.A.; Lablans, M.; Newe, S.; Hülsemann, J.L.; Neumaier, M.; Binder, H.; Renz, H.; Acker, T.; et al. Integrating clinical decision support systems for pharmacogenomic testing into clinical routine—A scoping review of designs of user-system interactions in recent system development. BMC Med. Inform. Decis. Mak. 2017, 17, 81. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. FDA Drug Safety Podcast: FDA Restricts Use of Prescription Codeine Pain and Cough Medicines and Tramadol Pain Medicines in Children; Recommends against Use in Breastfeeding Women. Available online: https://www.fda.gov/drugs/fda-drug-safety-podcasts/fda-drug-safety-podcast-fda-restricts-use-prescription-codeine-pain-and-cough-medicines-and-tramadol#:~:text=On%20April%2020%2C%202017%2C%20FDA,children%20younger%20than%2012%20years (accessed on 1 September 2023).
- Blake, M.J.; Gaedigk, A.; Pearce, R.E. Ontogeny of dextromethorphan O- and N-demethylation in the first year of life. Clin. Pharmacol. Ther. 2007, 81, 510–516. [Google Scholar] [CrossRef] [PubMed]
- Rowland, A.; Miners, J.O.; Mackenzie, P.I. The UDP-glucuronosyltransferases: Their role in drug metabolism and detoxification. Int. J. Biochem. Cell. Biol. 2013, 45, 1121–1132. [Google Scholar] [CrossRef]
- Holthe, M.; Rakvåg, T.N.; Klepstad, P.; Idle, J.R.; Kaasa, S.; Krokan, H.E.; Skorpen, F. Sequence variations in the UDPglucuronosyltransferase 2B7 (UGT2B7) gene: Identification of 10 novel single nucleotide polymorphisms (SNPs) and analysis of their relevance to morphine glucuronidation in cancer patients. Pharmacogenom. J. 2003, 3, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Udomuksorn, W.; Elliot, D.J.; Lewis, B.C.; Mackenzie, P.I.; Yoovathaworn, K.; Miners, J.O. Influence of mutations associated with Gilbert and Crigler–Najjar type II syndromes on the glucuronidation kinetics of bilirubin and other UDP-glucuronosyltransferase 1A substrates. Pharmacogenet Genom. 2007, 17, 1017–1029. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.-Y.; Cho, J.-Y.; Yu, K.-S.; Kim, J.-R.; Lim, K.; Sohn, D.-R.; Shin, S.-G.; Jang, I.-J. Pharmacokinetic and pharmacodynamic interaction of lorazepam and valproic acid in relation to UGT2B7 genetic polymorphism in healthy subjects. Clin. Pharmacol. Ther. 2008, 83, 595–600. [Google Scholar] [CrossRef]
- Krishnaswamy, S.; Hao, Q.; Duan, S.X.; Patten, C.J.; Von Moltke, L.L.; Greenblatt, D.J. Evaluation of 3-azido-3-deoxythymidine, morphine, and codeine as probe substrates for udp-glucuronosyltransferase 2B7 (UGT2B7) in human liver microsomes: Specificity and influence of the UGT2B7*2 polymorphism. Drug Metab. Disp. 2003, 31, 1125–1133. [Google Scholar]
- He, X.; Hesse, L.M.; Hazarika, S.; Masse, G.; Harmatz, J.S.; Greenblatt, D.J.; Court, M.H. Evidence for oxazepam as an in vivo probe of UGT2B15: Oxazepam clearance is reduced by UGT2B15 D85Y polymorphism but unaffected by UGT2B17 deletion. Br. J. Clin. Pharmacol. 2009, 68, 721–730. [Google Scholar] [CrossRef]
- Hulshof, E.C.; de With, M.; de Man, F.M.; Creemers, G.J.; Deiman, B.A.; Swen, J.J.; Houterman, S.; Koolen, S.L.; Bins, S.; Thijs, A.M.; et al. UGT1A1 genotype-guided dosing of irinotecan: A prospective safety and cost analysis in poor metaboliser patients. Eur. J. Cancer 2022, 162, 148–157. [Google Scholar] [CrossRef]
