
Targeting MutT Homolog 1 (MTH1) for Breast Cancer Suppression by Using a Novel MTH1 Inhibitor MA−24 with Tumor-Selective Toxicity

 ,
,
Abstract

1. Introduction
2. Results
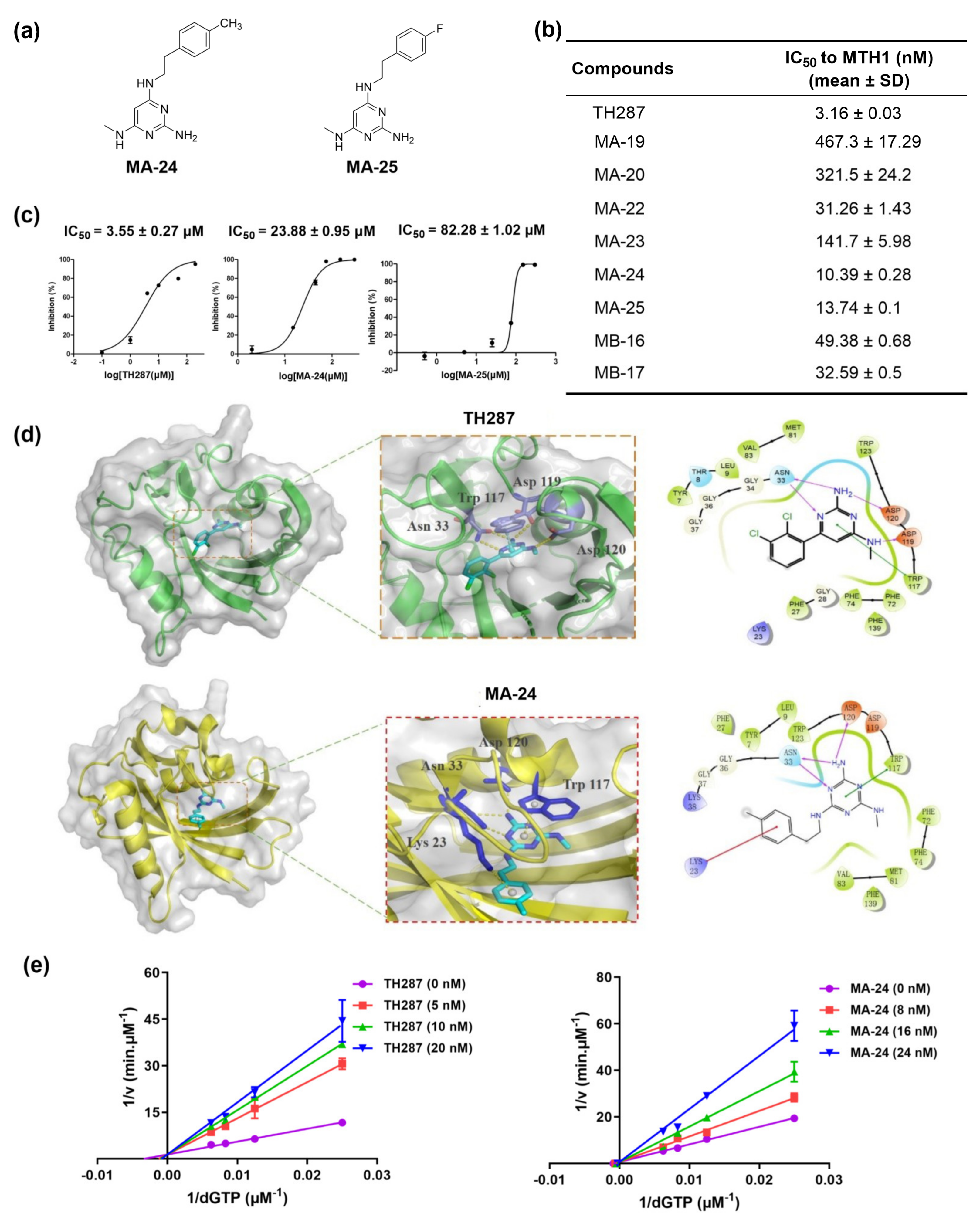
2.1. Identification of Promising MTH1 Inhibitors
2.2. MA−24 Reduces Cell Survival and Induces Cell Apoptosis
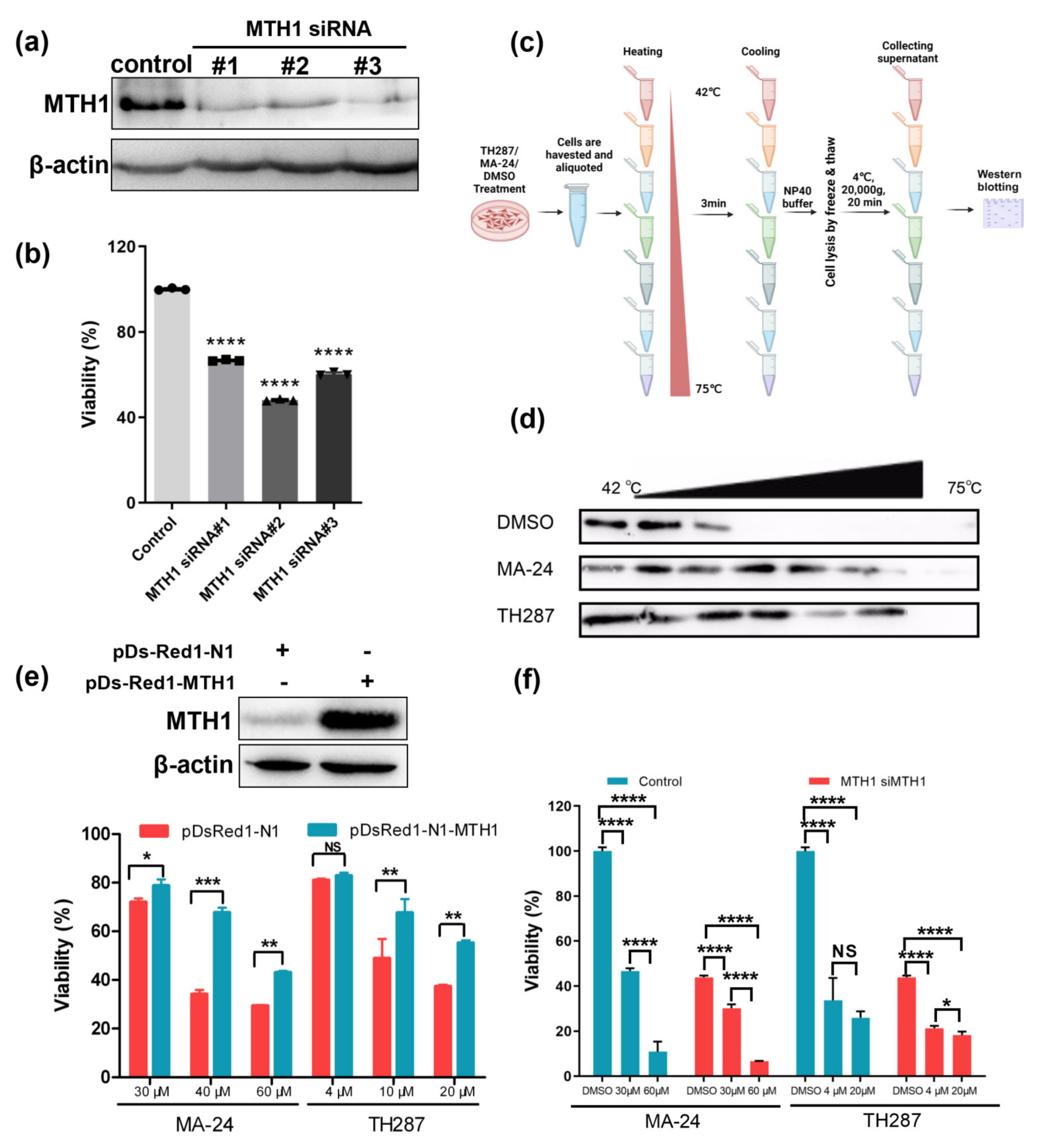
2.3. MTH1 as the Target of MA−24 Is a Major Determinant for the Survival of Cancer Cells
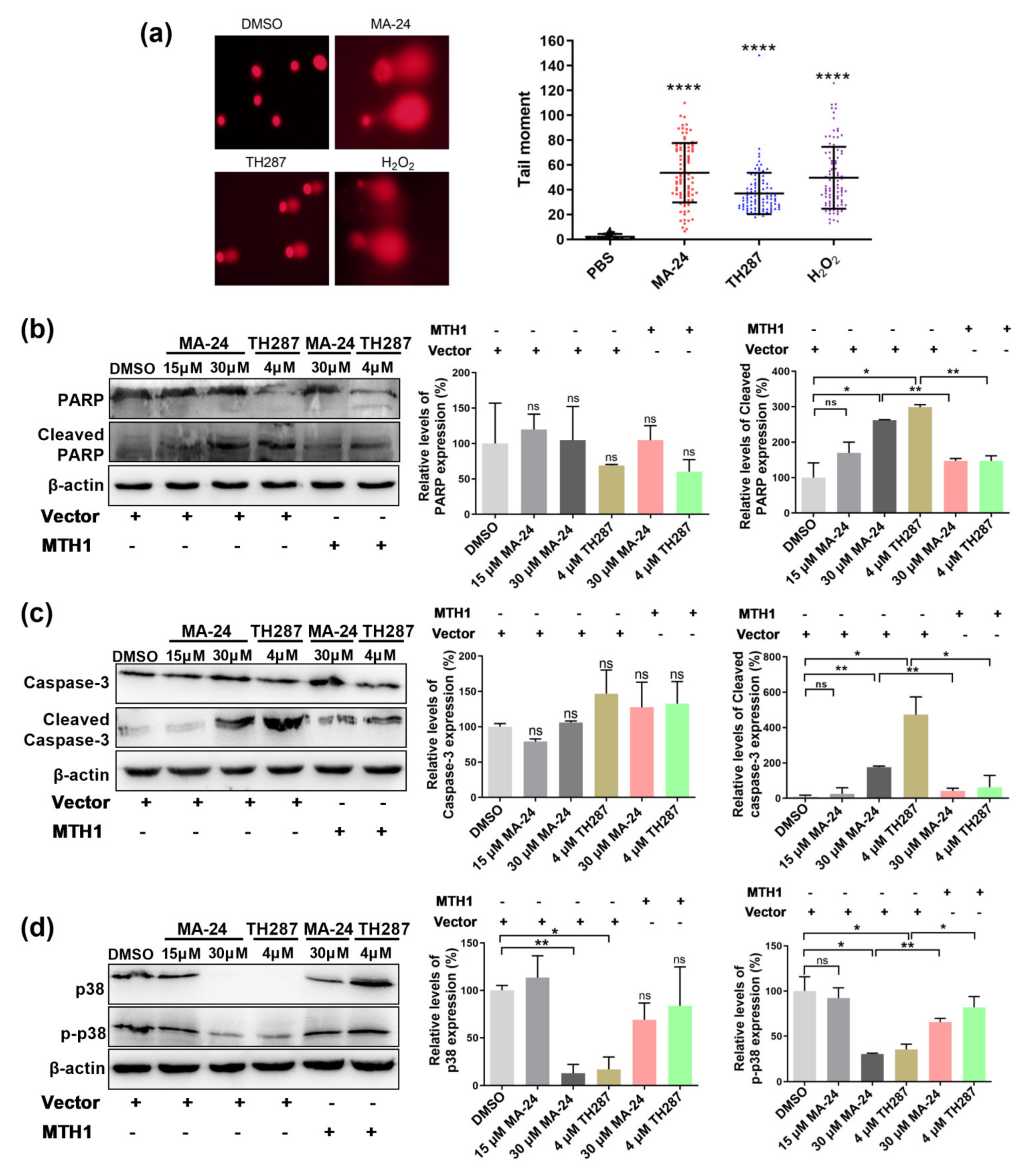
2.4. MA−24 Causes Breast Cancer Cell Death Referring to Multiple Cellular Signaling Pathways
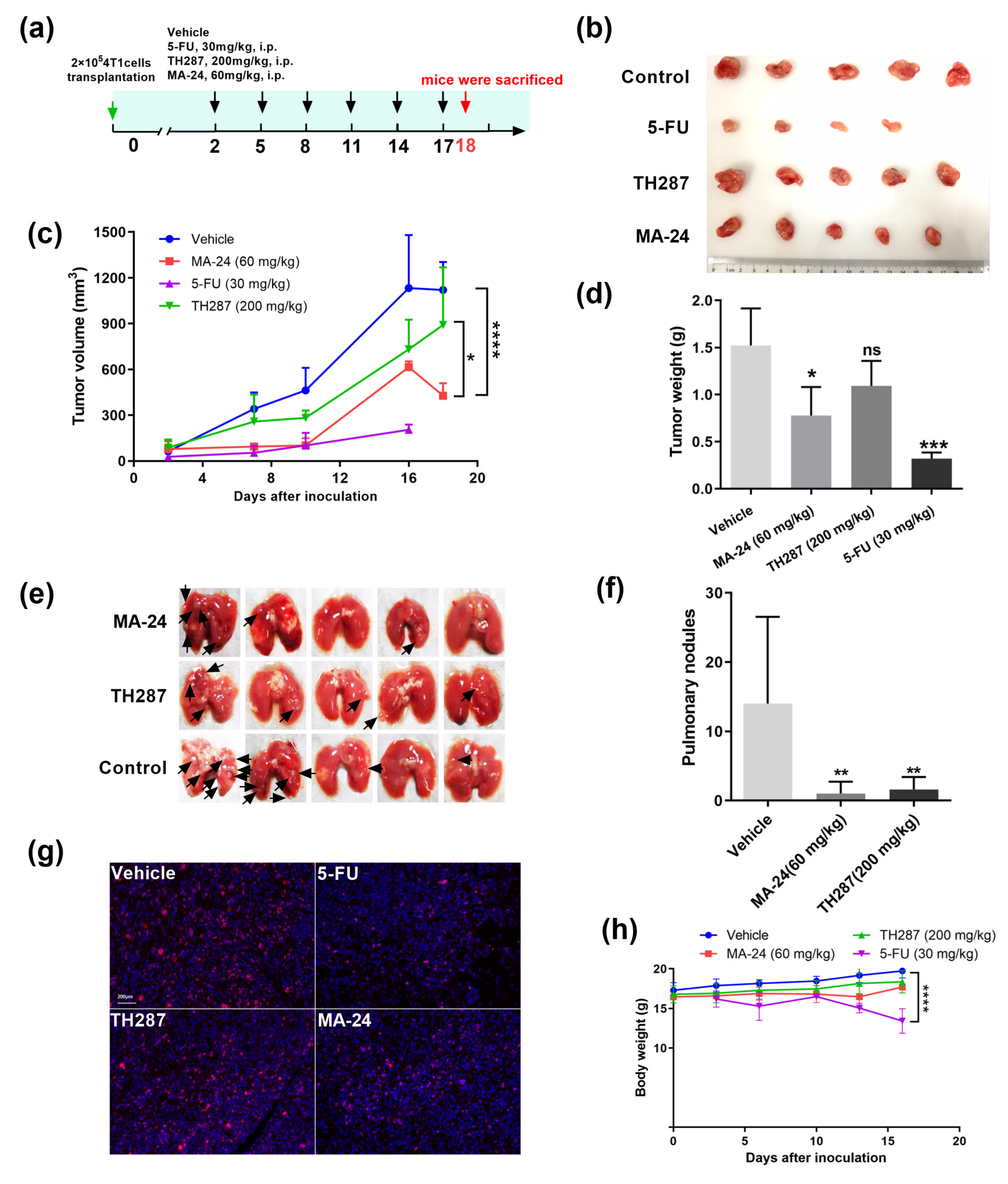
2.5. MA−24 Inhibited Tumor Growth and Lung Metastasis In Vivo
3. Discussion
4. Materials and Methods
4.1. Compounds
4.2. IC50 Determination
4.3. Cell Lines and Cell Culture
4.4. Cell Viability Assays
4.5. Annexin V/PI Apoptosis Assay
4.6. Comet Assay
4.7. Western Blot Analysis
4.8. Target Engagement Assay
4.9. RNA Interference
4.10. MTH1 Overexpression Studies
4.11. Animals and In Vivo Efficacy Studies
4.12. Molecular Docking Method
4.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Rivera, E.; Gomez, H. Chemotherapy resistance in metastatic breast cancer: The evolving role of ixabepilone. Breast Cancer Res. 2010, 12 (Suppl. S2), S2. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Kong, X.; Xuan, L.; Wang, Z.; Huang, Y. Prolactin and endocrine therapy resistance in breast cancer: The next potential hope for breast cancer treatment. J. Cell Mol. Med. 2021, 25, 10327–10348. