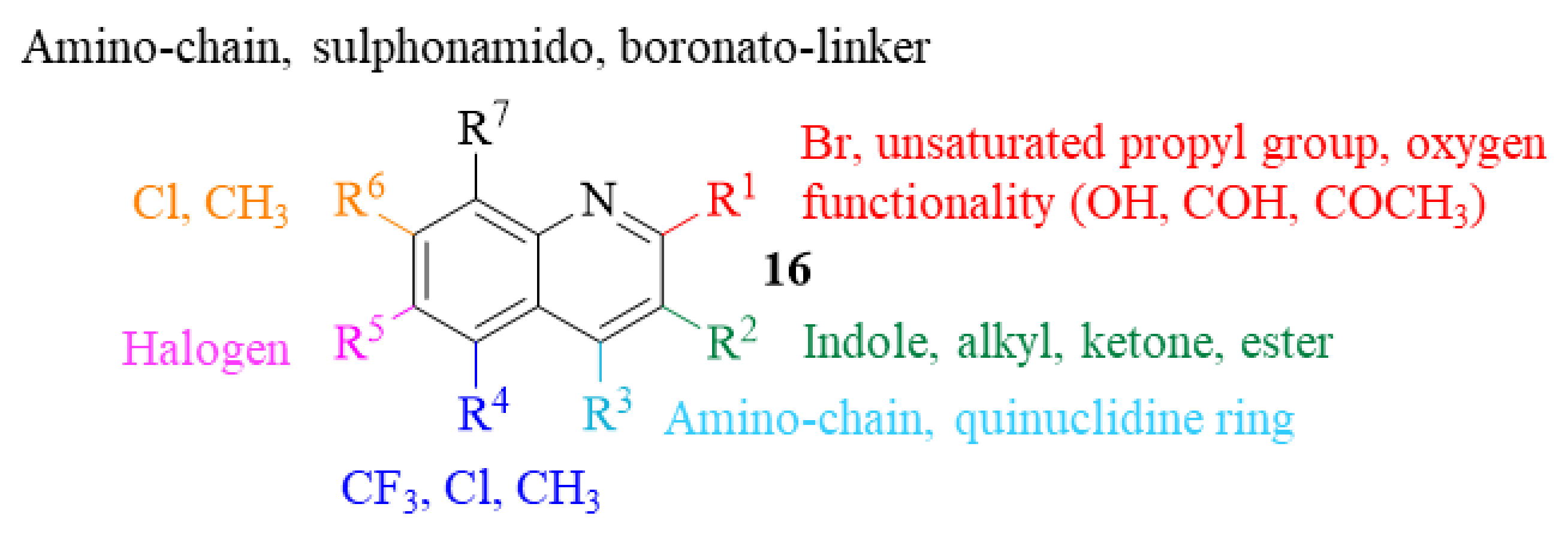
2.2. Quinoline Derivatives as Antileishmanial Agents Post-2013
Despite the huge amount of information available by mid-2013, the interest of many medicinal chemistry research groups in developing promising novel antileishmanial quinoline derivatives was still accentuated. This interest led numerous research groups to continue making significant contributions to developing antileishmanial treatments based on quinoline derivatives, as this subtopic will thoroughly depict.
In 2013, while developing a novel rapid drug screening assay for antileishmanial activity, Bringmann et al. synthesized and evaluated a series of forty-nine quinolinium salts against both
L. major promastigotes and amastigotes [
23]. Considering their effect on promastigotes, the results demonstrated that only twenty quinolinium salts from the entire series of evaluated compounds have considerable antileishmanial activities (IC
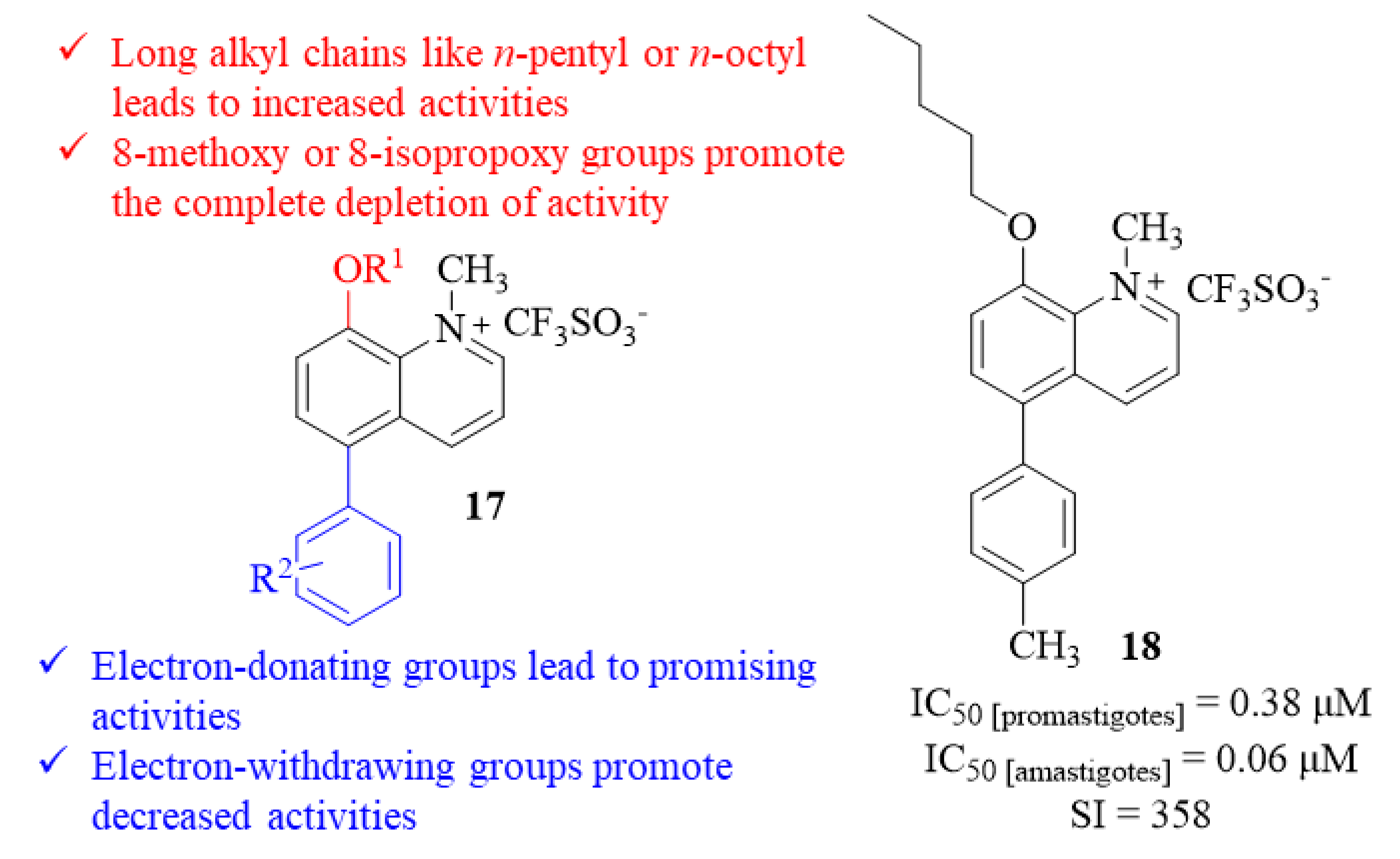
50 < 10 µM), with the remaining derivatives showing low to no activities. Based on the results, a SAR study was also delineated to understand the most critical structural moieties for the quinolinium salts’ antileishmanial properties. Introducing an 8-methoxy or an 8-isopropoxy function promotes the complete depletion of activity, while longer alkyl chains like pentyl or octyl clearly increase the compounds’ biological activity. The influence of the 5-substitutions was also considerably evaluated, particularly the effect of the functional group in a 5-aryl fragment. This evaluation demonstrated that an aryl fragment, containing an electron-donating substituent, promotes promising antileishmanial activities, while an aryl fragment with an electron-withdrawing substituent promotes low activity levels (
17,
Figure 5). Only seven of these twenty promising compounds were further evaluated against
L. major amastigotes infecting blood marrow-derived macrophages (BDMD). This final evaluation showed that six displayed high activity levels against intracellular amastigotes (IC
50 < 0.3 µM). Despite not being the most active compound against amastigotes, the potential of derivative
18 must be emphasized since it presents an IC
50 of 0.06 µM and a selectivity index of 358 (
Figure 5).
In the same year, through a screening program for new biologically active
N-heterocyclic compounds, Bompart et al. synthesized and evaluated a series of 2-aryl quinolines as promising antileishmanial agents against
L. braziliensis promastigotes and amastigotes [
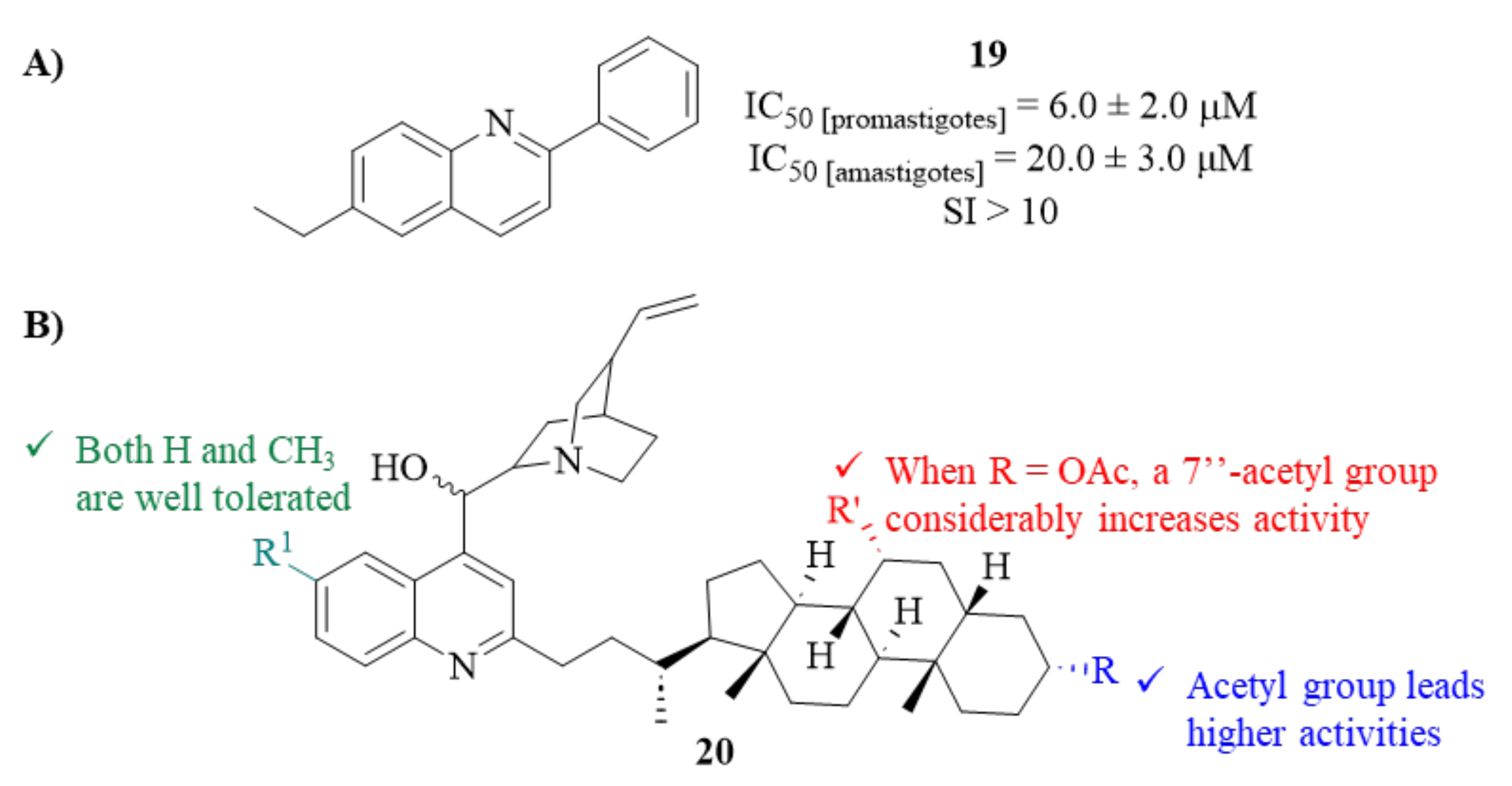
24]. From this series of seven derivatives, only one compound (
19,
Figure 6) demonstrates, simultaneously, considerable effects against promastigotes (IC
50 = 6.0 ± 2.0 µM) and amastigotes (IC
50 = 20 ± 2.0 µM), in addition to low levels of toxicity against BMDM macrophages (IC
50 > 200 µM). Following the promising profile of this derivative, its mechanism of action was also evaluated, particularly the evaluation of its effects on the parasite bioenergetics and sterol biosynthetic pathway. Considering its influence on the parasite bioenergetics, the results led this research group to suggest that this derivative’s cationic nature might activate an electrophoretic mechanism that compromises the parasite’s viability by promoting the failure of the mitochondrial potential. In turn, regarding the sterol biosynthetic pathway, this work was able to demonstrate an accumulation of squalene and a depletion of 5-dehydroepisterol in treated parasites, an effect that had already been described for miltefosine against
L. mexicana [
25].
Based on the principle of bioconjugation/hybridization, which allows the formation of hybrid compounds with the combined properties of their individual components by binding two or more active molecules, another research group prepared a series of sixteen new
Cinchona alkaloid-bile acid hybrids and evaluated them against
L. mexicana promastigotes (
20,
Figure 6) [
26]. The results demonstrated that, essentially, the entire compound series demonstrated considerable antileishmanial activity values (IC
50 < 20 µM). Interestingly, considering the group of acetylated molecules, one was able to verify that the presence of a 7″-acetyl group considerably increases the antileishmanial activity of these compounds (IC
50 from 18.08–20.04 to 4.62–5.50 µM). Considering the group of deacetylated derivatives, most of the derivatives showed the same range of activity levels (IC
50 = 5.29–6.96 µM), being even more active than the correspondent
cinchona alkaloid precursors. However, this series of molecules also revealed high levels of toxicity against normal human fibroblast cell lines (WI-38), originating SI > 1, which makes this type of compound unsuitable for use in further developments of antileishmanial drugs.
Following a previous study by Alain Fournet et al. [
27], where several structurally simple 2-substituted quinolines were described as having in vitro and in vivo antiparasitic properties, another research group decided to further develop this type of derivative. Using two of the already identified molecules as parent compounds, Gopinath et al. prepared a series of 2-substituted quinolines to address the limitations of this previous work, namely the weak in vitro potency and metabolic instability [
28]. Through several C-2 modifications on the quinoline ring, it was possible to verify that the two most active derivatives against
L. donovani intracellular amastigotes were the ones containing the prop-2-en-diol (IC
50 = 10.04 ± 1.4 µM) and prop-2-enfluoride (IC
50 = 6.68 ± 1.1 µM) groups. However, in addition to weak potency, this first group of derivatives also demonstrated low solubility levels and/or metabolic stability.
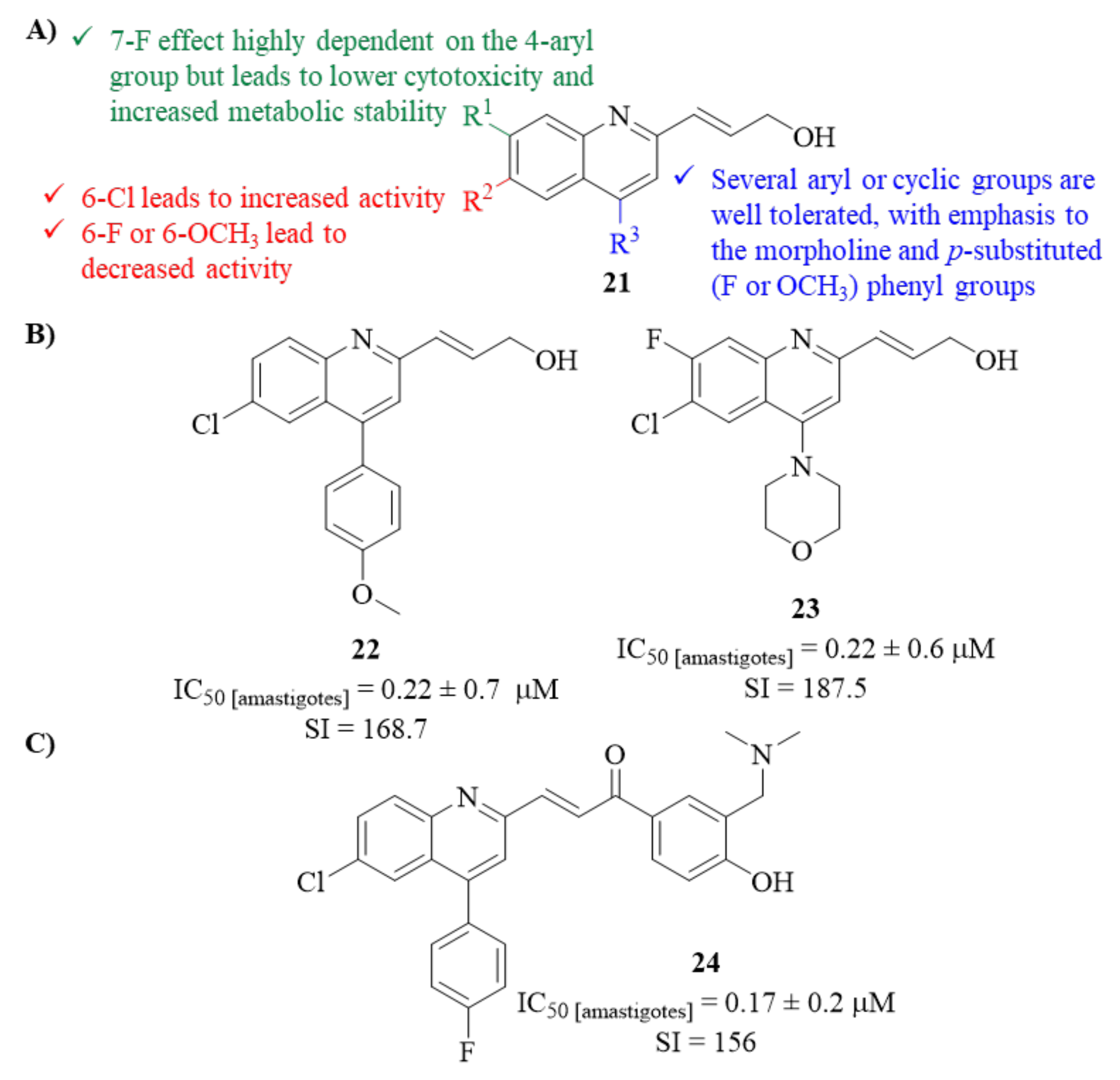
Then, using the prop-2-en-diol-contaning derivative as a core fragment, a second modification had to be performed to overcome these limitations, which was attempted by introducing chloro, fluoro, and methoxy substitution on the quinoline ring. The results demonstrated that considering C-6 modifications, only the introduction of a chloro atom led to a highly active derivative (IC50 = 0.86 ± 0.1 µM; SI = 33.59), while the introduction of both fluoro atom or methoxy group promoted a considerable activity decrease (IC50 = 17.9 ± 1.4 µM and IC50 = 32.38 ± 2.3 µM). Furthermore, while maintaining the 6-Cl atom, the introduction of a fluoro atom at either C-5 or C-7 led to a considerable loss of antileishmanial activity (IC50 = 4.1 ± 1.0 µM and IC50 = 14.35 ± 2.1 µM, respectively), with this effect being more accentuated with the introduction of a 7-F atom. Despite this activity loss, combining the 6-chloro and 7-fluoro atoms in the quinoline ring promoted lower cytotoxicity levels and significantly improved metabolic stability.
Finally, a further optimization process was performed by introducing several 4-aryl groups to assess the relevance of the substitution at this position to the in vitro potency against the parasite. Through this optimization, two derivatives emerged as promising antileishmanial agents, with high levels of activity and low levels of both cytotoxicity and metabolism (
22, IC
50 = 0.22 ± 0.7 µM; SI = 168.72 and
23, IC
50 = 0.22 ± 0.06 µM; SI = 187.5). Interestingly, during this process, one could verify that the influence of a 7-fluoro atom in the quinoline is highly dependent on the 4-aryl group introduced. For instance, considering derivative
22, introducing the 7-fluoro atom would considerably decrease the compound’s antileishmanial properties. In contrast, for derivative
23, the 7-fluoro atom seems crucial for the high levels of activity (
Figure 7).
