
Pharmacokinetics and Pharmacodynamics: A Comprehensive Analysis of the Absorption, Distribution, Metabolism, and Excretion of Psychiatric Drugs

 , ,
, ,  , and
, and
Abstract
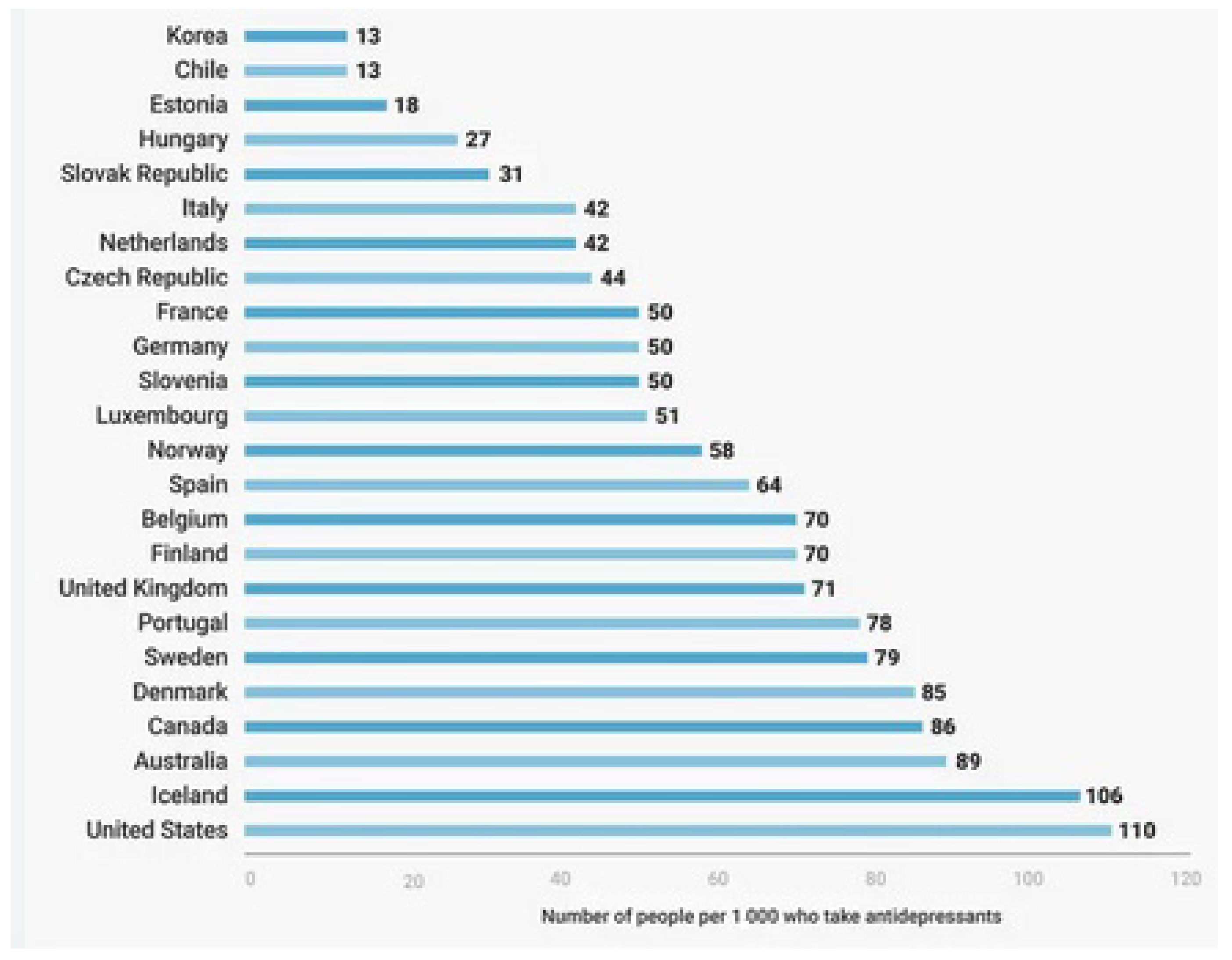
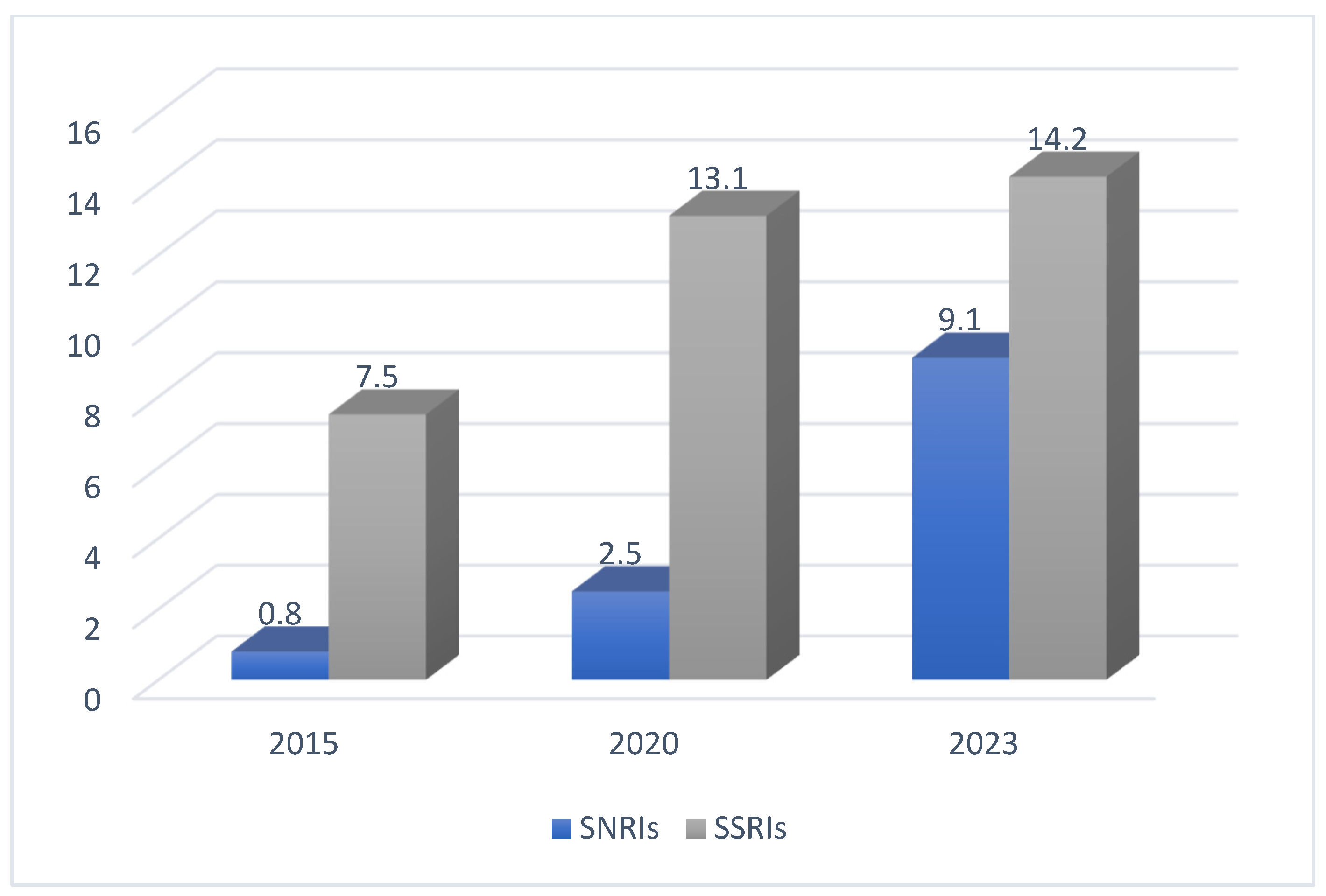
1. Introduction
2. Selective Serotonin Reuptake Inhibitors
2.1. Pharmacodynamics
2.1.1. Fluoxetine
2.1.2. Paroxetine
2.2. Pharmacokinetics
2.2.1. Absorption
Fluoxetine
Paroxetine
2.2.2. Distribution
Fluoxetine
Paroxetine
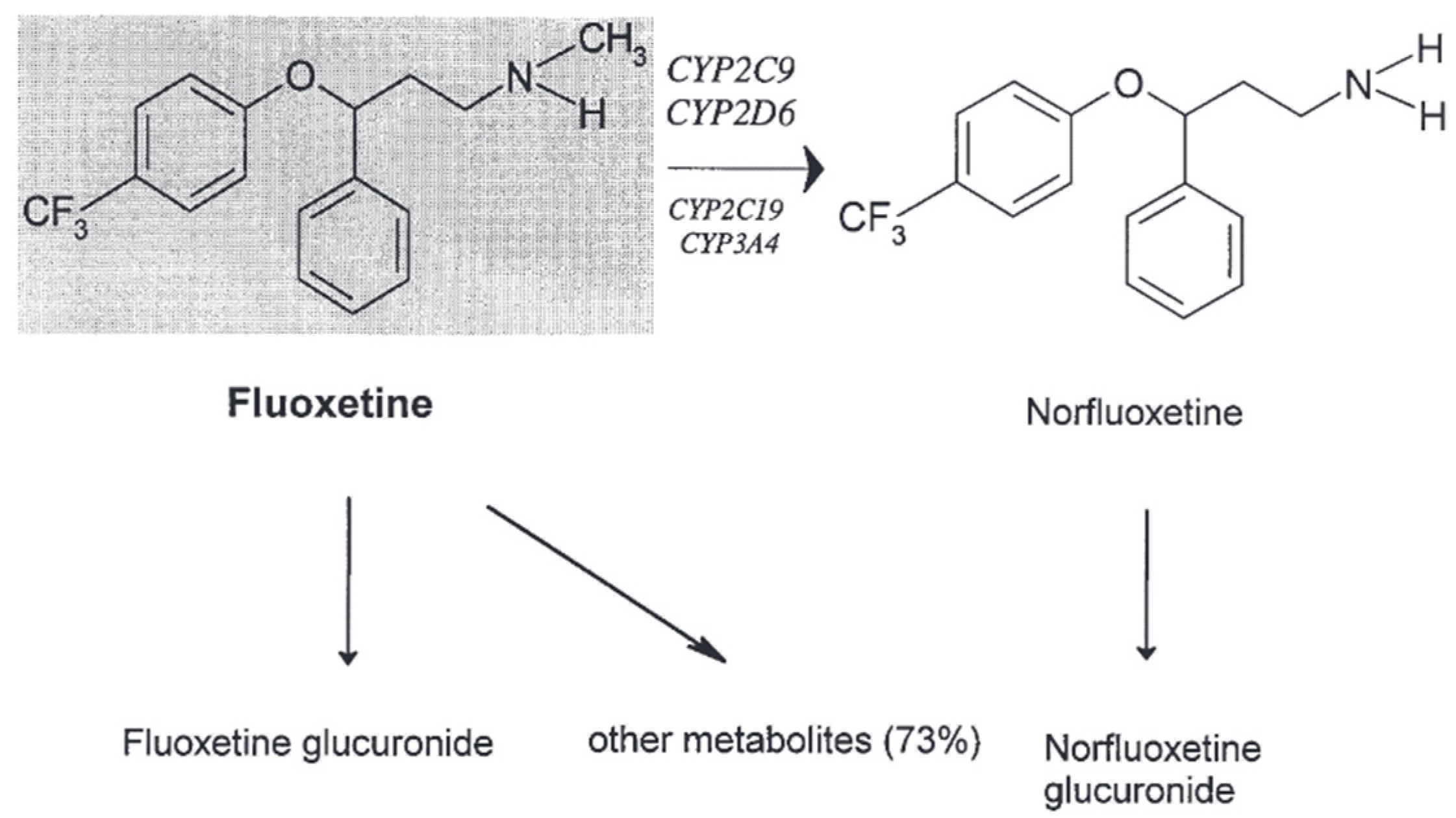
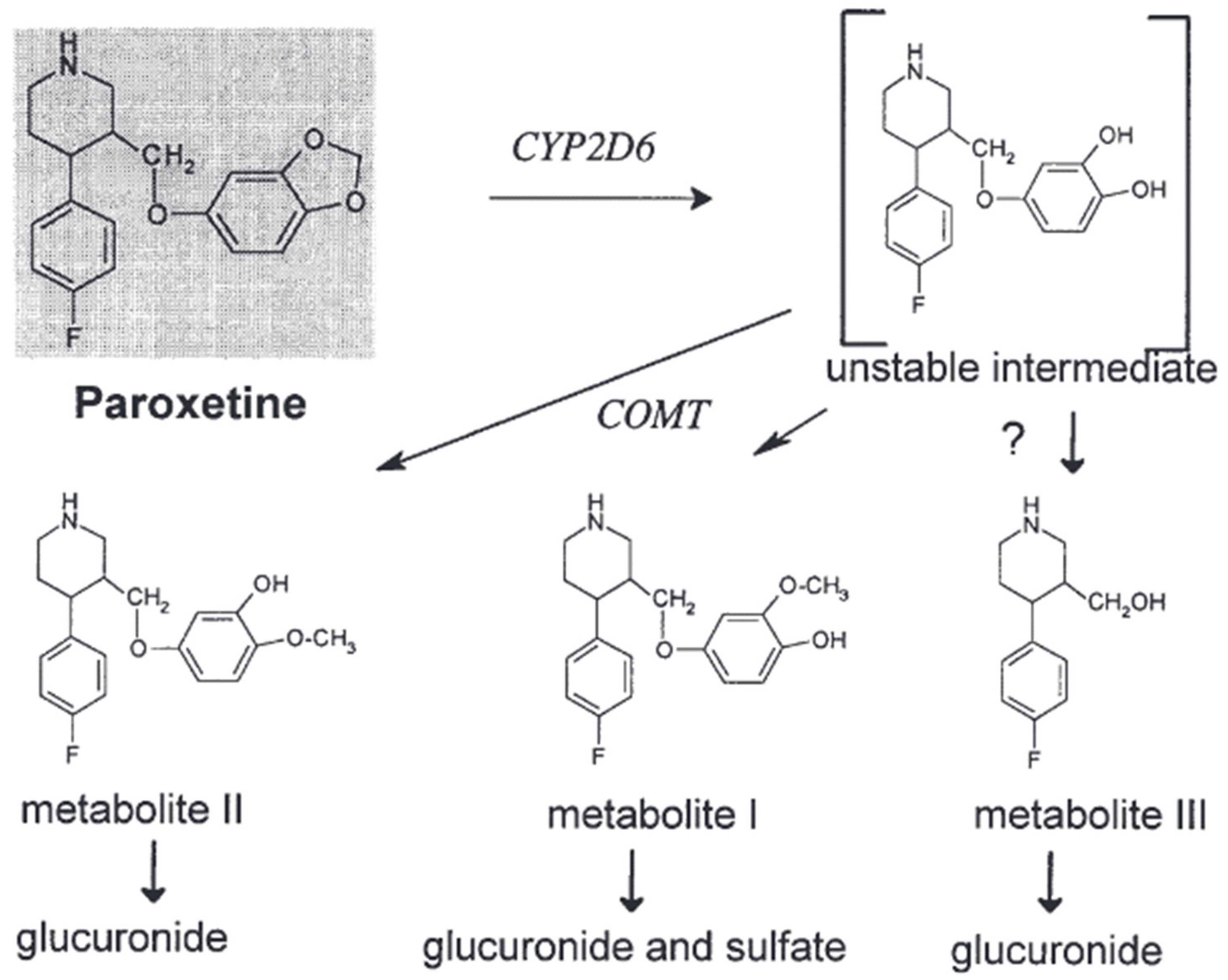
2.2.3. Metabolism
Fluoxetine
Paroxetine
2.2.4. Excretion
Fluoxetine
Paroxetine
3. Drug–Drug Interactions between SSRIs, SNRIs, and Other Drugs
4. Selective Norepinephrine Reuptake Inhibitor: Duloxetine
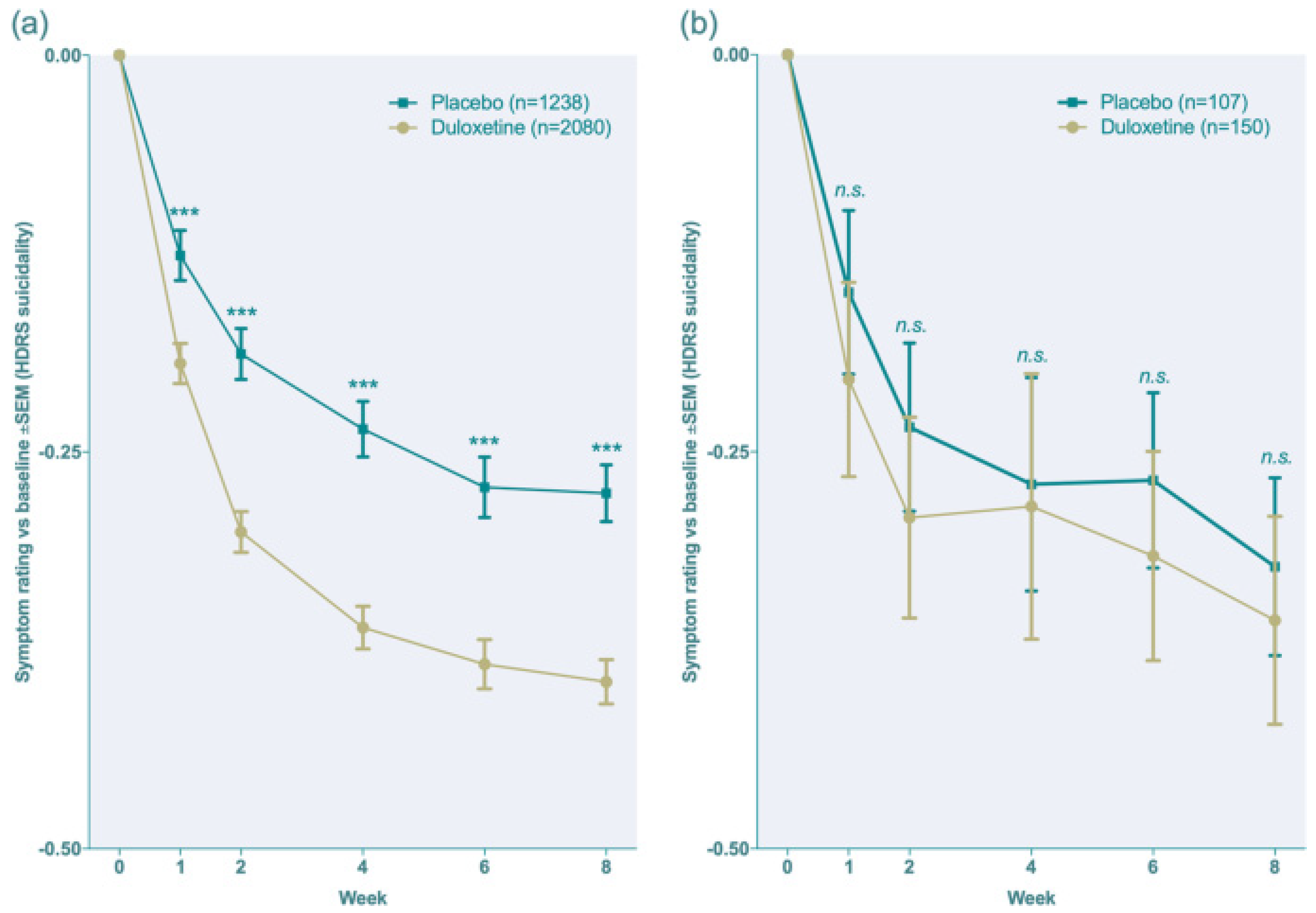
4.1. Pharmacodynamics
Duloxetine
4.2. Pharmacokinetics
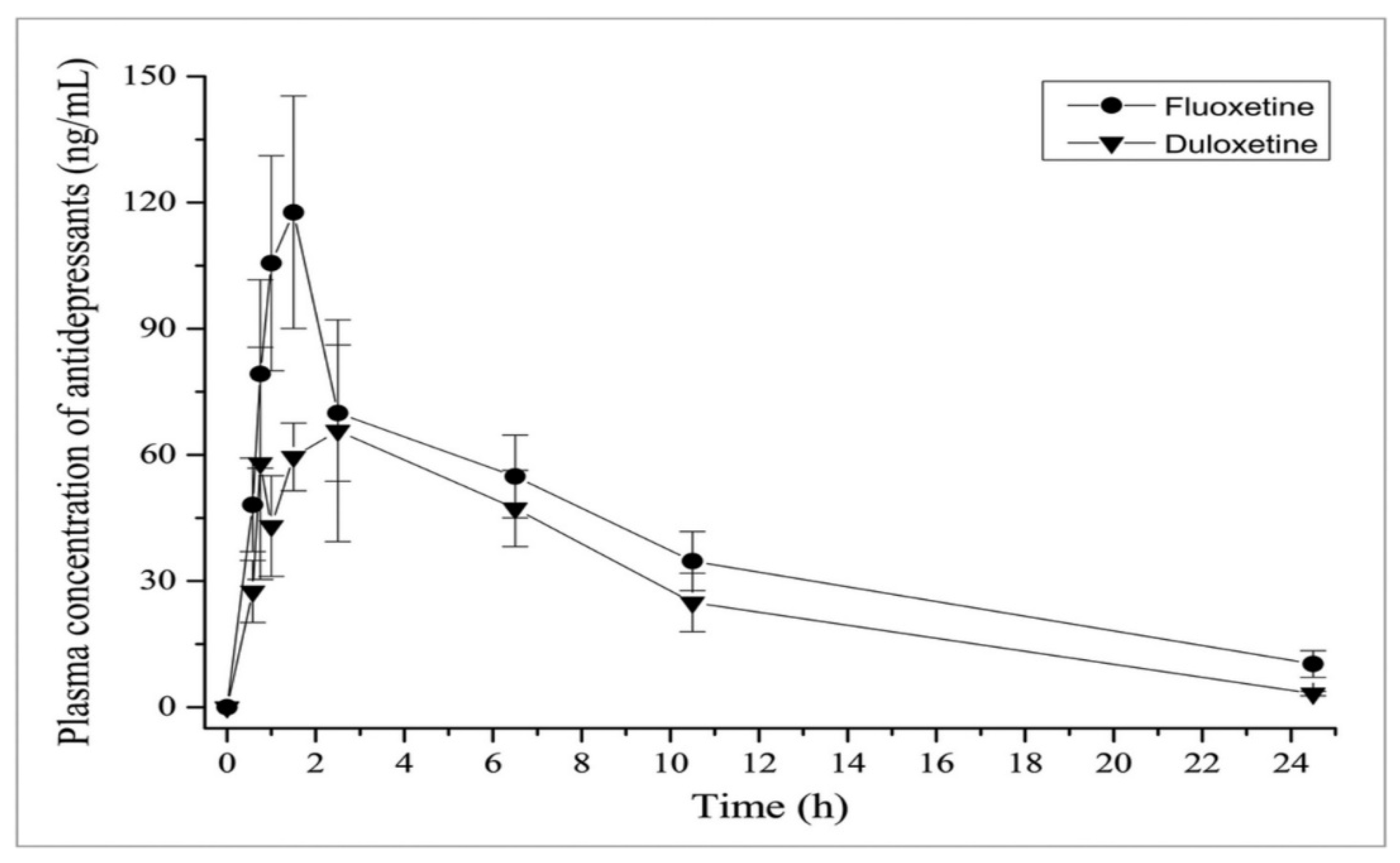
4.2.1. Absorption
Duloxetine
4.2.2. Distribution
Duloxetine
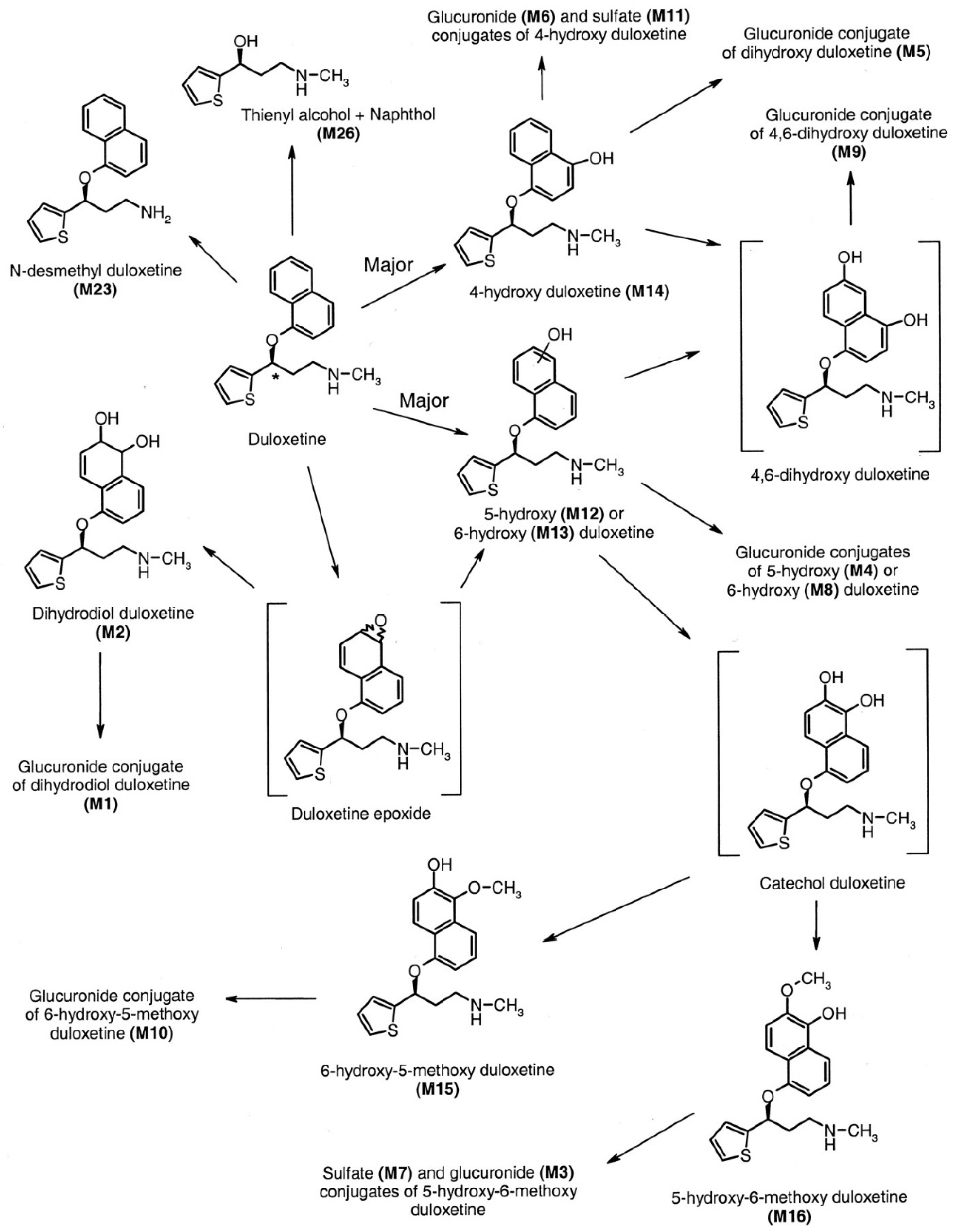
4.2.3. Metabolism
Duloxetine
4.2.4. Excretion
Duloxetine
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Christensen, M.K.; Lim, C.C.W.; Saha, S.; Plana-Ripoll, O.; Cannon, D.; Momen, N.C.; Whiteford, H.A.; Iburg, K.M.; McGrath, J.J. The Cost of Mental Disorders: A Systematic Review. Epidemiol. Psychiatr. Sci. 2020, 29, e161. [Google Scholar] [CrossRef]
- Ambwani, S.; Dutta, S.; Mishra, G.; Lal, H.; Singh, S.; Charan, J. Adverse Drug Reactions Associated with Drugs Prescribed in Psychiatry: A Retrospective Descriptive Analysis in a Tertiary Care Hospital. Cureus 2021, 13, e19493. [Google Scholar] [CrossRef] [PubMed]
- Malak, M.Z.; Al-amer, R.M.; Khalifeh, A.H.; Jacoub, S.M. Evaluation of Psychological Reactions among Teenage Married Girls in Palestinian Refugee Camps in Jordan. Soc. Psychiatry Psychiatr. Epidemiol. 2021, 56, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Ornoy, A.; Koren, G. Selective Serotonin Reuptake Inhibitor Use in Pregnant Women; Pharmacogenetics, Drug-Drug Interactions and Adverse Effects. Expert. Opin. Drug Metab. Toxicol. 2018, 14, 247–259. [Google Scholar] [CrossRef]