- Wong, M.; Behrendt, C.E.; Yu, W.; Bosserman, L.D.; Lavasani, S.M.; Patel, N.; Sedrak, M.S.; Stewart, D.B.; Waisman, J.R.; Yuan, Y.; et al. UGT1A1 *28/*28 genotype and risk of toxicity and disease progression in breast cancer patients treated with sacituzumab govitecan-hziy. J. Clin. Oncol. 2023, 41 (Suppl. S16), 1033–1036. [Google Scholar] [CrossRef]
- Singer, J.B.; Shou, Y.; Giles, F.; Kantarjian, H.M.; Hsu, Y.; Robeva, A.S.; Rae, P.; Weitzman, A.; Meyer, J.M.; Dugan, M.; et al. UGT1A1 promoter polymorphism increases risk of nilotinib-induced hyperbilirubinemia. Leukemia 2007, 21, 2311–2315. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.F.; Reck, B.H.; Xue, Z.; Huang, L.; Baker, K.L.; Chen, M.; Chen, E.P.; Ellens, H.E.; Mooser, V.E.; Cardon, L.R.; et al. Pazopanib-induced hyperbilirubinemia is associated with Gilbert’s syndrome UGT1A1 polymorphism. Br. J. Cancer 2010, 102, 1371–1377. [Google Scholar] [CrossRef] [PubMed]
- Bayoumy, A.B.; Crouwel, F.; Chanda, N.; Florin, T.H.; Buiter, H.J.; Mulder, C.J.; de Boer, N.K. Advances in Thiopurine Drug Delivery: The Current State-of-the-Art. Eur. J. Drug Metab. Pharmacokinet. 2021, 46, 743–758. [Google Scholar] [CrossRef]
- Yoshida, M.; Brown, S.A.; Moriyama, T.; Nishii, R.; Tsujimoto, S.I.; Yamada, Y.; Yoshida, K.; Shirai, R.; Osumi, T.; Utano, T.; et al. Low gNUDT15 expression levels due to biallelic NUDT15 variants and 6-mercaptopurine intolerance. Br. J. Haematol. 2022, 199, 270–276. [Google Scholar] [CrossRef]
- Guo, H.L.; Zhao, Y.T.; Wang, W.J.; Dong, N.; Hu, Y.H.; Zhang, Y.Y.; Chen, F.; Zhou, L.; Li, T. Optimizing thiopurine therapy in children with acute lymphoblastic leukemia: A promising “MINT” sequencing strategy and therapeutic “DNA-TG” monitoring. Front. Pharmacol. 2022, 13, 941182. [Google Scholar] [CrossRef]
- Gargallo-Puyuelo, C.J.; Laredo, V.; Gomollón, F. Thiopurines in Inflammatory Bowel Disease. How to Optimize Thiopurines in the Biologic Era? Front. Med. 2021, 8, 681907. [Google Scholar] [CrossRef]
- Asadov, C.; Aliyeva, G.; Mustafayeva, K. Thiopurine S-Methyltransferase as a Pharmacogenetic Biomarker: Significance of Testing and Review of Major Methods. Cardiovasc. Hematol. Agents Med. Chem. 2017, 15, 23–30. [Google Scholar] [CrossRef]
- Lennard, L. Implementation of TPMT testing. Br. J. Clin. Pharmacol. 2014, 77, 704–714. [Google Scholar] [CrossRef]
- Bruhn, O.; Cascorbi, I. Polymorphisms of the drug transporters ABCB1, ABCG2, ABCC2 and ABCC3 and their impact on drug bioavailability and clinical relevance. Expert Opin. Drug Metab. Toxicol. 2014, 10, 1337–1354. [Google Scholar] [CrossRef]
- Bruckmueller, H.; Cascorbi, I. ABCB1, ABCG2, ABCC1, ABCC2, and ABCC3 drug transporter polymorphisms and their impact on drug bioavailability: What is our current understanding? Expert Opin. Drug Metab. Toxicol. 2021, 17, 369–396. [Google Scholar] [CrossRef] [PubMed]
- Raj, G.M.; Mathaiyan, J.; Wyawahare, M.; Priyadarshini, R. Lack of effect of the SLC47A1 and SLC47A2 gene polymorphisms on the glycemic response to metformin in type 2 diabetes mellitus patients. Drug Metab. Pers. Ther. 2018, 33, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Cao, Y.; Chen, S.; Liu, Z.; Chen, S.; Guo, Y. Association of SLC22A1, SLC22A2, SLC47A1, and SLC47A2 Polymorphisms with Metformin Efficacy in Type 2 Diabetic Patients. Biomedicines 2022, 10, 2546. [Google Scholar] [CrossRef]