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y.; Jin, L.; Yan, X.G.; Sherwin, S.; Farrelly, M.; Zhang, Y.Y.; Liu, F.; Wang, C.Y.; Guo, S.T.; Yari, H.; et al. Reactive Oxygen Species Dictate the Apoptotic Response of Melanoma Cells to TH588. J. Investig. Dermatol. 2016, 136, 2277–2286. [Google Scholar] [CrossRef] [PubMed]
- Cheung, E.C.; Vousden, K.H. The role of ROS in tumour development and progression. Nat. Rev. Cancer 2022, 22, 280–297. [Google Scholar] [CrossRef] [PubMed]
- Funahashi, S.; Okazaki, Y.; Akatsuka, S.; Takahashi, T.; Sakumi, K.; Nakabeppu, Y.; Toyokuni, S. Mth1 deficiency provides longer survival upon intraperitoneal crocidolite injection in female mice. Free Radic. Res. 2020, 54, 195–205. [Google Scholar] [CrossRef]
- Helleday, T.; Rudd, S.G. Targeting the DNA damage response and repair in cancer through nucleotide metabolism. Mol. Oncol. 2022, 16, 3792–3810. [Google Scholar] [CrossRef]
- Gad, H.; Koolmeister, T.; Jemth, A.S.; Eshtad, S.; Jacques, S.A.; Ström, C.E.; Svensson, L.M.; Schultz, N.; Lundbäck, T.; Einarsdottir, B.O.; et al. MTH1 inhibition eradicates cancer by preventing sanitation of the dNTP pool. Nature 2014, 508, 215–221. [Google Scholar] [CrossRef]
- Scaletti, E.R.; Vallin, K.S.; Bräutigam, L.; Sarno, A.; Berglund, U.W.; Helleday, T.; Stenmark, P.; Jemth, A.S. MutT homologue 1 (MTH1) removes N6-methyl-dATP from the dNTP pool. J. Biol. Chem. 2020, 295, 4761–4772. [Google Scholar] [CrossRef]
