1. Introduction
Breast cancer stands as the most prevalent form of malignancy among women worldwide and accounts for 24.5% of all malignancies in the female population. Despite better diagnostic skills, it causes around 6.9% of cancer-related deaths. Until recently, lung cancer was considered the most commonly diagnosed type of cancer; however, in 2020, around 2.26 million new cases of breast cancer were recorded, surpassing the number of new lung cancer cases and accounting for 11.7% of all reported cancer cases [
1]. As it is a multifactorial disease, various factors such as genetic factors, population structure, environment, and lifestyle contribute to its occurrence [
2,
3]. The worldwide prevalence of breast cancer has risen due to changes in risk factors [
4].
If it is diagnosed at an early stage, the main goal of breast cancer treatment is to cure and prevent the risk of recurrence; thus, therapy involves a radical approach [
5]. In the metastatic form, on the other hand, the aim is to achieve control of the disease and as long as possible survival of the patient while maintaining quality of life [
6]. Several local and systemic methods are used for treatment. The most commonly used methods are surgical removal, radiation, chemo-, and targeted therapy. Targeted therapy encompasses endocrine treatment for breast cancer with positive hormone status and anti-HER2 therapy for HER2+ breast cancer [
7]. Innovative approaches showing promising results include immunotherapy, conjugated antibodies, and targeting metabolic pathways [
8].
Predominant histological forms of breast cancer include apocrine, medullary, cribriform, metaplastic, classic lobular, neuroendocrine, mucinous, pleomorphic lobular, and tubular carcinomas. In addition to these types, the most frequently diagnosed cases involve the nonspecific type of invasive ductal carcinoma. Presently, four molecular subgroups, luminal A, luminal B, HER2+, and triple-negative, are widely recognized and firmly established in clinical practice [
8,
9]. The term triple-negative breast cancer (TNBC) describes a subtype of breast cancer in which expression of the three primary breast tumor markers, including estrogen receptor (ER), progesterone receptor (PR), and HER2 protein, is absent. This form of breast cancer constitutes approximately 15 to 20% of all breast cancer cases and approximately 40% of high-grade tumors [
10]. TNBC is of particular interest to researchers as it poses a therapeutic challenge mainly due to its poor response to therapy and highly invasive nature [
11].
Apart from the initial diversity of neoplastic transformation, cancer also includes many other cell types in addition to tumor cells, such as adipocytes, fibroblasts, endothelial cells, and immune cells (macrophages and lymphocytes), as well as the extracellular matrix’s non-cellular constituents (fibronectin, collagen, hyaluronan, laminin, etc.). All of them create the network of the tumor microenvironment (TME) [
12]. Tumor cells and other components of the TME are in constant, reciprocal communication. Normally, the immune system acts as a defense mechanism against foreign entities, including tumors, aiming to prevent their growth and metastasis. However, many reports have shown that tumor-infiltrating immune cells can also be involved in tumor progression in many ways. Apart from well-known tumor immune evasion strategies, the TME architecture and extracellular matrix construction enables recruitment of tumor suppressors such as MDSCs and Tregs that orchestrate the establishment of local immunosuppression [
13,
14,
15]. Considering the tumor tissue as a never-healing wound, control points of immune response regulation support the entry of effector cells into the exhaustion state [
16]. Therefore, cancer therapy should not only aim to destroy tumor cells but also trigger the flow of events leading to refreshed antitumor immune response.
Despite many treatment options, recurrence and accompanying problems persist among patients, primarily due to many side effects, general toxicity, and aggressive behavior of tumors. Lately, the use of some types of phytotherapy in breast cancer patients has increased significantly [
17]. Medicinal plants are becoming more and more popular as they are recognized as efficient and affordable sources of new chemotherapeutic agents [
18]. Following the rule that nature is best complemented and balanced by nature itself, the potential of naturally occurring compounds to interact directly or indirectly with host cells opens up numerous opportunities to influence the tumor ecosystem. When it comes to the complex mixture of compounds in the organic extract, two types of interactions and the crosstalk between them may be crucial—a direct effect on tumor cells and the potential to affect the phenotype and function of immune cells. The immunogenic death of tumor cells induced by various plant polyphenols leads to the release of danger signals and the subsequent refreshment of the antitumor immune response through enhanced potential of antigen-presenting dendritic cells and effector T-cell responses [
16]. Finally, natural products can disrupt the inhibitory signals responsible for creating an immunosuppressive environment in tumors and refresh T-cell effector performance.
Among many plants that exert a variety of biological activities is
Alchemilla vulgaris, widely known as lady’s mantle, as many ethnomedicinal reports mention its well-known effects against disorders of the female reproductive system (pruritus vulvae, dysmenorrhea, heavy menstrual flow, menopausal complaints). In addition, its activity against reproductive system-related diseases (endometriosis, cysts, fibroids, and infertility) has been described [
19,
20,
21,
22,
23]. The anticancer activity of this plant has been reported against tumors of the female reproductive organs (A2780 and HeLa), as well as against human prostate cancer (PC-3), human breast cancer (MCF-7), human melanoma A375, human lung carcinoma A549, and human colorectal cancer (Caco2, HCT116) cell lines in vitro [
24,
25,
26]. Our previous research showed strong anticancer properties of ethanolic extract of
A. vulgaris against non-invasive and highly invasive murine melanoma [
27]. Based on ethnomedicinal data on female disorders and illnesses, the objective of this research was to evaluate the direct activity of
A. vulgaris extract against murine triple negative breast cancer cells (4T1) in an in vitro and in vivo setting, along with its effect on the tumor microenvironment in an ex vivo setting. The data obtained showed that in addition to the direct antitumor effect of the
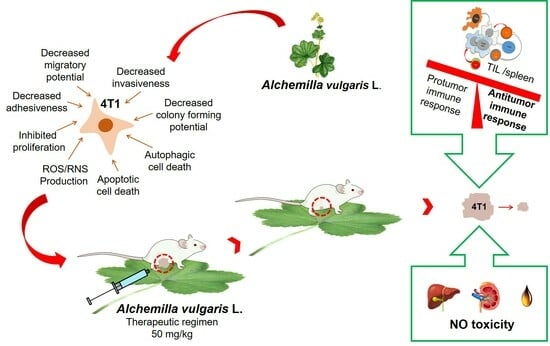
A. vulgaris extract realized through inhibition of cell division, apoptotic cell death and diminished metastatic features, tested extract exerted a strong impact on the systemic and local immune response. Considering all of the above points, we conclude that
A. vulgaris extract reduces tumor growth through a direct effect on the viability of tumor cells and reestablishment of an effective antitumor immune response.