- Van Den Driessche, G.; Fourches, D. Adverse drug reactions triggered by the common HLA-B*57:01 variant: A molecular docking study. J. Cheminform. 2017, 9, 13. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.-R.; Jee, Y.K.; Kim, Y.-S.; Lee, C.H.; Jung, J.-W.; Kim, S.-H.; Park, H.-W.; Chang, Y.-S.; Jang, I.-J.; Cho, S.-H.; et al. Positive and negative associations of HLA class I alleles with allopurinol-induced SCARs in Koreans. Pharm. Genom. 2011, 21, 303–307. [Google Scholar] [CrossRef] [PubMed]
- Jeiziner, C.; Wernli, U.; Suter, K.; Hersberger, K.E.; Meyer Zu Schwabedissen, H.E. HLA-associated adverse drug reactions—Scoping review. Clin. Transl. Sci. 2021, 14, 1648–1658. [Google Scholar] [CrossRef]
- Kloypan, C.; Koomdee, N.; Satapornpong, P.; Tempark, T.; Biswas, M.; Sukasem, C. A Comprehensive Review of HLA and Severe Cutaneous Adverse Drug Reactions: Implication for Clinical Pharmacogenomics and Precision Medicine. Pharmaceuticals 2021, 14, 1077. [Google Scholar] [CrossRef]
- Gerussi, A.; Natalini, A.; Antonangeli, F.; Mancuso, C.; Agostinetto, E.; Barisani, D.; Di Rosa, F.; Andrade, R.; Invernizzi, P. Immune-Mediated Drug-Induced Liver Injury: Immunogenetics and Experimental Models. Int. J. Mol. Sci. 2021, 22, 4557. [Google Scholar] [CrossRef]
- Redwood, A.J.; Pavlos, R.K.; White, K.D.; Phillips, E.J. HLAs: Key regulators of T-cell-mediated drug hypersensitivity. HLA 2018, 91, 3–16. [Google Scholar] [CrossRef]
- Jaruthamsophon, K.; Thomson, P.J.; Sukasem, C.; Naisbitt, D.J.; Pirmohamed, M. HLA Allele-Restricted Immune-Mediated Adverse Drug Reactions: Framework for Genetic Prediction. Annu. Rev. Pharmacol. Toxicol. 2022, 62, 509–529. [Google Scholar] [CrossRef]
- Vakrinou, A.; Bellampalli, R.; Gulcebi, M.I.; Martins Custodio, H.; Research Consortium, G.E.; Balestrini, S.; Sisodiya, S.M. Risk-conferring HLA variants in an epilepsy cohort: Benefits of multifaceted use of whole genome sequencing in clinical practice. J Neurol Neurosurg Psychiatry. 2023, 94, 887–892. [Google Scholar] [CrossRef] [PubMed]
- Mallal, S.; Phillips, E.; Carosi, G.; Molina, J.M.; Workman, C.; Tomažič, J.; Jägel-Guedes, E.; Rugina, S.; Kozyrev, O.; Cid, J.F.; et al. HLA-B*5701 screening for hypersensitivity to abacavir. N. Engl. J. Med. 2008, 358, 568–579. [Google Scholar] [CrossRef] [PubMed]
- Hung, S.-I.; Chung, W.-H.; Liou, L.-B.; Chu, C.-C.; Lin, M.; Huang, H.-P.; Lin, Y.-L.; Lan, J.-L.; Yang, L.-C.; Hong, H.-S.; et al. HLA-B*5801 allele as a genetic marker for severe cutaneous adverse reactions caused by allopurinol. Proc. Natl. Acad. Sci. USA 2005, 102, 4134–4139. [Google Scholar] [CrossRef] [PubMed]
- Lucena, M.I.; Molokhia, M.; Shen, Y.; Urban, T.J.; Aithal, G.P.; Andrade, R.J.; Day, C.P.; Ruiz-Cabello, F.; Donaldson, P.T.; Stephens, C.; et al. Susceptibility to amoxicillin-clavulanate-induced liver injury is influenced by multiple HLA class I and II alleles. Gastroenterology 2011, 141, 338–347. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Lin, J.-J.; Lu, C.-S.; Ong, C.-T.; Hsieh, P.F.; Yang, C.-C.; Tai, C.-T.; Wu, S.-L.; Lu, C.-H.; Hsu, Y.-C.; et al. Carbamazepine-induced toxic effects and HLA-B*1502 screening in Taiwan. N. Engl. J. Med. 2011, 364, 1126–1133. [Google Scholar] [CrossRef]