A year later, in 2014, the same research group decided to develop their lead optimization program further by synthesizing a novel series of seven chalcone-type derivatives, with compound
23 as the structural core [
29]. This novel series was evaluated for its in vitro antileishmanial activity against
L. donovani intracellular amastigotes. The results demonstrated that, when compared with miltefosine (IC
50 = 8.10 ± 0.60 µM; SI = 7), all the evaluated derivatives present higher levels of antileishmanial activity (IC
50 = 0.17–6.42 µM) and selectivity indexes (SI = 13–156). Furthermore, these new analogues demonstrated similar levels of metabolic stability but lower solubility levels than the precursor (
23,
Figure 6). Structurally, different combinations of the same fragments promoted different levels of antileishmanial activity, impairing the development of a proper SAR study. Nevertheless, considering the substitution on the chalcone fragment, it was possible to verify that the most suitable substituent was the 3′-[(dimethylamino)methyl]-4′-hydroxyphenyl. Finally, the most active compound against
L. donovani was compound
24 (
Figure 7), which possessed the combination of a 4-fluorophenyl in the quinoline ring and a 3′-[(dimethylamino)methyl]-4′-hydroxyphenyl in the chalcone fragment (IC
50 = 0.17 ± 0.02 µM; SI = 156).
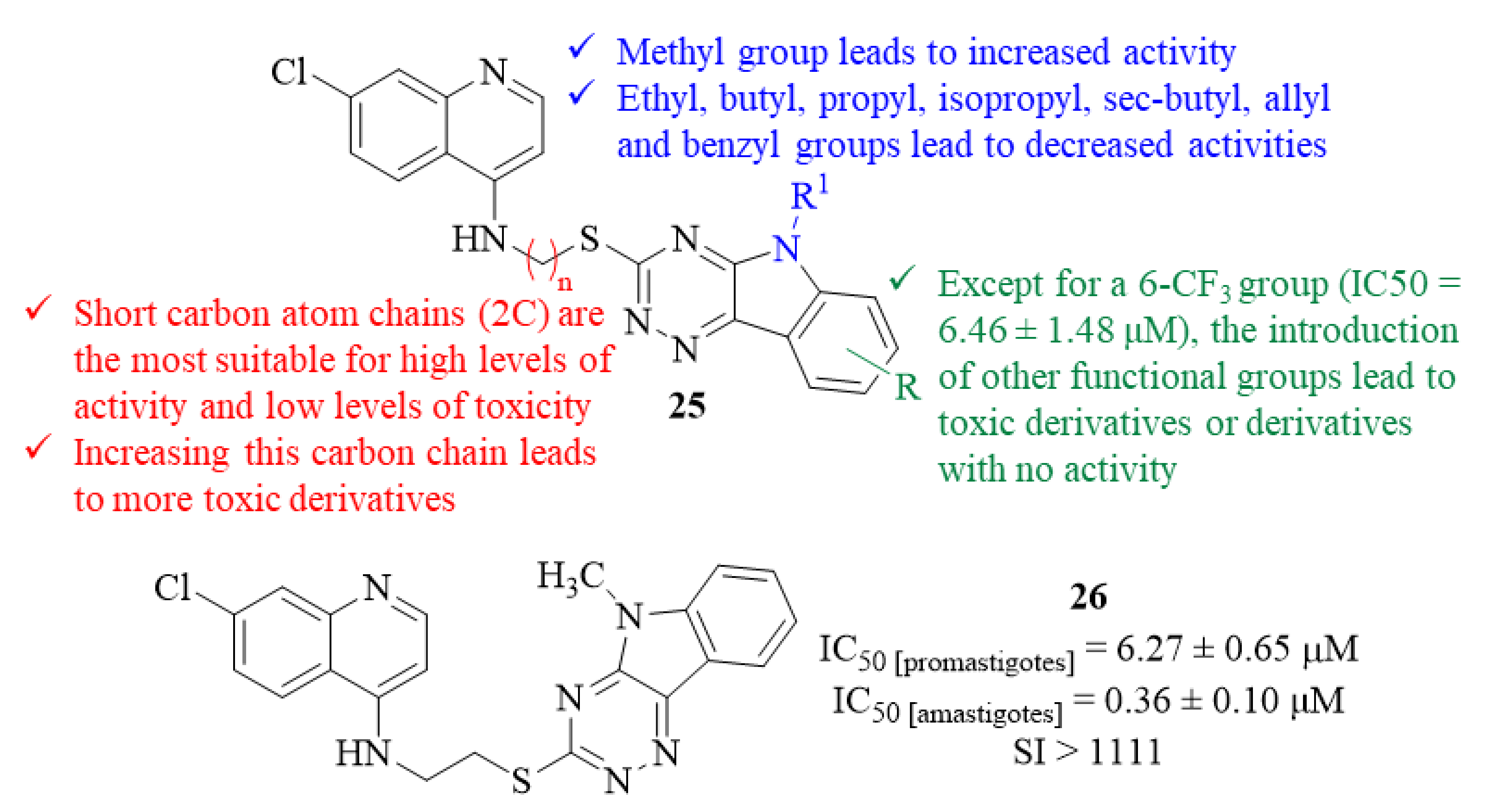
Still, in 2013, another research group decided to implement a hybridization approach for the development of novel antileishmanial agents against
L. donovani promastigotes and amastigotes, leading to the synthesis and evaluation of a series of nineteen triazino indole-quinoline hybrids [
30]. The antileishmanial evaluation of this series of derivatives, especially against the intracellular stage of the parasite, allowed the development of a SAR study with particular focus on three major structural features: (i) the chain length of the linker between the two pharmacophores; (ii) the
N-alkyl group introduced in the triazino indole fragment; and (iii) the substitution pattern on the indole’s aromatic ring (C-6 and C-8). Thus, considering the chain length of the linker, one could verify that derivatives with short carbon atom chains (2C) between the two pharmacophores were the most suitable compounds to be used as antileishmanial agents by simultaneously presenting high levels of activity (IC
50 = 0.36 ± 0.10–7.10 ± 1.27 µM) and considerable selectivity indexes (SI from 7 to >1111). Interestingly, increasing this carbon chain makes this type of derivative toxic to the J-774A.1 macrophage cells while maintaining considerable antileishmanial activity. Furthermore, considering the
N-alkyl group introduced in the triazino indole fragment, only the introduction of an
N-methyl group promoted an activity increase from 1.11 ± 0.19 µM to 0.36 ± 0.10 µM. In turn, the remaining functional groups introduced, such as ethyl, butyl, propyl, isopropyl, sec-butyl, allyl and benzyl, originated lower activity levels with IC
50 values as high as 29.48 ± 2.58 µM. Finally, regarding the substitution pattern on the indole’s aromatic ring (C-6 and C-8), except for the introduction of a 6-CF
3 group (IC
50 = 6.46 ± 1.48 µM), the introduction of any other functional group led to toxic derivatives or derivatives with no antileishmanial activity (
Figure 8).
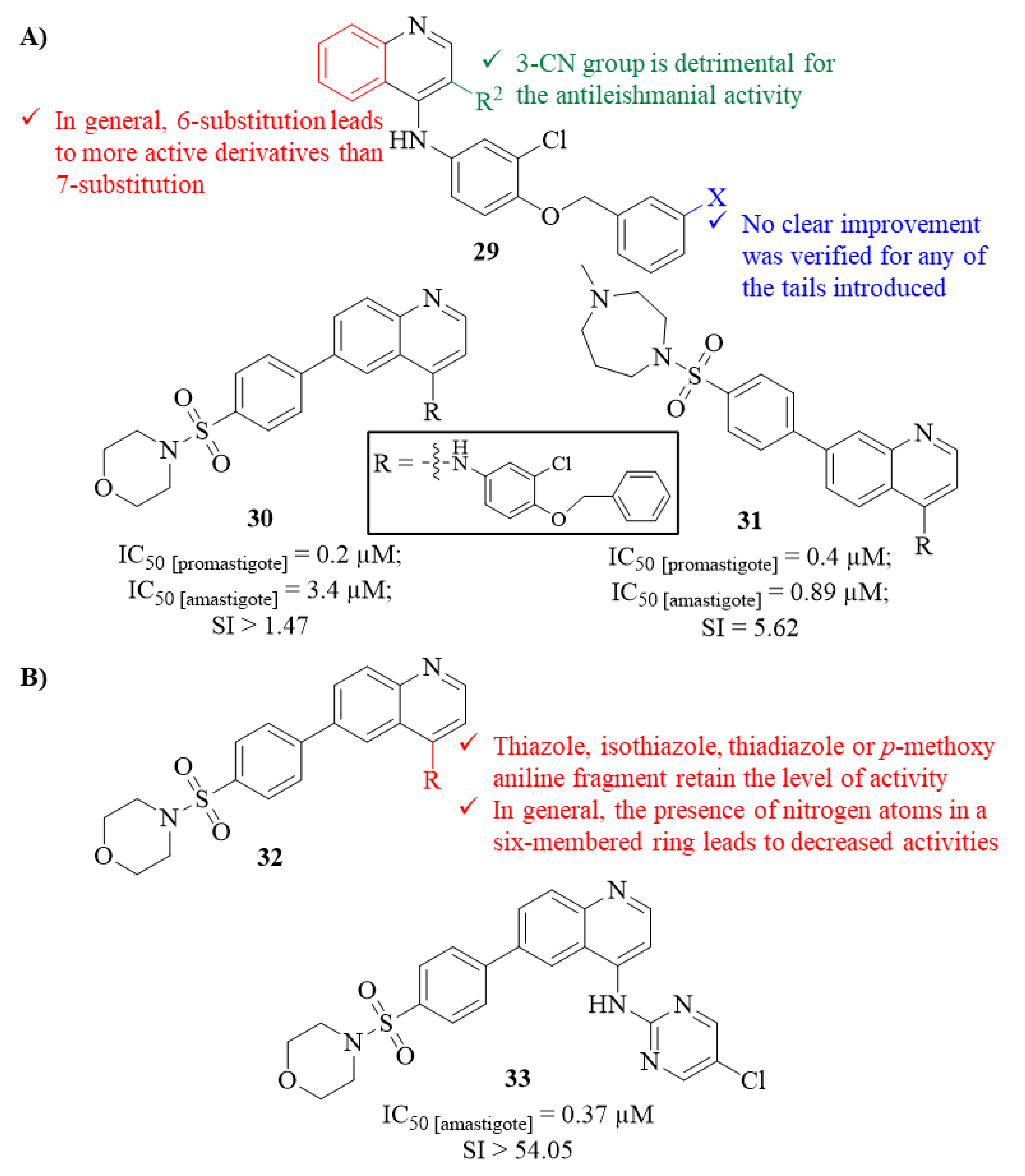
Two years later, in 2015, Devine et al. performed a considerable screening assay against several parasites, such as
T. cruzi (Chagas disease),
L. major (leishmaniasis), and
P. falciparum (malaria), evaluating numerous compounds from seven different scaffolds [
31]. From this entire library of compounds, and narrowing this work to the antileishmanial evaluation, two groups of quinoline derivatives must be highlighted for their antileishmanial potential against both stages of
L. major (
29,
Figure 9). The results demonstrated that several quinoline derivatives were able to impair the growth and survival of both promastigotes considerably (IC
50 0.2–4.1 µM) and amastigotes (IC
50 0.89–4.0 µM), with particular emphasis on the derivatives from the first group. The higher levels of antileishmanial activity observed for most of the derivatives from the first group, in comparison with the corresponding analogues from the second group, suggest that the presence of a 3-CN group is detrimental to the antileishmanial properties of this type of compound. Furthermore, by comparing the quinoline derivatives with the corresponding quinazoline and isoquinoline analogues, it was possible to verify that the sole presence of the N1 appears to be essential for their antipromastigote properties as both quinazoline (IC
50 = 0.50 µM) and isoquinoline (IC
50 > 15 µM) analogues present lower levels of activity than the quinoline correspondent (IC
50 = 0.20 µM). Considering the tail variations performed in these two groups of quinoline series, no clear improvement was verified for any of the tails introduced, suggesting that additional work should be performed to find the optimal core structure to promote higher activity levels. Finally, from this series of sixteen derivatives, analogues
30 (IC
50 [promastigote] = 0.2 µM and IC
50 [amastigote] = 3.4 µM) and
31 (IC
50 [promastigote] = 0.4 µM and IC
50 [amastigote] = 0.89 µM) should be highlighted as the most potent derivatives against promastigotes and amastigotes, respectively (
Figure 9).
Following the identification of derivative
31 as a lead molecule against leishmaniasis in 2017, this group focused on developing new analogues to improve its poor drug-like properties while maintaining or improving in vitro activity against
L. major intracellular amastigotes [
32]. For this optimization work, twenty-seven novel analogues were synthesized and evaluated against
L. major intracellular amastigotes with different functional groups introduced at C-4 of the quinoline scaffold. The results demonstrated that the analogues containing a thiazole, isothiazole, thiadiazole or
p-methoxy aniline fragment can maintain the antileishmanial activity in the same micromolar range (1.2−5.8 µM). In turn, apart from two analogues, the presence of nitrogen atoms in a six-membered ring considerably decreases the antileishmanial activity of this type of compound (
Figure 9). Interestingly, one of the exceptions emerged as the most active from this series of analogues, being the only compound able to achieve submicromolar antileishmanial activity (
33, IC
50 = 0.37 µM,
Figure 9).
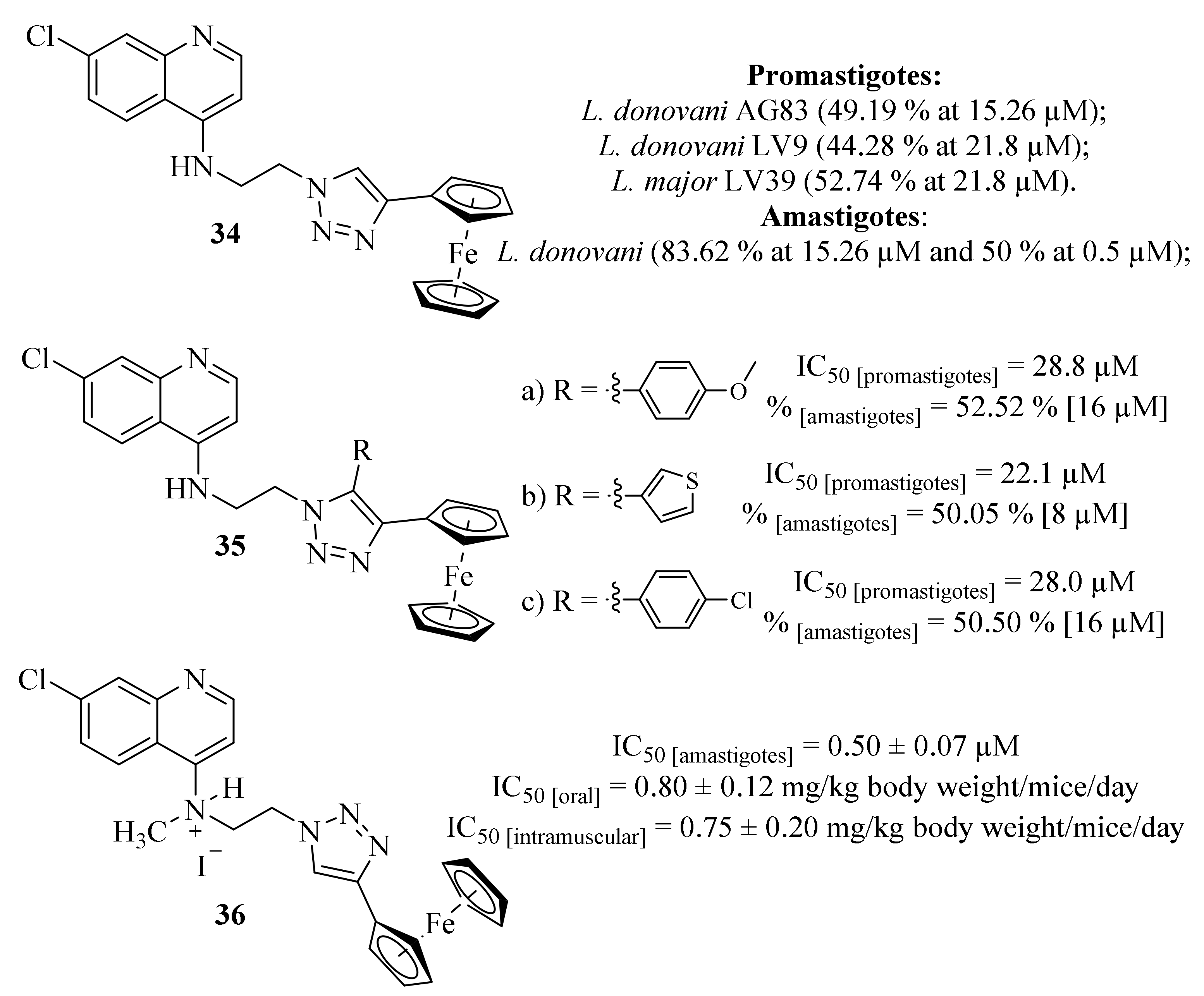
Also, in 2015, Yousuf et al. were particularly focused on the development of organometallic quinoline derivatives as novel antileishmanial agents through the synthesis and antileishmanial evaluation of ferrocenylquinoline (
34) against
L. donovani and
L. major (
Figure 10) [
33]. Considering its effects against
L. donovani and
L. major promastigotes, the results demonstrated that ferrocenylquinoline (
34) was able to inhibit the proliferation of the parasite in a dose-dependent manner, being more effective against
L. donovani AG83 (49.19% at 15.26 µM) and even more potent than miltefosine (IC
50 = 21 µM) against this particular species. Furthermore, ferrocenylquinoline (
34) was also able to inhibit the growth and proliferation of
L. donovani LV9 (44.28%) and
L. major LV39 (52.74%) at 21.8 µM. Regarding the amastigote stage of
L. donovani, this compound was also able to considerably affect the amastigotes in 50% at 0.5 µM, while also significantly increasing the level of NO in infected macrophages. This research group then identified a compound with promising antileishmanial properties, suggesting that these effects might be associated with its ability to induce parasite death by promoting oxidative stress and depolarizing mitochondrial membrane potential.
Following their work, this research group focused on synthesizing a series of thirteen ferrocenylquinoline derivatives as promising antileishmanial agents against
L. donovani promastigotes and amastigotes [
34]. From this series of thirteen derivatives, three of them presented promising antileishmanial activities against
L. donovani AG83 promastigotes (IC
50 [
35.a] = 28.7 µM, IC
50 [
35.b] = 22.1 µM and IC
50 [
35.c] = 28.0 µM), with the remaining derivatives presenting IC
50 values above 32 µM (
Figure 10). The higher antileishmanial activity of derivative
35.b might be related to the presence of the thiophene fragment since this type of nucleus provides conjugation for electron delocalization pathways more efficiently than phenyl groups, which may promote chain reactions and additional redox properties for these compounds. Then, the three most active compounds were selected for further evaluation, including their effects on cell cycle arrest and apoptosis and their effects against intracellular amastigotes and induction of NO. These further evaluations demonstrated that these derivatives appear to induce cell apoptosis on
L. donovani promastigotes as well as promote the generation of NO, which is considered the primary effector molecule of a pro-inflammatory response leading to the suppression of
L. donovani amastigote in the infected macrophages. Furthermore, these three derivatives were also demonstrated to be considerably effective against
L. donovani amastigotes, being able to inhibit its growth and proliferation by 52.51% [16 µM], 50.05% [8 µM] and 50.50% [16 µM], respectively.
A few years later, and following the promising results already described, this research group decided to compile the advantages of both quinoline and ferrocene scaffolds for the development of antileishmanial agents by synthesizing a novel series of four water-soluble ferrocenyl quinoline derivatives (
Figure 10) [
35]. In this work, the series of ferrocenylquinoline derivatives was evaluated for its effects against
L. donovani amastigotes, demonstrating that all derivatives present promising antileishmanial activities (IC
50 = 0.50 ± 0.07–5.05 ± 0.16 µM), with particular emphasis to derivative
36 (IC
50 = 0.50 ± 0.07 µM). Due to its in vitro activity, the derivative was further evaluated against an animal model of
L. donovani infection, being active through both oral and intramuscular administrations (IC
50 = 0.80 ± 0.12 mg/kg body weight/mice/day and IC
50 = 0.75 ± 0.20 mg/kg body weight/mice/day, respectively). From a mechanism of action perspective, the authors suggest that this compound’s activity is closely related to a critical interference in the parasite’s mechanism of defense against oxidative stress, particularly in the thiol redox pathway. In particular, it became clear that this molecule is capable of downregulating the expressions of the thiol-dependent enzymes transcriptionally and inhibiting the tyrosine reductase (TyrR) activity at a micromolar concentration. In addition to the inhibition of TyrR, this compound can also induce the host’s pro-inflammatory response, creating a dual effect against the intracellular parasite.
