
Clindamycin Derivatives: Unveiling New Prospects as Potential Antitumor Agents

Abstract
1. Introduction
2. Results
2.1. Intersection of Two Clindamycin Derivative Targets
2.2. Screening of the Antibacterial Targets of the Clindamycin Derivatives through the PubChem Database
2.3. Molecular Docking Simulation and Validation
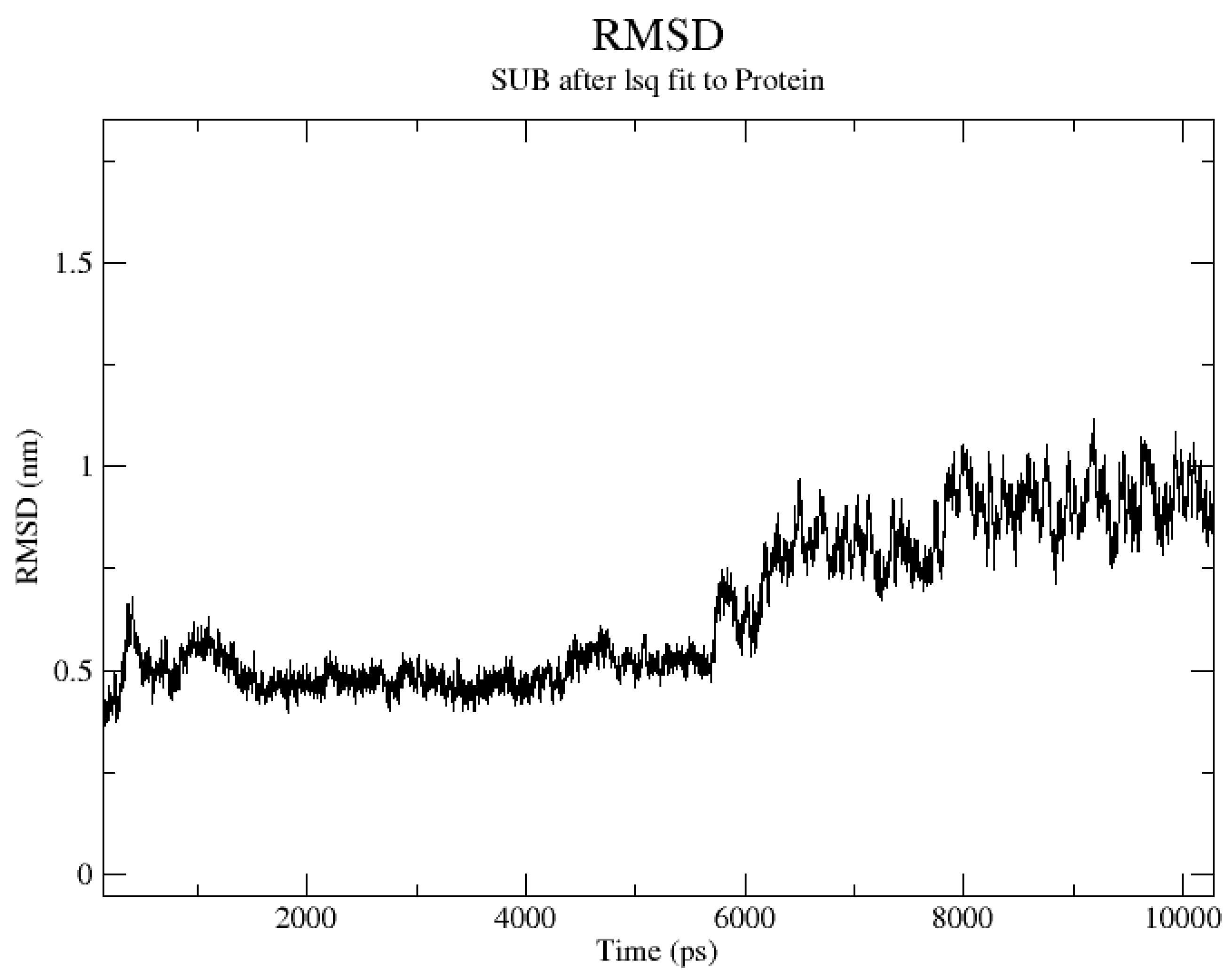
2.4. Stability of the Docked Complexes Studied via MD Simulation
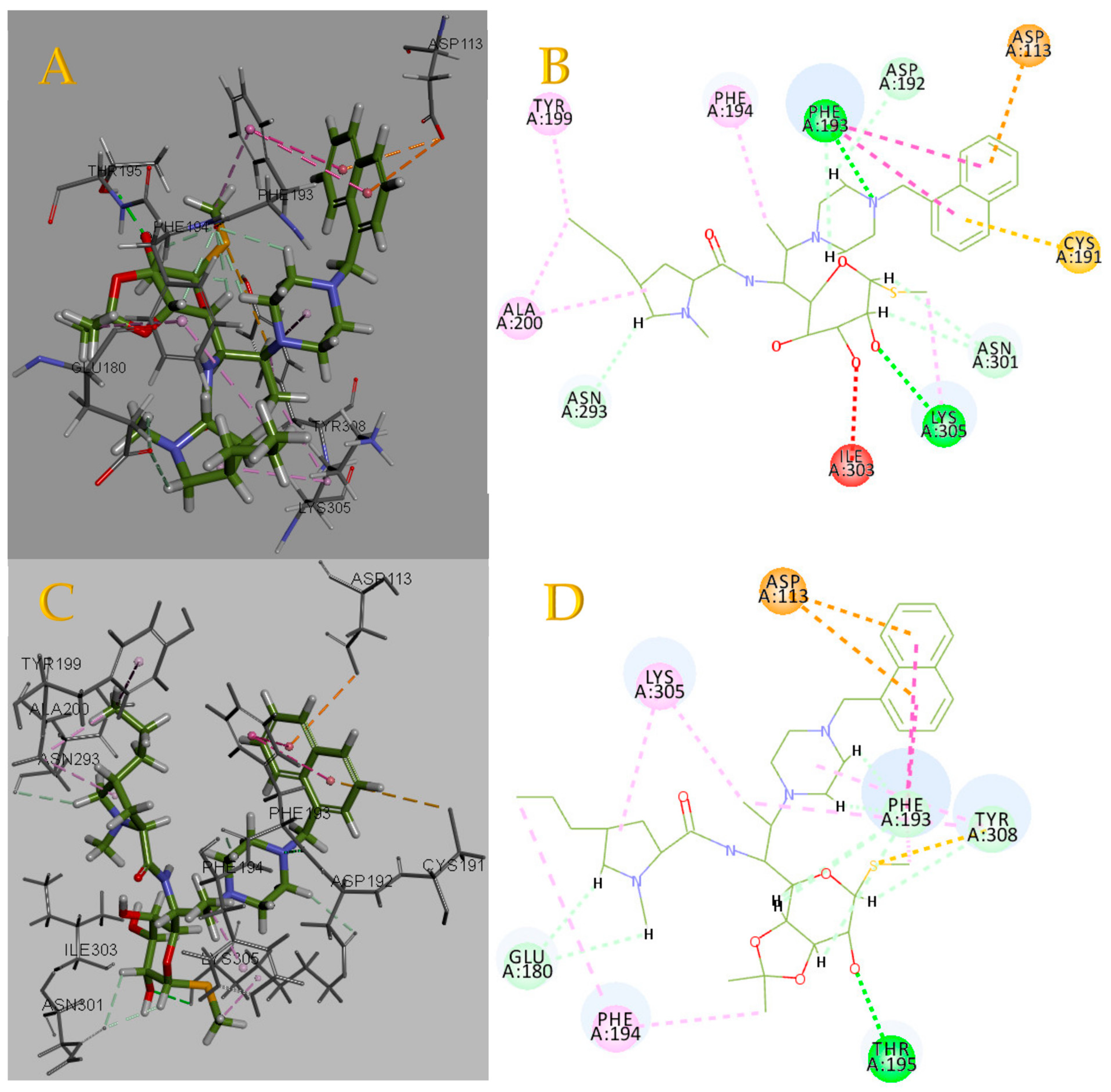
2.5. Binding Force Analysis
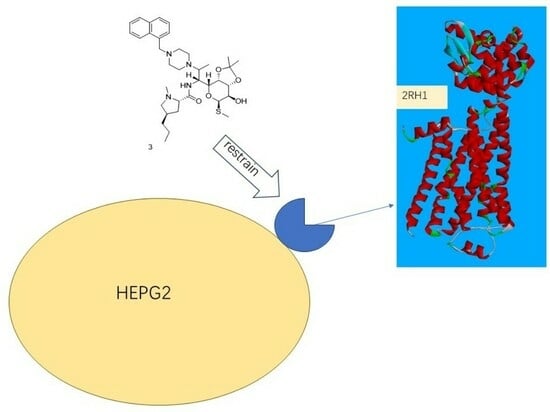
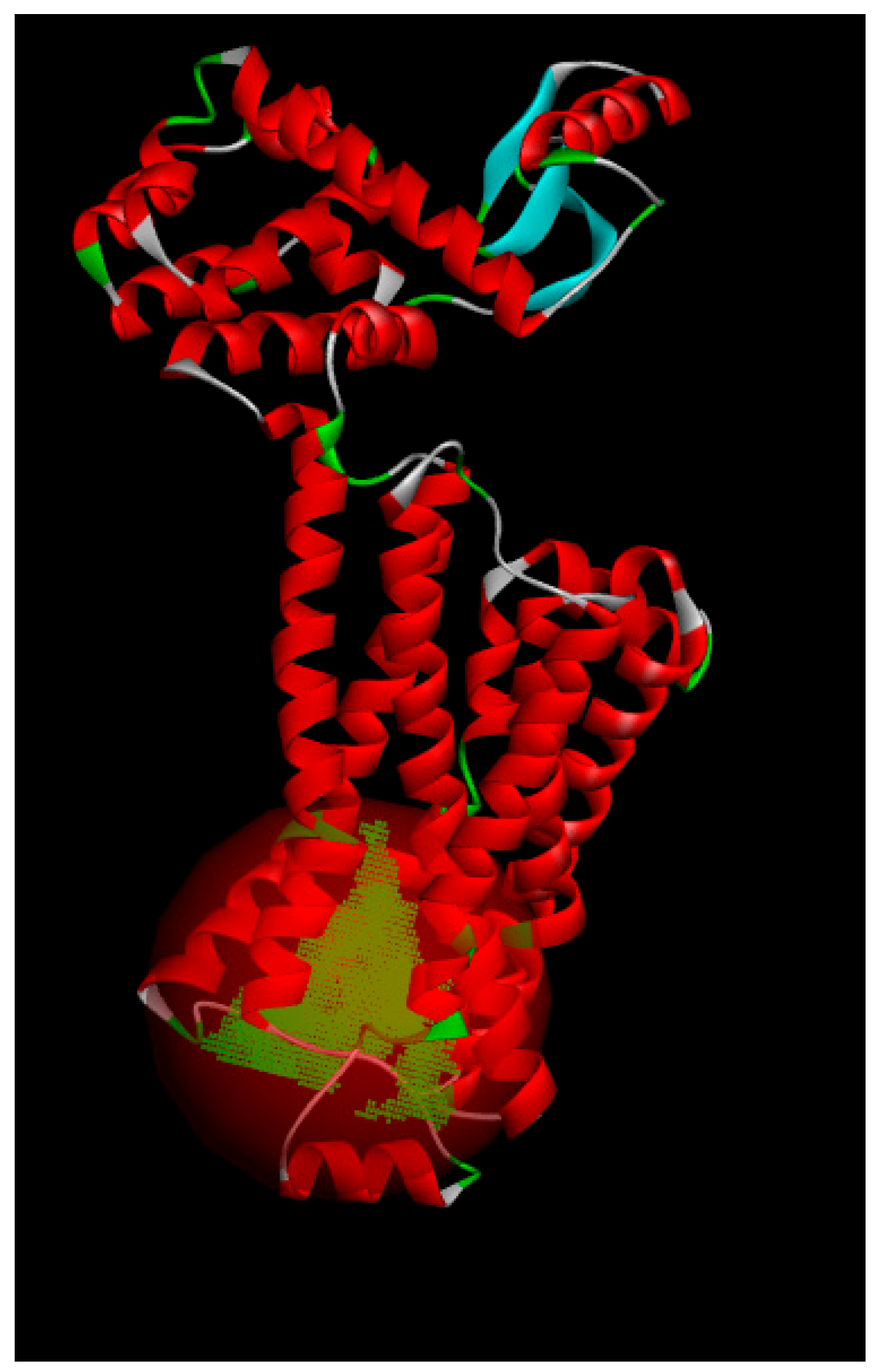
2.6. Protein Subcellular Localization
2.7. Biological Evaluation of Compounds
3. Materials and Methods
3.1. Intersection of Two Clindamycin Derivative Targets
3.2. Screening of the Antibacterial Targets of the Clindamycin Derivatives through the PubChem Database
3.3. Molecular Docking Simulation and Validation
3.4. Stability of the Docked Complexes Studied via MD Simulation
3.5. Binding Force Analysis
3.6. Protein Subcellular Localization
3.7. Biological Evaluation of Compounds
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Robinson, P.C.; Liew, D.F.; Tanner, H.L.; Grainger, J.R.; Dwek, R.A.; Reisler, R.B.; Steinman, L.; Feldmann, M.; Ho, L.-P.; Hussell, O.S. COVID-19 therapeutics: Challenges and directions for the future. Proc. Natl. Acad. Sci. USA 2022, 119, e2119893119. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Rousseau, B.; Qiu, K.; Huang, G.; Zhang, Y.; Su, H.; Le Bihan-Benjamin, C.; Khati, I.; Artz, O.; Foote, M.B. Killing tumor-associated bacteria with a liposomal antibiotic generates neoantigens that induce anti-tumor immune responses. Nat. Biotechnol. 