3. Discussion
Ethnobotanical data refers to the traditional knowledge and practices of various cultures regarding the use of plants and plant-based remedies for medicinal purposes. Throughout history, different societies have relied on inherited knowledge passed down from generation to generation for treatment of various ailments, including cancer. Breast cancer, being one of the foremost prevalent cancer types affecting women, is also the subject of research in the world of ethnobotanical medicine. Ethnobotanical data can provide valuable insights and serve as a starting point for further scientific research and the discovery of novel compounds that may have therapeutic potential and/or improve current protocols, mainly through the sensitization of tumor cells to radio- or chemotherapy, or refreshment of immune cell activities against the malignant phenotype [
28,
29,
30,
31]. Importantly, several plants are traditionally used in various cultures to treat breast cancer symptoms and improve overall well-being [
18,
29,
32,
33]. Some of them are sources of drugs that are successfully used in modern chemotherapy for breast cancer treatment, such as paclitaxel [
34,
35]. In general, rigorous scientific research has been necessary to confirm the safety and effectiveness of these plant-derived remedies against breast carcinoma.
The literature data report well-known effects of
Alchemilla vulgaris L. against female reproductive system disorders and related diseases, as well as its use in the treatment of diabetes, multiple sclerosis, anemia, skin rashes, hernias, ulcers, wounds, and inflammations [
19,
20,
21,
22,
36]. Our previous results showed the anticancer potential of
A. vulgaris extract against a panel of human cancer cell lines in vitro as well as against murine melanoma of different aggressiveness in vivo [
26,
27].
In the current research, the impact of
A. vulgaris ethanolic extract was investigated on a panel of breast cancer cell lines, starting from the human ER+ breast cancer cell line (MDA-MB-361) and continuing with TNBC cell lines of human (MDA-MB-468 and MDA-MB-231) and murine origin (4T1). In concordance with previously reported data,
A. vulgaris extract treatment dose-dependently decreased the cell viability of all tested cells observed with both MTT and SRB assays [
26,
27]. Selectivity toward the malignant phenotype has already been described with no effect against primary cells extracted from the peritoneal cavity of C57BL/6 mice [
26]. Concordantly, Ibrahim et al. reported that methanolic extract of
A. vulgaris root showed no influence on normal Vero cells [
25]. To allow transfer of the in vitro study to in vivo, the drug efficacy was further tested in detail on the highly aggressive breast cancer cell line 4T1, enabling the subsequent assessment of the extract’s antitumor potential in a syngeneic animal model. This approach covers multiple aspects of the extract’s influences on solid tumor formation and tumor microenvironmental features [
37]. To reach this goal, it was essential to define the main routes of direct influence of the extract in cell culture. Flow cytometric analysis and fluorescence microscopy showed that
A. vulgaris extract induced apoptosis of 4T1 cells accompanied by moderate caspase activation, which was in line with previous data showing a similar mode of action on human lung cancer cells (A549) and highly invasive murine melanoma cells (B16F10) [
26,
27]. The literature data report many natural compounds isolated from plants with apoptosis-inducing potential [