- Daly, A.K.; Donaldson, P.T.; Bhatnagar, P.; Shen, Y.; Pe’er, I.; Floratos, A.; Daly, M.J.; Goldstein, D.B.; John, S.; Nelson, M.R.; et al. HLA-B*5701 genotype is a major determinant of drug-induced liver injury due to flucloxacillin. Nat. Genet. 2009, 41, 816–819. [Google Scholar] [CrossRef]
- Phung, T.H.; Cong Duong, K.N.; Junio Gloria, M.A.; Nguyen, T.K. The association between HLA-B*15:02 and phenytoin-induced severe cutaneous adverse reactions: A meta-analysis. Pharmacogenomics 2022, 23, 49–59. [Google Scholar] [CrossRef]
- Pan, R.-Y.; Chu, M.-T.; Wang, C.-W.; Lee, Y.-S.; Lemonnier, F.; Michels, A.W.; Schutte, R.; Ostrov, D.A.; Chen, C.-B.; Phillips, E.J.; et al. Identification of drug-specific public TCR driving severe cutaneous adverse reactions. Nat. Commun. 2019, 10, 3569. [Google Scholar] [CrossRef]
- Caudle, K.E.; Klein, T.E.; Hoffman, J.M.; Muller, D.J.; Whirl-Carrillo, M.; Gong, L.; McDonagh, E.M.; Sangkuhl, K.; Thorn, C.F.; Schwab, M.; et al. Incorporation of pharmacogenomics into routine clinical practice: The Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline development process. Curr. Drug Metab. 2014, 15, 209–217. [Google Scholar] [CrossRef]
- McDermott, J.H.; Newman, W. Introduction to pharmacogenetics. Drug Ther. Bull. 2023, 61, 168–172. [Google Scholar] [CrossRef]
- Dunnenberger, H.M.; Crews, K.R.; Hoffman, J.M.; Caudle, K.E.; Broeckel, U.; Howard, S.C.; Hunkler, R.J.; Klein, T.E.; Evans, W.E.; Relling, M.V. Preemptive clinical pharmacogenetics implementation: Current programs in five US medical centers. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 89–106. [Google Scholar] [CrossRef] [PubMed]
- Hicks, J.K.; Stowe, D.; Willner, M.A.; Wai, M.; Daly, T.; Gordon, S.M.; Lashner, B.A.; Parikh, S.; White, R.; Teng, K.; et al. Implementation of Clinical Pharmacogenomics within a Large Health System: From Electronic Health Record Decision Support to Consultation Services. Pharmacotherapy 2016, 36, 940–948. [Google Scholar] [CrossRef] [PubMed]
- Cavallari, L.H.; Beitelshees, A.L.; Blake, K.V.; Dressler, L.G.; Duarte, J.D.; Elsey, A.; Eichmeyer, J.N.; Empey, P.E.; Franciosi, J.P.; Hicks, J.K.; et al. The IGNITE Pharmacogenetics Working Group: An Opportunity for Building Evidence with Pharmacogenetic Implementation in a Real-World Setting. Clin. Transl. Sci. 2017, 10, 143–146. [Google Scholar] [CrossRef] [PubMed]
- van der Wouden, C.H.; Cambon-Thomsen, A.; Cecchin, E.; Cheung, K.C.; Dávila-Fajardo, C.L.; Deneer, V.H.; Dolžan, V.; Ingelman-Sundberg, M.; Jönsson, S.; Karlsson, M.O.; et al. Implementing Pharmacogenomics in Europe: Design and Implementation Strategy of the Ubiquitous Pharmacogenomics Consortium. Clin. Pharmacol. Ther. 2017, 101, 341–358. [Google Scholar] [CrossRef]
- Petry, N.; Baye, J.; Aifaoui, A.; Wilke, R.A.; Lupu, R.A.; Savageau, J.; Gapp, B.; Massmann, A.; Hahn, D.; Hajek, C.; et al. Implementation of wide-scale pharmacogenetic c testing in primary care. Pharmacogenomics 2019, 20, 903–913. [Google Scholar] [CrossRef]
- Luzum, J.A.; Pakyz, R.E.; Elsey, A.R.; Haidar, C.E.; Peterson, J.F.; Whirl-Carrillo, M.; Handelman, S.K.; Palmer, K.; Pulley, J.M.; Beller, M.; et al. The Pharmacogenomics Research Network Translational Pharmacogenetics Program: Outcomes and Metrics of Pharmacogenetic Implementations across Diverse Healthcare Systems. Clin. Pharmacol. Ther. 2017, 102, 502–510. [Google Scholar] [CrossRef]