2023, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Zhang, Y.; Zhu, H. Structure–Activity Relationship Target Prediction Studies of Clindamycin Derivatives with Broad-Spectrum Bacteriostatic Antibacterial Properties. Molecules 2023, 28, 7357. [Google Scholar] [CrossRef] [PubMed]
- Childs-Disney, J.L.; Yang, X.; Gibaut, Q.M.; Tong, Y.; Batey, R.T.; Disney, M.D. Targeting RNA structures with small molecules. Nat. Rev. Drug Discov. 2022, 21, 736–762. [Google Scholar] [CrossRef] [PubMed]
- Adewumi, O.A.; Singh, V.; Singh, G.J. Phytochemistry Chemical composition, traditional uses and biological activities of artemisia species. J. Pharmacogn. Phytochem. 2020, 9, 1124–1140. [Google Scholar]
- Damavandi, M.S.; Shojaei, H.; Esfahani, B.N. The anticancer and antibacterial potential of bioactive secondary metabolites derived from bacterial endophytes in association with Artemisia absinthium. Sci. Rep. 2023, 13, 18473. [Google Scholar] [CrossRef]
- Liu, J.; Tang, H.; Xu, C.; Zhou, S.; Zhu, X.; Li, Y.; Prézeau, L.; Xu, T.; Pin, J.-P.; Rondard, P.J. Biased signaling due to oligomerization of the G protein-coupled platelet-activating factor receptor. Nat. Commun. 2022, 13, 6365. [Google Scholar] [CrossRef]
- Yang, D.; Zhou, Q.; Labroska, V.; Qin, S.; Darbalaei, S.; Wu, Y.; Yuliantie, E.; Xie, L.; Tao, H.; Cheng, J.J.; et al. G protein-coupled receptors: Structure-and function-based drug discovery. Signal Transduct. Target. Ther. 2021, 6, 7. [Google Scholar] [CrossRef]