38,
39,
40]. The significant autophagy observed in 4T1 cells exposed to
A. vulgaris extract was abolished with concomitant treatment with the autophagy inhibitor chloroquine, resulting in restored cell viability, verifying once more the contribution of this process to the tumor-cell-destructive activity of
A. vulgaris extract [
26,
27]. The strong oxidative burst provoked by the treatment may be responsible for the tumoricidal activities of the
A. vulgaris extract compounds’ mixture and their interplay.
The 4T1 cells are a well-known model considered to be one of the most formidable forms of breast cancer with strong potential to form distant metastases in various organs [
41,
42]. In line with our previous data,
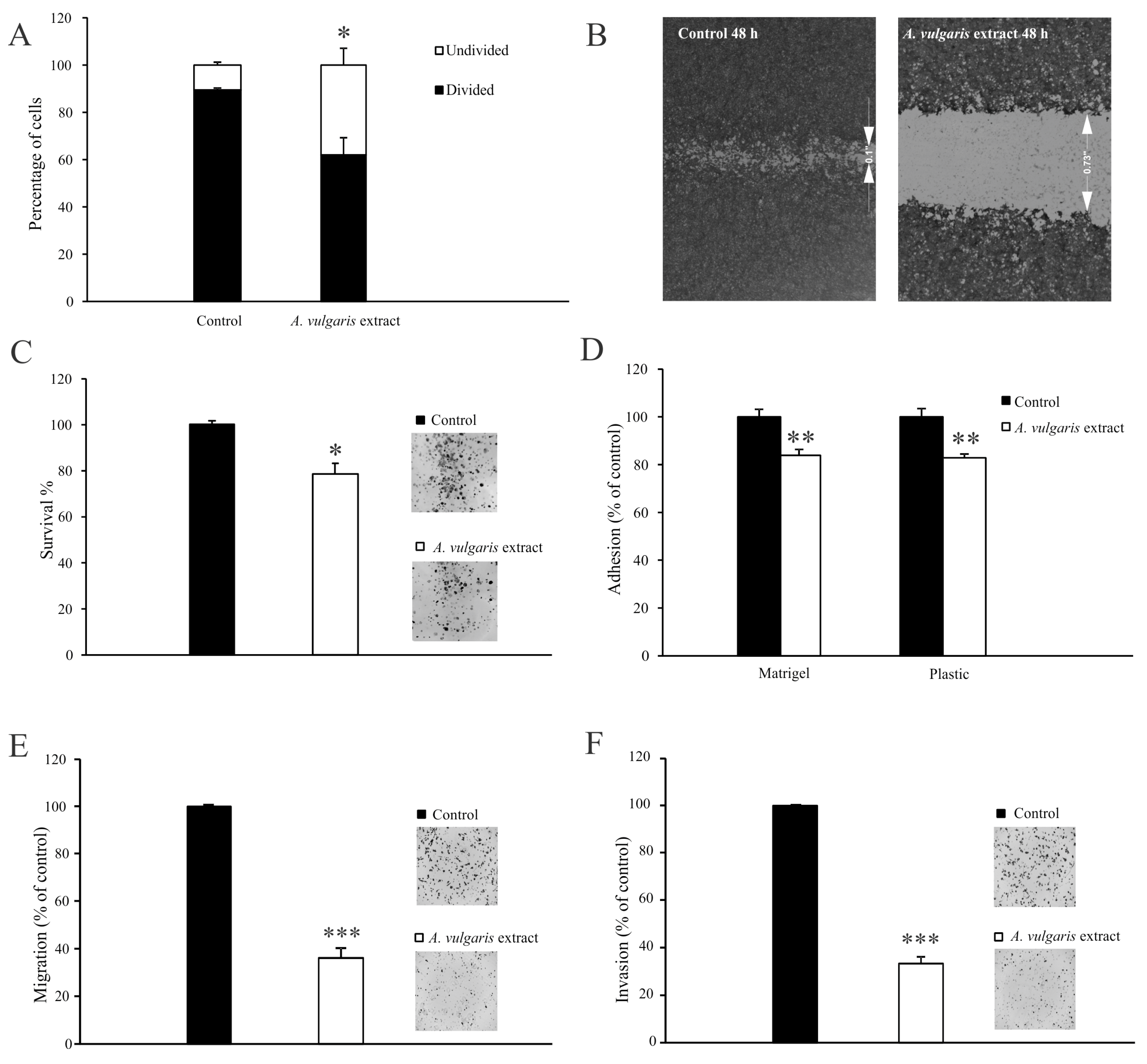
A. vulgaris extract treatment inhibited proliferation of 4T1 cells [
26,
27], and significantly reduced their ability to fill a gap or migrate through porous membranes when attracted by chemo-stimuli. In addition, the potential of 4T1 cells to pass the reconstructed basal membrane barrier was also considerably diminished, confirming that the treatment affected the invading capacity of cells, predicting decreased penetration of cells upon exposure to
A. vulgaris extract in surrounding tissues in vivo. Altogether, pretreatment with a subtoxic dose of
A. vulgaris extract, which did not affect the viability of 4T1 cells, markedly affected all features important for the metastatic process—adhesiveness, migration, invasion, and colony forming potential. Simultaneously, the relative expression of integrin β-1, FAK, vinculin, and α-SMA proteins was inhibited by the treatment. According to the literature, adhesion to the ECM is the key event in the initiation of the metastatic cascade [
43]. The proteins that connect the ECM to the intracellular cytoskeleton play an important role in adhesion, migration, and invasion. The literature supports the fact that the migratory potential of cells and the invasiveness of tumors depend on the ligation of integrin receptors and the transmission of a downstream signal that links the extracellular compartment to the intracellular cytoskeleton, thus enabling adequate response to dictate from the outside [
44,
45]. Proteins involved in this intimate contact are part of complex structures called focal adhesions, whose formation and separation are dynamically regulated during the process of cell migration. Inside the cell, the cytoskeleton represents a protein network that defines its morphology and behavior [
43,
46]. Overexpression of integrin β-1 is a poor prognostic factor for cancer patients [
47]. In contrast, negative regulation of integrin β-1 is a good sign and a desirable therapeutic response, as observed upon the exposure to
A. vulgaris extract, affecting every further step of the metastatic process. Focal adhesion kinase is the key regulator of survival, proliferation, migration, and invasion. This kinase transmits signals from focal adhesions to the cellular cytoskeletal machinery [
46]. It is well known that FAK overexpression is linked to an extensive range of human epithelial cancers. Furthermore, the level of FAK expression correlates with tumor invasive potential and poor prognosis [
48,
49]. Accordingly, in this study, continuous inhibition of FAK relative expression after
A. vulgaris treatment was detected. Vinculin is an important part of focal adhesion, and perturbations of vinculin–actin interaction impact cell motility, morphology, and adhesion [