- Spigset, O. Adverse Reactions of Selective Serotonin Reuptake Inhibitors: Reports from a Spontaneous Reporting System. Drug Saf. 1999, 20, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Maideen, N.M.P.; Rajkapoor, B.; Muthusamy, S.; Ramanathan, S.; Thangadurai, S.A.; Sughir, A.A. A Review on Pharmacokinetic and Pharmacodynamic Drug Interactions of Adrenergic β-Blockers with Clinically Relevant Drugs-An Overview. Curr. Drug Metab. 2021, 22, 672–682. [Google Scholar] [CrossRef]
- Oliveira, P.; Ribeiro, J.; Donato, H.; Madeira, N. Smoking and Antidepressants Pharmacokinetics: A Systematic Review. Ann. Gen. Psychiatry 2017, 16, 17. [Google Scholar] [CrossRef] [PubMed]
- Kuzin, M.; Schoretsanitis, G.; Haen, E.; Ridders, F.; Hiemke, C.; Gründer, G.; Paulzen, M. Pharmacokinetic Interactions between Clozapine and Sertraline in Smokers and Non-Smokers. Basic. Clin. Pharmacol. Toxicol. 2020, 127, 303–308. [Google Scholar] [CrossRef]
- Lochmann, D.; Richardson, T. Selective Serotonin Reuptake Inhibitors. In Handbook of Experimental Pharmacology; Springer New York LLC: New York, NY, USA, 2019; Volume 250, pp. 135–144. [Google Scholar]
- Mandrioli, R.; Protti, M.; Mercolini, L. New-Generation, Non-SSRI Antidepressants: Therapeutic Drug Monitoring and Pharmacological Interactions. Part 1: SNRIs, SMSs, SARIs. Curr. Med. Chem. 2017, 25, 772–792. [Google Scholar] [CrossRef]
- Huddart, R.; Hicks, J.K.; Ramsey, L.B.; Strawn, J.R.; Smith, D.M.; Bobonis Babilonia, M.; Altman, R.B.; Klein, T.E. PharmGKB Summary: Sertraline Pathway, Pharmacokinetics. Pharmacogenet Genom. 2020, 30, 26–33. [Google Scholar] [CrossRef]
- Lampropoulou, D.I.; Lioliou, K.; Zerva, E.; Madia, X.; Vardoulaki, D.; Aravantinos, G.; Filippou, D.; Gazouli, M. CDK4/6 Inhibitors and SSRIs/SNRIs: A Brief Review of Their Safety Profiles Focusing on Potential Drug Interactions. Top. Biomed. Res. Educ. 2023, 1, 24–32. [Google Scholar]
- Eugene, A.R. Optimizing Drug Selection in Psychopharmacology Based on 40 Significant CYP2C19- And CYP2D6-Biased Adverse Drug Reactions of Selective Serotonin Reuptake Inhibitors. PeerJ 2019, 7, e7860. [Google Scholar] [CrossRef]
- Kowalska, M.; Nowaczyk, J.; Fijałkowski, Ł.; Nowaczyk, A. Paroxetine—Overview of the Molecular Mechanisms of Action. Int. J. Mol. Sci. 2021, 22, 1662. [Google Scholar] [CrossRef]