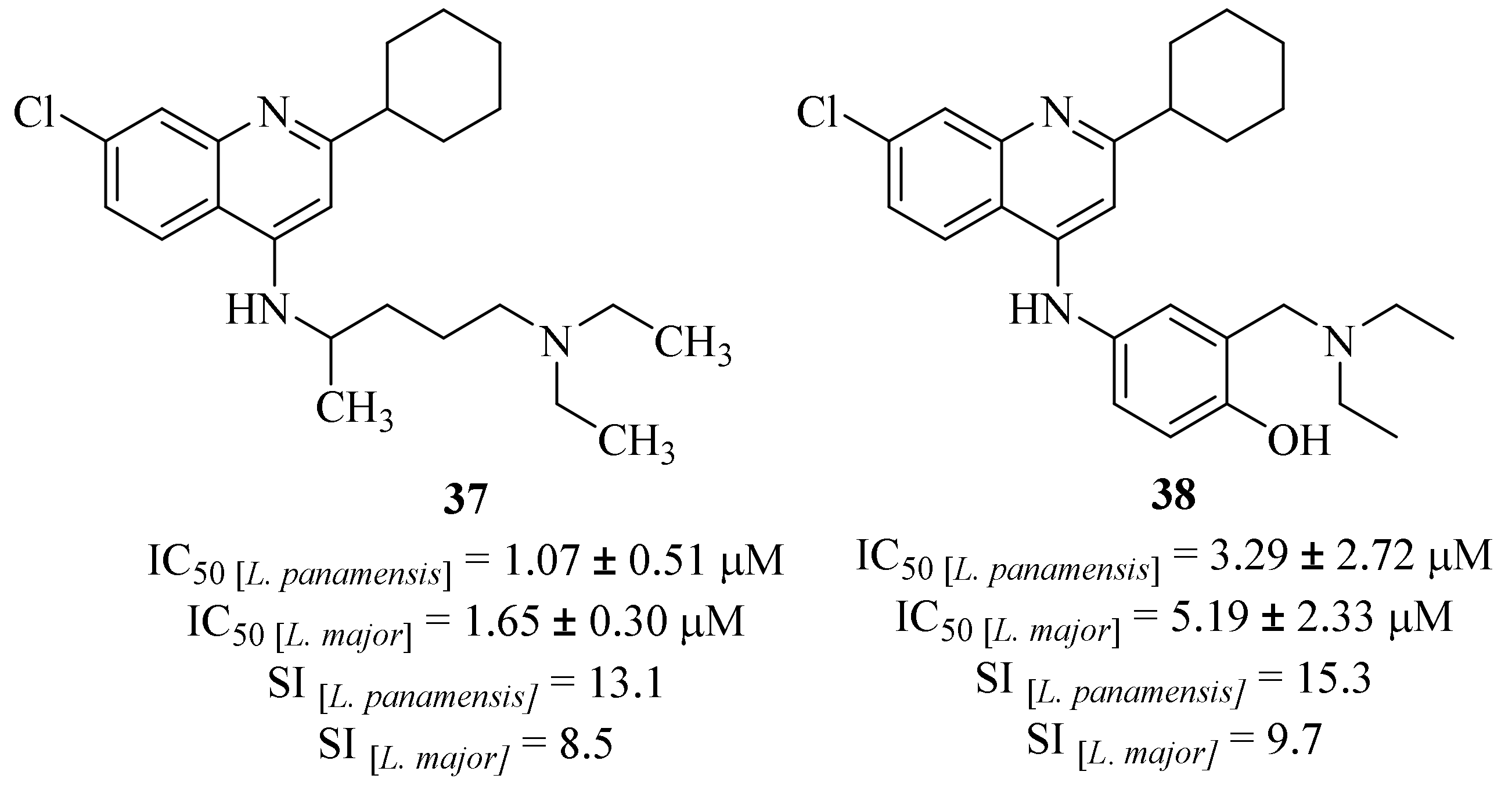
A year later, in 2016, Herrera et al. evaluated the antileishmanial effects of bi- and tricyclic
N-heterocycles against
L. panamensis and
L. major promastigotes and intracellular amastigotes, with particular emphasis on the quinoline derivatives evaluated [
36]. Interestingly, these quinoline derivatives were inactive against the promastigote stage of both
Leishmania species. However, two of the series of twenty evaluated quinolines (
37 and
38,
Figure 11) demonstrated promising activity levels against both
L. major and
L. panamensis intracellular amastigotes. In addition, both derivatives were more active against
L. panamensis than
L. major, with particular emphasis on derivative
37 for being the most active quinoline derivative (IC
50 [
L. panamensis] = 1.07 ± 0.51 µM and IC
50 [
L. major] = 1.65 ± 0.30 µM,
Figure 11). Further studies showed that these derivatives inhibit the production of IL-10 by macrophages infected with
Leishmania, suggesting that the compound-induced parasite-killing mechanism may be associated with the regulation of macrophage activation. Four years later, in 2020, the same research group evaluated derivative
37 in an animal model of
L. panamensis infection, which corroborated this compound’s potential against Leishmania and the compound-induced inhibition of IL-10 production by macrophages [
37].
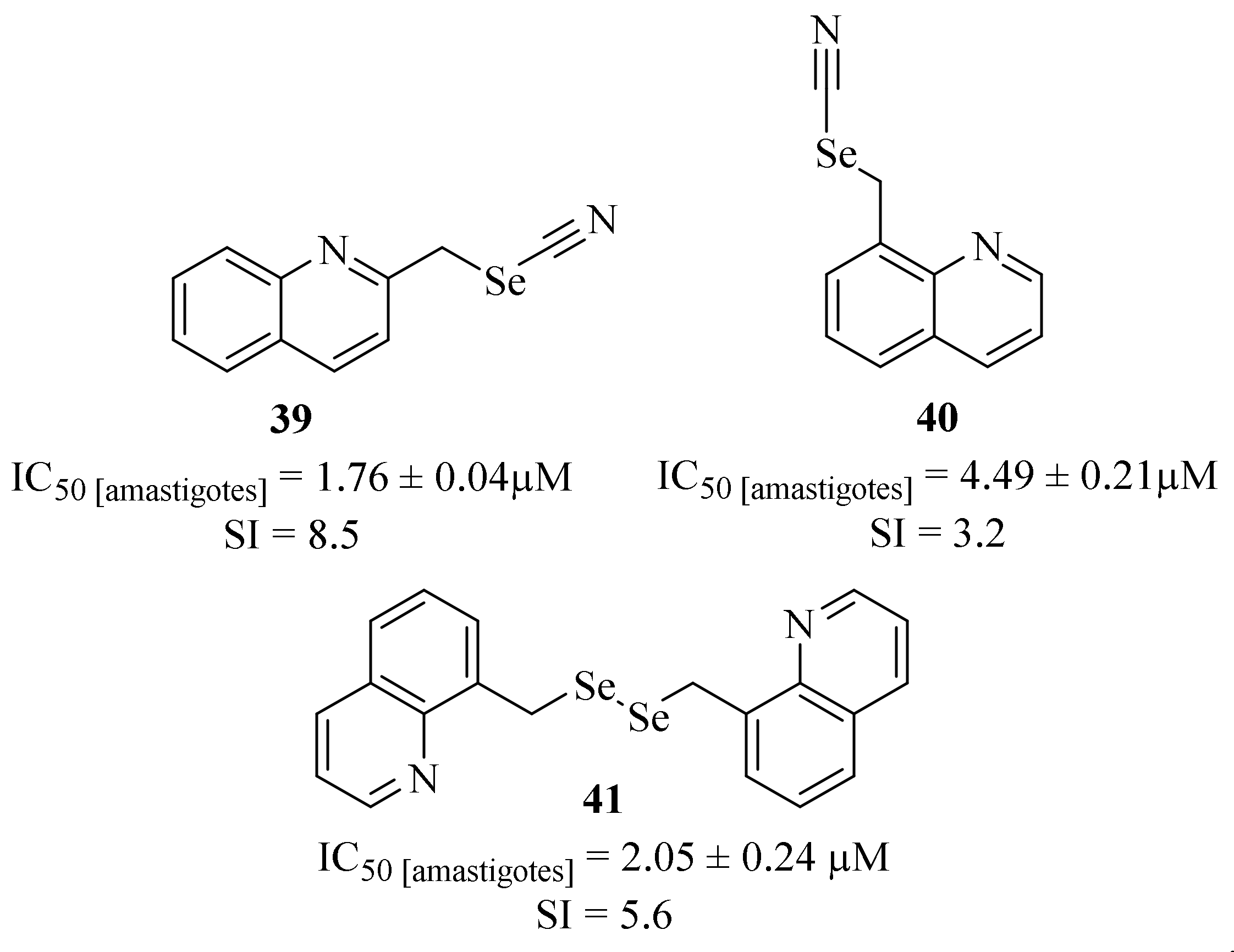
Still, in 2016, Baquedano et al. designed a series of new selenocyanates and diselenides containing several bioactive scaffolds as potential antileishmanial agents against
L. infantum axenic amastigotes, from which it is important to emphasize three quinoline-containing derivatives (
Figure 12) [
38]. Their results demonstrated that, even with a scarce number of derivatives, the quinoline-containing compounds present promising antileishmanial properties in addition to low levels of toxicity against THP-1 cells. Furthermore, it was also possible to verify that the position of the selenyl substitution in the quinoline scaffold considerably affects the compound’s antileishmanial activity, with the 2-substituted quinoline (
39) being significantly more active than the 8-substituted one (
40) (IC
50 = 4.49 ± 0.21 µM and 1.76 ± 0.04 µM, respectively). Finally, the dimerization of this type of compound can also promote significant improvements in terms of antileishmanial activity, with compound
41 (IC
50 = 2.05 ± 0.24 µM) being two times more active than the corresponding monomer (
40).
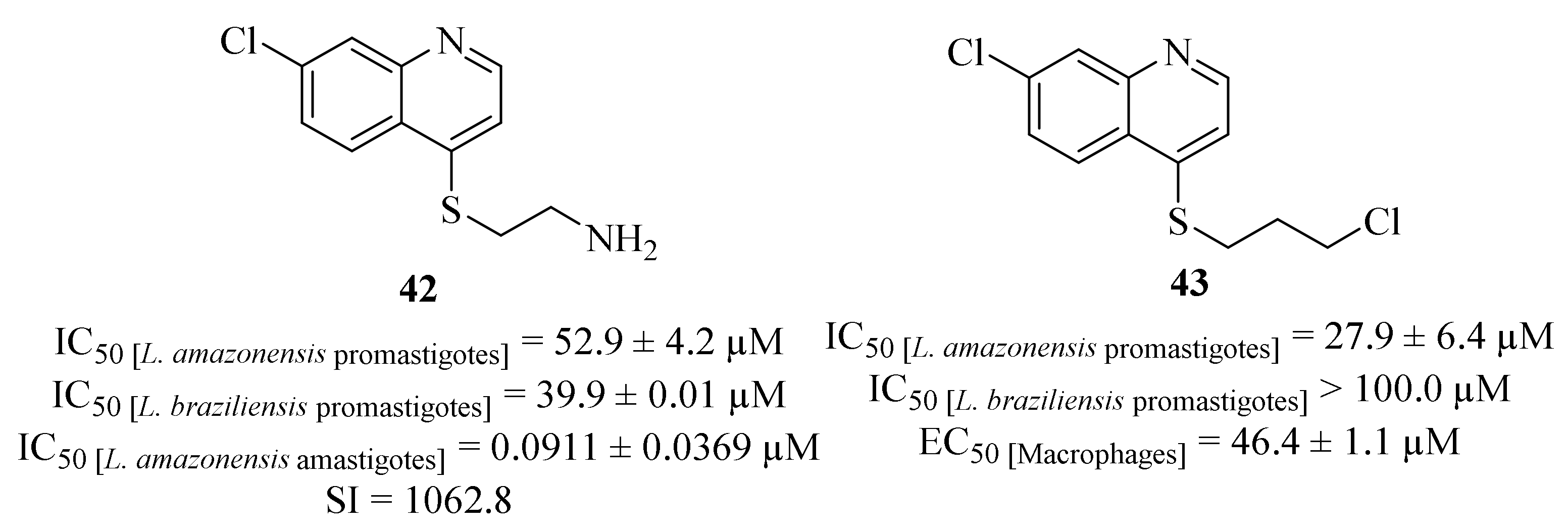
Another research group focused their efforts on the synthesis and evaluation of a series of 4-substituted quinoline derivatives against
L. amazonensis and
L. braziliensis promastigotes and
L. amazonensis amastigotes [
39]. Following some of their previous works [
40,
41] this novel series of derivatives contemplated quinoline derivatives in which the two amino groups of the side chain were replaced by sulfur, hydroxy or chloro substituents. Considering their effects against the promastigote stage of both
L. amazonensis and
L. braziliensis, it was possible to verify that only two derivatives (
42 and
43,
Figure 13) present moderate to significant levels of antileishmanial activity against
L. amazonensis (IC
50 = 52.9 ± 4.2 µM and IC
50 = 27.9 ± 6.4 µM, respectively), while only one of the derivatives (
42) was active against
L. braziliensis (IC
50 = 39.9 ± 0.01 µM). However, derivative
43 also demonstrates high levels of toxicity against murine macrophages (IC
50 = 46.4 ± 1.1 µM). In turn, against
L. amazonensis amastigotes, derivative
42 was the only one to show considerable efficiency, with an IC
50 = 0.0911 ± 0.0369 µM, being 139 times more active than the reference drug miltefosine (IC
50 = 12.7 ± 0.9 µM). Further studies also allowed the authors to suggest that the antileishmanial effects of this derivative (
42) might be associated with the induction of a high generation of ROS with low alterations of the mitochondrial membrane potential and without affecting plasma membrane, being mediated by mitochondrial oxidative stress.
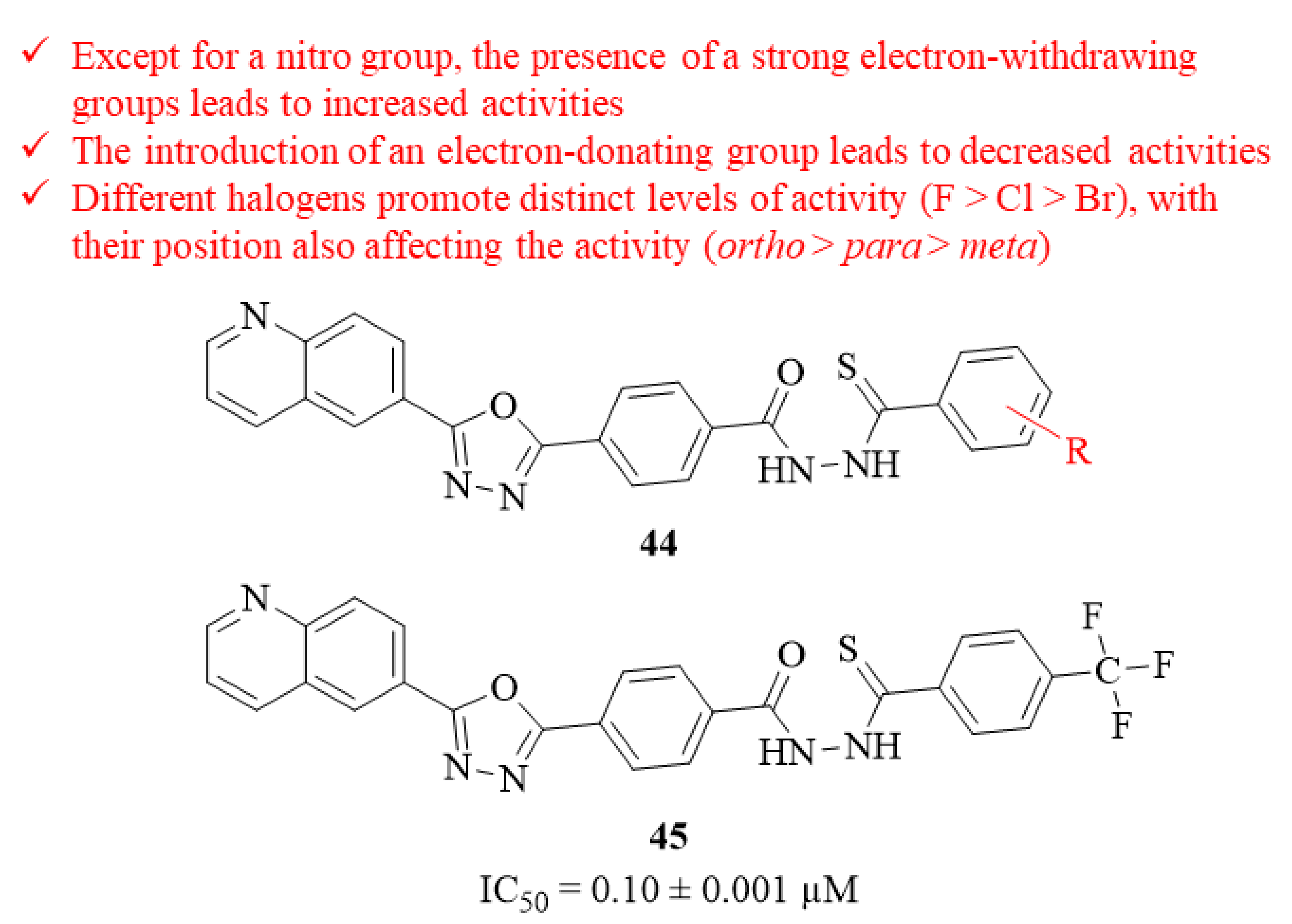
In the same year, a series of eighteen quinolinyl-oxadiazole thiosemicarbazide hybrids were designed and evaluated for its antileishmanial potential against
L. major intracellular amastigotes by Taha et al. [
42]. The results demonstrated that, from this series of eighteen hybrids, thirteen of them present significant levels antileishmanial activity (IC
50 = 0.10 ± 0.001–7.40 ± 0.41 µM), being comparable with the reference drug, pentamidine (IC
50 = 7.02 ± 0.09 µM). Structurally, it became clear that the presence of a strong electron-withdrawing group like the
p-trifluoromethyl group (
45, IC
50 = 0.10 ± 0.001 µM) considerably contributes to the antileishmanial properties of this type of compound (
Figure 14). However, the introduction of another type of electron-withdrawing group, like a nitro group, was also able to originate derivatives with poor antileishmanial activities (IC
50 = 4.98 ± 0.21 µM–8.70 ± 0.30 µM) In turn, the introduction of an electron-donating group, like a methoxyl group, promoted the lowest levels of activity (IC
50 = 7.40 ± 0.41 µM–18.12 ± 0.85 µM), suggesting that this type of functionalization might be detrimental to the antileishmanial properties of these compounds. Considering the halogenation pattern of the aromatic ring, one can assume that the presence of different halogen atoms promotes distinct levels of antileishmanial activity, with the fluoro substituted analogues being the most active derivatives (IC
50 = 0.15 ± 0.001–3.30 ± 0.01 µM), followed by the chlorinated (IC
50 = 0.72 ± 0.01–4.50 ± 0.15 µM) and the bromated ones (IC
50 = 6.9 ± 0.20 µM–21.40 ± 0.50 µM). Interestingly, the presence of a simple methyl group in the aromatic ring also promoted high levels of activity (IC
50 = 1.12 ± 0.01 µM), which might indicate that the introduction of alkyl chains would also be beneficial to the evaluated activity. Finally, the position of these halogens in the aromatic ring also affects its antileishmanial activity, with the
ortho-position being the optimal introduction, followed by the
para-position and the
meta-position.