50]. Its suppression upon exposure to the tested extract is tightly related to the diminished migratory and invasive properties of 4T1 cells. Finally,
A. vulgaris inhibits the expression of α-SMA, the protein associated with the aggressive phenotype and poor survival of patients with advanced forms of cancer, thus silencing the aggressive nature of 4T1 cells [
51,
52,
53].
The fact that a tumor is not a simple group of highly proliferating cells with an abnormal phenotype, but a multicellular structure which functions at an impressively organized level [
12,
54,
55], leads to the conclusion that any in vitro data are of limited value until they are confirmed by adequate in vivo screening. The greatest advantage of the syngeneic model relates to preserved constituents and activities in the TME, and the whole-body support in combating disease, including systemic immunity and non-immune cell activities relevant to the therapeutic response [
37]. Thus, the antitumor action of
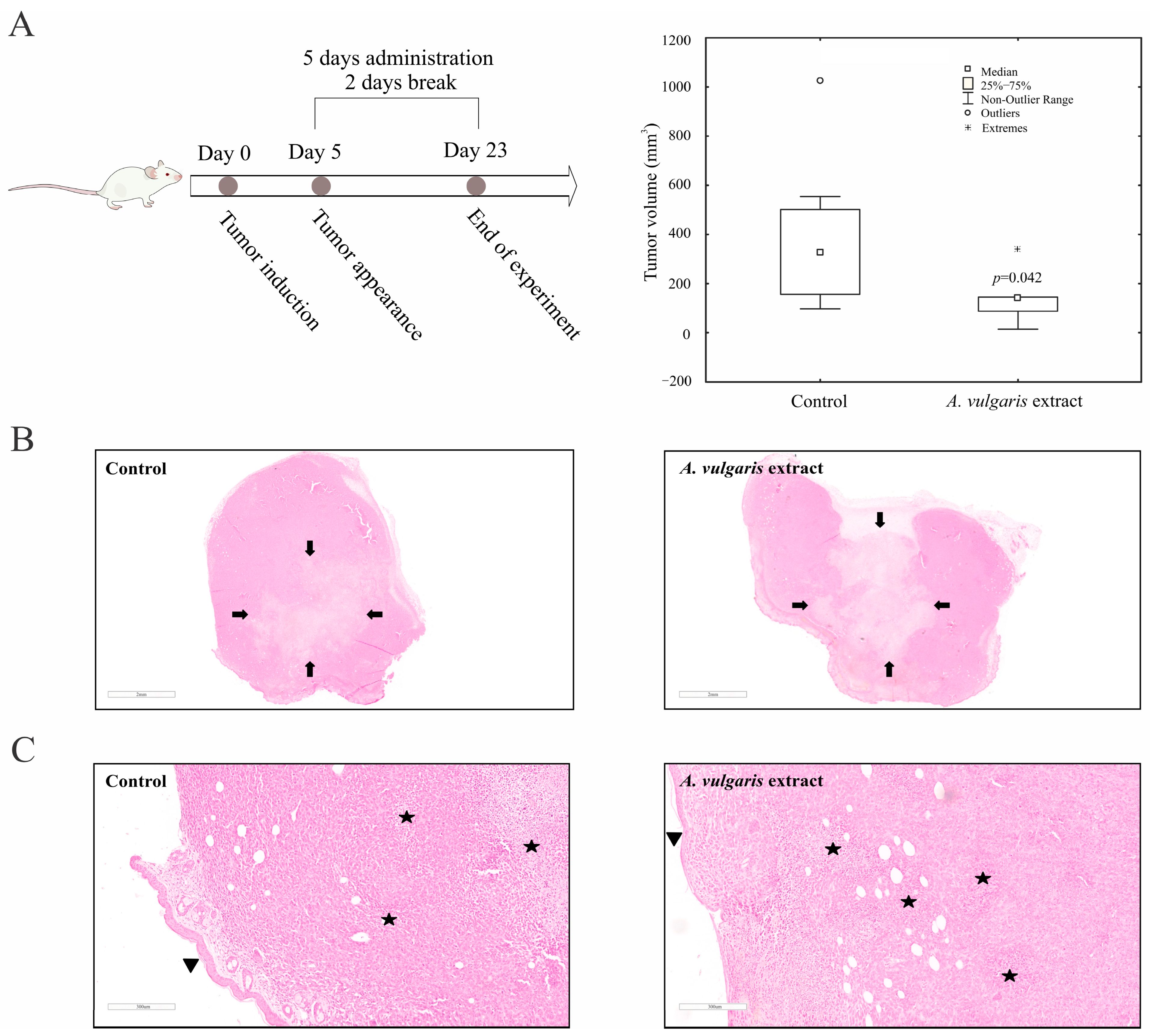
A. vulgaris extract was investigated in an orthotopic syngeneic mammary carcinoma model, as the most appropriate for simulating stage IV breast cancer behavior. The applied regime of the
A. vulgaris extract treatment led to a significant decrease in tumor growth. Similarly, the same effect was noted in non-invasive and highly invasive melanoma models in vivo [
27]. The TME, including the innate and acquired immune response, significantly contributed to the efficacy of the applied antitumor therapy [
56]. The assessment of the
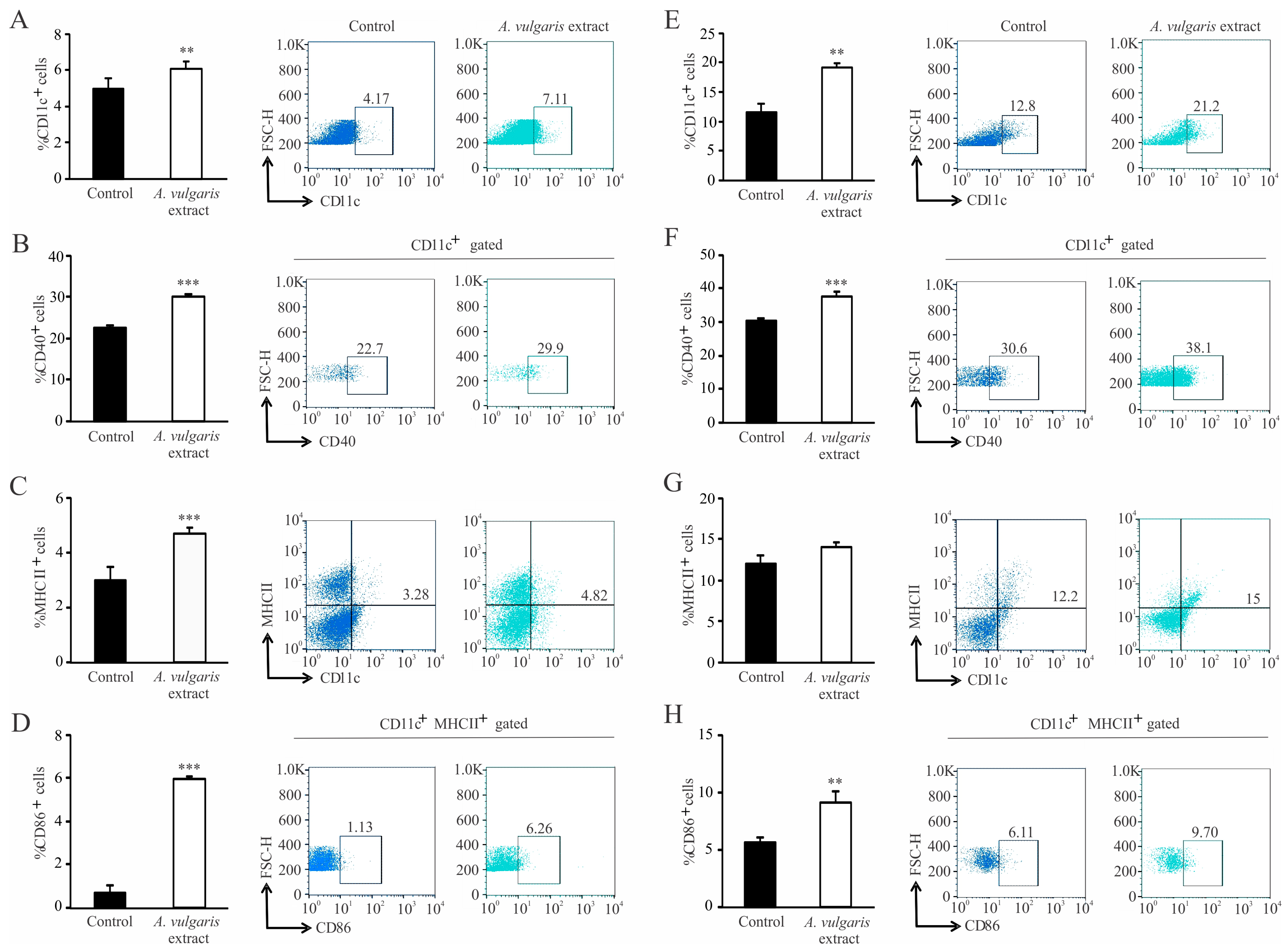
A. vulgaris extract’s influence on systemic and local immune cell activities in the TME of the 4T1 breast cancer model revealed multiple and well-orchestrated changes, affecting tumor progression and spread. Dendritic cells are professional antigen-presenting cells that trigger and shape the immune response [
57,
58]. Upon recognition of antigen, they interact with effector cells, such as NK, NKT cells, and T lymphocytes, and tend to navigate the latter course of overall immune response [
59,
60]. The importance of dendritic cells is also well illustrated in antitumor immune response [
61], as it is known that dendritic cells are the first cells to encounter tumor antigens, migrate towards lymph nodes, and provide activating signals for effector cells (“cancer-immunity cycle”) [
62]. Of course, various malignancies have developed defense mechanisms to escape recognition by dendritic cells, either by expressing inhibitory signals or by metabolic impairment [
63,
64]. The application of
A. vulgaris ethanolic extract significantly increases the percentage of dendritic cells and potently stimulates their maturation, which is illustrated by a remarkable elevation of CD40, CD86, and MHCII molecules. Identical changes in dendritic cell phenotype were observed in the spleen and in the tumor microenvironment. These results might imply that
A. vulgaris extract could be equally important for both the initiation and maintenance of antitumor immune response to breast carcinoma. As is already known, 4T1 breast cancer cells are considered to be weakly to moderately immunogenic, and as such, activation of cytotoxic lymphocytes might be hindered [
65]. Lately, the low number of dendritic cells and their immature forms in tumor tissue has been thought to be another hallmark of a weakly immunogenic tumor [
57]. The results presented in this study show that administration of