- Colombo, A.; Giordano, F.; Giorgetti, F.; Di Bernardo, I.; Bosi, M.F.; Varinelli, A.; Cafaro, R.; Pileri, P.; Cetin, I.; Clementi, E.; et al. Correlation between Pharmacokinetics and Pharmacogenetics of Selective Serotonin Reuptake Inhibitors and Selective Serotonin and Noradrenaline Reuptake Inhibitors and Maternal and Neonatal Outcomes: Results from a Naturalistic Study in Patients with Affec. Hum. Psychopharmacol. 2021, 36, e2772. [Google Scholar] [CrossRef]
- de Leon, J.; Spina, E. Possible Pharmacodynamic and Pharmacokinetic Drug-Drug Interactions That Are Likely to Be Clinically Relevant and/or Frequent in Bipolar Disorder. Curr. Psychiatry Rep. 2018, 20, 17. [Google Scholar] [CrossRef] [PubMed]
- Edinoff, A.N.; Fort, J.M.; Woo, J.J.; Causey, C.D.; Burroughs, C.R.; Cornett, E.M.; Kaye, A.M.; Kaye, A.D. Selective Serotonin Reuptake Inhibitors and Clozapine: Clinically Relevant Interactions and Considerations. Neurol. Int. 2021, 13, 445–463. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, E.K.; Howell, L.L. Pharmacokinetics of Fluoxetine in Rhesus Macaques Following Multiple Routes of Administration. Pharmacology 2011, 88, 44. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.T.; Biernacka, J.M.; Jenkins, G.; Rush, A.J.; Shinozaki, G.; Veldic, M.; Kung, S.; Bobo, W.V.; Hall-Flavin, D.K.; Weinshilboum, R.M.; et al. Pharmacokinetic-Pharmacodynamic Interaction Associated with Venlafaxine-XR Remission in Patients with Major Depressive Disorder with History of Citalopram / Escitalopram Treatment Failure. J. Affect. Disord. 2019, 246, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Kazmi, F.; Hensley, T.; Pope, C.; Funk, R.S.; Loewen, G.J.; Buckley, D.B.; Parkinson, A. Lysosomal Sequestration (Trapping) of Lipophilic Amine (Cationic Amphiphilic) Drugs in Immortalized Human Hepatocytes (Fa2N-4 Cells). Drug Metab. Dispos. 2013, 41, 897. [Google Scholar] [CrossRef] [PubMed]
- Bolo, N.R.; Hodé, Y.; Nédélec, J.F.; Lainé, E.; Wagner, G.; MacHer, J.P. Brain Pharmacokinetics and Tissue Distribution In Vivo of Fluvoxamine and Fluoxetine by Fluorine Magnetic Resonance Spectroscopy. Neuropsychopharmacology 2000, 23, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Vashistha, V.K.; Sethi, S.; Tyagi, I.; Das, D.K. Chirality of Antidepressive Drugs: An Overview of Stereoselectivity. Asian Biomed (Res. Rev. News) 2022, 16, 55. [Google Scholar] [CrossRef] [PubMed]