- Leon, K.; Cunningham, R.L.; Riback, J.A.; Feldman, E.; Li, J.; Sosnick, T.R.; Zhao, M.; Monk, K.R.; Araç, D.J. Structural basis for adhesion G protein-coupled receptor Gpr126 function. Nat. Commun. 2020, 11, 194. [Google Scholar] [CrossRef]
- Sandhu, M.; Cho, A.; Ma, N.; Mukhaleva, E.; Namkung, Y.; Lee, S.; Ghosh, S.; Lee, J.H.; Gloriam, D.E.; Laporte, S.A.; et al. Dynamic spatiotemporal determinants modulate GPCR: G protein coupling selectivity and promiscuity. Nat. Commun. 2022, 13, 7428. [Google Scholar] [CrossRef]
- Benemei, S. Molecular Agonists Mechanisms of 5-HT1F Receptor. In Novel Synthetic Drugs in Migraine; Springer Nature: Berlin/Heidelberg, Germany, 2022; Volume 73. [Google Scholar]
- Mir, M.A. Cytokine and Chemokine Networks in Cancer; Springer Nature: Berlin/Heidelberg, Germany, 2023. [Google Scholar]
- Zhuge, H.; Ge, Z.; Wang, J.; Yao, J.; He, J.; Wang, Y.; Wang, Y.; Tang, Y. The Tandem of Liquid Chromatography and Network Pharmacology for the Chemical Profiling of Pule’an Tablets and the Prediction of Mechanism of Action in Treating Prostatitis. Pharmaceuticals 2023, 17, 56. [Google Scholar] [CrossRef]
- Praveen, M.; Ullah, I.; Buendia, R.; Khan, I.A.; Sayed, M.G.; Kabir, R.; Bhat, M.A.; Yaseen, M. Exploring Potentilla nepalensis Phytoconstituents: Integrated Strategies of Network Pharmacology, Molecular Docking, Dynamic Simulations, and MMGBSA Analysis for Cancer Therapeutic Targets Discovery. Pharmaceuticals 2024, 17, 134. [Google Scholar] [CrossRef]