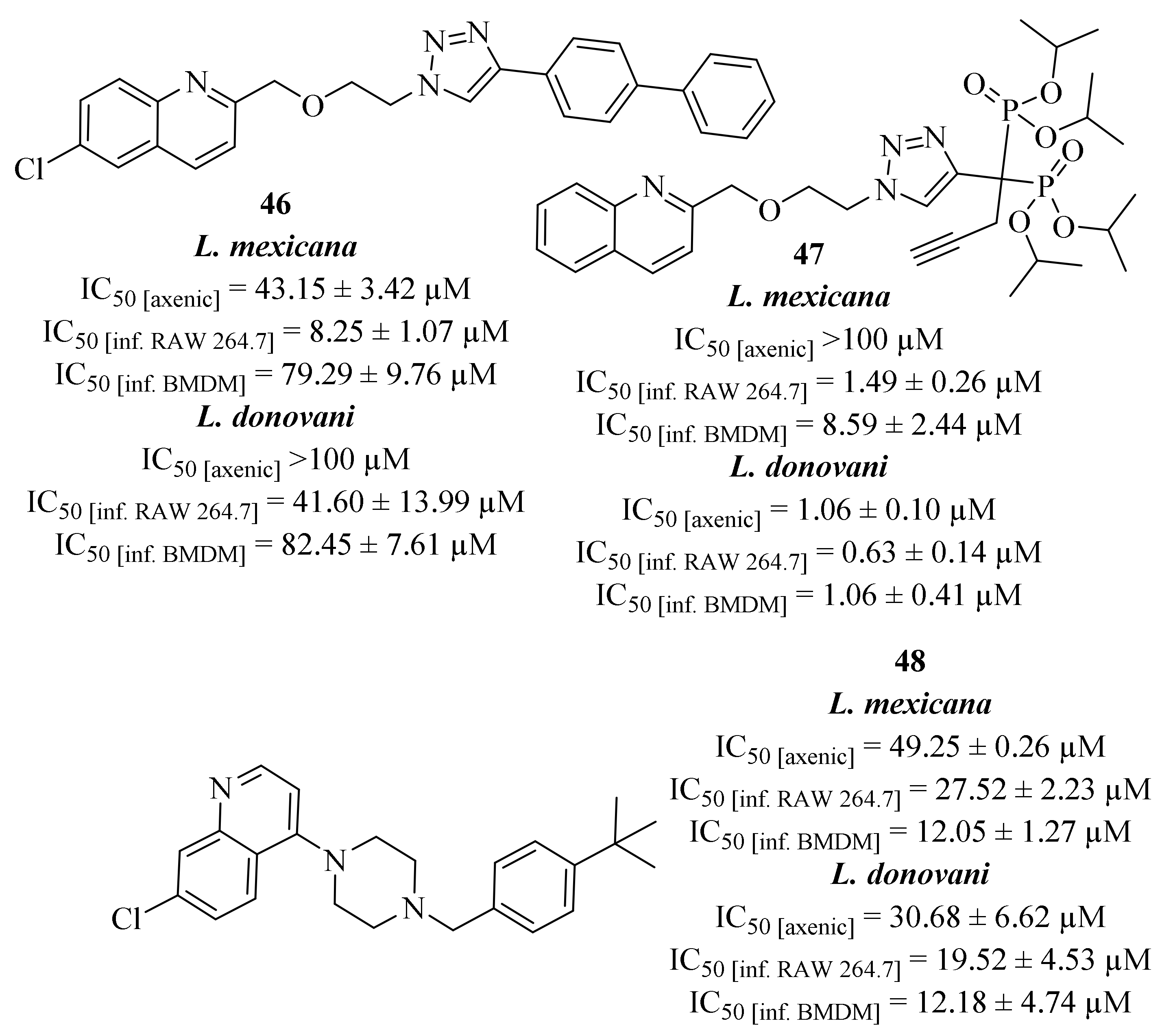
Following some of their previous works in which some 2-substituted quinolines were already identified as antileishmanial leads, [
27,
43] Mao et al. performed a preliminary molecular study that allowed them to suggest that the quinoline motif could replace the guanine group of GDP-mannose within the GDP-MP catalytic site [
44]. This evidence prompted them to design a series of one hundred GDP-MP competitive inhibitors, some containing the quinoline core in the inhibitor scaffold, and evaluate it against the pure enzyme (GDP-MP), from both
L. donovani and
L. mexicana, as well as against both parasite species. In a first evaluation, this series of derivatives was evaluated against two
Leishmania and one human GDP-MPs, with only eleven derivatives demonstrating IC
50 values below the screening concentration (100 µM). However, only five of them present considerable levels of affinity by exhibiting significant
Ki values on a leishmanial GDP-MP, being three of them quinoline-containing derivatives (
46–
48,
Figure 15). These three derivatives (
46–
48) were then evaluated against both axenic and intracellular amastigotes of
L. donovani and
L. mexicana on two cell host models, RAW264.7 macrophages and bone marrow derived macrophages (BMDM). Considering
L. donovani, derivative
47 presents the most interesting levels of antileishmanial activity, in both axenic (IC
50 = 1.06 ± 0.10 µM) and intracellular amastigotes (IC
50 [RAW 264.7] = 0.63 ± 0.14 µM and IC
50 [BMDM] = 1.06 ± 0.41 µM), while the remaining derivatives only demonstrate moderate levels of activity. In turn, considering
L. mexicana, the three derivatives (
46–
48) demonstrate moderate to no activity against axenic amastigotes but significant levels of activity against intracellular amastigotes. In particular, derivative
46 demonstrates high levels of activity against infected RAW 264.7 macrophages (IC
50 = 8.25 ± 1.07 µM), while derivative
48 only presents significant activity levels against infected BMDM (IC
50 = 12.05 ± 1.27 µM). Once again, derivative
47 was the most active derivative against the intracellular amastigotes (IC
50 [RAW 264.7] = 1.49 ± 0.26 µM and IC
50 [BMDM] = 8.59 ± 2.44 µM), highlighting this derivative as the most promising compound for further developments.
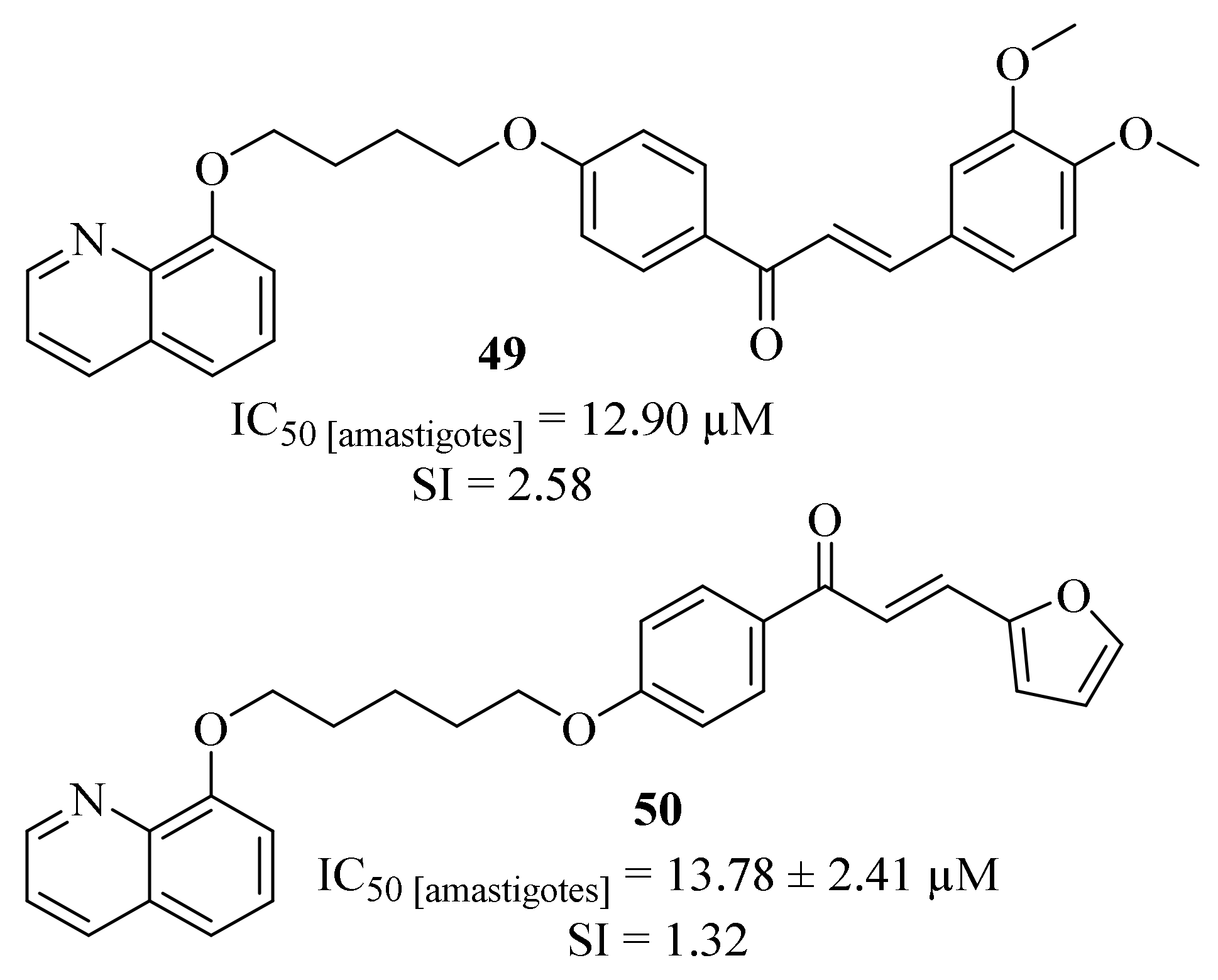
Still in 2017, Coa et al. designed a series of quinoline-chalcone and quinoline-chromone hybrids and evaluated their potential as antileishmanial agents against
L. (V) panamensis amastigotes [
45]. The results demonstrated that, from this series of eleven hybrids, four of them present significant levels of antileishmanial activity (EC
50 < 20 µM), with particular emphasis to compound
49 (IC
50 = 12.90 µM and SI = 2.58,
Figure 16). Even though there is no clear relation between the antileishmanial activity and the length of the alkyl linker, it was still possible to verify that this hybridization approach promotes a combined improvement for this type of hybrids when compared with the corresponding parent compounds. In particular, through hybridization, this research group was able to design molecules that present higher activity levels than parent chromone (IC
50 = 718.45 µM), while showing lower levels of cytotoxicity than the parent quinoline (IC
50 = 1.38 µM).
In the beginning of 2018, and following the biological potential of molecular hybridization, the same research group focused their efforts on the development of a new series of furanchalcone–quinoline hybrids, amongst other types of hybrids, (
Figure 16) and their evaluation against
L. (V) panamensis intracellular amastigotes [
46]. The results demonstrated that the newly synthesized furanchalcone–quinolines only present moderate levels of antileishmanial properties, with IC
50 = 13.78 ± 2.41–207.36 ± 14.98 µM. Once again, relationship between the antileishmanial activity and the length of the alkyl linker was not clear, with the highest activity being achieved with a five-carbon alkyl chain (
50, IC
50 = 13.78 ± 2.41 µM), being consistent with the results obtained in the previous study.
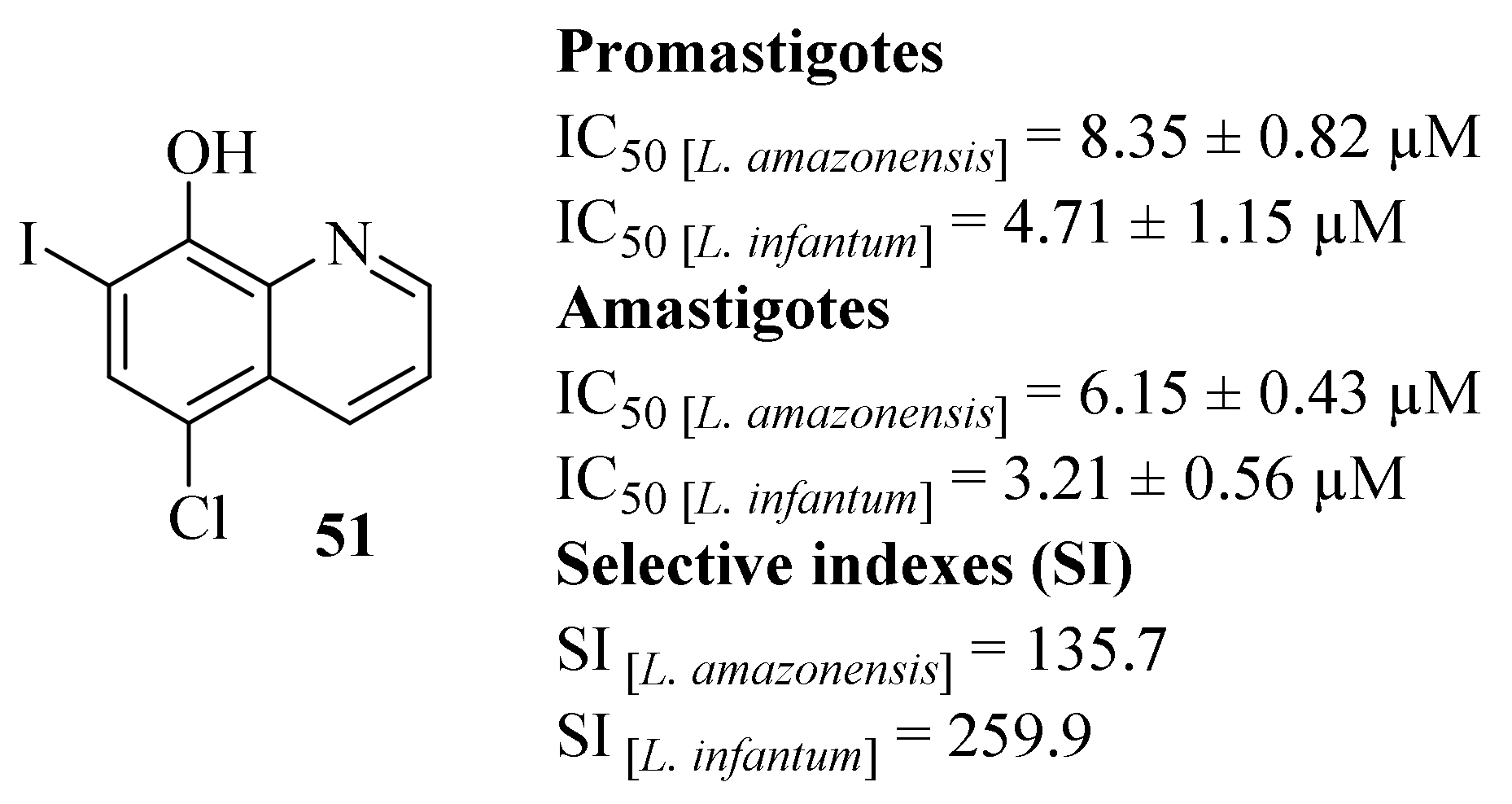
By the beginning of 2018, Tavares et al. decided to fully evaluate the compound 5-chloro-7-iodoquinolin-8-ol, also known as clioquinol (
51,
Figure 17) for its potential against
L. infantum and
L. amazonensis promastigotes and amastigotes, as well as its cytotoxicity effects against murine macrophages and human red cells [
47]. Considering its effects on
Leishmania promastigotes, clioquinol (
51) demonstrates promising antileishmanial properties against both
Leishmania species, with IC
50 = 8.35 ± 0.82 µM and 4.71 ± 1.15 µM against
L. amazonensis and
L. infantum, respectively. In turn, regarding
Leishmania axenic amastigotes, this compound also demonstrates high levels of activity, with IC
50 = 6.15 ± 0.43 µM (
L. amazonensis) and 3.21 ± 0.56 µM (
L. infantum), being more active against this second stage of the parasite. Comparing with the reference drug amphotericin B (AmB), even though clioquinol (
51) presents slightly lower levels of antileishmanial properties, this compound was considerably less toxic against murine macrophages, originating high SI values (135.7 for
L. amazonensis and 259.9 for
L. infantum). Furthermore, the compound’s mechanism of action was also evaluated against
L. amazonensis, allowing us to observe that the treatment of promastigotes with clioquinol (
51) induces changes in cell mobility and morphology. In particular, the effects promoted by the treatment with clioquinol (
51) include a significant cell volume reduction, alterations in the mitochondrial membrane potential and also the induction of oxidative stress, culminating in the rupture of the plasma membrane. Finally, the toxicity of this compound was also evaluated in BALB/c mice demonstrating that the administration of clioquinol (
51) presents no toxicity in this animal model, thus being considered safe for therapeutic usage and future developments for the treatment of
Leishmania-infected hosts.
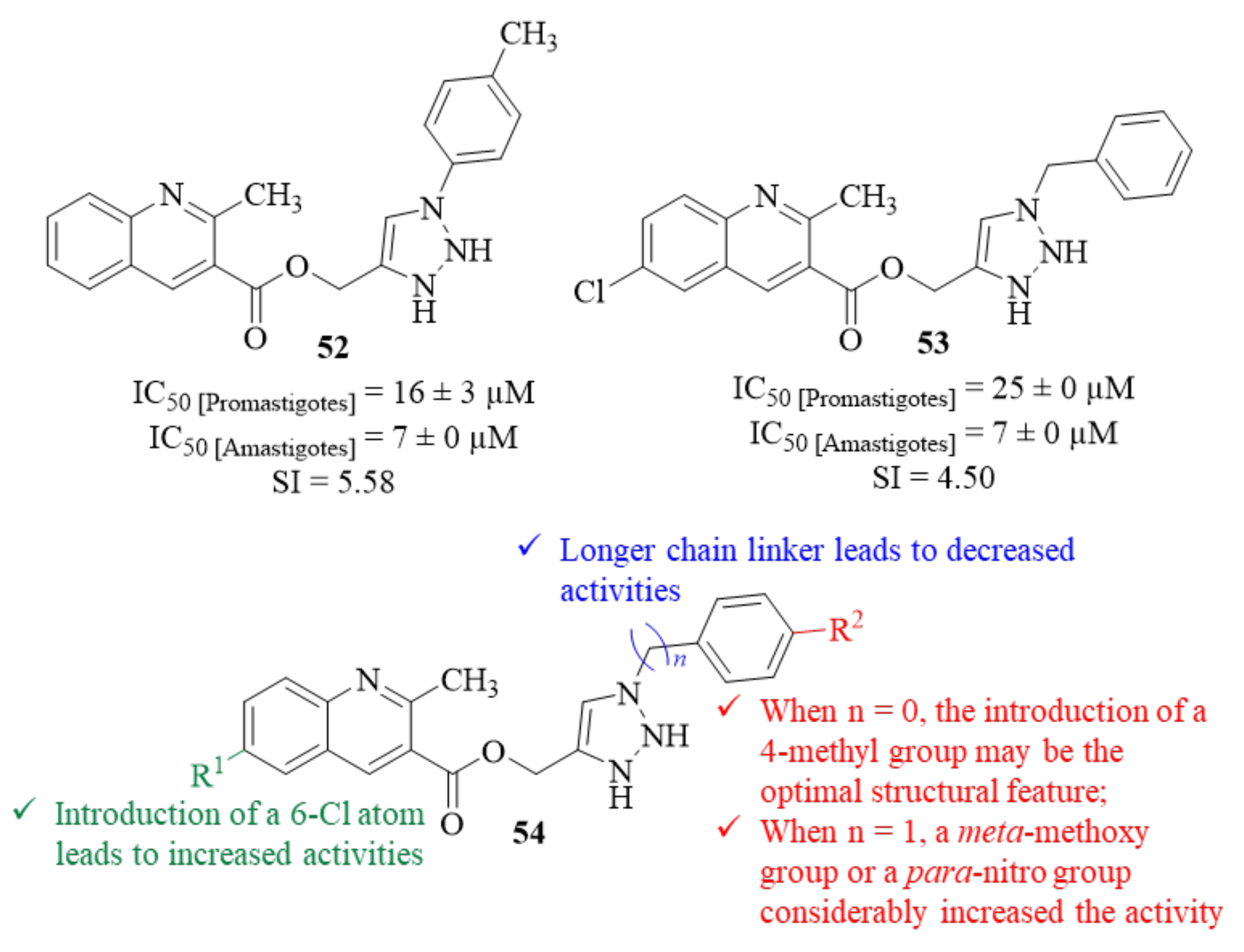
In the same year and based on previous reports on the antileishmanial effects of both quinoline and 1,2,3-triazole containing compounds, another research group followed the hybridization strategy to design a series of twenty-five quinoline-triazole hybrids [
48]. These hybrids were then evaluated for their potential as antileishmanial agents against
L. donovani promastigotes and amastigotes. Considering its effects on
Leishmania promastigotes, the results demonstrated that this entire series present moderate to considerable antileishmanial activities (IC
50 = 2.76–45.75 µM). However, regarding the intracellular amastigotes, only nine derivatives present considerable levels of antileishmanial activities, with particular emphasis on derivatives
52 and
53 (IC
50 = 7 ± 0 µM,
Figure 18) for presenting activity levels comparable to the reference drug miltefosine (IC
50 = 8 ± 2 µM). Structurally, the results indicated that the distance between the triazole and phenol groups has a considerable influence on these derivatives’ antileishmanial properties (54,
Figure 18). This suggestion is clearly corroborated by the fact that, for two derivatives with the same substitution pattern and only varying the linker length, the one with longer chain (
n = 1, IC
50 = 22 ± 0 µM) presents a considerable decrease of activity when compared with base derivative (
n = 0, IC
50 =7 ± 0 µM). Considering the substitution pattern introduced in phenol fragment, it was possible to verify that the introduction of a 4-methyl group may be the optimal structural feature for this type of activity for compounds with
n = 0, originating the most active compound against
L. donovani amastigotes (IC
50 =7 ± 0 µM). However, when
n = 1, the presence of this 4-methyl group seems to be less effective, leading to a significant decrease in activity (IC
50 =34 ± 1 µM). In this case, the replacement of the 4-methyl group by both an electron donating group (3-methoxy group) at
meta-position, or an electron-withdrawing group (4-nitro group) considerably increased the compound’s antileishmanial activity (IC
50 = 22 ± 0 µM and IC
50 = 18 ± 0 µM, respectively). Finally, the introduction of a 6-Cl atom in the quinoline fragment originates derivatives with higher antileishmanial activities than those without any substitution in this position. When evaluated in vivo by monitoring the parasite burden of a golden hamster’s spleen, only derivative
53 demonstrated promising activity against
L. donovani intracellular amastigotes, presenting consistent levels of activity up to day 28 post-treatment (37.81 ± 10.46% [7th day] and 46.89 ± 4.26% [28th day]), with derivative
52 only demonstrating moderate levels of activity 28 days (40.36 ± 6.05% [28th day]).