- Kawai, H.; Machida, M.; Ishibashi, T.; Kudo, N.; Kawashima, Y.; Mitsumoto, A. Chronopharmacological Analysis of Antidepressant Activity of a Dual-Action Serotonin Noradrenaline Reuptake Inhibitor (SNRI), Milnacipran, in Rats. Biol. Pharm. Bull. 2018, 41, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Spina, E.; Barbieri, M.A.; Cicala, G.; Bruno, A.; de Leon, J. Clinically Relevant Drug Interactions between Newer Antidepressants and Oral Anticoagulants. Expert. Opin. Drug Metab. Toxicol. 2020, 16, 31–44. [Google Scholar] [CrossRef]
- Tasker, T.C.G.; Bye, C.M.; Zussman, B.D.; Link, C.G.G. Paroxetine Plasma Levels: Lack of Correlation with Efficacy or Adverse Events. Acta Psychiatr. Scand. Suppl. 1989, 350, 152–155. [Google Scholar] [CrossRef]
- Hiemke, C.; Härtter, S. Pharmacokinetics of Selective Serotonin Reuptake Inhibitors. Pharmacol. Ther. 2000, 85, 11–28. [Google Scholar] [CrossRef]
- Irons, J. Fluvoxamine in the Treatment of Anxiety Disorders. Neuropsychiatr. Dis. Treat. 2005, 1, 289. [Google Scholar]
- Hiemke, C. Paroxetine: Pharmacokinetics and Pharmacodynamics. Fortschr. Neurol. Psychiatr. 1994, 62 (Suppl. S1), S2–S8. [Google Scholar] [CrossRef]
- Serretti, A.; Calati, R.; Massat, I.; Linotte, S.; Kasper, S.; Lecrubier, Y.; Sens-Espel, R.; Bollen, J.; Zohar, J.; Berlo, J.; et al. Cytochrome P450 CYP1A2, CYP2C9, CYP2C19 and CYP2D6 Genes Are Not Associated with Response and Remission in a Sample of Depressive Patients. Int. Clin. Psychopharmacol. 2009, 24, 250–256. [Google Scholar] [CrossRef]
- Fatunde, O.A.; Brown, S.A. The Role of CYP450 Drug Metabolism in Precision Cardio-Oncology. Int. J. Mol. Sci. 2020, 21, 604. [Google Scholar] [CrossRef]
- Hurst, M.; Lamb, H.M. Fluoxetine. CNS Drugs 2000, 14, 51–80. [Google Scholar] [CrossRef]
- Altamura, A.C.; Moro, A.R.; Percudani, M. Clinical Pharmacokinetics of Fluoxetine. Clin. Pharmacokinet. 1994, 26, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Malak, M.Z.; Shuhaiber, A.H.; Alsswey, A.; Tarawneh, A. Social Support as the Mediator for the Relationship between Internet Gaming Disorder and Psychological Problems among University Students. J. Psychiatr. Res. 2023, 164, 243–250. [Google Scholar] [CrossRef]