- Keretsu, S.; Bhujbal, S.P.; Cho, S.J. Rational approach toward COVID-19 main protease inhibitors via molecular docking, molecular dynamics simulation and free energy calculation. Sci. Rep. 2020, 10, 17716. [Google Scholar] [CrossRef]
- Opo, F.A.D.M.; Rahman, M.M.; Ahammad, F.; Ahmed, I.; Bhuiyan, M.A.; Asiri, A.M. Structure based pharmacophore modeling, virtual screening, molecular docking and ADMET approaches for identification of natural anti-cancer agents targeting XIAP protein. Sci. Rep. 2021, 11, 4049. [Google Scholar] [CrossRef]
- Fu, H.; Chen, H.; Blazhynska, M.; Goulard Coderc De Lacam, E.; Szczepaniak, F.; Pavlova, A.; Shao, X.; Gumbart, J.C.; Dehez, F.; Roux, B.; et al. Accurate determination of protein: Ligand standard binding free energies from molecular dynamics simulations. Nat. Protoc. 2022, 17, 1114–1141. [Google Scholar] [CrossRef]
- Zeng, Z.; Wodaczek, F.; Liu, K.; Stein, F.; Hutter, J.; Chen, J.; Cheng, B. Mechanistic insight on water dissociation on pristine low-index TiO2 surfaces from machine learning molecular dynamics simulations. Nat. Commun. 2023, 14, 6131. [Google Scholar] [CrossRef] [PubMed]
- Almontasser, A.; Al-Mufti, S.M.; Arya, R.K. Application of Nanofillers in Drug Delivery Industry. In Handbook of Nanofillers; Springer: Berlin/Heidelberg, Germany, 2023; pp. 1–41. [Google Scholar]
- Wang, H.; Yang, Y.; Zeng, Y.; Li, L. Ionic Liquid-Based Sensors for Fast Determination of Aromatic Compounds in the Environment. In Encyclopedia of Ionic Liquids; Springer: Berlin/Heidelberg, Germany, 2023; pp. 620–627. [Google Scholar]