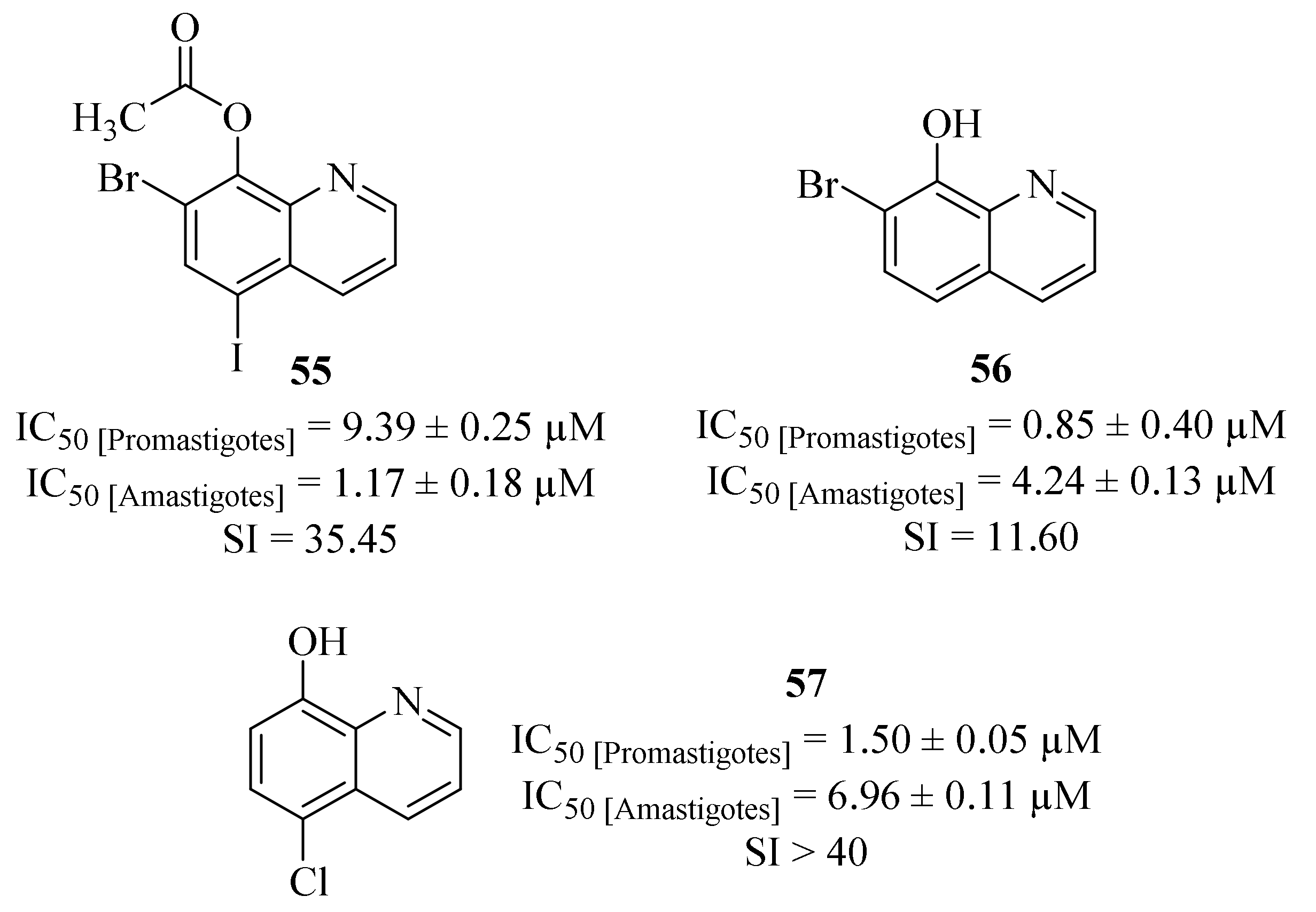
Another research group compiled a series of twenty-two quinoline derivatives, three commercially available and nineteen synthesized, and evaluated them against
L. (L.) amazonensis promastigotes and amastigotes, particularly a strain capable of inducing anergic diffuse cutaneous leishmaniasis [
49]. Considering its effects against
Leishmania promastigotes, all the evaluated derivatives demonstrate significant levels of antileishmanial activity (IC
50 < 10 µM), with seven of them being even more effective than the reference drug miltefosine (IC
50 = 7.88 ± 2.11. µM). In turn, regarding their potential against
Leishmania amastigotes, only eight derivatives were active against the intracellular stage of the parasite, presenting IC
50 values ranging from 1.17 ± 0.18 µM to 29.62 ± 1.43 µM, with most of them being more active than miltefosine (IC
50 = 31.36 ± 3.78 µM). From this series of derivatives, the most active compounds were derivatives
55 (IC
50 = 1.17 ± 0.18 µM),
56 (IC
50 = 4.24 ± 0.40 µM) and
57 (IC
50 = 6.96 ± 0.11 µM), with particular emphasis to derivative
57 for additionally presenting the highest SI value (SI > 40,
Figure 19). Unfortunately, the results obtained against
Leishmania amastigotes were not conclusive enough to establish a proper structure-antileishmanial activity relationship study for this type of compounds.
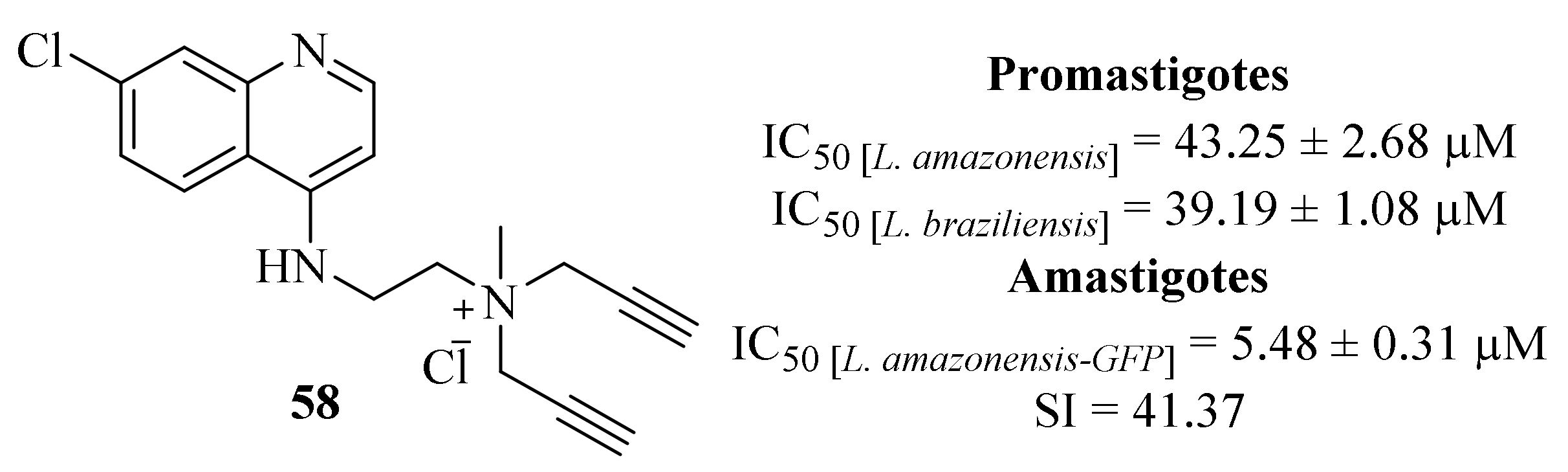
Still, in 2018, Calixto et al. synthesized a series of organic salts from active molecules, in a strategy intended to improve the biological and the physical-chemical properties of these compounds. Following a previous study in which some quinoline derivatives presented low levels of antileishmanial activities [
41], despite being considerably active against other protozoans, this research group focused their efforts on derivatizing these compounds into organic salts to achieve higher levels of antileishmanial activity against
L. amazonensis and
L. braziliensis promastigotes and amastigotes (
Figure 20) [
50]. Considering its effects against promastigotes, the results demonstrated that only one derivative (
58) is effective against both species of
Leishmania (IC
50 [
L. amazonensis] = 43.25 ± 2.68 µM and IC
50 [
L. brazilensis] = 39.19 ± 1.08 µM). Furthermore, this derivative (
58) was also the only active molecule against
L. amazonensis-GFP intracellular amastigotes (IC
50 = 5.48 ± 0.31 µM), a value similar to the observed against
L. amazonensis-Wild type amastigotes (IC
50 = 5.62 µM), while also presenting a low level of toxicity against murine macrophages (IC
50 = 226.70 ± 0.31 µM). In terms of mechanism of action, this compound (
58) induced a considerable reduction in the membrane potential and mitochondrial swelling, leading to its dysfunction and impairing the survival of the parasite. Furthermore, the treatment of promastigotes with this compound (
58) promoted several morphological modifications such as rounded bodies and reduction of cell volume, alterations usually associated with apoptosis-like cell death. Finally, the treatment with this compound (
58) also inhibits the formation of autophagic vacuoles while promoting the production of ROS, leading to an accelerated cell death.
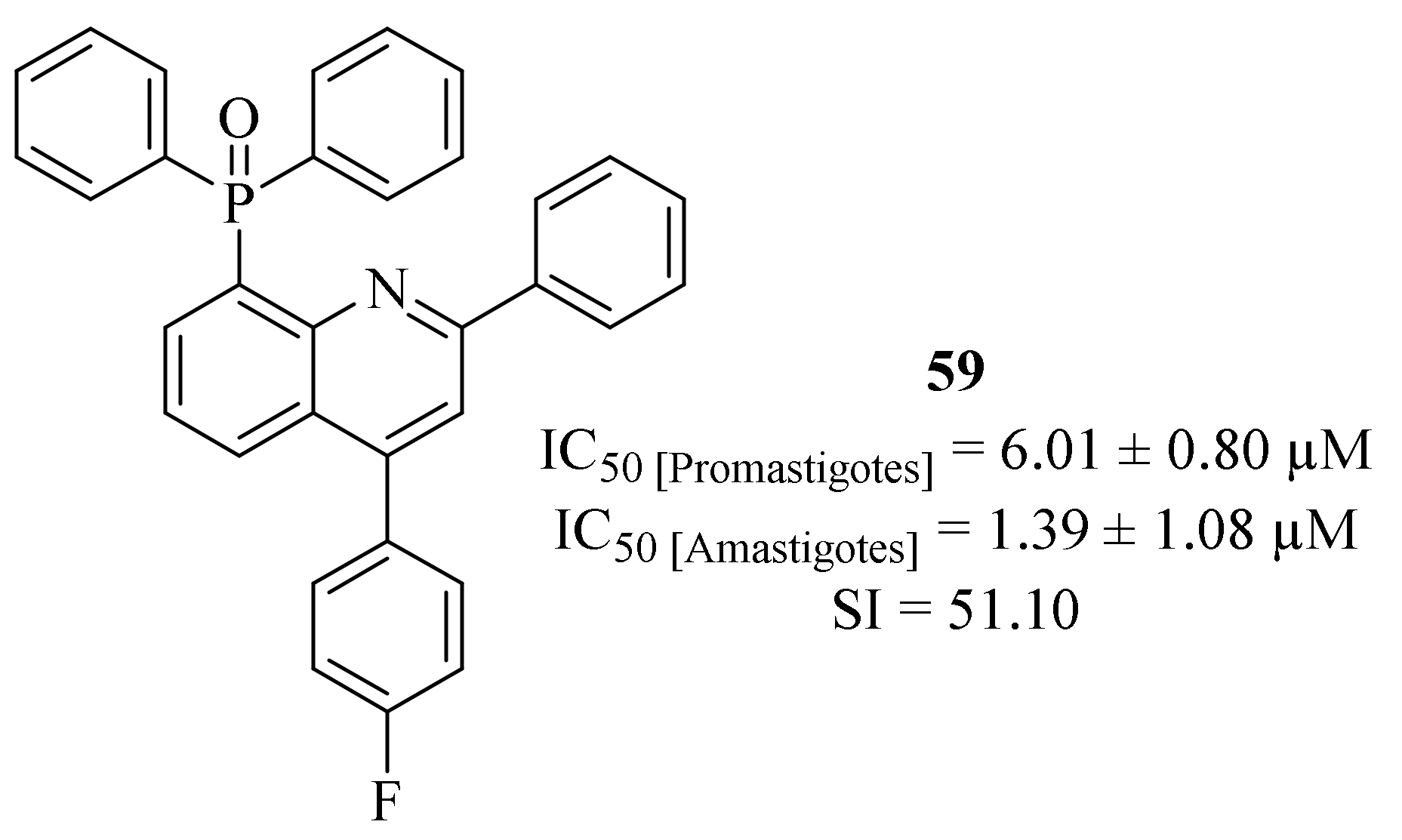
By the end of 2018, Tejería et al. synthesized a series of five quinoline derivatives containing phosphorus substituents such as phosphine, phosphine sulfide and phosphine oxide groups, and evaluated them against
L. infantum promastigotes and amastigotes [
51]. The results demonstrated that the quinoline derivatives containing phosphine oxide groups were considerably more active against both stages of the parasite than the ones containing phosphine sulfide groups, with the latter group presenting non considerable values of activity against intracellular amastigotes (IC
50 > 10 µM). The quinoline derivatives containing phosphine oxide groups presented significant levels of antileishmanial activity against both promastigotes (IC
50 = 2.33 ± 0.25–6.01 ± 0.80 µM) and intracellular amastigotes (IC
50 = 1.39 ± 1.08–4.14 ± 1.64 µM). In particular, it is important to highlight derivative
59 (
Figure 21) for, not only being the most active quinoline derivative against intracellular amastigotes (IC
50 = 1.39 ± 1.08 µM), but also for presenting the higher value of selectivity index (SI = 51.10).
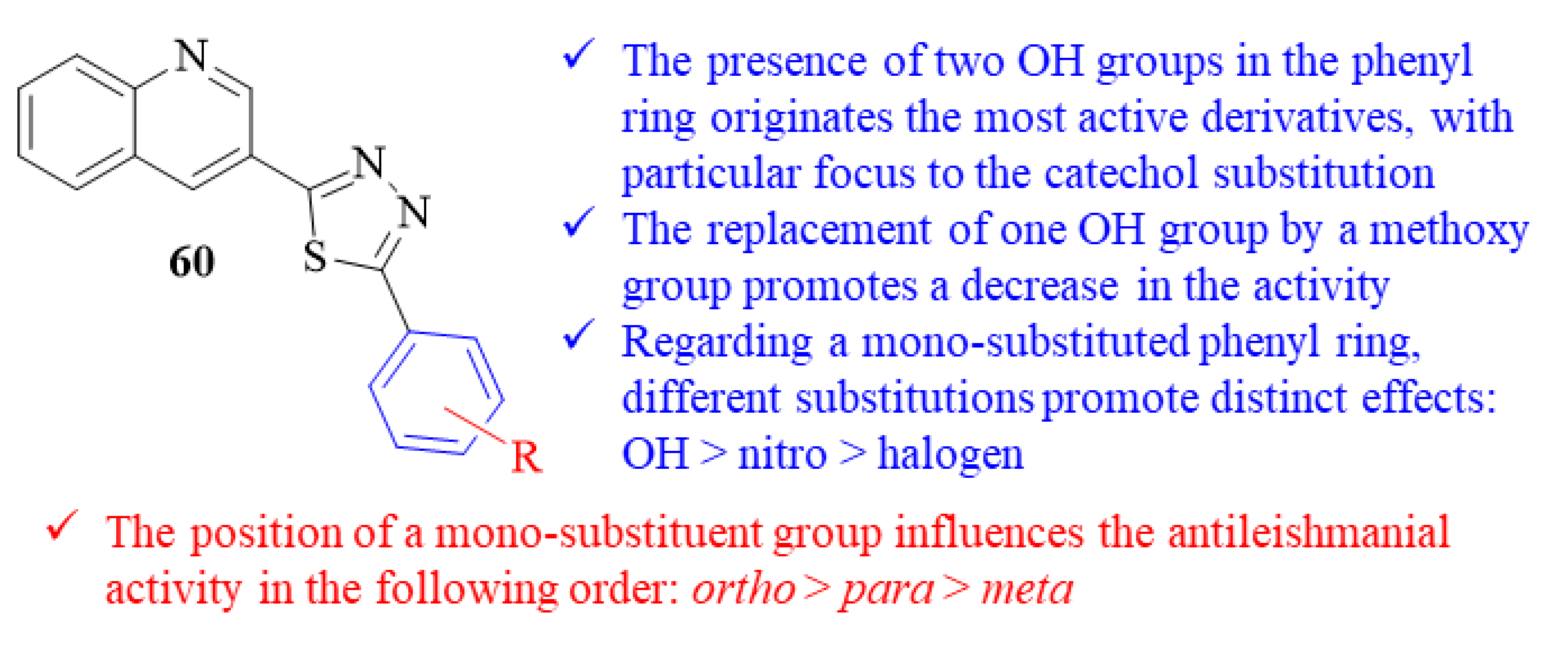
At the same time, another research group focused their efforts on designing a series of twenty quinoline-thiadiazole hybrids and evaluated them against
L. major intracellular amastigotes [
52]. From this series of twenty quinoline-thiadiazole hybrids, sixteen derivatives presented levels of antileishmanial activity comparable to the reference drug pentamidine (IC
50 = 7.02 ± 0.09 µM), with IC
50 values ranging from 0.04 ± 0.01 µM to 5.60 ± 0.21 µM. Structurally, the results demonstrated that the presence of two hydroxy groups in the phenyl ring originates the most active derivatives from the entire series (IC
50 = 0.04 ± 0.01–0.90 ± 0.10 µM), with particular focus to the one bearing a catechol substitution (
Figure 22). Furthermore, the replacement of one of these hydroxy groups by a methoxy group clearly promotes a decrease in the compounds’ antileishmanial activities (IC
50 = 2.10 ± 0.10–4.10 ± 0.20 µM). When it comes to derivatives with a mono-substituted phenyl ring, the presence of a hydroxy group originates derivatives with higher antileishmanial activities (IC
50 = 1.18 ± 0.10–3.40 ± 0.20 µM) than those bearing a nitro group (IC
50 = 4.68 ± 0.20–8.20 ± 0.35 µM) or a halogen atom (IC
50 = 0.98 ± 0.02–5.60 ± 0.21 µM). Interestingly, the position of these substitutions has a major influence on the compounds’ antileishmanial activities, with the results suggesting that the
ortho-position plays a vital role in this activity followed by the
meta- and
para-positions.