- Ahmed, W.; Lingner, J. PRDX1 and MTH1 cooperate to prevent ROS-mediated inhibition of telomerase. Genes Dev. 2018, 32, 658–669. [Google Scholar] [CrossRef]
- Jemth, A.S.; Gustafsson, R.; Bräutigam, L.; Henriksson, L.; Vallin, K.S.; Sarno, A.; Almlöf, I.; Homan, E.; Rasti, A.; Berglund, U.W.; et al. MutT homologue 1 (MTH1) catalyzes the hydrolysis of mutagenic O6-methyl-dGTP. Nucleic Acids Res. 2018, 46, 10888–10904. [Google Scholar] [CrossRef] [PubMed]
- Li, D.N.; Yang, C.C.; Li, J.; Yang, Q.-G.O.; Zeng, L.-T.; Fan, G.-Q.; Liu, T.-H.; Tian, X.-Y.; Wang, J.-J.; Zhang, H.; et al. The high expression of MTH1 and NUDT5 promotes tumor metastasis and indicates a poor prognosis in patients with non-small-cell lung cancer. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118895. [Google Scholar] [CrossRef] [PubMed]
- Kumagae, Y.; Hirahashi, M.; Takizawa, K.; Yamamoto, H.; Gushima, M.; Esaki, M.; Matsumoto, T.; Nakamura, M.; Kitazono, T.; Oda, Y. Overexpression of MTH1 and OGG1 proteins in ulcerative colitis-associated carcinogenesis. Oncol. Lett. 2018, 16, 1765–1776. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Song, W.; Zhou, Y.; Mao, F.; Lin, Y.; Guan, J.; Sun, Q. Expression and function of MutT homolog 1 in distinct subtypes of breast cancer. Oncol. Lett. 2017, 13, 2161–2168. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Gui, X.; Chu, X.; Sun, Y.; Zhang, S.; Tong, H.; Ju, W.; Li, Y.; Sun, Z.; Xu, M.; et al. MTH1 protects platelet mitochondria from oxidative damage and regulates platelet function and thrombosis. Nat. Commun. 2023, 14, 4829. [Google Scholar] [CrossRef] [PubMed]