- Huebner, T.; Steffens, M.; Scholl, C. Current status of the analytical validation of next generation sequencing applications for pharmacogenetic profiling. Mol. Biol. Rep. 2023, 50, 9587–9599. [Google Scholar] [CrossRef]
- Koch, B.C.; van Schaik, R.H.; van Gelder, T.; Mathijssen, R.H. Rotterdam Clinical Pharmacology Pharmacogenetics Group. Doubt about the feasibility of preemptive genotyping. Clin. Pharmacol. Ther. 2013, 93, 233. [Google Scholar]
- Wang, B.; Canestaro, W.J.; Choudhry, N.K. Clinical evidence supporting pharmacogenomic biomarker testing provided in US Food and Drug Administration drug labels. JAMA Intern. Med. 2014, 174, 1938–1944. [Google Scholar] [CrossRef]
- Swen, J.J.; van der Wouden, C.H.; Manson, L.E.; Abdullah-Koolmees, H.; Blagec, K.; Blagus, T.; Böhringer, S.; Cambon-Thomsen, A.; Cecchin, E.; Cheung, K.C.; et al. A 12-gene pharmacogenetic panel to prevent adverse drug reactions: An open-label, multicentre, controlled, cluster-randomised crossover implementation study. Lancet 2023, 401, 347–356. [Google Scholar] [CrossRef]
- Karamperis, K.; Koromina, M.; Papantoniou, P.; Skokou, M.; Kanellakis, F.; Mitropoulos, K.; Vozikis, A.; Müller, D.J.; Patrinos, G.P.; Mitropoulou, C. Economic evaluation in psychiatric pharmacogenomics: A systematic review. Pharmacogenom. J. 2021, 21, 533–541. [Google Scholar] [CrossRef] [PubMed]
- Luzum, J.A.; Petry, N.; Taylor, A.K.; Van Driest, S.L.; Dunnenberger, H.M.; Cavallari, L.H. Moving Pharmacogenetics into Practice: It’s All about the Evidence! Clin. Pharmacol. Ther. 2021, 110, 649–661. [Google Scholar] [CrossRef] [PubMed]
- Frueh, F.W. Back to the future: Why randomized controlled trials cannot be the answer to pharmacogenomics and personalized medicine. Pharmacogenomics 2009, 10, 1077–1081. [Google Scholar] [CrossRef] [PubMed]
- Visvanathan, K.; Hurley, P.; Bantug, E.; Brown, P.; Col, N.F.; Cuzick, J.; Davidson, N.E.; DeCensi, A.; Fabian, C.; Ford, L.; et al. Use of pharmacologic interventions for breast cancer risk reduction: American Society of Clinical Oncology clinical practice guideline. J. Clin. Oncol. 2013, 31, 2942–2962. [Google Scholar] [CrossRef]
- Morris, S.A.; Alsaidi, A.T.; Verbyla, A.; Cruz, A.; Macfarlane, C.; Bauer, J.; Patel, J.N. Cost Effectiveness of Pharmacogenetic Testing for Drugs with Clinical Pharmacogenetics Implementation Consortium (CPIC) Guidelines: A Systematic Review. Clin. Pharmacol. Ther. 2022, 112, 1318–1328. [Google Scholar] [CrossRef]
- Relling, M.V.; Altman, R.B.; Goetz, M.P.; Evans, W.E. Clinical implementation of pharmacogenomics: Overcoming genetic exceptionalism. Lancet Oncol. 2010, 11, 507–509. [Google Scholar] [CrossRef]
- Zineh, I.; Lesko, L.J. Pharmacogenetics in medicine: Barriers, critical factors and a framework for dialogue. Pers. Med. 2009, 6, 359–361. [Google Scholar] [CrossRef]
- Kabbani, D.; Akika, R.; Wahid, A.; Daly, A.K.; Cascorbi, I.; Zgheib, N.K. Pharmacogenomics in practice: A review and implementation guide. Front. Pharmacol. 2023, 14, 1189976. [Google Scholar] [CrossRef]