- Deodhar, M.; Al Rihani, S.B.; Darakjian, L.; Turgeon, J.; Michaud, V. Assessing the Mechanism of Fluoxetine-Mediated CYP2D6 Inhibition. Pharmaceutics 2021, 13, 148. [Google Scholar] [CrossRef]
- Boguta, P.; Juchnowicz, D.; Wróbel-Knybel, P.; Biała-Kędra, A.; Karakuła-Juchnowicz, H. Safety of Concomitant Treatment with Non-Vitamin K Oral Anticoagulants and SSRI/SNRI Antidepressants. Curr. Probl. Psychiatry 2018, 19, 267–278. [Google Scholar] [CrossRef]
- LLerena, A.; Dorado, P.; Berecz, R.; González, A.P.; Peñas-LLedó, E.M. Effect of CYP2D6 and CYP2C9 Genotypes on Fluoxetine and Norfluoxetine Plasma Concentrations during Steady-State Conditions. Eur. J. Clin. Pharmacol. 2004, 59, 869–873. [Google Scholar] [CrossRef]
- Mikami, A.; Ohtani, H.; Hori, S.; Sawada, Y. Pharmacokinetic Model Incorporating Mechanism-Based Inactivation of CYP2D6 Can Explain Both Non-Linear Kinetics and Drug Interactions of Paroxetine. Int. J. Clin. Pharmacol. Ther. 2013, 51, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Pasi, P.; Kröll, D.; Siegfried, A.; Sykora, M.; Wildisen, A.; Milone, C.; Milos, G.; Horka, L.; Fischli, S.; Henzen, C. Plasma Concentrations of SSRI/SNRI after Bariatric Surgery and the Effects on Depressive Symptoms. Front. Psychiatry 2023, 14, 1132112. [Google Scholar] [CrossRef] [PubMed]
- Strawn, J.R.; Vaughn, S.; Ramsey, L.B. Pediatric Psychopharmacology for Depressive and Anxiety Disorders. Focus 2022, 20, 184–190. [Google Scholar] [CrossRef]
- Rendić, S.P.; Crouch, R.D.; Guengerich, F.P. Roles of Selected Non-P450 Human Oxidoreductase Enzymes in Protective and Toxic Effects of Chemicals: Review and Compilation of Reactions. Arch. Toxicol. 2022, 96, 2145–2246. [Google Scholar] [CrossRef]
- Bourin, M.; Chue, P.; Guillon, Y. Paroxetine: A Review. CNS Drug Rev. 2001, 7, 25–47. [Google Scholar] [CrossRef]
- Kaye, C.M.; Haddock, R.E.; Langley, P.F.; Mellows, G.; Tasker, T.C.G.; Zussman, B.D.; Greb, W.H. A Review of the Metabolism and Pharmacokinetics of Paroxetine in Man. Acta Psychiatr. Scand. 1989, 80, 60–75. [Google Scholar] [CrossRef]
- Porcelli, S.; Drago, A.; Fabbri, C.; Gibiino, S.; Calati, R.; Serretti, A. Pharmacogenetics of Antidepressant Response. J. Psychiatry Neurosci. 2011, 36, 87–113. [Google Scholar] [CrossRef] [PubMed]