- Oka, S.; Leon, J.; Sakumi, K.; Abolhassani, N.; Sheng, Z.; Tsuchimoto, D.; LaFerla, M.F.; Nakabeppu, Y. MTH1 and OGG1 maintain a low level of 8-oxoguanine in Alzheimer’s brain, and prevent the progression of Alzheimer’s pathogenesis. Sci. Rep. 2021, 11, 5819. [Google Scholar] [CrossRef]
- Das, I.; Tuominen, R.; Helleday, T.; Hansson, J.; Warpman Berglund, U.; Egyházi Brage, S. Coexpression of MTH1 and PMS2 Is Associated with Advanced Disease and Disease Progression after Therapy in Melanoma. J. Investig. Dermatol. 2022, 142, 736–740.e736. [Google Scholar] [CrossRef] [PubMed]
- McPherson, L.A.; Troccoli, C.I.; Ji, D.; Bowles, A.E.; Gardiner, M.L.; Mohsen, M.G.; Nagathihalli, N.S.; Nguyen, D.M.; Robbins, D.J.; Merchant, N.B.; et al. Increased MTH1-specific 8-oxodGTPase activity is a hallmark of cancer in colon, lung and pancreatic tissue. DNA Repair 2019, 83, 102644. [Google Scholar] [CrossRef]
- Moukengue, B.; Brown, H.K.; Charrier, C.; Battaglia, S.; Baud’Huin, M.; Quillard, T.; Pham, T.M.; Pateras, I.S.; Gorgoulis, V.G.; Helleday, T.; et al. TH1579, MTH1 inhibitor, delays tumour growth and inhibits metastases development in osteosarcoma model. EBioMedicine 2020, 53, 102704. [Google Scholar] [CrossRef]
- Das, I.; Gad, H.; Bräutigam, L.; Pudelko, L.; Tuominen, R.; Höiom, V.; Almlöf, J.; Rajagopal, V.; Hansson, J.; Helleday, T.; et al. AXL and CAV-1 play a role for MTH1 inhibitor TH1579 sensitivity in cutaneous malignant melanoma. Cell Death Differ. 2020, 27, 2081–2098. [Google Scholar] [CrossRef]