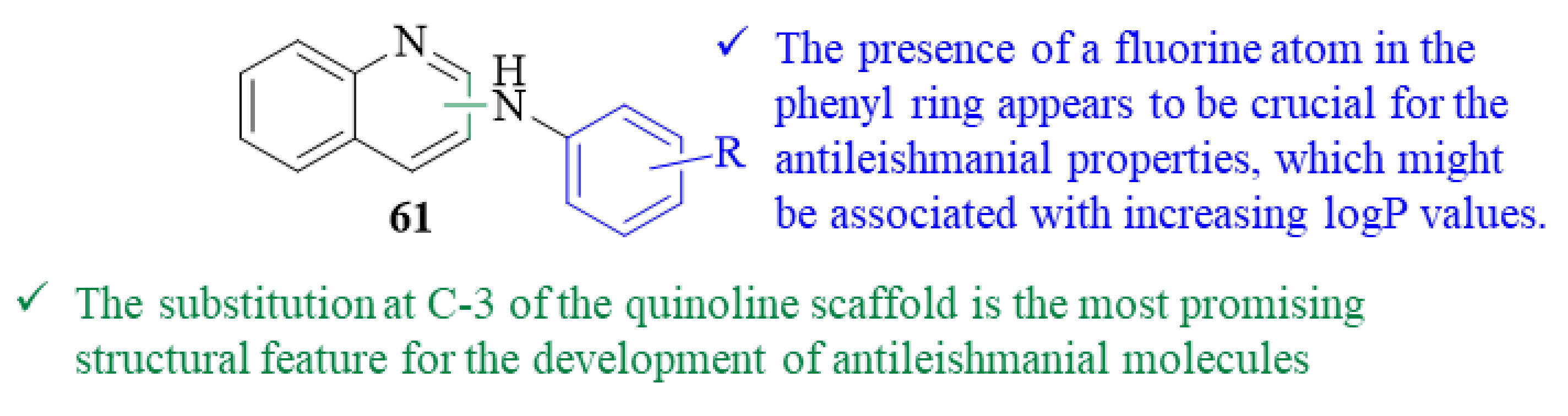
As part of a project intended to develop new safe chemotherapeutic agents against tropical diseases, Chanquia et al. synthesized a series of twelve aryl derivatives of 2- and 3-aminoquinoline and evaluated them for their antileishmanial potential against
L. mexicana promastigotes (61,
Figure 23) [
53]. After 6 days of incubation, the results demonstrated that four derivatives show moderate levels of antileishmanial activity by inhibiting the growth of the parasite, with IC
50 values ranging from 41.9 ± 0.8 µM to 98.1 ± 1.6 µM. The remaining eight derivatives showed no antileishmanial activity whatsoever, with IC
50 > 200 µM. Structurally, the most active compounds were the ones bearing a fluorine atom in the phenyl ring, suggesting that the presence of this type of substitution might be crucial for these compounds’ antileishmanial properties. The authors suggest that this influence might be associated with the improved logP value promoted by the fluorine atom, which may facilitate cell membrane permeation. Furthermore, since three of the four active molecules consist in 3-aminoquinoline derivatives, one might assume that the substitution at C-3 of the quinoline scaffold is the most promising structural feature for the development of antileishmanial molecules.
In the beginning of 2019, Abdelwahid et al. synthesized a series of fifteen quinoline-4-carboxylic acids and evaluated them for its potential as antileishmanial agents against
L. donovani promastigotes [
54]. The results demonstrated that, from this entire series of derivatives, five derivatives present moderate to weak antileishmanial activities, with IC
50 values ranging from 75.46 µM to 313.86 µM. Interestingly, one derivative (
63, IC
50 = 7.96 µM,
Figure 24) emerged as being two times more potent than AmB (IC
50 = 15.90 µM) and with an activity level comparable to the reference drug sodium stibogluconate (IC
50 = 8.85 µM). Structurally, it was possible to verify that the simple introduction of a 6-nitro group in the quinoline scaffold leads to a complete depletion of the compound’s antileishmanial activity (IC
50 = 7.96–925.93 µM). However, when considering 2-phenyl-4-carboxylic acid quinoline derivatives, the effect promoted by the 6-nitro group appears to be dependent on the substitution pattern on the phenyl fragment. In particular, in the presence of an EDG, like hydroxy or methoxy groups, the introduction of the 6-nitro group has a weak to no effect on the antileishmanial activity (IC
50 = 791.65–683.28 µM and IC
50 = 741.14–311.45 µM, respectively). In turn, in the presence of a 4′-bromide atom in the phenyl fragment, the same 6-nitro introduction promotes a significant improvement in the compound’s antileishmanial properties (IC
50 = 313.86–46.07 µM). Also, in these 2-phenyl quinoline-4-carboxylic acid derivatives, the introduction of a 6-bromide atom has moderate to significant effects in the compounds’ antileishmanial activity (From IC
50 = 313.86–741.14 µM to IC
50 = 75.46–313.81 µM). Finally, when a naphthalene fragment at C-2 of the quinoline scaffold, the introduction of a 6-bromide atom leads to a considerable loss of activity (IC
50 = 96.85–509.49 µM).
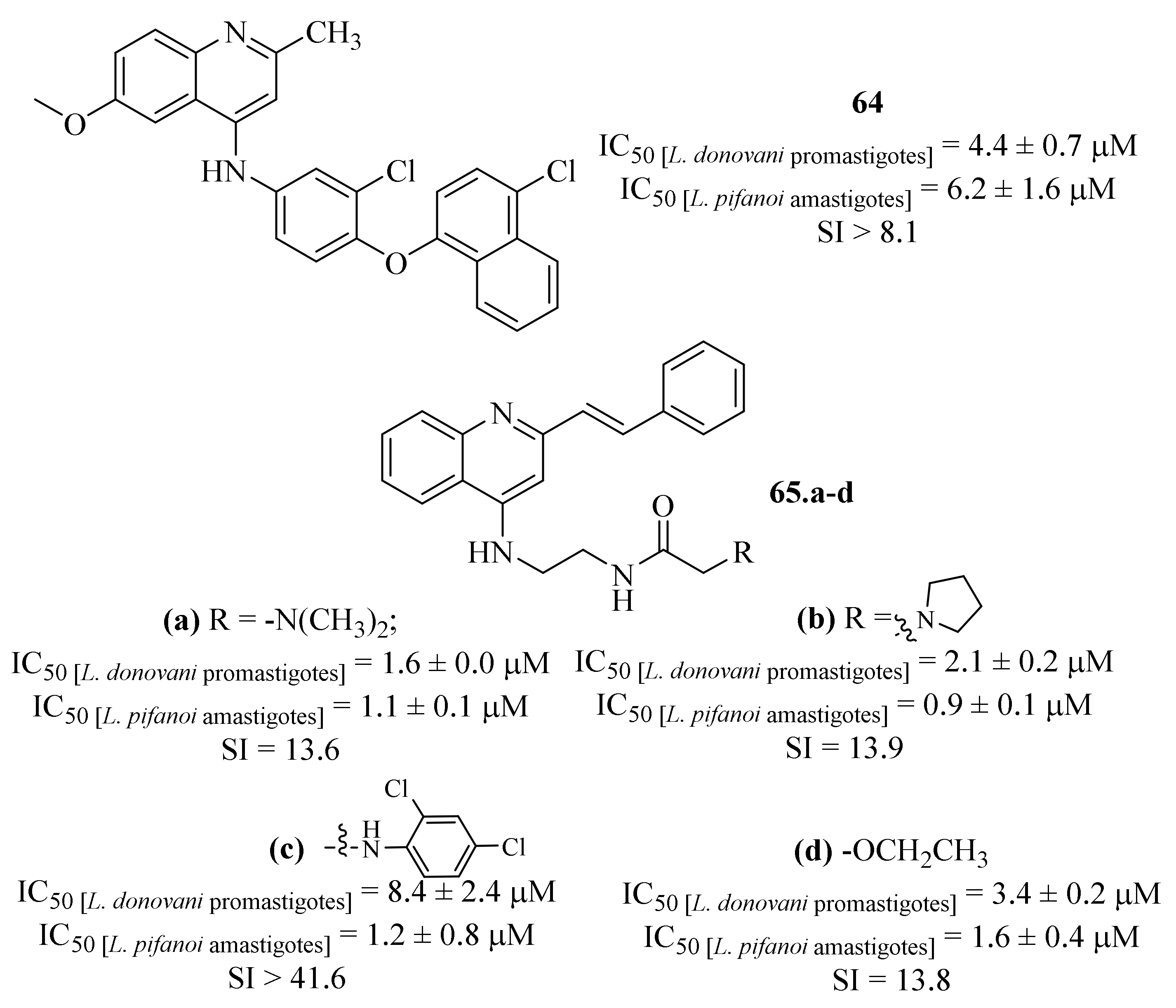
Based on the widely known potential of quinoline derivatives as antileishmanial agents, another research group designed a new class of 4-aminostyrylquinolines and evaluated them against
L. donovani promastigotes and
L. pifanoi amastigotes [
55]. Furthermore, some other derivatives were also synthesized and evaluated to serve as a control group, allowing a deeper understanding of the 4-aminostyrylquinolines’ structural influences. Considering their effects against
L. donovani promastigotes, the results demonstrated that, except for 2-styrylquinoline, all the evaluated molecules present considerable antileishmanial properties, with IC
50 values ranging from 0.2 ± 0.0 µM to 35.1 ± 4.6 µM. Interestingly, most of the evaluated compounds were more potent than an already marketed antileishmanial quinoline (
64,
Figure 25) From this preliminary evaluation, it was already possible to verify the importance of a 2-styryl group since the removal of this particular structural feature leads to a considerable loss of antileishmanial activity (IC
50 = 0.5 ± 0.1–10.9 ± 2.2 µM). Regarding the
Leishmania amastigotes, once again most of the evaluated molecules present promising antileishmanial properties (IC
50 = 0.9 ± 0.1–13.4 ± 3.8 µM), with particular emphasis to six derivatives that presented IC
50 values below 1.5 µM. From these six most promising anti-amastigote derivatives, and considering their toxicity levels against J774 cells, four of them must be highlighted by presenting both promising antileishmanial properties and high levels of SI (
65.a-
d,
Figure 25) Mechanistically, the authors demonstrated that these compounds’ antileishmanial activity is closely related with their effect on the parasite’s mitochondria, particularly by promoting mitochondrial dysfunction.
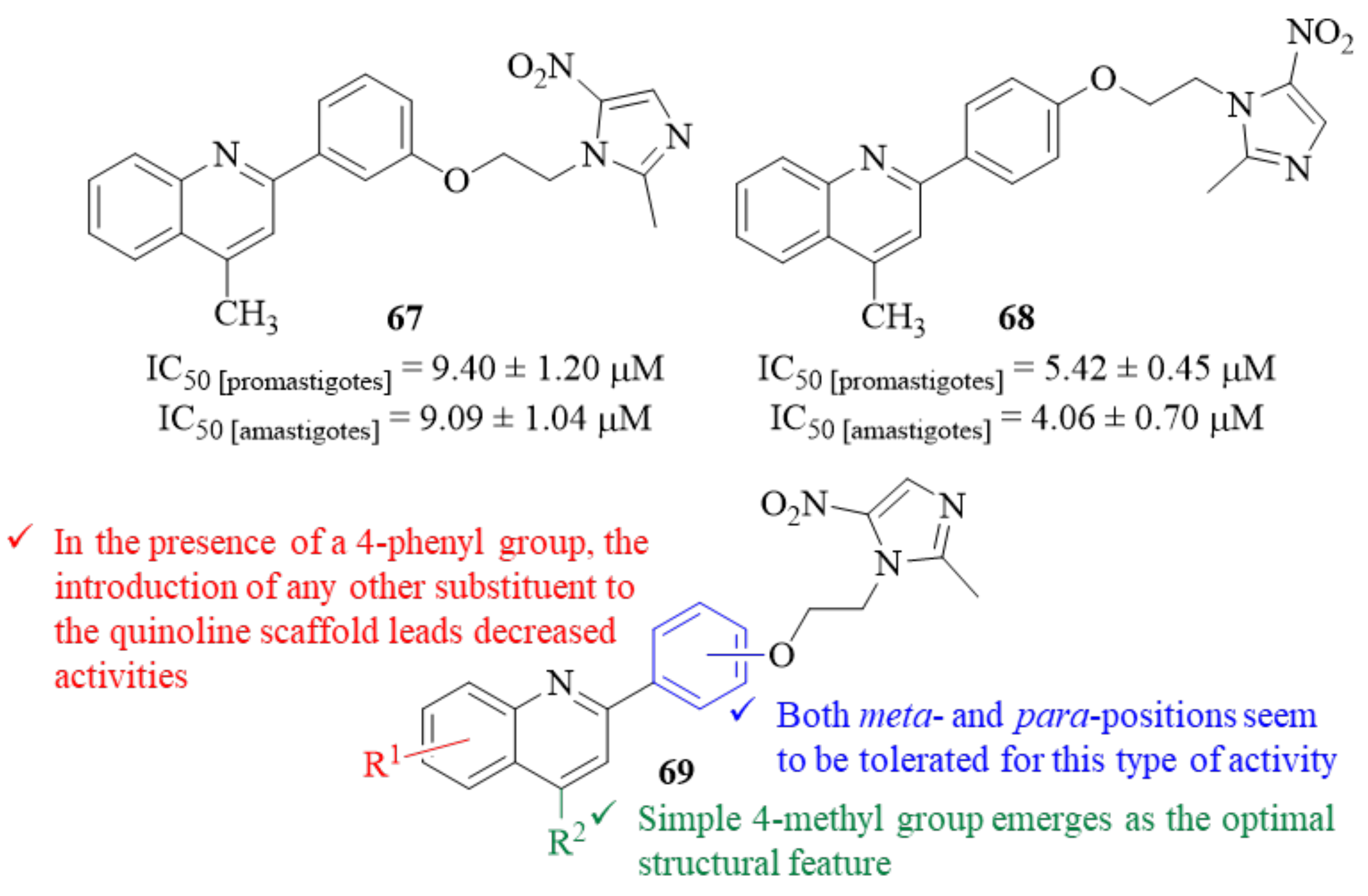
Still, in 2019, and in an attempt to identify novel chemical scaffolds for the development of antileishmanial molecules, Upadhyay et al. developed a series of thirteen quinoline-metronidazole hybrids and evaluated them against
L. donovani promastigotes and amastigotes [
56]. The results demonstrated that, from this series of thirteen hybrids, only two derivatives (
67 and
68,
Figure 26) exhibit significant activity levels against both promastigotes and amastigotes in the preliminary screening at 50 µM and 25 µM. Based on this, the IC
50 concentrations of these derivatives were also determined, with derivative
68 being the most active quinoline-metronidazole hybrid (IC
50 [promastigotes] = 5.30 ± 0.65 µM and IC
50 [amastigotes] = 4.06 ± 0.70 µM). Structurally, it was possible to verify that, regarding the substitution pattern in the quinoline fragment, a simple 4-methyl group emerges as the optimal structural feature for these compounds’ antileishmanial activity (
69,
Figure 26). Furthermore, in the presence of a 4-phenyl group, the introduction of any other substituent to the quinoline scaffold leads to a considerable decrease of activity. Regarding the hybridization position in the 2-phenyl group, both
meta- and
para-positions seem to be tolerated for this type of activity, originating compounds with similar activity levels (
67 and
68). Focusing on the potential of these two derivatives (
67 and
68), further in vivo evaluation was performed in a BALB/c model of VL, demonstrating that derivative
68 is much more effective in clearing parasite burden from both liver and spleen (>62% at 25 mg/kg) than derivative
67 (>50% at 50 mg/kg dose). Finally, in terms of mechanism of action, the authors demonstrated that derivative
68 is able to kill the parasite by inducing an apoptotic cascade, that begins with the disturbance of the mitochondria’s membrane potential, and also by promoting ROS and NO generation.
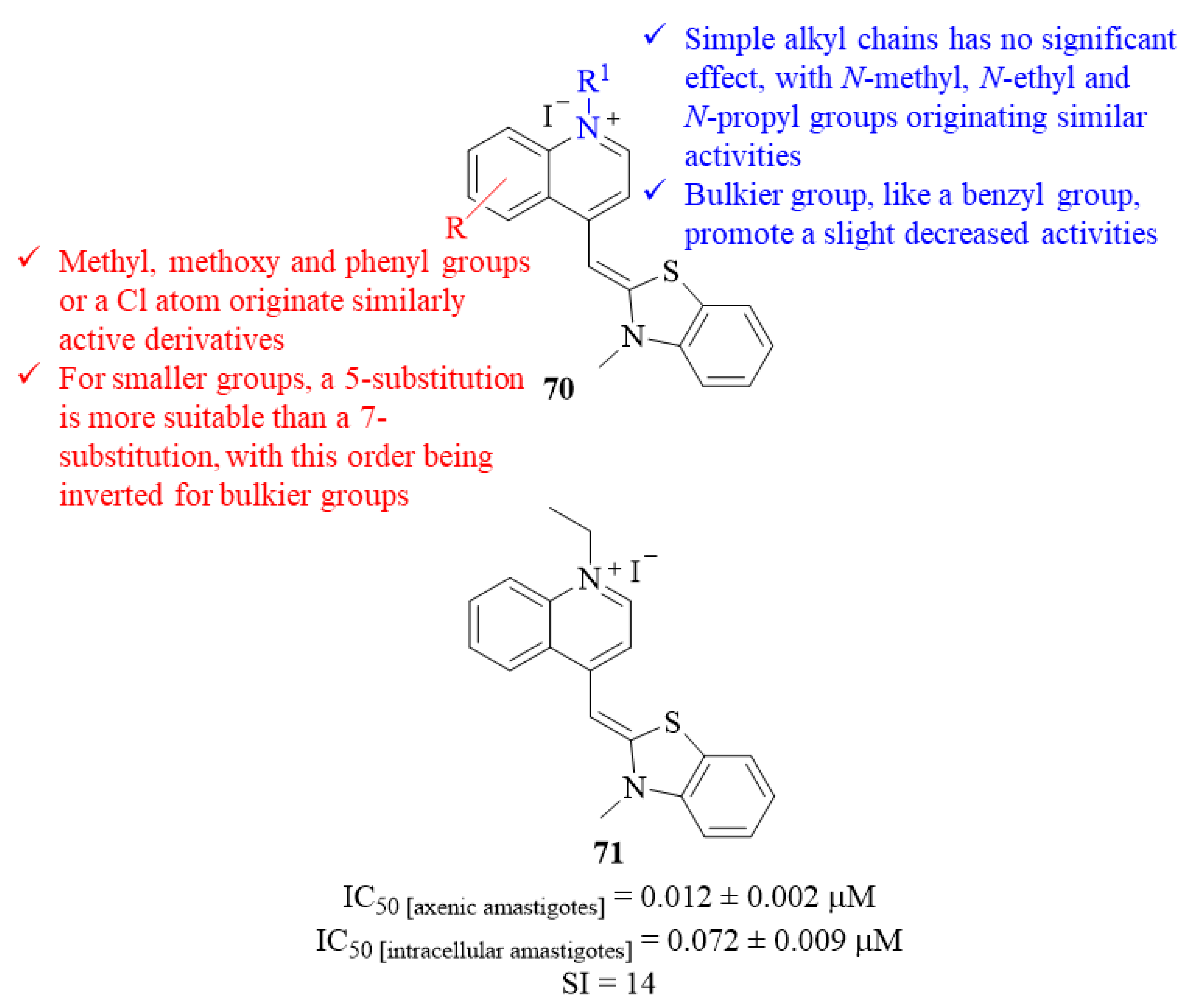
Another research group, following the previously reported potential of cyanine compounds, focused their efforts on the development of a series of twenty-one thiazole orange analogs, and evaluated them against
L. donovani axenic amastigotes [
57]. The results demonstrated that these compounds are considerably active, presenting IC
50 = 0.012 ± 0.002–0.042 ± 0.010 µM. Structurally, it was possible to verify that the introduction of simple alkyl chains in the nitrogen atom of the quinoline fragment has no significant effect on the compound’s antileishmanial activity, with
N-methyl (IC
50 = 0.014 ± 0.000 µM),
N-ethyl (IC
50 = 0.012 ± 0.002 µM) and
N-propyl (IC
50 = 0.013 ± 0.001 µM) groups originating derivatives with similar activities (
70,
Figure 27). In turn, the introduction of a bulkier group, like a benzyl group, promotes a slight decrease in the compound’s antileishmanial activity (IC
50 = 0.026 ± 0.004 µM). While maintaining the
N-methyl group, several modifications were also performed in the phenyl ring to understand the effects of different substituent groups and their position in the ring. The introduction of these different functional groups, such as methyl, methoxy and phenyl groups or a chlorine atom, had no considerable effect on the compounds’ antileishmanial properties originating derivatives with similar ranges of activity. However, it was still possible to understand that, for smaller groups, a 5-substitution originates the most active derivatives and a 7-substitution to the weaker derivatives, with this order being inverted for bulkier groups like a phenyl group. The relevance of both quinoline and benzothiazole fragments was assessed by replacing each of these fragments. These final modifications showed that replacing the benzothiazole fragment for a thiazole ring or the quinoline fragment for a pyridine ring leads to a considerable decrease in the compounds’ antileishmanial activities (IC
50 = 0.014 ± 0.000–0.160 ± 0.010 µM and IC
50 = 0.140 ± 0.030 µM, respectively). Finally, the most active compound (
71,
Figure 27) was also evaluated against
L. donovani intracellular amastigotes presenting a lower but still considerable level of activity (IC
50 = 0.072 ± 0.009 µM), being comparable to the activity presented by the reference drug AmB (IC
50 = 0.045 ± 0.009 µM).