A. vulgaris extract might raise the immunogenicity of a tumor, making the antitumor immune response more efficient. There is growing evidence that naturally occurring compounds are potent inducers of immunogenic cell death, triggering the release of danger-associated molecules, and therefore serve as immunoadjuvants [
16]. This cascade of events amplifies the two branches of the immune response, innate and adaptive. The strong immunogenic outcome of treatment with
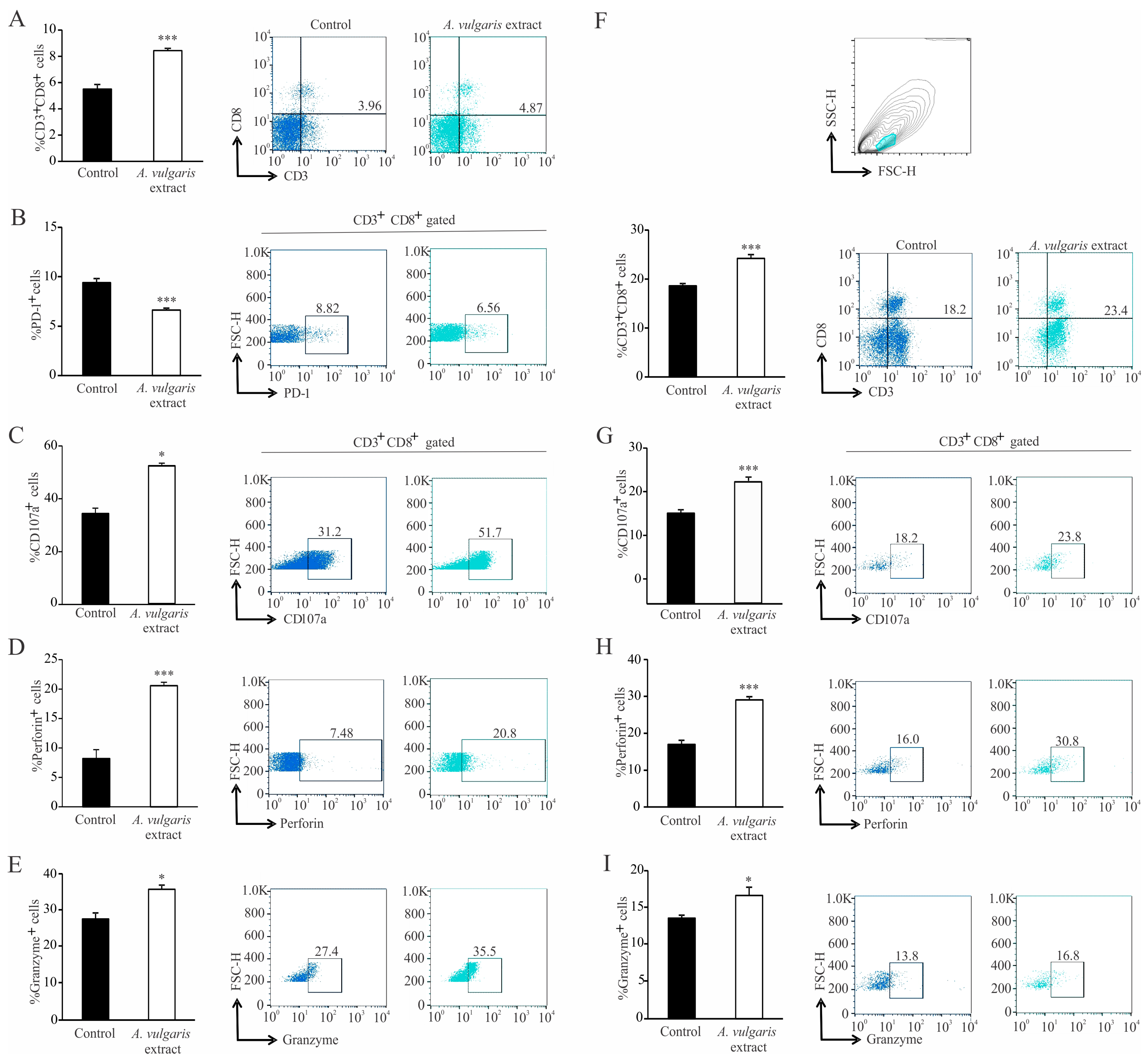
A. vulgaris extract can be related to the complexity of its content and the pleotropic effect of its bioactive ingredients. When it comes to cytotoxic T lymphocytes, the administration of
A. vulgaris extract increases the percentage of CD3
+CD8
+ cells in the spleen and tumor tissue. In addition to increased frequency of cytotoxic T lymphocytes in the spleen and tumor microenvironment, the phenotype of these cells is also altered to a more cytotoxic one, as illustrated by significant increment of the degranulation marker CD107a followed by significant increase in perforin and granzyme production. In addition, the expression of the inhibitory marker, PD-1, is significantly lower in splenic cytotoxic T lymphocytes, which implies a positive effect of the
A. vulgaris extract on the activation of splenic cytotoxic T lymphocytes. Histopathological analysis showed enhanced tumor necrosis in the primary tumor of
A. vulgaris-treated mice. The existence of tumor necrosis may straighten an antitumor immune response [
66], as necrosis promotes the maturation of dendritic cells [
67,
68].
A. vulgaris extract may stimulate antitumor immunity in at least two ways, by directly activating dendritic and cytotoxic T cells, and indirectly via tumor necrosis facilitation of dendritic cells’ maturation, then further activating cytotoxic T lymphocytes.
4. Materials and Methods
4.1. Reagents and Cells
The reagents and the cells were supplied by the manufacturers listed below: Capricorn Scientific GmbH (Hessen, Germany)—fetal bovine serum (FBS) and culture medium RPMI-1640; Sigma (St. Louis, MO, USA)—ethylenediamine tetraacetic acid (EDTA), phosphate-buffered saline (PBS), Triton X-100, dimethyl sulfoxide (DMSO), Fluoromount-G, Trypsin, RNase, carboxyfluoresceindiacetate succinimidyl ester (CFSE), propidium iodide (PI), trisaminomethane hydrochloride (TRIS HCl), dithiothreitol (DTT), glycerol, phenylmethylsulfonyl fluoride (PMSF), sulforhodamine B (SRB), chloroquine, and acridine orange (AO); Biowest (Riverside, MO, USA)—crystal violet (CV); BD (Pharmingen, San Diego, CA, USA, SAD)—annexin V-FITC (AnnV); BD Bioscience (Bedford, MA, USA)—Matrigel®; Thermo Fisher Scientific (Waltham, MA, USA)—dihydrorhodamine 123 (DHR123); Fresenius Kabi AG Germany (Bad Homburg, Germany)—paclitaxel (PCT); Biological Industries (Cromwell, CT, USA)—the penicillin–streptomycin solution; Serva (Heidelberg, Germany)—paraformaldehyde (PFA) and N,N,NN-tetramethylethylenedi (TEMED); AppliChem (St. Louis, MO, USA)—bovine serum albumin (BSA) and 3-(4,5 dimethythiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT); AppliChem (Darmstadt, Germany)—Tween 20 and sodium dodecyl sulfate (SDS); R&D Systems (Minneapolis, MN, USA)—ApoStat; Lach-Ner (Neratovice, Czechia)—sodium hydroxide (NaOH). American Type Culture Collection (ATCC, Manassas, VA, USA)—human breast adenocarcinoma (MDA-MB-231, MDA-MB-468, and MDA-MB-361).
Mouse breast cancer (4T1) cells were provided as a gift from Prof. Dr. Ludger Wessjohan from the Leibniz Institute of Plant Biochemistry, Halle (Saale), Germany. This cell line was primarily purchased from ATCC.