- Huber, K.V.; Salah, E.; Radic, B.; Gridling, M.; Elkins, J.M.; Stukalov, A.; Jemth, A.-S.; Göktürk, C.; Sanjiv, K.; Strömberg, K.; et al. Stereospecific targeting of MTH1 by (S)-crizotinib as an anticancer strategy. Nature 2014, 508, 222–227. [Google Scholar] [CrossRef]
- Imtiyaz, K.; Rahmani, A.H.; Alsahli, M.A.; Almatroodi, S.A.; Alam Rizvi, M.M. Fisetin induces apoptosis in human skin cancer cells through downregulating MTH1. J. Biomol. Struct. Dyn. 2022, 41, 7339–7353. [Google Scholar] [CrossRef]
- Wahi, D.; Soni, D.; Grover, A. A Double-Edged Sword: The Anti-Cancer Effects of Emodin by Inhibiting the Redox-Protective Protein MTH1 and Augmenting ROS in NSCLC. J. Cancer 2021, 12, 652–681. [Google Scholar] [CrossRef]
- Shi, H.; Ishikawa, R.; Heh, C.H.; Sasaki, S.; Taniguchi, Y. Development of MTH1-Binding Nucleotide Analogs Based on 7,8-Dihalogenated 7-Deaza-dG Derivatives. Int. J. Mol. Sci. 2021, 22, 1274. [Google Scholar] [CrossRef]
- Kettle, J.G.; Alwan, H.; Bista, M.; Breed, J.; Davies, N.L.; Eckersley, K.; Fillery, S.; Foote, K.M.; Goodwin, L.; Jones, D.R.; et al. Potent and Selective Inhibitors of MTH1 Probe Its Role in Cancer Cell Survival. J. Med. Chem. 2016, 59, 2346–2361. [Google Scholar] [CrossRef]
- Coskun, E.; Singh, N.; Scanlan, L.D.; Jaruga, P.; Doak, S.H.; Dizdaroglu, M.; Nelson, B.C. Inhibition of human APE1 and MTH1 DNA repair proteins by dextran-coated γ-Fe2O3 ultrasmall superparamagnetic iron oxide nanoparticles. Nonomedicine 2022, 17, 2011–2021. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T. Cancer phenotypic lethality, exemplified by the non-essential MTH1 enzyme being required for cancer survival. Ann. Oncol. 2014, 25, 1253–1255. [Google Scholar] [CrossRef] [PubMed]
- Magkouta, S.F.; Vaitsi, P.C.; Iliopoulou, M.P.; Pappas, A.G.; Kosti, C.N.; Psarra, K.; Kalomenidis, I.T. MTH1 Inhibition Alleviates Immune Suppression and Enhances the Efficacy of Anti-PD-L1 Immunotherapy in Experimental Mesothelioma. Cancers 2023, 15, 4962. [Google Scholar] [CrossRef] [PubMed]
- Pompsch, M.; Vogel, J.; Classen, F.; Kranz, P.; Iliakis, G.; Riffkin, H.; Brockmeier, U.; Metzen, E. The presumed MTH1-inhibitor TH588 sensitizes colorectal carcinoma cells to ionizing radiation in hypoxia. BMC Cancer 2018, 18, 1190. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.J.; Chen, Y.; Li, Z.H.; Peng, S.Y.; Sun, Y.; Zhang, X.Z. Augment of Oxidative Damage with Enhanced Photodynamic Process and MTH1 Inhibition for Tumor Therapy. Nano Lett. 2019, 19, 5568–5576. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, L.; Huang, Y.; Liu, B.; Chi, H.; Shi, L.; Zhang, W.; Li, G.; Niu, Y.; Zhu, X. Synergistic therapy of chemotherapeutic drugs and MTH1 inhibitors using a pH-sensitive polymeric delivery system for oral squamous cell carcinoma. Biomater. Sci. 2017, 5, 2068–2078. [Google Scholar] [CrossRef]
- Saleh, A.; Gökturk, C.; Warpman-Berglund, U.; Helleday, T.; Granelli, I. Development and validation of method for TH588 and TH287, potent MTH1 inhibitors and new anti-cancer agents, for pharmacokinetic studies in mice plasma. J. Pharm. Biomed. Anal. 2015, 104, 1–11. [Google Scholar] [CrossRef]