- Karas Kuželički, N.; Prodan Žitnik, I.; Gurwitz, D.; Llerena, A.; Cascorbi, I.; Siest, S.; Simmaco, M.; Ansari, M.; Pazzagli, M.; Di Resta, C.; et al. Pharmacogenomics education in medical and pharmacy schools: Conclusions of a global survey. Pharmacogenomics 2019, 20, 643–657. [Google Scholar]
- El Rouby, N.; Lima, J.J.; Johnson, J.A. Proton pump inhibitors: From CYP2C19 pharmacogenetics to precision medicine. Expert Opin. Drug Metab. Toxicol. 2018, 14, 447–460. [Google Scholar] [CrossRef]
- Ward, R.M.; Tammara, B.; Sullivan, S.E.; Stewart, D.L.; Rath, N.; Meng, X.; Maguire, M.K.; Comer, G.M. Single-dose, multiple-dose, and population pharmacokinetics of pantoprazole in neonates and preterm infants with a clinical diagnosis of gastroesophageal reflux disease (GERD). Eur. J. Clin. Pharmacol. 2010, 66, 555–561. [Google Scholar] [CrossRef]
- Koukouritaki, S.B.; Manro, J.R.; Marsh, S.A.; Stevens, J.C.; Rettie, A.E.; McCarver, D.G.; Hines, R.N. Developmental expression of human hepatic CYP2C9 and CYP2C19. J. Pharmacol. Exp. Ther. 2004, 308, 965–974. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Phase 1 Enzymes | Phase 2 Enzymes |
|---|---|
| Cytochrome P450 Monooxygenase (CYP) | Uridine diphosphate glucuronosyl Transferase (UDPGT) |
| Flavin-containing Monooxygenase | Sulfo transferase (ST) |
| Esterase | N-Acetyl transferase (NAT |
| Alcohol Dehydrogenase (ADH) | Glutathione S-Transferase (GST) |
| Aldehyde Dehydrogenase (ALDH) | Methyl Transferase |
| Monoamine Oxidase (MAO) | Amino Acid Conjugation |
| Drug | Therapeutic Area | Clinical Impact |
|---|---|---|
| S-warfarin | Cardiovascular diseases | When usual doses of warfarin are used, in PMs, the risk of internal bleeding is greatly increased. Drug dose should be established according to CYP2C9 polymorphism genotype. IMs should use 65% of the standard initial dose; PMs 20%. Drug monitoring is recommended to establish maintaining doses. |
| Phenytoin | Neurology | PMs are at greater risk of developing CNS adverse effects as well as serious cutaneous adverse reactions when given usual dosages of phenytoin. It is recommended to start with recommended doses and reduce maintaining doses by about 50%. |
| Some NSAIDs (ibuprofen, celecoxib, meloxicam, piroxicam, flurbiprofen, mefenamic acid) | Diseases with inflammation | In PMs, increased risk of gastrointestinal ulcers, serious cardiovascular events, hypertension, acute renal failure, and worsening of preexisting heart failure. In these patients, it is recommended to initiate treatment at 25–50% of the traditional dose or use NSAIDs not metabolized by CYP2C9 (acetylsalicylic acid, ketorolac, naproxen, sulindac). |
| Some hypoglycemic drugs (glipizide, tolbutamide) | Diabetology | These drugs are a substrate of the genetically polymorphic enzyme CYP2C9. However, the pronounced differences in pharmacokinetics due to the variants did not significantly affect plasma insulin and glucose concentrations. No dose variations are needed. |
| Drug | Therapeutic Area | Clinical Impact |
|---|---|---|
| Diazepam | Neurology and psychiatry | In PMs, standard doses can lead to increased risk of sedation and unconsciousness. Plasma half-life of the drug is about up to six times longer than in individuals homozygous for wild-type CYP2C19 genotype. However, modification of dosage is not required unless drugs that inhibit CYP2C19 gene expression are given at the same time. |