- Robo, M.T.; Hayes, R.L.; Ding, X.; Pulawski, B.; Vilseck, J.Z. Fast free energy estimates from λ-dynamics with bias-updated Gibbs sampling. Nat. Commun. 2023, 14, 8515. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Chen, L.; Yin, D.; Yang, Z.; Feng, J.; Sun, Q.; Lai, L.; Guo, X. Visualizing single-molecule conformational transition and binding dynamics of intrinsically disordered proteins. Nat. Commun. 2023, 14, 5203. [Google Scholar] [CrossRef]
- Kruse, M.; Altattan, B.; Laux, E.-M.; Grasse, N.; Heinig, L.; Möser, C.; Smith, D.M.; Hölzel, R. Characterization of binding interactions of SARS-CoV-2 spike protein and DNA-peptide nanostructures. Sci. Rep. 2022, 12, 12828. [Google Scholar] [CrossRef]
- Kobayashi, H.; Cheveralls, K.C.; Leonetti, M.D.; Royer, L.A. Self-supervised deep learning encodes high-resolution features of protein subcellular localization. Nat. Methods 2022, 19, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Lin, R.; Yu, H.; Liu, M.; Xing, E.; Wang, W.; Zhang, F.; Zhao, D.; Li, X. Versatile synthesis of metal-compound based mesoporous Janus nanoparticles. Nat. Commun. 2023, 14, 4249. [Google Scholar] [CrossRef] [PubMed]
- Mehany, M.M.; Hammam, O.A.; Selim, A.A.; Sayed, G.H.; Anwer, K.E. Novel pyridine bearing pentose moiety-based anticancer agents: Design, synthesis, radioiodination and bioassessments. Sci. Rep. 2024, 14, 2738. [Google Scholar] [CrossRef] [PubMed]
- Sroor, F.M.; Mahrous, K.F.; El-Kader, H.A.M.A.; Othman, A.M.; Ibrahim, N.S. Impact of trifluoromethyl and sulfonyl groups on the biological activity of novel aryl-urea derivatives: Synthesis, in-vitro, in-silico and SAR studies. Sci. Rep. 2023, 13, 17560. [Google Scholar] [CrossRef] [PubMed]