All cell lines used in this study were routinely maintained in HEPES-buffered RPMI-1640 medium. The medium was supplemented with 2 mM L-glutamine, 0.01% sodium pyruvate, penicillin, and streptomycin (100 units/mL and 100 μg/mL, respectively) and 10% heat-inactivated FBS. Cells were stored in the incubator (37 °C, 5% CO2). For viability determination in 96-well plates, cells were seeded in the following densities: 4T1, MDA-MB-361, and MDA-MB-468 cells (4 × 103 cells/well), and MDA-MB-231 cells (7 × 103 cells/well). For flow cytometry, 4T1 cells were seeded in 6-well plates at a density of 1 × 105 cells/well.
The detailed chemical composition of the ethanolic extract of
Alchemilla vulgaris L. used in this study and the extraction process was as previously reported [
26]. The plant material was collected from the species’ natural habitat—the moderately humid hilly-mountainous grassland in the south-eastern part of Serbia. The above-mentioned plant parts were collected in the flowering phase. Typical samples were taxonomically identified and stored as voucher specimens (No. RS-120718-1). The plant material was air-dried and extracted for two hours in hot ethanol. The chemical composition of the solid, vacuum-evaporated extracts was determined by UHPLC-HRMS. A total of 45 compounds were identified, mainly belonging to the flavonol and flavone glycosides with a dominance of quercetin and kaempferol derivatives [
26]. The ethanolic stock solution of
A. vulgaris was dissolved in pure DMSO (100%) at a concentration of 200 mg/mL. The stock solution was prepared fresh before every usage and the final DMSO concentration in the working solutions did not exceed 0.1% (in vitro) or 4% (in vivo) at the highest concentration.
4.2. Animals
The mice used in this study (BALB/C strain, six to eight weeks old, female) were obtained from the Institute for Biological Research “Siniša Stanković”—National Institute of the Republic of Serbia, University of Belgrade. The animals were kept in the animal facility under standard laboratory conditions (free of specific pathogens, ad libitum food and water consumption). The handling of the mice and the protocols used in the in vivo study were aligned with the rules of European Community guidelines (EEC Directive of 1986; 86/609/EEC). Additionally, the local Institutional Animal Care and Use Committee (IACUC) authorized all study protocols. Finally, the experimental protocols were authorized and granted by the national licensing committee at the Department of the Animal Welfare, Veterinary Directorate, of the Ministry of Agriculture, Forestry and Water Management of the Republic of Serbia (permission No. 323-07-12008/2020-05).
4.3. Colorimetric Assays for Cellular Viability
Mitochondrial dehydrogenase activity was assessed through the reduction of MTT to formazan. All cells were first seeded overnight and afterward exposed to
A. vulgaris extract and PCT (as a positive control). After the incubation period (72 h), the cells were washed twice with 200 μL PBS and cultivated in the presence of MTT solution with a final concentration of 0.5 mg/mL and kept in the incubator (37 °C) until the solution turned from yellow to brown. Following this, the dye was removed and DMSO was added to dissolve the formed formazan. Using an automatic reader for microtiter plates (LKB 5060-006, LKB, Vienna, Austria), the absorbance was measured at 540/670 nm. Cell viability was then expressed as a percentage relative to the control (untreated cells) [
26]. All experiments were repeated three independent times.
For the SRB assay [
26], following treatment with
A. vulgaris extract and PCT (as a positive control) for 72 h, cells underwent PBS washing before fixation with 10% TCA for 2 h at 4 °C. Subsequently, cells were rinsed in distilled water and stained with a 0.4% SRB solution for 30 min at RT. After the incubation period, the dye was dissolved in 1% acetic acid, washed, and left to dry overnight. Finally, the dye was then dissolved in 10 mM tris buffer for 20 min. The absorbance was measured using an automatic reader for microtiter plates (LKB 5060-006, LKB, Vienna, Austria) at 540/670 nm. The cell viability was expressed as a percentage of control, non-treated cells, which was arbitrarily set to 100%. All experiments were repeated three independent times. Dose–response curves were constructed through non-linear regression analysis in GraphPad Prism 10.1.2 software (San Diego, CA, USA). IC
50 values, defined as 50% reduction of cell viability, were calculated using SigmaPlot 15 software and Microsoft Excel 2013 with the four-parametric logistic function.
Combined treatment with A. vulgaris extract and chloroquine (autophagy inhibitor) was carried out to evaluate the nature of the detected autophagy. The 4T1 cells were seeded overnight and treated with an IC50 concentration of A. vulgaris extract (45 μg/mL) and chloroquine (20 µM) simultaneously. After 72 h incubation, cell viability was evaluated using the SRB assay.
4.4. Detection of Apoptosis (Ann/PI Staining)
In order to detect apoptosis, 4T1 cells were seeded overnight and treated with an IC
50 concentration of
A. vulgaris extract (45 μg/mL). After 72 h incubation, the cells were trypsinized and stained with annexin V-FITC/PI (15 μg/mL) for 15 min at RT in the dark. Afterward, annV-binding buffer (ABB) was used for resuspension of the 4T1 cells. Finally, the cells were analyzed using flow cytometry (CyFlow
® Space Partec and PartecFloMax
® 2.52 software (Munster, Germany)) [
69].
4.5. Activation of Caspases
For the detection of caspase activation, 4T1 cells were seeded overnight and exposed to an IC
50 concentration of
A. vulgaris extract (45 μg/mL). After the incubation period (72 h), the trypsinized cells were stained with pan-caspase inhibitor (apostat) and incubated for 30 min at 37 °C. Afterward, cells were then washed, resuspended in PBS, and analyzed with the CyFlow
® Space Partec flow cytometer using PartecFloMax
® 2.52 software (Munster, Germany) [
69].
Alternately, 4T1 cells were seeded in a four-chamber slide (Millicell® EZ SLIDES, Merck Millipore, Burlington, MA, USA) at 5 × 103 cells/well density and left to adhere overnight, followed by exposure to A. vulgaris extract (IC50 concentration) and 72 h incubation. Finally, cells were stained via apostat staining and analyzed using fluorescence microscopy at 400× magnification with a Leica DM4 B with DFC7000 T microscope camera (Leica Microsystems CMS GmbH, Wetzlar, Germany).
4.6. Detection of Autophagy
The 4T1 cells were seeded overnight and exposed to an IC
50 concentration of
A. vulgaris extract (45 μg/mL). After the 72 h incubation period, the cells were stained for 15 min at 37 °C with 10 µM of AO solution. The dye was removed by washing with PBS. Finally, cells were resuspended using PBS and analyzed with the CyFlow
® Space Partec flow cytometer using PartecFloMax
® 2.52 software (Munster, Germany) [
69].