- Buoli, M.; Caldiroli, A.; Serati, M. Pharmacokinetic Evaluation of Pregabalin for the Treatment of Generalized Anxiety Disorder. Expert. Opin. Drug Metab. Toxicol. 2017, 13, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Armah, F.A.; Henneh, I.T.; Amponsah, I.K.; Biney, R.P.; Malcolm, F.; Alake, J.; Ahlidja, W.; Ahmed, M.A.; Adokoh, C.K.; Adukpo, G.E.; et al. Antidepressant and Anxiolytic Effects and Subacute Toxicity of the Aerial Parts of Psychotria Ankasensis J.B.Hall (Rubiaceae) in Murine Models. Evid.-Based Complement. Altern. Med. 2021, 2021, 5543320. [Google Scholar] [CrossRef]
- Gillman, P.K. Tricyclic Antidepressant Pharmacology and Therapeutic Drug Interactions Updated. Br. J. Pharmacol. 2007, 151, 737. [Google Scholar] [CrossRef] [PubMed]
- Edinoff, A.N.; Akuly, H.A.; Hanna, T.A.; Ochoa, C.O.; Patti, S.J.; Ghaffar, Y.A.; Kaye, A.D.; Viswanath, O.; Urits, I.; Boyer, A.G.; et al. Selective Serotonin Reuptake Inhibitors and Adverse Effects: A Narrative Review. Neurol. Int. 2021, 13, 387–401. [Google Scholar] [CrossRef]
- Perrotta, C.; Giordano, F.; Colombo, A.; Carnovale, C.; Castiglioni, M.; Di Bernardo, I.; Giorgetti, F.; Pileri, P.; Clementi, E.; Viganò, C. Postpartum Bleeding in Pregnant Women Receiving SSRIs/SNRIs: New Insights from a Descriptive Observational Study and an Analysis of Data from the FAERS Database. Clin. Ther. 2019, 41, 1755–1766. [Google Scholar] [CrossRef] [PubMed]
- Pinninti, N.R.; de Leon, J. Interaction of Sertraline with Clozapine. J. Clin. Psychopharmacol. 1997, 17, 119–120. [Google Scholar] [CrossRef]
- Mrazek, D.A.; Biernacka, J.M.; O’Kane, D.J.; Black, J.L.; Cunningham, J.M.; Drews, M.S.; Snyder, K.A.; Stevens, S.R.; Rush, A.J.; Weinshilboum, R.M. CYP2C19 Variation and Citalopram Response. Pharmacogenet Genomics 2011, 21, 1–9. [Google Scholar] [CrossRef]
- Hiemke, C. Consensus Guideline Based Therapeutic Drug Monitoring (TDM) in Psychiatry and Neurology. Curr. Drug Deliv. 2016, 13, 353–361. [Google Scholar] [CrossRef]
- Spina, E.; Trifirò, G.; Caraci, F. Clinically Significant Drug Interactions with Newer Antidepressants. CNS Drugs 2012, 26, 39–67. [Google Scholar] [CrossRef] [PubMed]