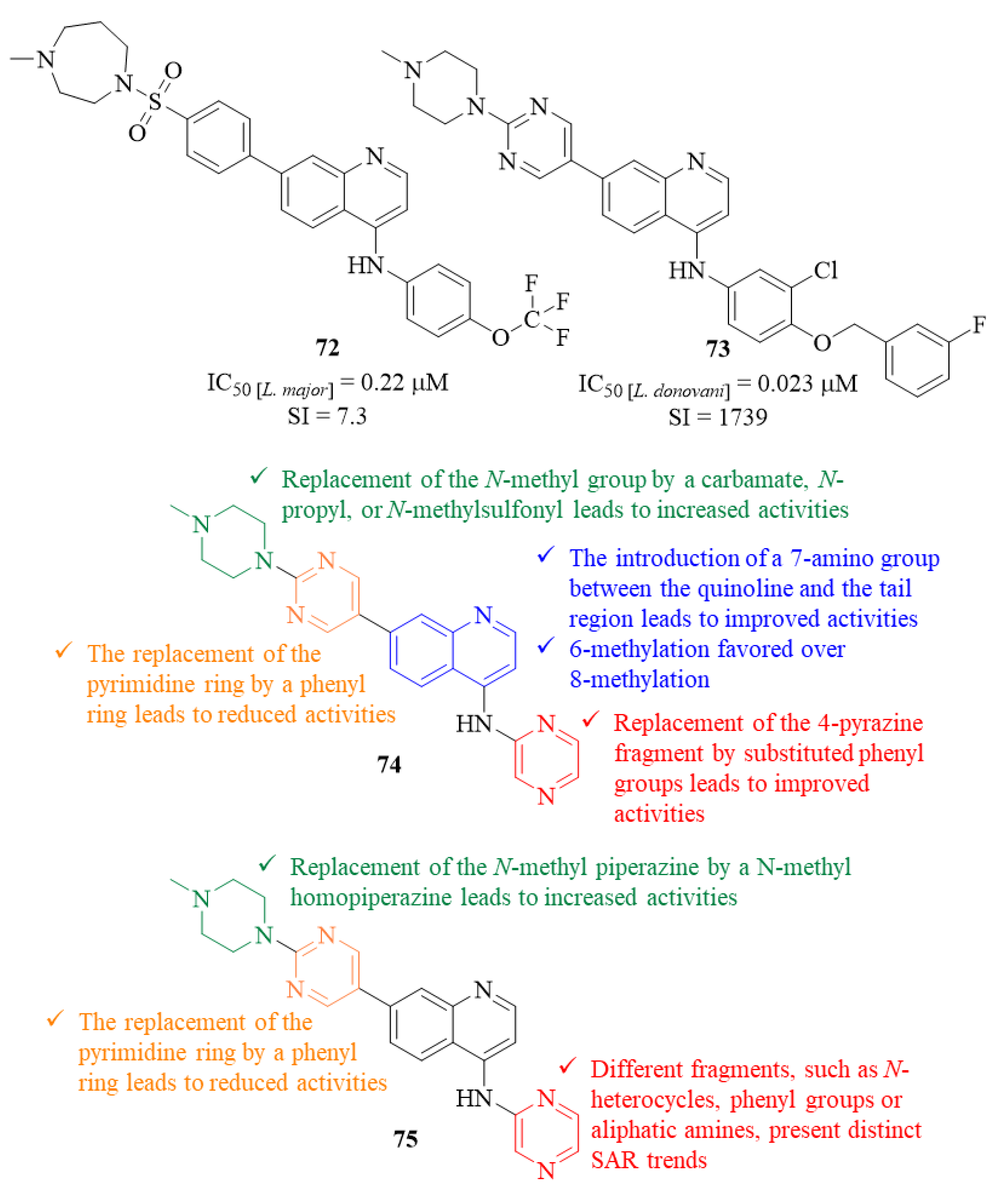
In the beginning of 2020, and following a target repurposing and parasite-hopping approach, Singh et al. developed a series of quinoline derivatives originated from the quinoline derivatives originated from the reoptimization of lapatinib to NEU-1953 and further optimizations of the latter [
58,
59,
60]. This entire series of quinoline derivatives was then evaluated for its antileishmanial potential against
L. major and
L. donovani intracellular amastigotes [
61]. Considering the effects against
L. major, several derivatives present considerable levels of antileishmanial activity, with particular emphasis on the most active derivative (
72,
Figure 28) that exhibited an IC
50 = 0.22 µM and a SI of 7.3. Structurally, for compounds containing a 4-pyrazine, the introduction of a 7-amino group between the quinoline and the tail region originates derivatives with significantly improved activities (IC
50 > 15–1.7-5.3 µM). Furthermore, in the tail region, the replacement of the pyrimidine ring by a phenyl ring leads to a reduced level of antileishmanial activity (IC
50 = 1.7–7.9 µM). Additionally, the replacement of the terminal
N-methyl group by a carbamate (IC
50 = 0.35 µM), a
N-propyl (IC
50 = 4.3 µM), or
N-methylsulfonyl (IC
50 = 0.73 µM) originates a significant increase of these compounds’ antileishmanial activities. Interestingly, the methylation of the quinoline fragment provides different effects depending on the position in which the methyl group was introduced, with a 6-methylation resulting in an improved activity (IC
50 = 1.5 µM) and an 8-methylation originating a complete loss of activity (IC
50 > 24 µM). Finally, the replacement of the 4-pyrazine fragment by substituted anilines resulted in derivatives with improved antileishmanial activities, particularly 3′-chloro-4′-methoxyaniline (IC
50 = 1.6 µM) and 4-(trifluoromethoxy)aniline (IC
50 = 0.22 µM).
Following the positive hits provided by the evaluation against L. major, a selection of analogs was further evaluated against L. donovani, leading to distinct activity trends. The results demonstrated that this additional evaluation identified three molecules with low micromolar inhibition (IC50 < 10 µM) and four with submicromolar activity levels (IC50 < 1 µM), with particular emphasis on derivative 73 that presents an IC50 = 0.023 µM and a SI of 1739. Structurally, considering the head fragment, the replacement of the 4-pyrazine ring by a tetrahydropyran originates a derivative with a significantly improved activity (IC50 > 15–4.0 µM). Furthermore, the combination of two distinct modifications, in this case the replacement of the piperazine with the homopiperazine and the pyrazine with 4-(trifluoromethoxy)aniline, led to the formation of one of the most active compounds from this series (IC50 = 0.085 µM).
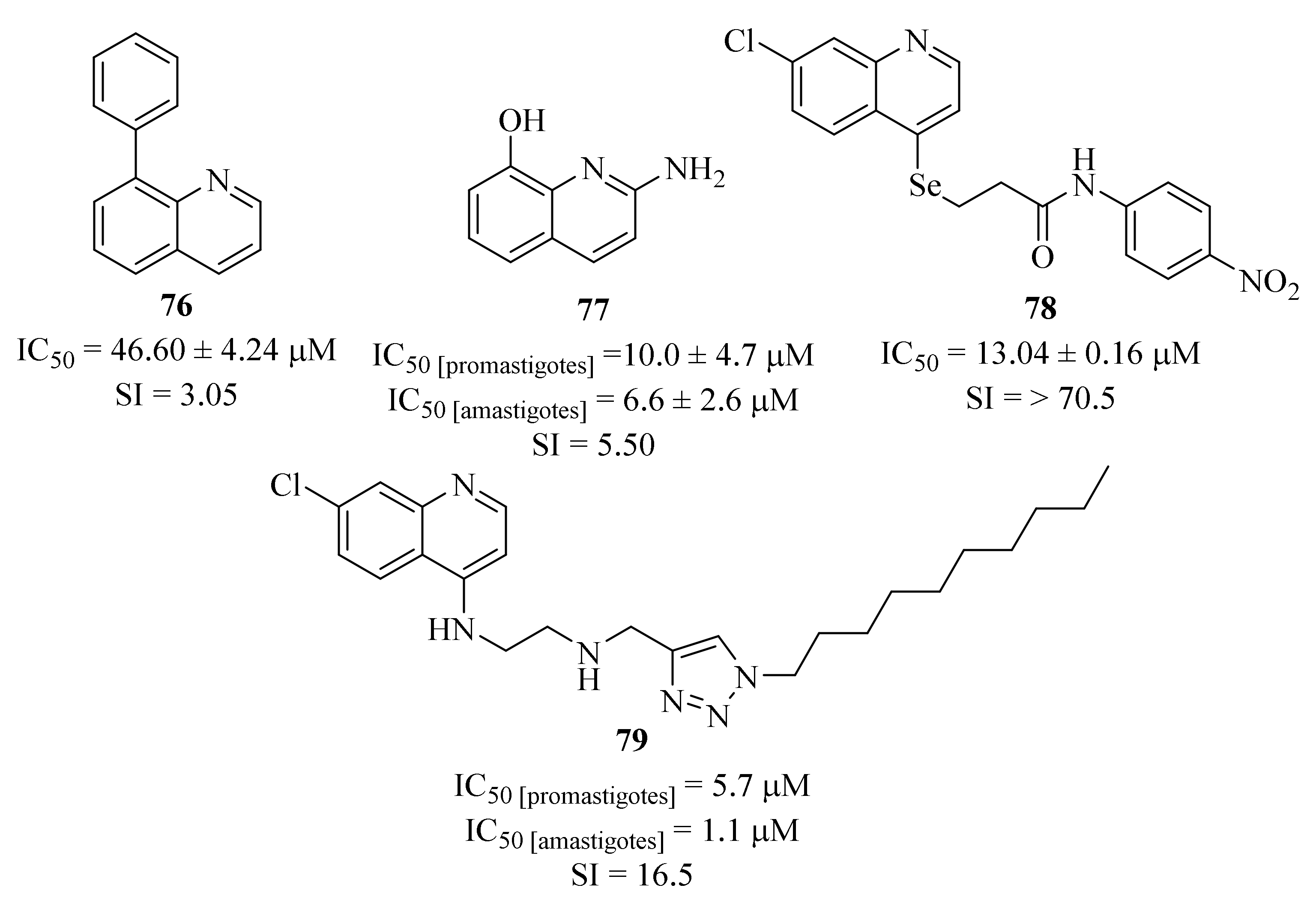
In the same year, another research group developed a series of eleven quinoline-biphenyl hybrids in the search for new therapeutic alternatives for the treatment of cutaneous leishmaniasis and evaluated them against
L. (V) panamensis intracellular amastigotes [
62]. The results demonstrated that, from the eleven derivatives, only five were moderately active against intracellular amastigotes (IC
50 < 50 µM), with particular emphasis on derivative
76 (
Figure 29) that presents an antileishmanial activity comparable with the reference drug meglumine antimoniate (IC
50 = 46.60 ± 4.24 µM and 25.69 ± 5.74 µM, respectively). Structurally, it was possible to verify that the introduction of any substitution pattern in the phenyl ring would lead to a considerable decrease in the antileishmanial activity. Nevertheless, one could still assess that the effect promoted by the introduction of a methyl group is most accentuated that the one induced by a hydroxy group.
By the end of 2020, Suarez et al. used in silico techniques to identify synthetic quinoline alkaloids with a structure similar to the natural product
N-methyl-8-methoxyflindersine and evaluated them against
L. (V.) panamensis promastigotes and amastigotes [
63]. The results demonstrated that, from the entire list of evaluated derivatives, only one (
77,
Figure 29) presents considerable levels of antileishmanial activity (IC
50 [promastigotes] = 10.0 ± 4.7 µM and IC
50 [amastigotes] = 6.6 ± 2.6 µM) and low levels of toxicity (SI = 5.5). This compound was further evaluated in an animal model of
L. (V.) panamensis infection, demonstrating a 50% cure rate in addition to neutrophil and macrophage migration. In terms of mechanism of action, this compound is able to induce cell apoptosis in both promastigotes and intracellular amastigotes, leading to the inhibition of parasitic growth and development.
In 2021, and following their previous success in identifying new candidates for antileishmanial drug development, Huang et al. performed a virtual screening on a series of selenide-derived quinoline derivatives, synthesized the most likely to be active and evaluated them against
L. amazonensis [
64]. The biological evaluation of these compounds corroborates the prediction of the virtual screening, with the derivatives predicted to be active demonstrating promising antileishmanial activity levels (IC
50 = 13.04 ± 0.16–171.11 ± 14.10 µM), with particular emphasis to derivative
78 (IC
50 = 13.04 ± 0.16 µM).
In the same year, another research group focused their efforts on the synthesis of a novel series of quinoline-triazole hybrids and evaluated them against
L. amazonensis promastigotes and intracellular amastigotes [
65]. Considering both promastigotes and amastigotes, the results demonstrated that only one derivative (
79,
Figure 29) presents promising antileishmanial properties with an IC
50 values of 5.7 µM and 1.1 µM against promastigotes and amastigotes, respectively. In terms of mechanism of action, this quinoline derivative exerts its antileishmanial effects by inducing a pronounced reduction of the mitochondrial membrane potential, leading to the disruption of its function. Furthermore, this disruption accelerates the generation of ROS and can also culminate in the collapse of the bioenergetic metabolism of the parasite. In conclusion, this derivative induces modifications to biochemical processes through the interference in the bioenergetic system and plasma membrane permeabilization, with consequent activation of apoptosis-like and necrosis processes, culminating in cell death.
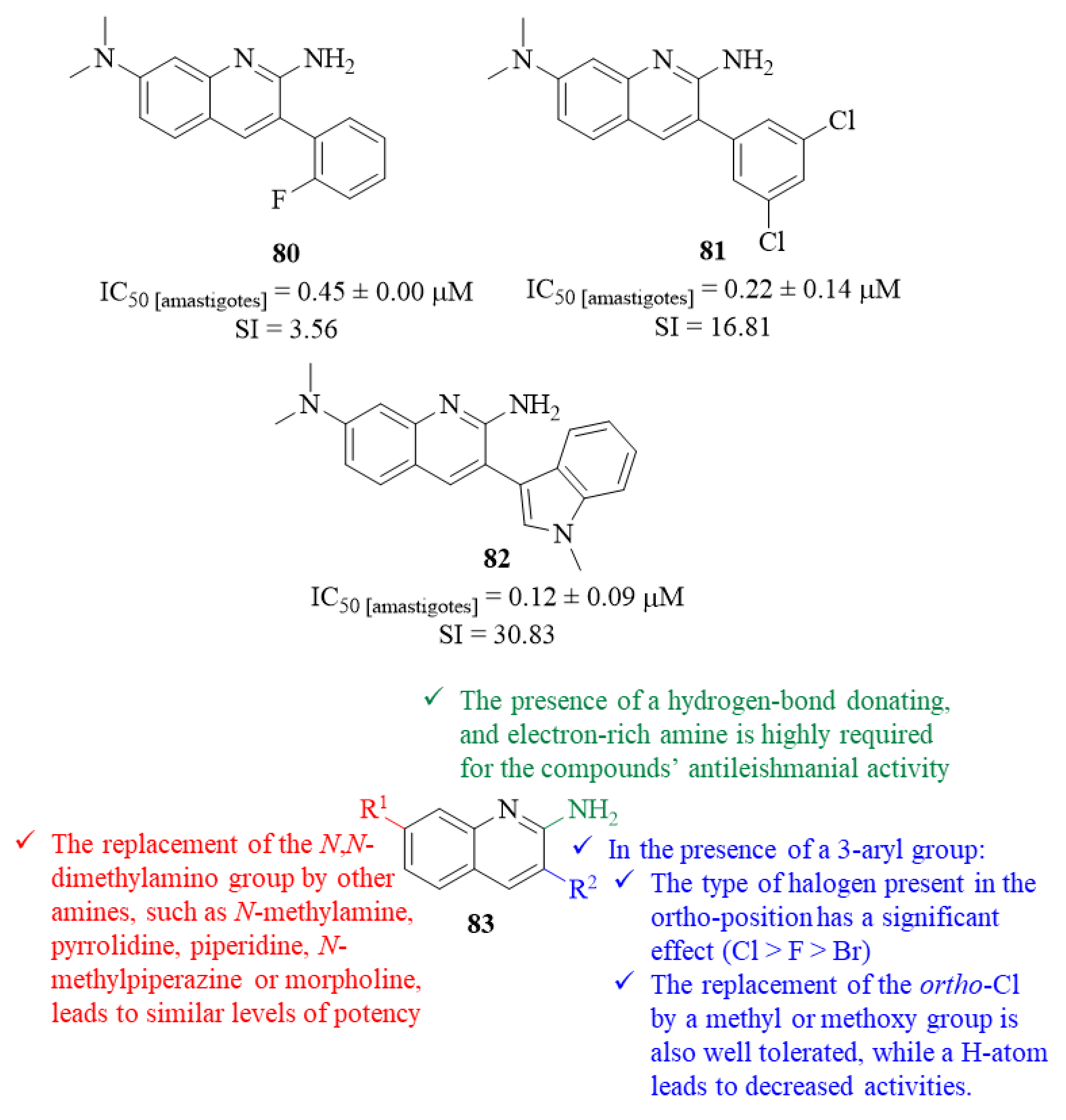
Hammill et al. continued their previous work by developing a novel series of 3-arylquinoline derivatives and evaluating them for their antileishmanial potential against
L. mexicana intracellular amastigotes [
66]. In this work, a phenotypic high-throughput screening was performed to identify novel antileishmanial leads, with more than 100 molecules being evaluated, leading to further dose–response assays for the most promising antileishmanial agents. To develop the most complete SAR study possible, this research group adopted a strategy based on the modification of four specific structural features, namely the importance of the quinoline scaffold itself and of the substituent group introduced at C-2 and C-7 positions and 3-aryl group, in comparison with derivative
80 (
Figure 30).
Considering the quinoline scaffold, the results demonstrated that the quinoline fragment is crucial for the compound’s antileishmanial activity since the introduction of additional nitrogen atoms into the quinoline scaffold, its replacement with other bicyclic heterocycles and its fusion to an additional heterocyclic ring originates weaker derivatives (IC
50 > 1.0 µM) than
80 (IC
50 = 0.45 ± 0.00 µM). Maintaining the same quinoline scaffold fragment, a series of modifications were performed at C-2 to provide a deeper understanding about the optimal structural features to introduce to this quinoline position. From these modifications, one can verify that the replacement of the 2-amino group by any other structural feature originates weaker derivatives (IC
50 > 1.0 µM), suggesting that the presence of a hydrogen-bond donating and electron-rich amine at this position is highly required for the compounds’ antileishmanial properties (
Figure 30).