Additionally, 4T1 cells were analyzed via fluorescence microscopy after treatment with an IC50 concentration of A. vulgaris extract. Cells were seeded overnight in the four-chamber slide (Millicell® EZ SLIDES, Merck Millipore, Burlington, MA, USA) at 5 × 103 cells/well density and exposed to A. vulgaris extract for 72 h. After the incubation period, the treatment was discarded and the cells were washed with PBS followed by a 15 min incubation with AO staining at a final concentration of 10 µM. Finally, the cells were washed with PBS and evaluated with fluorescence microscopy at 400× magnification using the Leica DM4 B with DFC7000 T microscope camera (Leica Microsystems CMS GmbH, Wetzlar, Germany).
4.7. Detection of Cell Proliferation
The 4T1 cells were prestained with CFSE (1 μM) and kept in the incubator for 10 min at 37 °C. After the incubation period, 10% FBS-RPMI medium was added and the cells were centrifuged, washed with PBS, resuspended in medium, seeded, and exposed to an IC
50 concentration of
A. vulgaris extract (45 μg/mL). After the 72 h incubation period, the 4T1 cells were washed with PBS, trypsinized, and analyzed with the CyFlow
® Space Partec flow cytometer using PartecFloMax
® 2.52 software (Munster, Germany) [
69].
4.8. Assessment of Reactive Oxygen and Nitrogen Species (ROS/RNS) Production
Using redox-sensitive dye DHR123, production of ROS/RNS was detected. Prior to seeding, 4T1 cells were stained with DHR123 (1 μM) and incubated for 20 min at 37 °C. Afterward, cells were then exposed to an IC
50 concentration of
A. vulgaris extract. Following the 72 h incubation period, the cells were washed with PBS, trypsinized, and analyzed with the CyFlow
® Space Partec flow cytometer using PartecFloMax
® 2.52 software (Munster, Germany) [
26].
In order to detect via fluorescence microscopy the production of ROR/RNS in 4T1 cells after exposure to A. vulgaris extract (IC50 concentration), cells were prestained with DHR123 to a final concentration of 1 µM, seeded in a four-chamber slide (Millicell® EZ SLIDES, Merck Millipore, Burlington, MA, USA) at 5 × 103 cells/well density, left to adhere overnight, and treated with A. vulgaris extract for 72 h. Finally, cells were washed with PBS and analyzed at 400× magnification using a Leica DM4 B with DFC7000 T microscope camera (Leica Microsystems CMS GmbH, Wetzlar, Germany).
4.9. Staining with Propidium Iodide (PI) on Chamber Slides
For fluorescence microscopy analysis, 4T1 cells were seeded overnight on sterile 4-chamber glass microscope slides (Millicell
® EZ SLIDES, Merck Millipore, Burlington, MA, USA) at a density of 3 × 10
4 cells/well in 400 µL medium. Afterward, the 4T1 cells were then exposed to an IC
50 concentration of
A. vulgaris extract and incubated for 72 h. After incubation, the supernatant was discarded from the chambers, the cells were then washed with PBS, and they were then fixed in 4% PFA for 15 min at RT. Thereafter, the cells were washed in PBS several times, and stained with a solution of 50 μg/mL PI, 0.1 mM EDTA pH 8.0, 85 μg/mL RNase, and 0.1% Triton X-100 in PBS for 1–2 min. Finally, fluorescent mounting medium (Fluoromount-G) was used for coating the slides. A Zeiss AxioObserver Z1 inverted fluorescence microscope (Carl Zeiss AG, Oberkochen, Germany) was used for the analysis of the formed slides at 400 × magnification [
70].
4.10. Clonogenic Assay—Colony Forming Assay
The assessment of the ability of 4T1 cells to form colonies in vitro after treatment with a subtoxic concentration of
A. vulgaris extract involved evaluation using a clonogenic (colony formation) assay. The 4T1 cells were grown in a monolayer in 75 cm
2 flasks, exposed to a subtoxic concentration (22.5 μg/mL) of
A. vulgaris extract, and incubated for 72 h. Following the incubation period, the cells were trypsinized, counted, and seeded at a low number of cells per well (1 × 10
3 cells/well) in 6-well plates. The 4T1 cells were allowed to grow and form colonies for 7 days, with the medium exchange on the fourth day. After the incubation period, the medium was discarded, and the formed colonies were washed using PBS and then fixed (4% PFA for 30 min at RT). Colonies were finally stained with 0.02% CV solution at RT for 15 min. Afterwards, cells were washed twice with PBS to remove the remaining dye. Colony quantification was performed using ImageJ analysis 1.51j8 software. In order to calculate the planting efficiency (PE) and the surviving fraction (SF), the mean value of the number of colonies obtained from three wells was used. PE, which is a measure of the number of colonies formed from individual cells, was calculated as the number of colonies counted divided by the number of planted cells × 100. SF was calculated as PE of treated cells divided by the PE of control cells × 100 and graphically represented as a percentage [
69].
4.11. Wound Healing (Scratch) Assay
The scratch test, in which cells exhibit bidirectional migration from the edges of a scratch wound, was used for assessment of the cells’ motility. After reaching a confluence of 4T1 cells in the 6-well plates, a scratch was drawn with a sterile pipette tip (200 μL) across the center of the wells to make a clear wound area. After creating the wound, cells were washed using PBS several times in order to remove damaged cells and then exposed to a subtoxic dose (22.5 μg/mL) of
A. vulgaris extract for 48 h. Following the incubation period, the cells were washed, fixed with 4% PFA for 30 min at RT, stained with 0.02% of CV solution for 15 min, and digitally photographed with a Nikon stereomicroscope (SMZ800N, Nikon Instruments Inc., Melville, NY, USA) at 6× magnification [
69].
4.12. Assay for Quantification of Cell Adhesion In Vitro
For the assessment of cell adhesion in vitro, 4T1 cells were seeded in 75 cm
2 flasks and treated with a subtoxic dose (22.5 μg/mL) of
A. vulgaris extract. At 72 h, cells were trypsinized, harvested, and left for 30 min at RT to reconstruct cell membranes. Sterile 96-well plates were pre-coated with 20 μg/mL of Matrigel
® (50 μL/well) and left at 4 °C overnight to polymerize. Gel-coated wells were washed 3 times with PBS, and then the cells were seeded (3 × 10
4 cells/well). In parallel, cell adhesion was estimated on plastic (96-well plates not coated with Matrigel
®). Thereafter, the cells were kept at 37 °C for 1 h to adhere. Cells that did not attach in this time frame were removed by PBS washing, while the number of remaining adherent cells was determined by SRB assay. Relative cell adhesion was assessed by measuring the absorbance at 570/670 nm using an automatic microtiter plate reader [
69].