- Gul, N.; Karlsson, J.; Tängemo, C.; Linsefors, S.; Tuyizere, S.; Perkins, R.; Ala, C.; Zou, Z.; Larsson, E.; Bergö, M.O.; et al. The MTH1 inhibitor TH588 is a microtubule-modulating agent that eliminates cancer cells by activating the mitotic surveillance pathway. Sci. Rep. 2019, 9, 14667. [Google Scholar] [CrossRef] [PubMed]
- Berglund, U.W.; Sanjiv, K.; Gad, H.; Kalderen, C.; Koolmeister, T.; Pham, T.; Gokturk, C.; Jafari, R.; Maddalo, G.; Seashore-Ludlow, B.; et al. Validation and development of MTH1 inhibitors for treatment of cancer. Ann. Oncol. 2016, 27, 2275–2283. [Google Scholar] [CrossRef]
- Zhang, L.; Misiara, L.; Samaranayake, G.J.; Sharma, N.; Nguyen, D.M.; Tahara, Y.-K.; Kool, E.T.; Rai, P. OGG1 co-inhibition antagonizes the tumor-inhibitory effects of targeting MTH1. Redox Biol. 2021, 40, 101848. [Google Scholar] [CrossRef] [PubMed]
- Sanjiv, K.; Calderón-Montaño, J.M.; Pham, T.M.; Erkers, T.; Tsuber, V.; Almlöf, I.; Höglund, A.; Heshmati, Y.; Seashore-Ludlow, B.; Danda, A.N.; et al. MTH1 Inhibitor TH1579 Induces Oxidative DNA Damage and Mitotic Arrest in Acute Myeloid Leukemia. Cancer Res. 2021, 81, 5733–5744. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, Z.-H.; Dang, Y.-M.; Li, D.-N.; Liu, Z.; Dai, D.-P.; Cai, J.-P. MTH1 suppression enhances the stemness of MCF7 through upregulation of STAT3. Free Radic. Biol. Med. 2022, 188, 447–458. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds (Concentration: 200 μM) | Inhibition (%) (Mean ± SD) |