| Proton pump inhibitors | Gastroenterology | Increased and decreased drug effectiveness in PMs and EMs, respectively. |
| Clopidrogel | Cardiology | In PMs, drug activity is reduced, leading to increased risk of cardiovascular events. |
| Voriconazole | Infectious diseases | In PMs, standard doses can lead to increased incidence of severe adverse events. In these patients, alternative drugs or use of lower doses with careful monitoring of plasma levels are recommended. |
| Drug | Genetic Marker | Associated Manifestations |
|---|---|---|
| Abacavir [80] | HLA-B*57:01 | Development within 6 months from starting therapy. Symptoms are fever, rash, nausea, vomiting, diarrhea or abdominal pain, and fatigue and malaise. Occasionally, respiratory symptoms are prominent and pneumonia occurs. Frequency of polymorphism is about 14% in Caucasian, 12.6% in Asian, 2.6% in South American, 2.2% in Mexican, and 1% in African populations. All patients should be screened for the genetic variation prior to initiating or reinitiating therapy with abacavir, unless patients have a previously documented HLA-B*57:01allele assessment. |
| Allopurinol [81] | HLA-B*58:01 | DRESS, SJS/TEN. Common among Asian subpopulations, notably in individuals of Korean, Han-Chinese, or Thai descent. Presently, the FDA-approved drug label does not discuss HLA-B genotype. Testing for the HLA–B*58:01 allele prior to starting allopurinol is conditionally recommended for individuals of Southeast Asian descent (e.g., Han Chinese, Korean, Thai) and for African American individuals, over not testing for the HLA-B*58:01 allele. Universal testing for the HLA-B*58:01allele prior to starting allopurinol is conditionally recommended against in individuals of other ethnic or racial background over testing for the HLA-B*58:01allele. |
| Amoxicillin-clavulanate [83] | HLA-DRB1*-15.01 | Drug-induced liver injury, mainly a transaminase increase. |
| Carbamazepine [84] | HLA-B*15:02 HLA-B*31:01 | The clinical manifestations can vary widely, ranging from a mild skin rash, such as MPE and EEM minor, to severe diseases such as EEM major, SJS, TEN, DRESS, and AGEP. HLA-B*15.02 has been found mostly in Asian people but not in Caucasian patients. HLA-B*31:01is prevalent globally, particularly in indigenous populations of the Americas (Argentina 28.8%, Mexico 10.1%, the USA 7.8%, Nicaragua 6.7%, and Chile). Values of about 8% in Asia and varying from <1% to about 6% in Europe. FDA-approved labeling recommends HLA-B*15.02 screening before CBZ therapy in patients of Asian ancestry. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Principi, N.; Petropulacos, K.; Esposito, S. Impact of Pharmacogenomics in Clinical Practice. Pharmaceuticals 2023, 16, 1596. https://doi.org/10.3390/ph16111596
Principi N, Petropulacos K, Esposito S. Impact of Pharmacogenomics in Clinical Practice. Pharmaceuticals. 2023; 16(11):1596. https://doi.org/10.3390/ph16111596
Chicago/Turabian StylePrincipi, Nicola, Kyriakoula Petropulacos, and Susanna Esposito. 2023. "Impact of Pharmacogenomics in Clinical Practice" Pharmaceuticals 16, no. 11: 1596. https://doi.org/10.3390/ph16111596
APA StylePrincipi, N., Petropulacos, K., & Esposito, S. (2023). Impact of Pharmacogenomics in Clinical Practice. Pharmaceuticals, 16(11), 1596. https://doi.org/10.3390/ph16111596