4.13. Assays for Quantification of Cell Migration and Invasion In Vitro
For quantitative assessment of the influence of
A. vulgaris extract treatment on the migration and invasive potential of 4T1 cells in vitro, transmigration and invasive assays were used. For these purposes, inserts with a diameter of 6.4 mm and a pore diameter of 8 μm were placed in sterile 24-well plates. In order to conduct the invasion assay, the upper membrane surface of these inserts was covered with a thin layer of Matrigel
® (500 μg/mL in PBS, 20 μL/membrane) that was allowed to polymerize at 4 °C overnight, while for the migration assay, the membrane remained uncovered with Matrigel
®. The 4T1 cells were grown in 75 cm
2 flasks and exposed to a subtoxic dose of
A. vulgaris extract (22.5 μg/mL) for 72 h. Before the 4T1 cells were seeded, the polymerized gel-coated inserts were washed twice with PBS. After incubation, the cells were trypsinized, then 7 × 10
4 cells were resuspended in 200 μL of 0.1% BSA-RPMI medium and seeded in the upper part of each well. As a chemoattractant for the cells, 10% FBS-RPMI was used, which was poured from the outside of the chambers, precisely into the wells of the cell culture plate. The 24-well plates were further incubated at 37 °C for 3 h to assess invasiveness and migration. Cells that did not migrate through the pores of the membrane (in the migration assay) or the extracellular matrix layer (in the invasion assay) were meticulously eliminated using cotton swabs. Conversely, invading or migrating cells underwent fixation with 4% PFA for 10 min and were then stained with a 0.02% CV solution for 20 min at RT. Following staining, the cells were washed with H
2O, air-dried, and subsequently, the membranes were detached from the chambers and mounted on microscope slides. Cells were observed and digitally photographed using the ZOE fluorescent cell imager (Bio-Rad Laboratories, Hercules, CA, USA). The average cell count from 5 randomly chosen independent fields on each membrane is shown. Each experiment was conducted in triplicate [
69].
4.14. Induction of 4T1 Cells and Experimental Treatment In Vivo
In order to evaluate the effect of A. vulgaris extract in vivo, tumors were induced by orthotopic implantation of 4T1 cells (2 × 104 in 50 μL PBS) into the fourth right mammary fat pad of female mice of the BALB/C strain. Upon tumor appearance (10 days after tumor induction) the mice were treated with A. vulgaris extract by i.p. injection (50 mg/kg body weight in 4% DMSO/PBS), with a regimen of 5 days treatment, 2 days break. The control group underwent treatment with a vehicle (4% DMSO/PBS). The animals were sacrificed 23 days after tumor implantation, the tumors were collected, and their volume was calculated using the formula: (length × width2) × 0.52.
4.15. Histopathology
After 23 days of in vivo experiment, animals were sacrificed via cervical dislocation and tumors, kidneys, and livers were macroscopically examined, isolated, and fixed in 10% buffered formalin of neutral pH value, for 24 h. All tissues were afterwards sectioned through the largest tissue plane and additionally fixed for 24 h. Tissue processing was carried out in an automated tissue processor (Milestone SRL LOGOS ONE, Sorisole, BG, Italy). Afterwards, using the embedding console (SAKURA Tissue-Tek TEC 5, Sakura Finetek, Torrance, CA, USA), tissues were embedded in paraffin blocks. The thickness of tissue slices was 4 µm and was obtained with a microtome LEICA RM 2245 (Leica Biosystems, Nussloch, Germany). Then, the tissue slices were mounted on glass slides and hematoxylin–eosin (H/E) staining was applied using the MYREVA SS-30H automated slide stainer (Especialidades Médicas MYR, S.L., Tarragona, Spain). For histopathological analysis of slides, an Olympus BX43 microscope (OLYMPUS EUROPA HOLDING GMBH, Hamburg, Germany) was used. For analysis and documentation purposes, a Leica Aperio AT2 slide scanner (Leica Biosystems, Nussloch GmbH, Germany) was additionally used. Furthermore, for morphometrical analysis of virtual slides, Leica AperioImageScope (version 12.4.6, Leica Biosystems, Nussloch GmbH, Germany) and FIJI-ImageJ 1.51j8 software were used [
69].
4.16. Western Blot Analysis
The 4T1 cells were treated with the IC
50 concentration of
A. vulgaris extract for 6, 12, 18, 24, 48, and 72 h periods. After incubation, the cells were lysed with a protein lysis buffer which contained 62.5 mM Tris–HCL pH 6.8, 10% glycerol, 50 mM dithiothreitol, and 2%
w/
v SDS. Lowry’s method was used for measuring protein content. Afterwards, cell lysates (30 μg) were separated on a 12% SDS–polyacrylamide gel with a protein molecular weight marker (prestained PageRuler ladder (Thermo Fisher Scientific, Waltham, MA, USA)). Proteins were then transferred to a polyvinylidene difluoride (PVDF) membrane using a semi-dry transfer (Fastblot B43; BioRad, Göttingen, Germany). After transfer, the membranes underwent blocking with 5% BSA in PBS containing 0.1% Tween 20 for 1 h at RT and were then probed with a primary antibody overnight at 4 °C. Following washing, the membranes were incubated with secondary antibodies (goat anti-rabbit IgG-HRP, and bovine anti-mouse IgG-HRP (Santa Cruz Biotechnology, Dallas, TX, USA)) for 1 h [
69]. The presence of bands was confirmed with an iBright™ FL1500 Imaging System (Thermo Fisher Scientific, Waltham, MA, USA) and analyzed using iBright Analysis 5.2.1 Software (Thermo Fisher Scientific, Waltham, MA, USA). In this study, the following primary antibodies were used: β-actin (Abcam, Cambridge, UK), integrin β-1, FAK, vinculin, and α-smooth muscle actin (α-SMA) (Elabscience Biotechnology Inc., Huston, TX, USA).
4.17. Preparation of Single-Cell Suspensions and Flow Cytometric Analysis
In order to obtain the single-cell suspensions of the spleen, mechanical dispersion was used. At the same time, as previously described [
71], enzymatic digestion was used in order to obtain the single-cell suspensions of primary tumors. Anti-mouse antibodies labeled with fluorochrome and specific for CD11c, CD40, MHCII, CD86, CD3, CD8, PD-1, or isotype-matched controls (BD Pharmingen, NJ/Invitrogen, Carlsbad, CA, USA) were used. For intracellular staining, phorbol 12-myristate 13-acetate (50 ng/mL, Sigma-Aldrich, St. Louis, MO, USA), ionomycin (500 ng/mL, Sigma-Aldrich), and GolgyStop (BD Pharmingen, Franklin Lakes, NJ, USA) were used for stimulation for 4 h. Finally, cells were stained with anti-mouse antibodies labeled with fluorochrome specific for perforin, CD107a, and granzyme (BD Pharmingen/ BioLegend/eBiosciences), and 2 × 10
4 to 5 × 10
4 cells were acquired for FACS analysis. Flow cytometry was carried out using a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA, USA), and the data were analyzed with FlowJo 10.0.7r2 Software.
4.18. Statistical Analysis
The statistical package Statistica 12 (Informer Technologies, Inc., Los Angeles, CA, USA) was used for data analysis. The significance between groups was evaluated by Student t-test or alternatively by one-way ANOVA followed by Tukey’s honest significant difference (HSD) post hoc test, and two-sided p values of less than 0.05 were considered to indicate statistical significance. For in vivo experiments, a non-parametric Mann–Whitney U test was used.


 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}