- El-Fakharany, Z.S.; Nissan, Y.M.; Sedky, N.K.; Arafa, R.K.; Abou-Seri, S.M. New proapoptotic chemotherapeutic agents based on the quinolone-3-carboxamide scaffold acting by VEGFR-2 inhibition. Sci. Rep. 2023, 13, 11346. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Common Name | PDB ID | Uniprot ID | ChEMBL ID | Target Class |
|---|---|---|---|---|---|
| Serotonin 1a (5-HT1a) receptor | HTR1A | 7E2X | P08908 | CHEMBL214 | Family A G protein-coupled receptor |
| C-C chemokine receptor type 1 | CCR1 | 7VL8 | P32246 | CHEMBL2413 | |
| Nociceptin receptor | OPRL1 | 4EA3 | P41146 | CHEMBL2014 | |
| Serotonin 2a (5-HT2a) receptor | HTR2A | 6WHA, 7VOD, 6A93, 6WGT | P28223 | CHEMBL224 | |
| Alpha-1a adrenergic receptor | ADRA1A | 8THK | P35348 | CHEMBL229 | |
| Histamine H1 receptor | HRH1 | 3RZE | P35367 | CHEMBL231 | |
| C-C chemokine receptor type 3 | CCR3 | 4ZYA, 5XIX | P51677 | CHEMBL3473 | |
| Serotonin 1d (5-HT1d) receptor | HTR1D | 7E32 | P28221 | CHEMBL1983 | |
| Serotonin 1b (5-HT1b) receptor | HTR1B | 4IAQ | P28222 | CHEMBL1898 | |
| Adrenergic receptor beta | ADRB2 | 2RH1 | P07550 | CHEMBL210 | |
| Serotonin 4 (5-HT4) receptor | HTR4 | 7XT8 | Q13639 | CHEMBL1875 | |
| Alpha-2a adrenergic receptor | ADRA2A | 7W7E | P08913 | CHEMBL1867 |
| PDB ID | Lib Dock Score | |
|---|---|---|
| 3 | 3e | |
| 7VL8 | 127.398 | 126.845 |
| 4EA3 | 152.611 | 149.226 |
| 7VOD | 143.659 | 159.64 |
| 6WHA | 143.333 | 157.689 |
| 8THK | 157.21 | 160.324 |
| 5XIX | 107.145 | 127.742 |
| 7E32 | 118.02 | 115.01 |
| 6A93 | 106.887 | 159.495 |
| 4IAQ | 155.758 | 159.28 |
| 2RH1 | 164.678 | 184.081 |