|---|---|
| MA−19 | 37.8 ± 8.8 |
| MA−20 | 44.2 ± 8.4 |
| MA−21 | 28.1 ± 1.5 |
| MA−22 | 30.3 ± 4.1 |
| MA−23 | 11.8 ± 4.5 |
| MA−24 | 99.6 ± 0.4 |
| MA−25 | 100.0 ± 0.1 |
| MB−16 | 52.1 ± 2.9 |
| MB−17 | 27.3 ± 4.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, N.; Ma, J.; Hu, Y.; Di, R.; Wang, L.; Zhang, X.; Lai, Y.; Liu, Y. Targeting MutT Homolog 1 (MTH1) for Breast Cancer Suppression by Using a Novel MTH1 Inhibitor MA−24 with Tumor-Selective Toxicity. Pharmaceuticals 2024, 17, 291. https://doi.org/10.3390/ph17030291
Kang N, Ma J, Hu Y, Di R, Wang L, Zhang X, Lai Y, Liu Y. Targeting MutT Homolog 1 (MTH1) for Breast Cancer Suppression by Using a Novel MTH1 Inhibitor MA−24 with Tumor-Selective Toxicity. Pharmaceuticals. 2024; 17(3):291. https://doi.org/10.3390/ph17030291
Chicago/Turabian StyleKang, Nannan, Jun Ma, Yuling Hu, Rongrong Di, Lei Wang, Xuanling Zhang, Yisheng Lai, and Yu Liu. 2024. "Targeting MutT Homolog 1 (MTH1) for Breast Cancer Suppression by Using a Novel MTH1 Inhibitor MA−24 with Tumor-Selective Toxicity" Pharmaceuticals 17, no. 3: 291. https://doi.org/10.3390/ph17030291
APA StyleKang, N., Ma, J., Hu, Y., Di, R., Wang, L., Zhang, X., Lai, Y., & Liu, Y. (2024). Targeting MutT Homolog 1 (MTH1) for Breast Cancer Suppression by Using a Novel MTH1 Inhibitor MA−24 with Tumor-Selective Toxicity. Pharmaceuticals, 17(3), 291. https://doi.org/10.3390/ph17030291