- Perahia, D.G.; Bangs, M.E.; Zhang, Q.; Cheng, Y.; Ahl, J.; Frakes, E.P.; Adams, M.J.; Martinez, J.M. The Risk of Bleeding with Duloxetine Treatment in Patients Who Use Nonsteroidal Anti-Inflammatory Drugs (NSAIDs): Analysis of Placebo-Controlled Trials and Post-Marketing Adverse Event Reports. Drug Healthc. Patient Saf. 2013, 5, 211–219. [Google Scholar] [CrossRef]
- Ungvari, Z.; Tarantini, S.; Yabluchanskiy, A.; Csiszar, A. Potential Adverse Cardiovascular Effects of Treatment with Fluoxetine and Other Selective Serotonin Reuptake Inhibitors (SSRIs) in Patients With Geriatric Depression: Implications for Atherogenesis and Cerebromicrovascular Dysregulation. Front. Genet. 2019, 10, 455576. [Google Scholar] [CrossRef] [PubMed]
- Strawn, J.R.; Poweleit, E.A.; Ramsey, L.B. CYP2C19-Guided Escitalopram and Sertraline Dosing in Pediatric Patients: A Pharmacokinetic Modeling Study. J. Child. Adolesc. Psychopharmacol. 2019, 29, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Laux, G. Serotonin Reuptake Inhibitors: Citalopram, Escitalopram, Fluoxetine, Fluvoxamine, Paroxetine, and Sertraline. In NeuroPsychopharmacotherapy; Springer International Publishing: Berlin/Heidelberg, Germany, 2022; pp. 1257–1269. ISBN 9783030620592. [Google Scholar]
- Citrome, L. Levomilnacipran for Major Depressive Disorder: A Systematic Review of the Efficacy and Safety Profile for This Newly Approved Antidepressant—What Is the Number Needed to Treat, Number Needed to Harm and Likelihood to Be Helped or Harmed? Int. J. Clin. Pract. 2013, 67, 1089–1104. [Google Scholar] [CrossRef]
- McIntyre, R.S. The Role of New Antidepressants in Clinical Practice in Canada: A Brief Review of Vortioxetine, Levomilnacipran ER, and Vilazodone. Neuropsychiatr. Dis. Treat. 2017, 13, 2913–2919. [Google Scholar] [CrossRef]
- Sloan, G.; Alam, U.; Selvarajah, D.; Tesfaye, S. The Treatment of Painful Diabetic Neuropathy. Curr. Diabetes Rev. 2022, 18, e070721194556. [Google Scholar] [CrossRef]
- Hsu, E.S. Acute and Chronic Pain Management in Fibromyalgia: Updates on Pharmacotherapy. Am. J. Ther. 2011, 18, 487–509. [Google Scholar] [CrossRef]