| Compound | Gibbs Free Energy Values | Dissociation Constant |
|---|---|---|
| 3 | −23.8668 | 1108.10 |
| 3e | −6.2632 | 28.57 |
| Drug | Protein | Location (k = 23) | PDB ID |
|---|---|---|---|
| Compound 3 | Adrenergic receptor beta | 52.2%: plasma membrane | 2RH1 |
| 26.1%: endoplasmic reticulum | |||
| 8.7%: vacuolar | |||
| 4.3%: mitochondrial | |||
| 4.3%: nuclear | |||
| 4.3%: Golgi |
| Drug | 0 μg/mL | 2.5 μg/mL | 5 μg/mL | 10 μg/mL |
|---|---|---|---|---|
| Compound 3e | 100 ± 0.05 | 101.57 ± 2.48 | 105.41 ± 1.53 ** | 70.95 ± 2.60 ** |
| Compound 3 | 100 ± 0.05 | 69.98 ± 3.51 ** | 64.26 ± 1.18 ** | 41.75 ± 3.27 ** |
| 5-FU | 100 ± 0.05 | 47.72 ± 3.59 ** | 50.28 ± 3.87 ** | 58.78 ± 6.97 ** |
| blank control | 100 ± 0.05 | 100 ± 0.05 | 100 ± 0.05 | 100 ± 0.05 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jia, Y.; Zhang, Y.; Zhu, H. Clindamycin Derivatives: Unveiling New Prospects as Potential Antitumor Agents. Pharmaceuticals 2024, 17, 276. https://doi.org/10.3390/ph17030276
Jia Y, Zhang Y, Zhu H. Clindamycin Derivatives: Unveiling New Prospects as Potential Antitumor Agents. Pharmaceuticals. 2024; 17(3):276. https://doi.org/10.3390/ph17030276
Chicago/Turabian StyleJia, Yiduo, Yinmeng Zhang, and Hong Zhu. 2024. "Clindamycin Derivatives: Unveiling New Prospects as Potential Antitumor Agents" Pharmaceuticals 17, no. 3: 276. https://doi.org/10.3390/ph17030276
APA StyleJia, Y., Zhang, Y., & Zhu, H. (2024). Clindamycin Derivatives: Unveiling New Prospects as Potential Antitumor Agents. Pharmaceuticals, 17(3), 276. https://doi.org/10.3390/ph17030276