Regarding the influence of the 3-aryl group, while maintaining a 2-amino group, the results demonstrated that the type of halogen present in the
ortho-position has a significant effect on the compounds’ antileishmanial properties, with an
ortho-Cl atom originating the most active derivative (IC
50 = 0.28 ± 0.07 µM), followed by
ortho-F (IC
50 = 0.45 ± 0.00 µM) and
ortho-Br (IC
50 = 0.61 ± 0.02 µM). However, the position of this Cl atom in the aryl ring seems to not affect the compounds’ antileishmanial properties, with all the derivatives presenting similar levels of activity (IC
50 = 0.36 ± 0.04 µM for
meta-Cl and IC
50 = 0.40 ± 0.03 µM for
para-Cl). In turn, the introduction of a second Cl atom promotes a wider range of antileishmanial activity levels, IC
50 = 0.22 ± 0.14–0.76 ± 0.09 µM, with the derivative containing two
meta-Cl atoms being the most active compound from this group (
81, IC
50 = 0.22 ± 0.14 µM, SI = 17). The replacement of the
ortho-Cl by a methyl or methoxy group does not affect the antileishmanial properties (IC
50 = 0.33 ± 0.02–0.30 ± 0.09 µM, respectively), while a hydrogen atom originates a considerably less active derivative (EC
50 = 0.63 ± 0.17 µM). Based on the concerns that derivative
81 could have chemical properties that might limit its in vivo potential (fairly high hydrophobicity with cLogP = 4.86), the 3- dichlorophenyl ring was also replaced by a series of different heterocycles in an attempt to improve the compound’s chemical properties while retaining potency and selectivity. This optimization demonstrated that compounds containing five-membered heterocycles were considerably less effective (IC
50 > 1.00 µM), while isosteric six membered heterocycles originate derivatives with similar antileishmanial activity levels (IC
50 = 0.33 ± 0.03–0.87 ± 0.17 µM). The presence of more sterically encumbered bicyclic heterocycles was also well tolerated (IC
50 = 0.12 ± 0.09–0.90 ± 0.84 µM), with derivative
82 being the most active from the group (IC
50 = 0.12 ± 0.09 µM), suggesting that the potential target presents a deep and flexible hydrophobic pocket in this region (
Figure 30).
Finally, while maintaining either the 3,5-dicholorophenyl ring or the N-methyl indole fragment, some modifications were also performed at 7-(N,N-dimethylamino) group to fully understand the steric and electronic tolerances of this specific position. The results demonstrated that the replacement of the N,N-dimethylamino group by other amines, such as N-methylamine, pyrrolidine, piperidine, N-methylpiperazine or morpholine, originates derivatives with similar levels of potency (IC50 = 0.37 ± 0.06–0.71 ± 0.33 µM for the 3,5-dicholorophenyl ring and IC50 = 0.16 ± 0.28–0.64 ± 0.15 µM for the N-methyl indole fragment). This evidence reveals that it is possible to modify this position in order to improve the physicochemical properties while retaining both potency and selectivity.
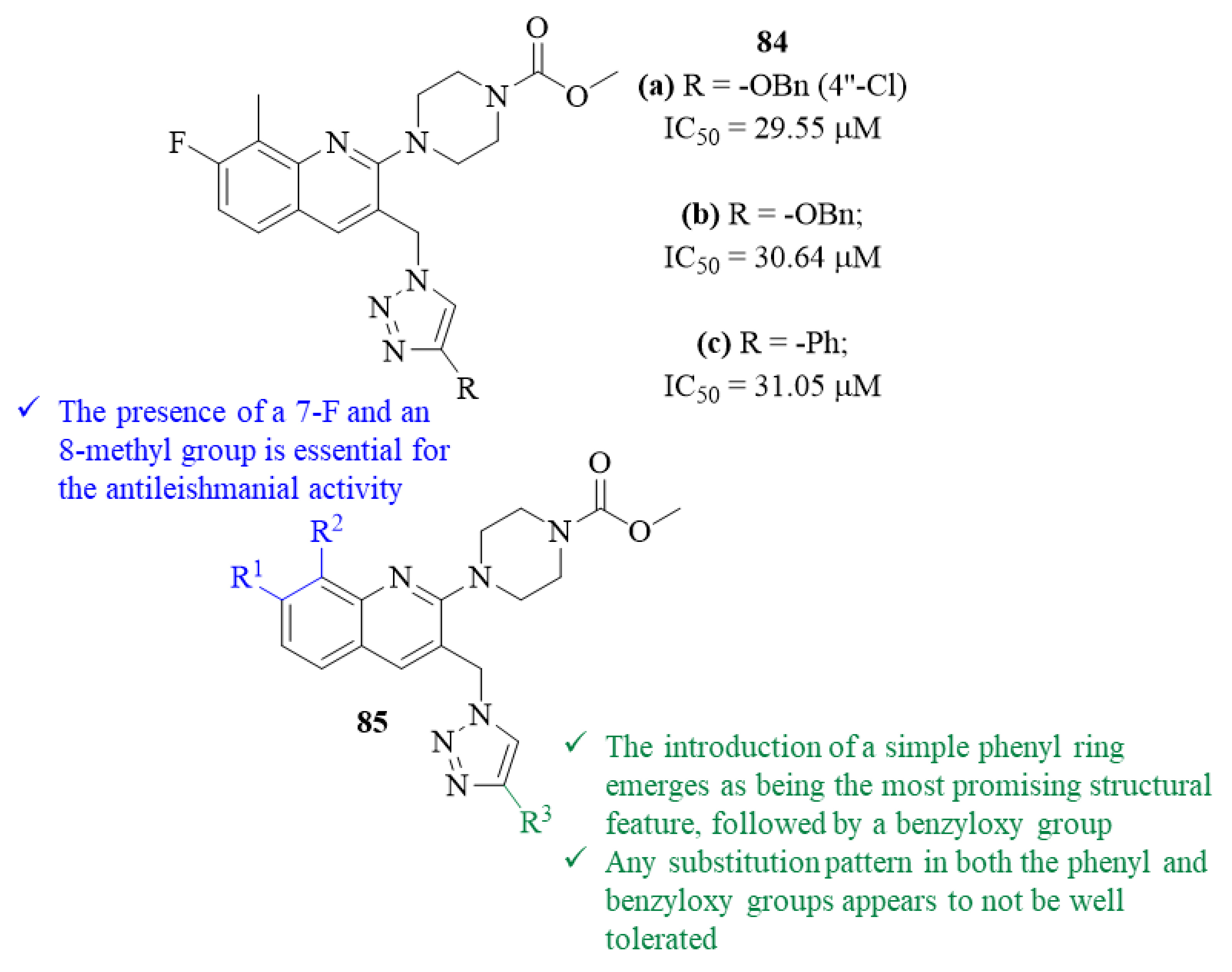
Still, in 2021, another research group developed a series of twelve novel quinoline-1,2,3-triazole hybrids and evaluated them against
L. donovani promastigotes, followed by molecular docking studies against
L. major pteridine reductase (
Lm-PTR1) [
67]. From their results, this research group was able to verify that three of the derivatives present significant levels of antileishmanial activity, with IC
50 values ranging from 29.55 µM to 31.05 µM, similar to the reference drug AmB. Structurally, the results also indicated that the substitution pattern on the quinoline scaffold has a considerable effect on the compounds’ antileishmanial properties. In particular, it was possible to verify that the presence of a 7-F and an 8-methyl group is essential for the compounds’ antileishmanial activity, with the three most active derivatives containing this substitution pattern (
Figure 31). Furthermore, the functional group introduced at C-4′ of the triazole ring also seems to significantly affect the antileishmanial properties of this type of compounds. In this case, the introduction of a 4″-chloro-benzyloxy group emerges as being the most promising structural feature, originating the most active compound from this series (
84.a, IC
50 = 29.55 µM), followed by the introduction of an unsubstituted benzyloxy group (
84.b, IC
50 = 30.64 µM) and a simple phenyl ring (
84.c, IC
50 = 31.05 µM). Except for a 4″-Cl atom in the benzyloxy group, the presence of any substitution pattern in both the phenyl and benzyloxy groups appears to not be well tolerated, originating derivatives with low to no activity whatsoever. In terms of mechanism of action, based on the molecular docking studies, the authors suggest that this might be associated with their effect on the enzyme PTR1, since all the derivatives demonstrate a significant binding affinity to the enzyme, with the theoretical results following the same pattern as the experimental ones.
One year later, in 2022, Sabt et al. developed a series of twenty quinoline-isatin hybrids and evaluated them for their antileishmanial activity against both
L. major promastigotes and amastigotes (
Figure 32) [
67]. Considering their activity against promastigotes, the results demonstrated that all the evaluated derivatives present considerable levels of antileishmanial activity with IC
50 values ranging from 0.51 ± 0.06 µM to 5.95 ± 0.28 µM, being more active than the reference drug miltefosine (IC
50 = 7.90 ± 0.26 µM). Structurally, it was possible to verify that the derivatives containing a
N-unsubstituted isatin fragment constitute the group of most active derivatives from this series, IC
50 = 0.51 ± 0.06–1.10 ± 0.14 µM, being more active than the parent hydrazine (IC
50 = 3.00 ± 0.34 µM). In turn, the introduction of substituent groups at
N-position leads to a considerable decrease in the compounds’ antileishmanial activities, when compared to their unsubstituted congeners (IC
50 = 1.74 ± 0.12–5.95 ± 0.28 µM). The reduction of antileishmanial activity promoted by the
N-substitution of the isatin core is also dependent on the functional group introduced, with a benzyl group causing a more accentuated decrease, while alkyl groups promote only a slight reduction. Finally, the substitution pattern of the
N-unsubstituted isatin core also seems to have a significant effect on the compounds’ antileishmanial potential, with a 5-Br originating the most active derivative (IC
50 = 0.51 ± 0.34 µM) followed by 5-F (IC
50 = 0.56 ± 0.22 µM), 5-CF
3 (IC
50 = 0.67 ± 0.34 µM) and 5-Cl (IC
50 = 0.67 ± 0.16 µM). Considering
L. major amastigotes, the results showed that, except for one derivative, all the evaluated compounds were more active against this stage of the parasite than miltefosine (IC
50 = 8.08 ± 0.22 µM), with IC
50 values ranging from 0.60 ± 0.04 µM to 8.29 ± 0.32 µM. Structurally, the
N-substitution of the isatin core promotes the same type of effect as observed against the promastigote stage, a decrease of the compounds’ antileishmanial activities, with the
N-unsubstituted derivatives being the most active compounds against amastigotes (IC
50 = 0.60 ± 0.04–2.45 ± 0.28 µM). This evidence, observed against both stages of the parasite, suggests that the presence of a group with the ability to form H-bonding interactions might be crucial for these compounds’ antileishmanial properties. In terms of mechanism of action, this research group evaluated the effects of the most active compounds against parasites supplemented with folic and folinic acids to assess if these compounds were acting against the parasite’s folate pathway. By adding folic and folinic acids, it was possible to verify the almost complete depletion of antileishmanial activity, similar to what happens with a known Lm-PTR1 inhibitor (trimethoprim). This fact confirms the anti-folate mechanism of this type of molecules through the inhibition of DHFR-TS and PTR1.
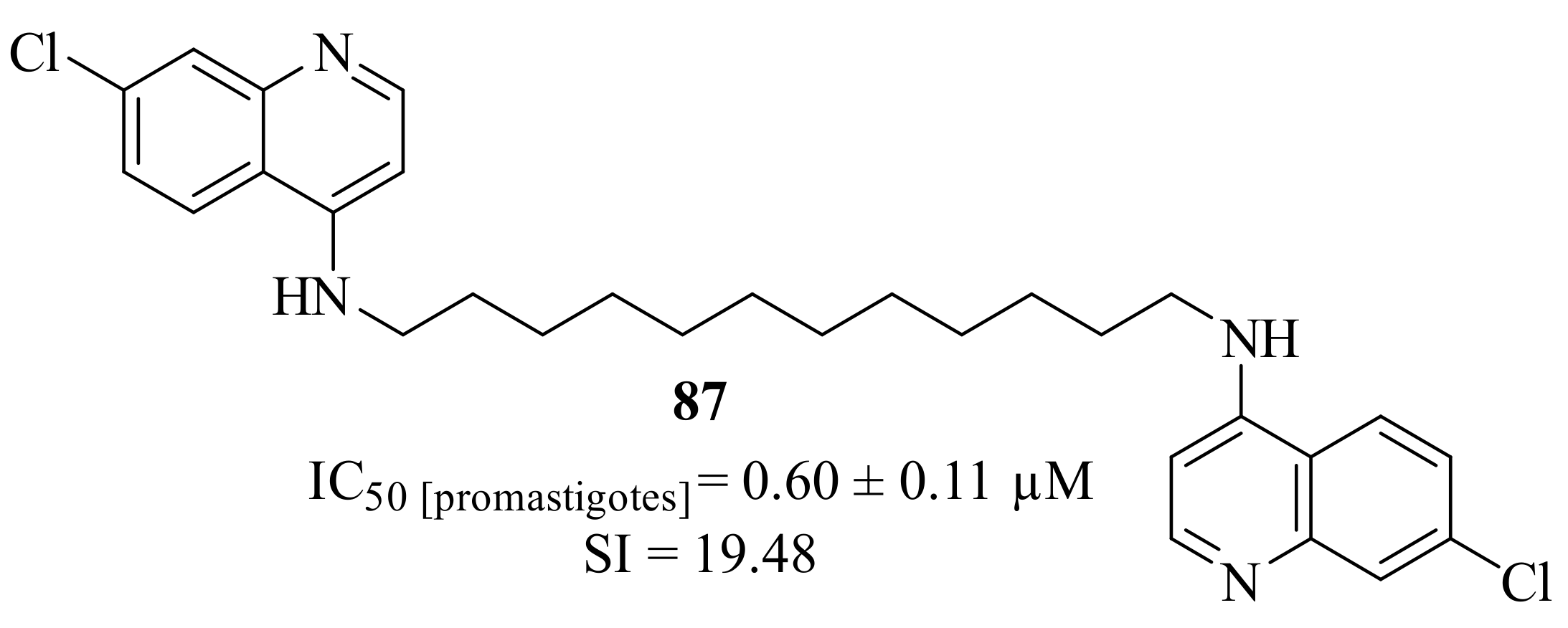
In the beginning of 2023, Silva et al. developed a series of 1,2,3,4-tetrahydroacridines, based on a virtual screening performed against the enzyme
S-adenosylmethionine decarboxylase, and evaluated them against
L. infantum promastigotes [
68]. From their results, a SAR study was structured and it was possible to verify that the length of the alkyl chain has a significant effect on the compounds’ antileishmanial activity, especially in the case of dimers. Based on this evidence, and in the fact that this series of 1,2,3,4-tetrahydroacridines present high levels of toxicity, this research group decided to replace the tetrahydroacridine scaffold by a 7-chloroquinoline core. This replacement allowed the retention of the promising antileishmanial properties while considerably decreasing the compound’s toxicity, resulting in derivative
87 (
Figure 33).
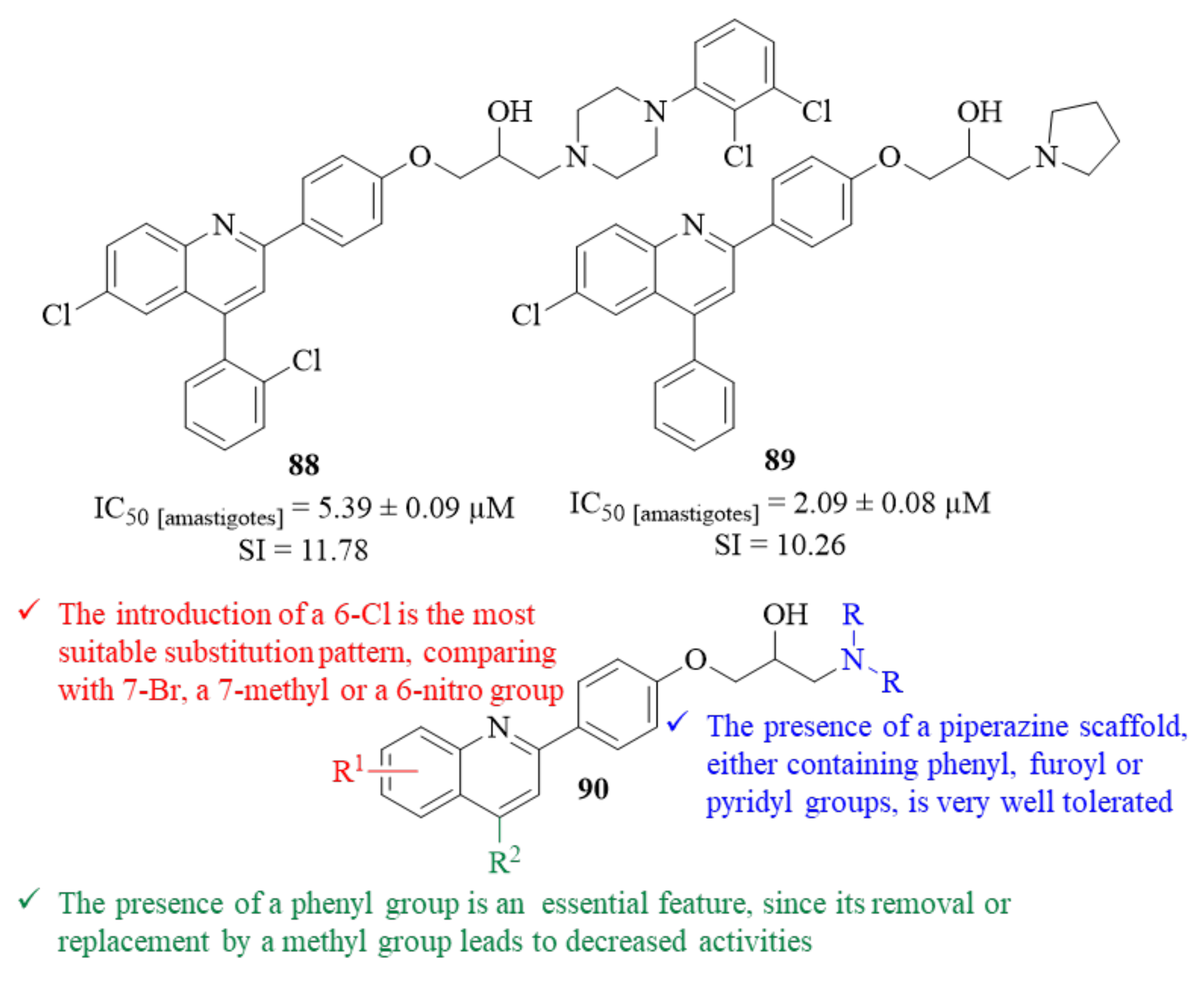
By the end of 2023, another research group synthesized a series of thirty-five quinoline-piperazine/pyrrolidine hybrids and evaluated them against
L. donovani intracellular amastigotes [
69]. This work was based on the hypothesis that, by conjugating the quinoline moiety with piperazine/pyrrolidine scaffold, one might be able to design effective antileishmanial agents, considering the widely known potential of quinoline derivatives. The results demonstrated that, from this series of thirty-five hybrids, only twelve molecules present moderate to significant levels of antileishmanial activity, with IC
50 values ranging from 2.09 ± 0.08 µM to 8.90 ± 0.18 µM. This series of hybrids can be divided into two distinct groups, twenty-four quinoline-piperazine derivatives and seven quinoline-pyrrolidine derivatives. Considering the first group, six derivatives emerged as being active against the amastigotes, IC
50 = 5.39 ± 0.06–8.22 ± 0.15 µM. Structurally, it was possible to verify that the introduction of a 6-Cl atom into the quinoline scaffold is the most suitable substitution pattern, comparing with compounds containing 7-Br atom, a 7-methyl or a 6-nitro group (
Figure 34). In addition, this introduction is not only responsible for retaining or increasing the compound’s antileishmanial activity but also for decreasing their toxicity, originating derivatives with improved SI values. Furthermore, regarding the C-4 of the quinoline scaffold, it became clear that the presence of a phenyl group is essential for the compounds’ antileishmanial properties, since its removal or replacement by a methyl group leads to inactive molecules at the evaluated concentrations. Finally, the presence of a piperazine scaffold as the terminal amino fragment, either containing phenyl, furoyl or pyridyl groups, is very well tolerated, leading to derivatives with moderate antileishmanial properties. In terms of the second group, the results demonstrated that five derivatives display moderate to significant levels of antileishmanial activity, with IC
50 values ranging from 2.09 ± 0.08 µM to 8.89 ± 0.18 µM. Structurally, the introduction of a 6-Cl atom into the quinoline scaffold, similar to the first group, leads to an increase in the compounds’ antileishmanial activity while decreasing their toxicity (
Figure 34). Following the promising in vitro results of derivatives
88 (IC
50 = 5.39 ± 0.09 µM, SI = 11.78) and
89 (IC
50 = 2.09 ± 0.09 µM, SI = 10.26), these compounds were selected for further in vivo antileishmanial evaluation. In this in vivo assay against
L. donovani infected hamsters, both compounds continued to present significant levels of antileishmanial activity, with particular emphasis on derivative
88 for being able to considerably inhibit the
leishmania parasite burden on the hamsters’ spleen (56.32 ± 4.23% at 50 mg/kg).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}