- Poweleit, E.A.; Cinibulk, M.A.; Novotny, S.A.; Wagner-Schuman, M.; Ramsey, L.B.; Strawn, J.R. Selective Serotonin Reuptake Inhibitor Pharmacokinetics During Pregnancy: Clinical and Research Implications. Front. Pharmacol. 2022, 13, 833217. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SSRI (Trade name) | 1A2 | 2C9/10 | 2C19 | 2D6 | 3A3/4 |
| Citalopram (Celexa) | − | − | − | ++ | − |
| Escitalopram (Lexapro) | − | − | − | ++ | − |
| Fluoxetine (Prozac) | − | ++ | ++ | +++ | + |
| Fluvoxamine (Luvox) | +++ | +++ | +++ | − | ++ |
| Sertraline (Zoloft) | − | − | − | − | |
| Paroxetine (Paxil) | − | − | − | − | − |
| SNRIs | 1A2 | 2C9/10 | 2C19 | 2D6 | 3A3/4 |
| Duloxetine (Cymbalta) | − | − | − | ++ | − |
| Venlafaxine (Effexor ER) | − | − | − | − | − |
| Newer Antidepressants | 1A2 | 2C9/10 | 2C19 | 2D6 | 3A3/4 |
| Bupropion (Wellbutrin) | ?? | ?? | ?? | +++ | ?? |
| Nefazodone (Serzone) | − | − | − | − | +++ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zakaraya, Z.; Abu Assab, M.; Tamimi, L.N.; Karameh, N.; Hailat, M.; Al-Omari, L.; Abu Dayyih, W.; Alasasfeh, O.; Awad, M.; Awad, R. Pharmacokinetics and Pharmacodynamics: A Comprehensive Analysis of the Absorption, Distribution, Metabolism, and Excretion of Psychiatric Drugs. Pharmaceuticals 2024, 17, 280. https://doi.org/10.3390/ph17030280
Zakaraya Z, Abu Assab M, Tamimi LN, Karameh N, Hailat M, Al-Omari L, Abu Dayyih W, Alasasfeh O, Awad M, Awad R. Pharmacokinetics and Pharmacodynamics: A Comprehensive Analysis of the Absorption, Distribution, Metabolism, and Excretion of Psychiatric Drugs. Pharmaceuticals. 2024; 17(3):280. https://doi.org/10.3390/ph17030280
Chicago/Turabian StyleZakaraya, Zainab, Mohammad Abu Assab, Lina N. Tamimi, Nida Karameh, Mohammad Hailat, Laila Al-Omari, Wael Abu Dayyih, Omar Alasasfeh, Mohammad Awad, and Riad Awad. 2024. "Pharmacokinetics and Pharmacodynamics: A Comprehensive Analysis of the Absorption, Distribution, Metabolism, and Excretion of Psychiatric Drugs" Pharmaceuticals 17, no. 3: 280. https://doi.org/10.3390/ph17030280
APA StyleZakaraya, Z., Abu Assab, M., Tamimi, L. N., Karameh, N., Hailat, M., Al-Omari, L., Abu Dayyih, W., Alasasfeh, O., Awad, M., & Awad, R. (2024). Pharmacokinetics and Pharmacodynamics: A Comprehensive Analysis of the Absorption, Distribution, Metabolism, and Excretion of Psychiatric Drugs. Pharmaceuticals, 17(3), 280. https://doi.org/10.3390/ph17030280